

Abstract
The transient receptor potential type V5 channel (TRPV5) is a Ca2+-selective TRP channel important for epithelial Ca2+ transport. Intracellular Mg2+ causes a fast voltage-dependent block of the TRPV5 channel by binding to the selectivity filter. Here, we report that intracellular Mg2+ binding to the selectivity filter of TRPV5 also causes a slower reversible conformational change leading to channel closure. We further report that PIP2 activates TRPV5. Activation of TRPV5 by PIP2 is independent of Mg2+. Yet, PIP2 decreases sensitivity of the channel to the Mg2+-induced slow inhibition. Mutation of aspartate-542, a critical Mg2+-binding site in the selectivity filter, abolishes Mg2+-induced slow inhibition. PIP2 has no effects on Mg2+-induced voltage-dependent block. Thus, PIP2 prevents the Mg2+-induced conformational change without affecting Mg2+ binding to the selectivity filter. Hydrolysis of PIP2 via receptor activation of phospholipase C sensitizes TRPV5 to the Mg2+-induced slow inhibition. These results provide a novel mechanism for regulation of TRP channels by phospholipase C–activating hormones via alteration of the sensitivity to intracellular Mg2+.
INTRODUCTION
Transient receptor potential (TRP) channels are widespread and play many important functions, ranging from thermal, tactile, taste, osmolar and fluid flow sensing, and embryo development to epithelial Ca2+ and Mg2+ transport (Hoenderop et al., 2002; Montell et al., 2002; Clapham, 2003). They are classified into TRPC, TRPV, TRPM, TRPP, TRPML, TRPA, and TRPN subfamilies (Hoenderop et al., 2002; Montell et al., 2002; Clapham, 2003). The TRPV subfamily is named after the first mammalian member of the subfamily, vanilloid receptor 1 (Montell et al., 2002; Clapham, 2003). TRPV5 and TRPV6 are highly Ca2+-selective TRP channels that mediate trans-epithelial Ca2+ transport in kidney and intestine (Hoenderop et al., 2002).
Intracellular Mg2+ is a cofactor for many enzymes and controls inward rectification of ion channels by causing voltage-dependent block of outward currents (Romani and Scarpa, 2000; Hille, 2001). Recently, intracellular Mg2+ has also been reported to regulate the activity of several TRP channels including TRPM6, TRPM7, and TRPV6 via a mechanism different from voltage-dependent block (Nadler et al., 2001; Runnels et al., 2001; Voets et al., 2003, 2004a). The intracellular free Mg2+ concentration in most mammalian cells is between 0.5 and 1 mM and changes only slightly in response to physiological stimuli (Romani and Scarpa, 2000). TRPM7 is permeable to Mg2+ (Schmitz et al., 2003). It is believed that Mg2+ ions entering cells through TRPM7 feedback on the channel to regulate its activity (Schmitz et al., 2003). TRPV6 does not conduct Mg2+ in physiological conditions. The physiological role of intracellular Mg2+ regulation of TRPV6 is unknown.
PIP2 directly regulates inward rectifying K+ and other channels (Hilgemann et al., 2001). How membrane lipids alter channel function is an area of intense research interests. For many ion channels, direct interaction with PIP2 stabilizes channels in certain conformations (Hilgemann et al., 2001). By stabilization in one conformation, PIP2 modulates the responses of ion channels to regulators, such as GTP binding proteins, intracellular pH, Na+, ATP, etc. (Huang et al., 1998; Shyng and Nichols, 1998; Sui et al., 1998; Leung et al., 2000).
In this report, we show that intracellular Mg2+ causes a fast voltage-dependent block and a slower inhibition of TRPV5. We further show that PIP2 activates TRPV5 and decreases sensitivity of the channel to the Mg2+-induced slow inhibition. Hydrolysis of PIP2 via receptor activation of phospholipase C increases the sensitivity of TRPV5 to the Mg2+-induced slow inhibition. These results provide a novel mechanism for regulation of TRP channels by PLC-activating hormones by increasing the sensitivity to intracellular Mg2+.
MATERIALS AND METHODS
Molecular Biology and Cell Culture
Nucleotide coding sequence of cDNAs for rabbit TRPV5 in pCDNA3 mammalian expression vector (Yeh et al., 2003) was used as template for site-directed mutagenesis using a commercial mutagenesis kit (QuickChange; Stratagene). Mutations were confirmed by sequencing. CHO-K1 clone (from American Type Culture Collection) were cultured in F12-K medium (GIBCO-BRL) containing 10% FCS. Cells (at ∼50% confluence) were cotransfected with cDNA for pEGFP (1 μg) plus cDNAs for wild-type or mutant TRPV5 (10 μg per 35 mM dish for cell-attached and inside-out recording; 3 μg per dish for whole-cell recording) using lipofectamine-plus transfection kits (GIBCO-BRL) as previously described (Yeh et al., 2003, 2005). About 24–48 h after transfection, cells were dissociated by limited trypsin treatment and placed in a chamber for recording. Transfected cells were identified using epi-fluorescent microscopy for recordings.
Electrophysiological Recordings
Cell-attached, inside-out, and whole-cell recordings were performed using an Axopatch 200B patch-clamp amplifier (Axon Instruments) as described previously (Yeh et al., 2003, 2005). For cell-attached recordings, the pipette and bath solution contained (in mM) 140 NaAsp (sodium aspartate), 10 NaCl, 1 EDTA, 10 HEPES (pH 7.4), and 140 KAsp, 10 NaCl, 1 EDTA, 10 HEPES (pH 7.4), respectively. For cell-attached recordings shown in Fig. 8 A, the pipette solution contained (in mM) 140 NaCl, 1 MgCl2, 1 CaCl2, and 10 HEPES (pH 7.4). For inside-out recordings, membrane patches were excised into a bath solution containing (in mM) 140 NaAsp, 10 NaCl, 10 HEPES (pH 7.4), 10 EDTA, and various amounts of MgCl2 titrated to the desired concentrations of ionized Mg2+ as specified. Bath solution was perfused at ∼200 μl per second. The volume of perfusion chamber and connecting tubing is ∼100 μl. Thus, it takes 2–3 s to exchange the bath solution. Concentrations of ionized Mg2+ were calculated using a computer program based on algorithms by Fabiato (Fabiato, 1988) (www.stanford.edu/~cpatton/webmaxc/webmaxcE.htm). For whole-cell recordings, the initial pipette and bath solution contained (in mM) 140 NaAsp, 10 NaCl, 10 HEPES (pH 7.4), 10 EDTA, and 140 NaAsp, 10 NaCl, 1 EDTA, 10 HEPES (pH 7.4), respectively. For whole-cell recordings shown in Fig. 9 B, the bath solution contained (in mM) 140 NaCl, 1 MgCl2, 1 CaCl2, and 10 HEPES (pH 7.4). Intracellular Mg2+ in whole-cell recording was altered by intrapipette perfusion of solutions containing (in mM) 140 NaAsp, 10 NaCl, 10 HEPES (pH 7.4), 10 EDTA, and various amount of MgCl2 titrated to the desired concentrations of ionized Mg2+ as specified. The intrapipette perfusion was performed via a capillary quartz tubing (flamed and pulled manually to outer and inner diameter ∼50 and 20 μm, respectively) placed within ∼200 μm of the tip of the opening (Hilgemann and Lu, 1998). Voltage protocol for each experiment was described in the individual figure. Currents were low-pass filtered at 1 kHz using an 8-pole Bessel filter, sampled every 0.1 ms (10 kHz) with Digidata-1300 interface and stored directly onto computer hard disk using pCLAMP9 software. Data were transferred to compact discs for long-term storage.
Figure 8.
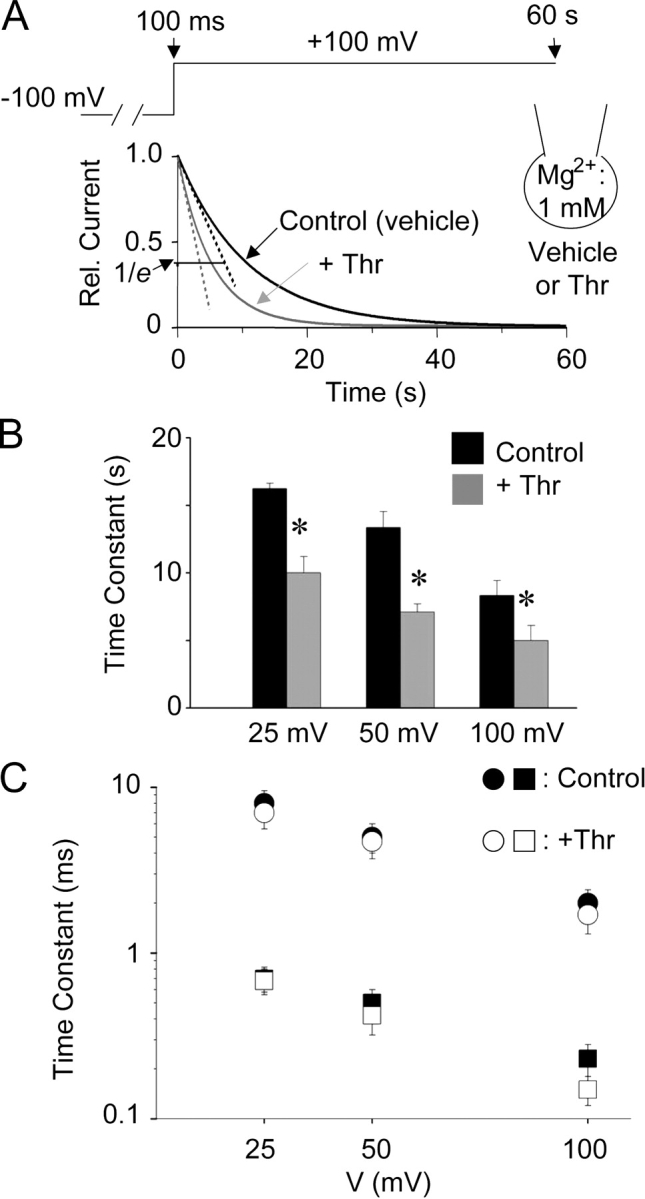
Effects of PIP2 hydrolysis on Mg2+-induced slow inhibition (A and B) and voltage-dependent block (C). (A) Whole-cell currents (between 100 ms and 60 s; at 1 mM intracellular Mg2+) in response to voltage jump from −100 mV holding potential to depolarized test potentials were measured as in Fig. 6 C. Cells were incubated with vehicle (Control) or thrombin + WMN (+Thr) in the extracellular bath for 10 min before patch-clamped for whole-cell recordings. Shown are representative single exponential fits to currents for Control (black curve) or +Thr (gray curve). Relative current level corresponding to 1/e (e, the base of natural logarithms) of the maximal current is shown. (B) Time constants of Mg2+-induced slow inhibition at +25, +50, +100 mV for Control and +Thr. * indicates P < 0.05 +Thr vs. Control. (C) Time constants of Mg2+-induced voltage-dependent block (as in Fig. 6, A and B) at +25, +50, +100 mV for Control and +Thr.
Figure 9.

Effect of PIP2 hydrolysis on the Mg2+-induced slow inhibition. (A) Effect of thrombin plus/minus wortmannin on TRPV5 in cell-attached recording. The top left panel shows cell-attached configuration for recording TRPV5-mediated Ca2+ currents. See text for the pipette solution. Voltage stimuli (step to −100 mV for 40 ms and ramp from −100 to +100 mV ramp over 200 ms) were applied every 2 s from +50 mV holding potential. Holding at +50 mV prevents Ca2+ entry and the Ca2+-dependent inactivation. Thrombin (Thr, 1 U/ml) was applied to the patch membrane by intrapipette perfusion. WMN (10 μM) or vehicle (0.5% DMSO) was applied to the extracellular bath in cells expressing TRPV5 5–10 min before addition of thrombin. The bottom panel shows inward currents (at −100 mV) normalized to the initial current (I/Imax). The top right panel shows ramp I-V curves at indicated time points. (B) Dose–response curve of inhibition by Mg2+ before and after membrane PIP2 hydrolysis. Whole-cell TRPV5-mediated Ca2+ currents were recorded in a bath solution containing (in mM) 140 NaCl, 1 MgCl2, 1 CaCl2, and 10 HEPES (pH 7.4). Intracellular Mg2+ (0 to 3 mM) was altered by intrapipette perfusion. Cells expressing TRPV5 were incubated with a bath solution containing either vehicle (closed circles) or WMN + Thr (open circles) for 5–10 min before patching for whole-cell recording. Voltage protocol and holding potential are as in the cell-attached experiment.
Data Analysis
Dose–response curves for inhibition of TRPV5 by intracellular Mg2+ and concentration of Mg2+ for half-maximal inhibition (IC50) were obtained by fitting relative currents (normalized to maximal currents; I/Imax) at different Mg2+ concentrations using the Sigma-Plot program according to equation I/Imax = IC50/IC50 + [Mg2+]. The fractional electrical distance of the Mg2+ binding site from the outer surface of the membrane (δ) was calculated according to equation IC50(V) = IC50(0)exp (2δFV/RT) (Nilius et al., 2000), where IC50(V) and IC50(0) represent IC50 at the test voltage V and at 0 mV, respectively, and R, T, and F have their usual meanings. Current traces were fitted to a single or double exponential curve (as indicated) to analyze time constant (τ) for inhibition of currents by Mg2+. The on-rate constant (Kon) for slow inhibition by Mg2+ was calculated according to the equation Kon = 1/τ[Mg2+]. Data are shown as mean ± SEM of number of observations. Statistical comparison was made using unpaired Student's t test.
RESULTS
Multiple Effects of Intracellular Mg2+ on TRPV5: Slow Reversible Inhibition, Fast Voltage-dependent Block, and “Run-down” via Reduction of PIP2
Effect One: Slow Reversible Inhibition
We studied TRPV5 channels expressed in Chinese hamster ovary (CHO) cells by cell-attached and inside-out patch-clamp recording (Fig. 1 A). In cell-attached recording, TRPV5-expressing CHO cells exhibited the characteristic strongly inwardly rectifying Na+ currents (Nilius et al., 2001; Hoenderop et al., 2003; Yeh et al., 2003) (Fig. 1 B, C/A). Inward (closed circles) and outward currents (open circles) both increased markedly when inside-out patch membranes containing TRPV5 channels were excised (Fig. 1 B, I/O) into divalent-free (DVF) bath solutions. Exchanging DVF solutions with one that contains 1 mM Mg2+ caused inhibition of TRPV5 currents over tens of seconds (Fig. 1, B and C). Currents recovered upon reintroduction of DVF solutions. TRPV5 currents increased by a similar extent when inside-out membranes were excised into Mg2+-free bath solutions containing 0, 200, or 1,000 nM Ca2+ (unpublished data). Thus, most (if not all) of the increase in currents upon excision of inside-out membranes into DVF solutions is due to loss of inhibition by intracellular Mg2+. We refer to this inhibition by intracellular Mg2+ occurring over tens of seconds as “slow reversible inhibition” to distinguish it from the much faster (also reversible) voltage-dependent block described below.
Figure 1.
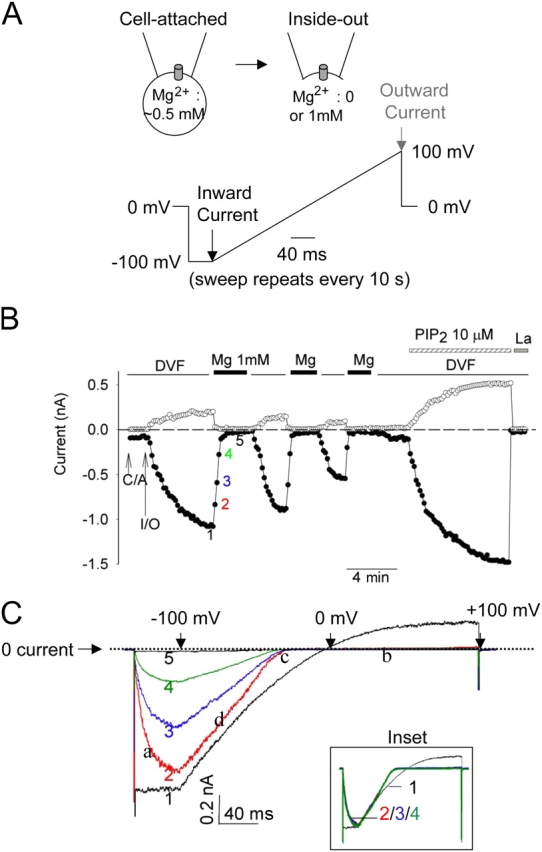
Multiple effects of intracellular Mg2+ on TRPV5. (A) Configuration and voltage protocol of recording. Voltage stimuli were applied every 10 s. Black and gray arrows indicate time points when inward and outward currents shown in Fig. 1 B were taken, respectively. (B) Intracellular Mg2+ causes a fast reversible voltage-dependent block, a slow reversible inhibition, and an irreversible run-down of TRPV5 in inside-out membranes. See text for details. Inside-out patches were bathed in either DVF or 1 mM Mg2+-containing solution. C/A and I/O indicate cell-attached and inside-out. Application of PIP2 and 1 mM La3+ (La) in DVF solution are indicated. Time interval between data points (shown in circles) is 10 s. As described previously by us (Yeh et al., 2003, 2005), expression of TRPV5 in CHO cells is robust. We estimated that under our experimental condition each excised patch contains ∼50–150 channels. PIP2 reactivated run-down TRPV5 channels (i.e., currents recovered to >50% of the level before run-down) in 54 out of 62 recordings. We did not observe significant background currents in inside-out membranes of mock-transfected cells either before or after PIP2 (inward [−100 mV] and outward [+100 mV] currents: −87 ± 32 pA and 117 ± 28 pA, respectively, before PIP2 vs. −135 ± 41 pA and 156 ± 48 pA, respectively, after PIP2; mean ± SEM, n = 6). (C) Ramp I-V curves of currents at indicated time points in B. Scale bars for X (40 ms) and Y axis (0.2 nA) are shown. Dotted line indicates 0 current. In the X axis, the time points corresponding to ramp potentials −100, 0, and +100 mV (from left to right) are indicated by three downward arrows, respectively. Inset shows I-V curves scaled up to the same peak current level.
Effect Two: Voltage-dependent Block and Distinction from the “Slow Reversible Inhibition”
Intracellular free Mg2+ also causes reversible voltage-dependent block of TRPV5 occurring over a relatively much faster time course (in millisecond time scale; see Hoenderop et al., 2003; Voets et al., 2003). The distinction between the above slow reversible inhibition occurring over tens of seconds and the voltage-dependent block by Mg2+ is rather evident from our results (see ramp I-V curves in Fig. 1 C). First, as mentioned above, inward currents shown in Fig. 1 B were measured at the end of 40-ms hyperpolarization (−100 mV) step (indicated by black arrow in voltage ramp protocol in Fig. 1 A). At these time points, Mg2+ ions were completely dissociated (from channels) by the hyperpolarization (Fig. 1 C) and thus did not exert voltage-dependent block on channels. Second, the kinetics of voltage-dependent block distinguishes itself from the slow reversible inhibition. The Mg2+-induced voltage-dependent block of TRPV5 was evident by the characteristic unblock during the −100 mV hyperpolarization step (indicated by “a” in Fig. 1 C), block of outward current (“b”), shift of reversal potential toward more negative membrane potential (“c”), and steeper I-V near the reversal potential (“d”) in the current–voltage (I-V) relationship curves (Fig. 1 C). The scaled-up I-V curves of currents from 10, 20, and 30 s after application of Mg2+ (time point 2, 3, and 4, respectively; time point 1 is immediately before application of Mg2+) were superimposable (Fig. 1 C, inset), indicating that voltage-dependent block reached its maximal effect immediately after application of intracellular Mg2+ while slow reversible inhibition gradually reached its maximum over 30–40 s (Fig. 1, B and C). Thus, intracellular Mg2+ causes distinct fast voltage-dependent block (occurring in ms time scale) and slower reversible inhibition of TRPV5 (in seconds to tens of seconds) (see Fig. 6 for time constant measurements for both processes).
Figure 6.

Kinetics of Mg2+-induced voltage-dependent block (A and B) and slow inhibition (C and D). (A) Whole-cell recording was performed at fixed intracellular Mg2+ (1 mM). Holding potential was −100 mV. Test pulse to +100 mV is illustrated. Time course of outward current (at +100 mV) from peak current (∼1 ms after test potential) to the current level at 50 ms is shown. Dotted line represents double exponential fit to the current. (B) Fast (squares) and slow (circles) exponential time constant of decrease of outward current from peak current (∼1 ms) to 50 ms of test potentials (+25, +50, +100 mV, respectively). (C) Time course of decrease of outward current from 100 ms to 60 s after test potential to +100 mV. Dotted line represents single exponential fit to the current. (D) Exponential time constants of decrease of outward current between 100 ms and 60 s of test potentials (+25, +50, and +100 mV, respectively).
Effect Three: “Run-down” via Reduction of PIP2
Membrane phospholipid PIP2 regulates function of many ion channels and transporters (Hilgemann et al., 2001). In inside-out membranes, a prolonged exposure of the cytoplasmic face to Mg2+ activates Mg2+-dependent lipid phosphatases and depletes PIP2 (Huang et al., 1998; Liu and Qin, 2005). Depletion of PIP2 causes an irreversible loss of activity (“run-down”) of channels that are regulated by PIP2 (Huang et al., 1998). For TRPV5, though each short exposure to Mg2+ solution caused reversible inhibition, repetitive exposures to 1 mM Mg2+ solutions led to progressive irreversible run-down of currents eventually (Fig. 1 B). Application of exogenous PIP2 to inside-out membranes reactivated TRPV5 after its irreversible run-down by Mg2+ (Fig. 1 B), suggesting that irreversible run-down of TRPV5 by intracellular Mg2+ is due to loss of PIP2 in the membrane and mechanistically different from the reversible voltage-dependent block and “slow inhibition” by Mg2+. Activation of a Ca2+-sensitive PLC by intracellular Ca2+ depletes PIP2 and leads to desensitization of TRPM8 channels (Rohács et al., 2005). We found that U-37122 (an inhibitor of PLC, 5–10 μM) did not prevent run-down of TRPV5 caused by prolonged exposure to Mg2+ (unpublished data), supporting that Mg2+-induced run-down is caused by activation of lipid phosphatase(s) rather than by activation of some Mg2+-sensitive PLC.
A monoclonal antibody against PIP2 (Fukami et al., 1988) interferes with PIP2 regulation of ion channels presumably by sequestering PIP2 in the membranes (Huang et al., 1998). Application of anti-PIP2 antibody to the cytoplasmic face of inside-out membranes decreased TRPV5 currents (Fig. 2 A). Currents recovered partially after extensive washout of the antibody (Fig. 2 B). Polylysine, with its positively charged residues, has also been used to inhibit the activity of PIP2-activated ion channels through competing PIP2–channel interaction (Huang et al., 1998; Oliver et al., 2004). We found that application of polylysine (average molecular weight 1 kD; 30 μg/ml) to the inside-out membranes inhibited TRPV5 by 89 ± 9% in 3 min (n = 5; unpublished data). Similar to application of PIP2, application of Mg-ATP after irreversible run-down in Mg2+ solutions reactivated TRPV5 (Fig. 2 B). Mg-ATP presumably works by activating lipid kinases, including phosphoinositide 4-kinase to regenerate PIP2 in the membranes (Huang et al., 1998; Hilgemann et al., 2001). Consistent with this idea, the reversal by Mg-ATP was blunted by an inhibitor of phosphoinositide 4-kinase wortmannin (Nakanishi et al., 1995) (Fig. 2 B, WMN). Furthermore, Na-ATP did not reactivate TRPV5 (unpublished data). Fig. 2 C shows I-V curves of currents at several indicated time points. These results provide compelling evidence for a critical role of membrane PIP2 in the function of TRPV5.
Figure 2.
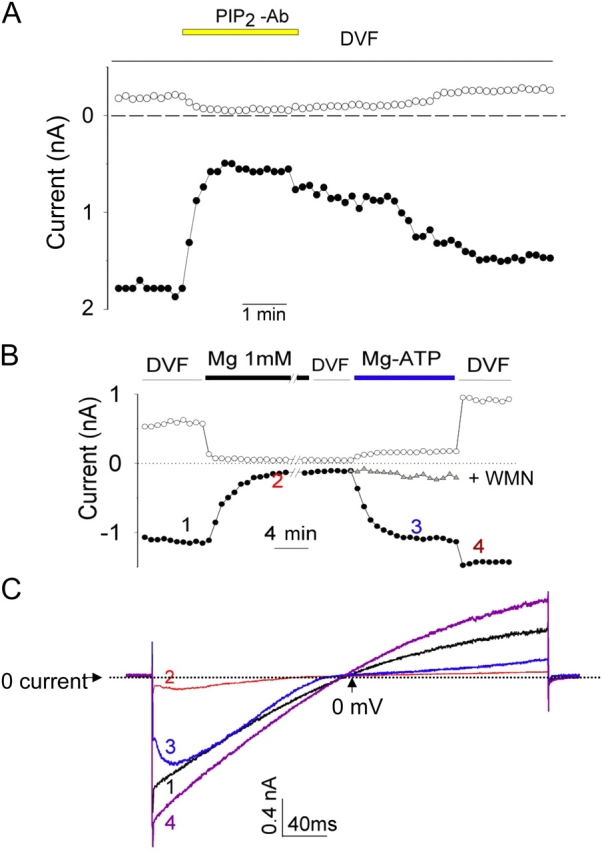
PIP2 is critical for TRPV5 channel activity. (A) Anti-PIP2 antibody reduces TRPV5 currents in inside-out patches. Experiments were repeated five times with similar results. The average (mean ± SEM) inhibition by anti-PIP2 antibody was 88 ± 13% and 79 ± 14% for outward and inward currents, respectively. (B) A representative experiment showing long exposure of inside-out patches to Mg2+ causes run-down of TRPV5, which can be subsequently reactivated by Mg-ATP. // indicates 10 min break. The inhibition by Mg2+ shown here appears to be slower compared with that in Fig. 1 B. This variability is likely due to differences in the configuration and accessibility of excised membranes to perfused bath solutions. Closed and open circles are currents at −100 and +100 mV, respectively. Similar results were observed in six separate experiments. In some experiments, Mg-ATP was applied together with WMN (10 μM). Gray triangles illustrate one such recording receiving Mg-ATP + WMN superimposed on one receiving the Mg-ATP alone. Similar results were observed in four separate experiments. Dimethyl sulfoxide (DMSO, vehicle for WMN, 0.5%) did not prevent reactivation by Mg-ATP (n = 4, not depicted). Of note is that wortmannin did not prevent recovery of currents from transient inhibition by Mg2+ (not depicted), indicating that the slow reversible inhibition is not due to regeneration of PIP2 from lipid kinases. (C) I-V curves of currents at indicated time points in B. In this experiment, voltage ramp from −100 to +100 mV was applied without a 40-ms step at −100 mV as indicated in Fig. 1 A. Dotted line indicates 0 current. In the X axis, the time point corresponding to ramp potential 0 mV is indicated by upward arrow. The characteristic voltage-dependent block in trace 3 is due to free (ionized) Mg2+ (0.13 mM) present in the Mg-ATP solution.
Application of PIP2 to Inside-out Membranes Releases Mg2+-induced Slow Reversible Inhibition
Activation of inward-rectifier K+ and other channels by PIP2 modulates the regulation of channels by other signaling pathways (Hilgemann et al., 2001). We examined whether PIP2 activation of TRPV5 alters its sensitivity to Mg2+-induced slow inhibition. We examined dose–response relationships for the intracellular Mg2+-induced slow reversible inhibition of TRPV5 in excised inside-out membranes before and after application of PIP2. As before, after reaching maximal currents in DVF solutions in inside-out membranes, TRPV5 currents were completely inhibited by a solution containing 1 mM Mg2+ (Fig. 3 A). Currents recovered in DVF solutions. The concentration of Mg2+ for half-maximal inhibition (IC50) of the inward currents of TRPV5 was 0.11 ± 0.03 mM (mean ± SEM, n = 7; Fig. 3 C, closed circles). Application of exogenous PIP2 to inside-out membranes in DVF solutions increased TRPV5 currents by 2.4 ± 0.3–fold (n = 8; see Fig. 3 A for representative experiment), indicating a submaximal concentration of PIP2 present in the excised membranes. The application of PIP2 reduced the sensitivity of TRPV5 to the slow reversible inhibition by intracellular Mg2+. After PIP2, the inward currents of TRPV5 were partially inhibited by 1 mM Mg2+ but completely inhibited by 10 mM Mg2+ (Fig. 3 A). Currents recovered in the DVF solution (Fig. 3 A). The IC50 for the slow inhibition by Mg2+ was increased by ∼28-fold to 3.11 ± 0.25 mM (n = 8; Fig. 3 C, open circles) after application of exogenous PIP2. The Mg2+-induced voltage-dependent block remains distinguishable from the slow reversible inhibition even after application of exogenous PIP2. As shown by the scaled-up I-V curves (Fig. 3 B), voltage-dependent block was complete in 1 mM Mg2+ in the presence of PIP2 (i.e., I-V curves at time point 2 and 3 are superimposable, Fig. 3 B). Yet, Mg2+-induced slow inhibition requires 10 mM Mg2+ over 30–40 s to reach its maximum (i.e., more inhibition occurred from time point 2 to time point 3 in Fig. 3 A).
Figure 3.
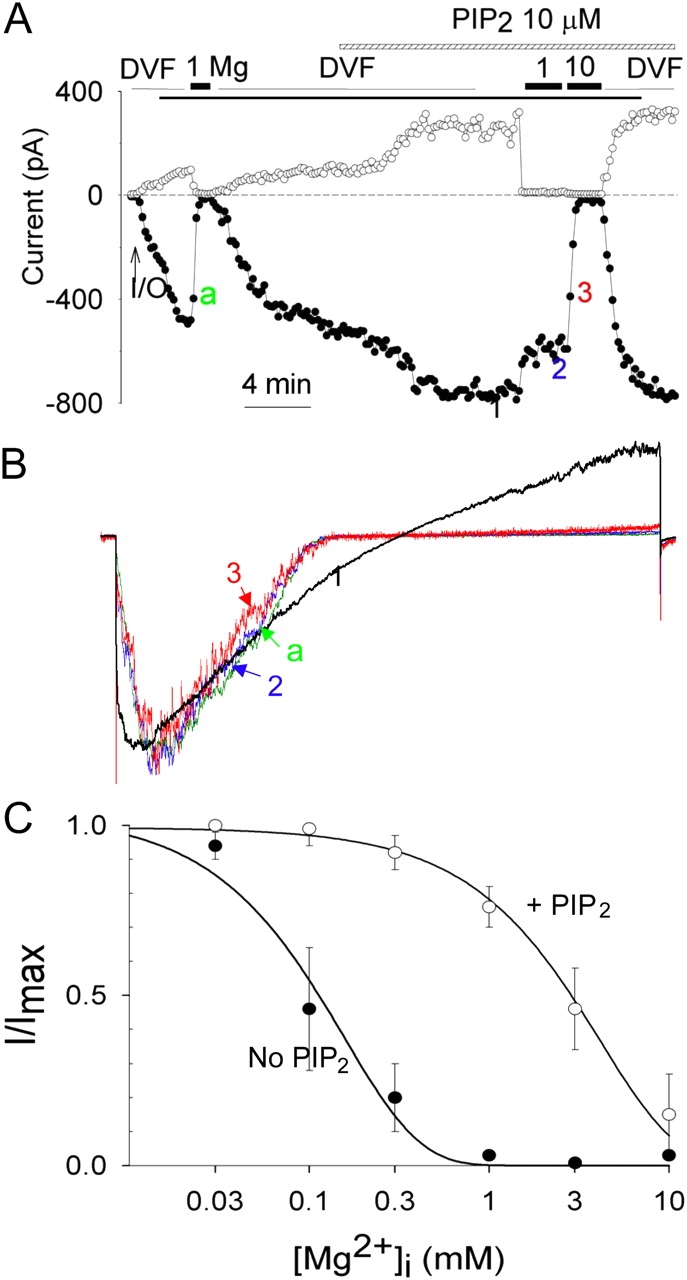
PIP2 decreases the sensitivity to Mg2+-induced slow inhibition but not the voltage-dependent block. (A) Application of PIP2 in inside-out patches increases TRPV5 currents and reduces the sensitivity to inhibition by Mg2+. Voltage ramp protocol is same as in Fig. 1. Holding potential between ramps is 0 mV. (B) Scaled-up I-V curves of currents at indicated time points in A. (C) Dose–response curves of inhibition by Mg2+ in inside-out patches before (no PIP2) and after application of PIP2 (+PIP2). IC50 was obtained by fitting relative inward currents (inward currents at −100 mV normalized to maximal currents; I/Imax) at different Mg2+ concentrations according to the equation I/Imax = IC50/IC50 + [Mg2+].
Effect of Intracellular Mg2+ on TRPV5 in Whole Cell
The concentration of PIP2 in the excised membranes is likely considerably lower than in the plasma membranes in vivo (Nasuhoglu et al., 2002). We further examined regulation of TRPV5 by intracellular Mg2+ using whole-cell recording (Fig. 4 A), which resembles physiological plasma membrane conditions better than the inside-out membranes. Strongly inwardly rectifying Na+ currents were observed immediately after establishment of whole-cell recording in TRPV5-expressing cells (time point 1 in Fig. 4 B and trace 1 in Fig. 4 C) but not in mock-transfected CHO cells (Fig. 4 D). Dialysis of the intracellular space with a DVF solution led to a large increase of whole-cell TRPV5 currents (Fig. 4, B and C). As reported previously (Hoenderop et al., 2003; Yeh et al., 2003), whole-cell TRPV5 currents exhibited slight outward rectification at membrane potentials >+50 mV in the presence of extracellular EDTA (Fig. 4 C, trace 3). Perfusion of the intracellular space with a solution containing 1 mM Mg2+ caused a large reversible inhibition on whole-cell TRPV5 currents (Fig. 4, B and C). Unlike in the inside-out recordings, run-down of TRPV5 by Mg2+ is rather infrequent in whole-cell recordings (>90% of experiments show >80% recovery upon changing from Mg2+ to DVF solutions) despite an average longer exposure of the cytoplasmic face of plasma membrane to Mg2+. Thus, run-down of TRPV5 via activation of lipid phosphatases (as observed in excised inside-out patches) is likely not a physiological action of intracellular Mg2+ (as in the conditions of whole-cell recording). Intracellular Mg2+ at concentrations ranging from 0.1 to 3 mM caused a dose-dependent inhibition of whole-cell TRPV5 currents (Fig. 4 E). The IC50 for intracellular Mg2+ inhibition of TRPV5 channels in whole-cell recordings was 0.29 ± 0.02 mM (n = 11; Fig. 4 F). This IC50 is between that for inside-out membranes before PIP2 (IC50: 0.11 mM) and after PIP2 (IC50: 3.11 mM), suggesting that PIP2 content in whole cell membranes is intermediate between the latter two inside-out membrane conditions.
Figure 4.

Regulation of TRPV5 by intracellular Mg2+ in whole-cell recording. (A) Alteration of intracellular Mg2+ in whole-cell recording using intrapipette perfusion. (B) Intracellular perfusion of Mg2+ (1 mM) in whole-cell recording inhibits TRPV5 reversibly. The Y axis is current density (nA/pF). Time bar for the X axis represents 4 min. (C) I-V curves of currents at indicated time points in B. (D) Time control of intracellular Mg2+ perfusion in mock-transfected CHO cells (transfected with GFP plasmid alone). The Y axis is current density (pA/pF). Time bar for the X axis represents 4 min. (E) Dose-dependent inhibition of whole-cell TRPV5 currents by Mg2+ (0–3 mM). Each experiment at a given Mg2+ concentration was performed as in Fig. 4 B and inward currents were scaled to the same maximal (current at DVF solution; I/Imax = 1) and minimal level (current after La3+; I/Imax = 0). (F) Dose–response curve of Mg2+ inhibition in whole-cell recording (closed squares) vs. in inside-out patches before (closed circles; no PIP2) and after PIP2 (open circles; +PIP2). IC50 was calculated according to the equation I/Imax = IC50 /IC50 + [Mg2+]. I/Imax = relative currents normalized to maximal currents. The two curves for inside-out patches before and after PIP2 are from Fig. 3 C.
Regulation of ion channels including inward rectifier K+ and TRP channels by PIP2 involves direct interaction with positively charged amino acids in the channels (Hilgemann et al., 2001; Prescott and Julius, 2003; Rohács et al., 2005). Recently, Rohács et al. reported that arginine-606 of the rabbit TRPV5 (corresponding to arginine-599 of the rat TRPV5) is critical for interaction with PIP2 (Rohács et al., 2005). We found that R606Q mutant was more sensitive to inhibition by intracellular Mg2+ in whole-cell recordings (IC50: 0.03 ± 0.01 mM for R606Q [n = 4] vs. 0.29 ± 0.02 mM for wild type, P < 0.05; not depicted in Fig. 4). These results further support that PIP2 activation of TRPV5 releases intracellular Mg2+ inhibition of the channel.
Mg2+-induced Slow Reversible Inhibition Is Voltage Dependent
To examine membrane voltage sensitivity, whole-cell TRPV5 currents were measured in intracellular DVF solution and in a solution containing 0.3 mM Mg2+ at different holding potentials. The fraction of TRPV5 currents in 0.3 mM intracellular Mg2+ relative to maximal currents in DVF solution (I/Imax) increased with increasing membrane hyperpolarization (Fig. 5 A). Voltage sensitivity for the intracellular Mg2+-induced slow reversible inhibition of TRPV5 is further supported by the finding in experiments using whole-cell recording that hyperpolarization of membrane holding potentials from 0 to −50 mV increased IC50 for Mg2+ from 0.29 to 0.79 mM (Fig. 5 B). Voltage dependency of inhibition by Mg2+ can be viewed as Mg2+ binds to inhibit the channel within the membrane electrical field. The fractional electrical distance of the binding site from the outer surface of the membrane (δ) can be calculated according to equation IC50(V) = IC50(0)exp(2δFV/RT) (Nilius et al., 2000), where IC50(V) and IC50(0) represent IC50 at the test voltage V and at 0 mV, respectively, and R, T, and F have their usual meanings. Our results reveal that Mg2+-induced slow inhibition of TRPV5 is owing to Mg2+ binding inside the channel at δ = 0.26.
Figure 5.

Voltage dependency of the Mg2+-induced slow reversible inhibition. (A) Whole-cell currents were recorded in DVF and 0.3 mM intracellular Mg2+ at different holding potentials (from +50 to −100 mV) as in Fig. 4 B. Relative currents (I/Imax, current at 0.3 mM Mg2+/current at DVF) were plotted against holding potentials (Vh). (B) Dose–response curve of inhibition by Mg2+ at 0 and −50 mV holding potentials. Dose–response curves were performed as in Fig. 4 B.
Time Constant Measurement Provides Further Support for Voltage Dependency for Mg2+-induced Slow Inhibition and its Distinction from Voltage-dependent Block
Up to now, our studies examine the regulation of TRPV5 by intracellular Mg2+ by perfusion of solutions containing different concentration of Mg2+. To avoid variability in the rate of perfusion and measure time constants for the Mg2+-induced voltage-dependent block and the slow inhibition, we recorded whole-cell currents using voltage jump protocol at fixed intracellular Mg2+ concentration (1 mM) (Fig. 6 A). Membrane potential was held at −100 mV. At this hyperpolarized potential, TRPV5 is fully open (not inhibited by intracellular Mg2+) and conducts inward current (see Fig. 1). Stepping from −100 to +100 mV resulted in a fast (i.e., occurring in ms and reaching maximal inhibition in ∼50 ms) time-dependent inhibition of the channel (shown as decrease of outward current in Fig. 6 A). This fast inhibition of currents (known as the Mg2+-induced voltage-dependent block) represents the time course of binding of intracellular Mg 2+ into the pore and contributes to inward rectification of currents through TRPV5 and TRPV6 channels (Hoenderop et al., 2003; Voets et al., 2003). As shown by others (Voets et al., 2003), the time course of voltage-dependent block could be fitted by two exponential terms. Fig. 6 B shows the two exponential time constants for voltage-dependent block at +25, +50, and +100 mV, respectively. Beyond the fast voltage-dependent block occurring within the first 50 ms, outward currents further decreased in a much slower time course (occurring in seconds and reaching minimal current level in <60 s) (Fig. 6 C). The mean exponential time constant (τ) for the “Mg2+-induced slow inhibition” at +100 mV was 8.3 ± 1.1 s (mean ± SEM, n = 4) (Fig. 6 D). The on-rate constant (Kon) for slow inhibition by Mg2+ at +100 mV (Kon = 1/τ [Mg2+]) was estimated at 120 ± 14 s−1M−1 (mean ± SEM, n = 4). Outward currents remained relatively stable beyond the first 50 ms in the absence of intracellular Mg2+ (current level at 60 s = 91 ± 8% of that at 100 ms, n = 4; not depicted in Fig. 6). Consistent with voltage dependency of Mg2+-induced slow inhibition, time constant at +50 and +25 mV were 13.4 ± 1.2 s and 16.2 ± 0.4 s, respectively (Fig. 6 D, each P < 0.05 vs. time constant holding at +100 mV). These time constant values at +100, +50, and +25 mV are in agreement with the earlier observation that Mg2+-induced slow inhibition occurred over 30–40 s at 0 mV (see Fig. 1; taking into consideration reduced PIP2 content in inside-out patches and probably a few seconds of delay in delivery of Mg2+ by perfusion).
Mutation of Aspartate-542 Abolishes Mg2+-induced Slow Reversible Inhibition
Acidic amino acids located in solvent-accessible region between the second intracellular loop (2nd IC loop) and the proximal region of the intracellular COOH terminus (CT) of TRPV5 may be involved in the binding and slow reversible inhibition by intracellular Mg2+ (Fig. 7 A). We mutated seven acidic amino acids within this region, one at a time, to neutral amino acid asparagine or glutamine and tested the sensitivity of each mutant to inhibition by Mg2+ in whole-cell recordings. We found that neutralization of aspartate-542 (D542N) in the putative pore region completely abolished the slow reversible inhibition of TRPV5 by intracellular Mg2+ (Fig. 7, B and C). In voltage jump experiment, currents through D542N mutant remained unchanged over 60 s at +100 mV (current level at 60 s = 89 ± 15% of that at 100 ms, n = 3; not depicted in Fig. 7), confirming that aspartate-542 is critical for Mg2+-induced slow inhibition. Neutralization of other aspartate or glutamate residues in this region had no significant effects on the slow reversible inhibition of TRPV5 by Mg2+ (Fig. 7 D).
Figure 7.

Identification of amino acid of TRPV5 involved in Mg2+-induced slow inhibition. (A) Membrane topology of amino acids between the fourth transmembrane (TM4) segment and the proximal COOH terminus (Ct) of TRPV5. Amino acids in the preselectivity filter region likely form a pore helix structure similar to that of KcsA (Dodier et al., 2004; Voets et al., 2004b). The location of amino acids mutated is shown. (B) A representative experiment of D542N mutant (in whole-cell recording) showing no inhibition by 1 mM Mg2+. (C) Dose–response curves of inhibition by Mg2+ for D542N mutant and wild-type (WT) TRPV5. (D) Average inhibition by Mg2+ for WT and each mutant (mean ± SEM, n = 5–11 each). * indicates P < 0.05 vs. WT.
Aspartate-542 is believed to be a part of the selectivity filter of TRPV5 and a binding site for Mg2+-induced voltage-dependent block (Nilius et al., 2001; Hoenderop et al., 2003). Our findings indicate that Mg2+ binding to aspartate-542 not only causes voltage-dependent block but also the slow inhibition. This conclusion is further supported by the fact that the fractional electrical distance for the slow inhibition we measured in this study (δ = 0.26) is similar to that measured for voltage-dependent block (δ = 0.31) (Nilius et al., 2000).
Reduction of PIP2 Enhances Mg2+-induced Slow Reversible Inhibition of TRPV5 without Affecting Mg2+ Binding
To investigate the mechanism for PIP2 modulation of Mg2+-induced slow reversible inhibition of TRPV5, we tested whether PIP2 affects Mg2+ binding. The kinetics of Mg2+-induced slow inhibition and voltage-dependent block were studied using voltage jump experiments as in Fig. 6. Whole-cell recordings were performed in the presence of intracellular Mg2+ (1 mM). The extracellular bath solution contained thrombin + wortmannin (WMN) or vehicle. WMN inhibits phosphoinositide 4-kinase (a rate-limiting enzyme for synthesis of PIP2 from phosphatidylinositol and phosphatidylinositol-4-phosphate) to allow reduction of membrane PIP2 following receptor-mediated hydrolysis of PIP2 (Nakanishi et al., 1995). Fig. 8 A shows representative single exponential fit to current traces in response to voltage jump from −100 to +100 mV in the presence of thrombin + WMN (labeled as Thr) or vehicle (Control). As expected for PIP2 releasing Mg2+-induced slow inhibition, reduction of membrane of PIP2 by incubating with thrombin and WMN decreased the time constants for inhibition at depolarized membrane potentials (Fig. 8 B, P < 0.05, +Thr vs. Control for all potentials). Interestingly, reduction of membrane PIP2 had no effects on the time constants for Mg2+-induced voltage-dependent block to the channel (Fig. 8 C; time constants measured as in Fig. 6 B).
Enhancement of Mg2+-induced Slow Inhibition as a Mechanism for PLC-coupled Receptor Regulation TRPV5-mediated Ca2+ Entry
The physiological function of TRPV5 is to allow Ca2+ entry through the apical membrane for transepithelial transport of Ca2+ in kidney and intestine (Hoenderop et al., 2002). We next examined the effects of PIP2 hydrolysis induced by receptor activation of PLC on TRPV5 functioning as a Ca2+-permeable channel. In the following cell-attached recordings, the pipette solution contained (in mM) 140 NaCl, 1 MgCl2, and 1 CaCl2. Because of the anomalous mole fraction behavior, TRPV5 conducts Ca2+ exclusively in this physiological solution (Vennekens et al., 2001). CHO cells express endogenous receptors for thrombin (Dickenson and Hill, 1997). Activation of PLC by endogenous thrombin receptors in CHO cells was verified by the release of Ca2+ from intracellular stores (unpublished data). Thrombin was applied to the patch membrane via intrapipette perfusion (Fig. 9 A). The bath solution contained vehicle or wortmannin.
In cell-attached recording, application of thrombin (Thr) in the absence of WMN decreased TRPV5 Ca2+ currents transiently (by 43 ± 8% in 90 s, n = 5, P < 0.05 vs. control without thrombin) (Fig. 9 A). Presumably due to desensitization of receptors and de novo synthesis of PIP2 via phosphoinositide 4-kinase, TRPV5 currents recovered at least partially over 3–4 min in the absence of wortmannin. In the presence of wortmannin, thrombin caused a persistent reduction of TRPV5 currents by 85 ± 5% in 90 s (n = 6, P < 0.05 vs. thrombin alone). As controls, TRPV5 currents did not change significantly without thrombin (decreased by 8 ± 7% over 90 s, n = 4; WMN alone in Fig. 9 A). The effects of PIP2 hydrolysis on dose-dependent inhibition by Mg2+ of TRPV5-mediated Ca2+ currents were examined by whole-cell recording (Fig. 9 B). The IC50 for Mg2+ inhibition of TRPV5-mediated Ca2+ currents were 0.61 ± 0.12 mM (n = 5) and 0.11 ± 0.08 (n = 5) for control cells (treated with vehicle) and cells treated with thrombin and wortmannin, respectively (P < 0.05). The concentration of the intracellular free Mg2+ in the cell-attached recording is estimated at ∼0.5 mM (Romani and Scarpa, 2000). These results support the hypothesis that an increase in sensitivity to intracellular Mg2+ is important for physiological regulation of TRPV5-mediated Ca2+ entry by PLC-activating hormones.
DISCUSSION
Many TRP channels are downstream of G protein–coupled receptors and PLC (for review see Clapham, 2003; Putney, 2003). Thus, TRP channels may be regulated by G protein–coupled receptors via inositol-1,4,5-trisphosphate and/or diacylglycerol, products of PIP2 breakdown catalyzed by PLC (Clapham, 2003; Putney, 2003). Several TRP channels are also directly regulated by PIP2 (Chuang et al., 2001; Hardie, et al., 2001; Runnels et al., 2002; Liu and Liman, 2003; Liu and Qin, 2005; Rohács et al., 2005). Therefore, an additional mechanism for PLC to control TRP channels is via reduction of PIP2 content (Chuang et al., 2001; Runnels et al., 2002; Liu and Qin, 2005). However, the reduction of PIP2 in the plasma membrane from physiological stimulation of PLC may not be sufficient to alter the activity of channels if the affinity of channels for PIP2 is relatively high (Kobrinsky et al., 2000). In the present study, we find that PIP2 activates TRPV5 and that activation of TRPV5 by PIP2 decreases the sensitivity of TRPV5 to inhibition by intracellular Mg2+ (see working model in Fig. 10). Hydrolysis of PIP2 by receptor activation of PLC increases the sensitivity to the inhibition by Mg2+. These results provide a novel mechanism for regulation of TRP channels by PLC-activating hormones by increasing the sensitivity to intracellular Mg2+.
Figure 10.

A working model showing the relationships of three different effects of Mg2+ on TRPV5. Binding of intracellular Mg2+ to D542 of TRPV5 causes a fast (in ms time scale) voltage-dependent block. Prolonged binding to the same site by Mg2+ (e.g., 10 s) leads to slow inhibition, which is likely due to conformational changes of the channel. The slow inhibition is also voltage dependent. Membrane PIP2 stabilizes TRPV5 in open conformation. PIP2 interaction with TRPV5 does not affect Mg2+ binding but prevents Mg2+-induced conformational changes. Mg2+ causes run-down by activating lipid phosphatases. However, run-down is probably not a physiological effect of Mg2+ as it occurs predominantly in excised inside-out membranes. Hydrolysis of PIP2 by activating PLC-coupled receptors sensitizes TRPV5 to inhibition by intracellular Mg2+. * indicates that time constant for Mg2+-induced slow inhibition of TRPV5 is shorter in PIP2-reduced (e.g., 5 s) than in PIP2-repleted state (e.g., 10 s). For comparison, Mg-induced slow inhibition has also been reported for TRPV6, TRPM6, and TRPM7 (Nadler et al., 2001; Runnels et al., 2001; Voets et al., 2003, 2004a). Mg2+-induced voltage-dependent block contributes to inward rectification of many channels, including inward-rectifier K+ channels and TRPV5 and 6. Mg2+ would presumably cause run-down on all PIP2-regulated ion channels.
The cytosolic free Mg2+ concentration in most mammalian cells is between 0.5 and 1 mM (Romani and Scarpa, 2000). The IC50 for the slow inhibition (0.79 mM) at −50 mV (near the physiological resting membrane potential) suggests that intracellular Mg2+ is an important physiological regulator of TRPV5 function in vivo. The intracellular free Mg2+ concentration changes only slightly in response to physiological stimuli (Romani and Scarpa, 2000). However, the dramatic shift in Mg2+ sensitivity by PIP2 will provide amplification for regulation of TRPV5 by intracellular Mg2+ and PLC-activating hormones. One such example is the inhibition of calcium transport in the distal renal tubules by prostaglandin E2 through activation of phospholipase C via EP1 receptors (Hoenderop et al., 2002).
Besides TRPV5 and TRPV6, intracellular Mg2+ has recently been recognized as an important regulator of TRPM6 and TRPM7 channels (Nadler et al., 2001; Runnels et al., 2001; Voets et al., 2004a). TRPM7 (also known as magnesium-inhibited cation channel [MIC]; Kozak et al., 2002; Prakriya and Lewis, 2002) is a ubiquitous channel important for cellular uptake of magnesium and cell viability (Schmitz et al., 2003). Magnesium ions entering cells through TRPM7 feedback on the channel to regulate its activity (Schmitz et al., 2003). TRPM7 is also regulated by PIP2 (Runnels et al., 2002). Whether PIP2 also modulates Mg2+ sensitivity for TRPM7 is unknown at present.
TRPV5 does not conduct Mg2+ in physiological conditions. However, TRPM6 (a close homology of TRPM7) is a Mg2+-permeable channel responsible for magnesium transport in epithelial tissues (Schlingmann et al., 2002; Walder et al., 2002; Voets et al., 2004a). Both TRPV5 and TRPM6 are present in the distal convoluted tubule (DCT) cells (Hoenderop et al., 2002; Schlingmann et al., 2002; Walder et al., 2002; Voets et al., 2004a), which are key cells responsible for transcellular reabsorption of magnesium and calcium in the kidney (Suki et al., 2000). Many diseases with reduced Mg2+ transport in the DCT exhibit increased Ca2+ transport. These diseases include genetic diseases such as familial hypomagnesemia with hypocalciuria and Gitelman's disease (Warnock, 2002) and acquired diseases such as cis-platinum toxicity (Mavichak et al., 1988). Our present finding that intracellular Mg2+ negatively regulates TRPV5 raises the possibility that a decrease in Mg2+ influx through TRPM6 leads to an increase in Ca2+ influx through TRPV5/TRPV6 in DCT cells, providing a possible molecular explanation for this disease phenotype. It should be noted that patients with mutations of TRPM6 develop hypocalcemia (Schlingmann et al., 2002; Walder et al., 2002), rather than hypercalcemia as would be expected from an increase in Ca2+ reabsorption in DCT cells alone. Hypocalcemia in these patients is believed secondary to parathyroid failure from severe magnesium wasting and hypomagnesemia (Anast et al., 1972).
Intracellular Mg2+ regulates inward rectification of ion channels including inward rectifier K+ channels and TRPV5 and TRPV6 channels via voltage-dependent block (Lu and MacKinnon, 1994; Hoenderop et al., 2003; Voets et al., 2003). In the present study, we report that intracellular Mg2+ regulates TRPV5 additionally via a mechanism likely involving conformational change of the protein (Fig. 10). We refer to this additional inhibition by Mg2+ as slow reversible inhibition to distinguish from the faster voltage-dependent block. A similar dual regulation of TRPV6 by intracellular Mg2+ via voltage-dependent block and a slower inhibition has recently been reported (Voets et al., 2003).
Aspartate-542 of TRPV5 is a part of the selectivity filter critical for permeation of Ca2+ ions and a binding site for Mg2+-induced voltage-dependent block of Ca2+ and Na+ currents (Nilius et al., 2001; Hoenderop et al., 2003). Our findings suggest that binding of intracellular Mg2+ to aspartate-542 in the selectivity filter inhibits TRPV5 via at least two separate but interrelated mechanisms. The first mechanism is by voltage-dependent block. In this mechanism, Mg2+ ions bind and occlude the channel pore, preventing ion permeation. This inhibition by blocking of channel pore occurs as soon as Mg2+ enters and binds the selectivity filter driven by membrane depolarization and reverses quickly by membrane hyperpolarization. This is similar to voltage-dependent block of inwardly rectifying K+ channels and other channels by Mg2+, polyamines, and other charged blockers (Hille, 2001). Binding of Mg2+ to the selectivity filter of TRPV5 for a longer duration, however, leads to the second mechanism of inhibition: the slow reversible inhibition. The slow reversible inhibition develops over 30–40 s and is not reversed by transient unbinding of Mg2+ ions during intermittent membrane hyperpolarization from voltage ramp applied every 10 s The exact mechanism for the slow inhibition remains unknown, but likely involves conformational change(s) of the channel protein.
Our results also shed light on the mechanism by which PIP2 alters the sensitivity of TRPV5 to slow inhibition by Mg2+. PIP2 activates many ion channels directly by affecting their structure (Hilgemann et al., 2001). We find that PIP2 activates TRPV5 in the absence of intracellular Mg2+ (Fig. 1 B). PIP2 also regulates D542N, a Mg2+ binding site mutant (unpublished data). Thus, activation of TRPV5 by PIP2 does not require its regulation by intracellular Mg2+. However, activation of TRPV5 by PIP2 decreases the sensitivity of TRPV5 for intracellular Mg2+-induced slow inhibition. PIP2 does so without affecting Mg2+ binding to TRPV5 (Fig. 8 C). We suggest that PIP2 desensitizes TRPV5 to Mg2+-induced slow inhibition by stabilizing channel in an open conformation and prevents the conformational change induced by binding of Mg2+.
It is known that ion binding may alter the structure of selectivity filter of ion channels. For example, occupancy of K+ ions in the selectivity filter slows the rate of C-type inactivation of voltage-gated K+ channels (Yellen, 1998). The crystal structure of the bacterial KcsA K+ channel reveals that presence of multiple K+ ions in the selectivity filter stabilizes the full open conformation (Zhou and MacKinnon, 2003). Our present study provides an example of conformational changes of ion channels caused by binding of a pore blocker.
Acknowledgments
We thank Dr. D. Hilgemann for discussion and for help in setting up intrapipette perfusion and Drs. Hilgemann, Baum, and Igarashi for critical reading of the manuscript.
This work is supported by National Institutes of Health grants DK-20543, DK-54368, and DK-59530 and an Established Investigator Award (0440019N) from American Heart Association. C.L. Huang is holder of the Jacob Lemann Professorship in Calcium Transport of the University of Texas Southwestern Medical Center at Dallas.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: DCT, distal convoluted tubule; DVF, divalent-free; TRP, transient receptor potential; WMN, wortmannin.
References
- Anast, C.S., J.M. Mohs, S.L. Kaplan, and T.W. Burns. 1972. Evidence for parathyroid failure in magnesium deficiency. Science. 177:606–608. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E. 2003. TRP channels as cellular sensors. Nature. 426:517–524. [DOI] [PubMed] [Google Scholar]
- Chuang, H.H., E.D. Prescott, H. Kong, S. Shields, S.E. Jordt, A.I. Basbaum, M.V. Chao, and D. Julius. 2001. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 411:957–962. [DOI] [PubMed] [Google Scholar]
- Dickenson, J.M., and S.J. Hill. 1997. Transfected adenosine A1 receptor-mediated modulation of thrombin-stimulated phospholipase C and phospholipase A2 activity in CHO cells. Eur. J. Pharmacol. 321:77–86. [DOI] [PubMed] [Google Scholar]
- Dodier, Y., U. Banderali, H. Klein, O. Topalak, O. Dafi, M. Simoes, G. Bernatchez, R. Sauve, and L. Parent. 2004. Outer pore topology of the ECaC-TRPV5 channel by cysteine scan mutagenesis. J. Biol. Chem. 279:6853–6862. [DOI] [PubMed] [Google Scholar]
- Fabiato, A. 1988. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 157:378–417. [DOI] [PubMed] [Google Scholar]
- Fukami, K., K. Matsuoka, O. Nakanishi, A. Yamakawa, S. Kawai, and T. Takenawa. 1988. Antibody to phosphatidylinositol 4,5-bisphosphate inhibits oncogene-induced mitogenesis. Proc. Natl. Acad. Sci. USA. 85:9057–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie, R.C., P. Raghu, S. Moore, M. Juusola, R.A. Baines, and S.T. Sweeney. 2001. Calcium influx via TRP channels is required to maintain PIP2 levels in Drosophila photoreceptors. Neuron. 30:149–159. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Third edition. Sinauer Associates, Sunderland, MA. 814 pp.
- Hilgemann, D.W., and C.C. Lu. 1998. Giant membrane patches: improvements and applications. Methods Enzymol. 293:267–280. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., S. Feng, and C. Nasuhoglu. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE. 111:RE19–RE21. [DOI] [PubMed] [Google Scholar]
- Hoenderop, J.G., B. Nilius, and R.J. Bindels. 2002. Molecular mechanism of active Ca2+ reabsorption in the distal nephron. Annu. Rev. Physiol. 64:529–549. [DOI] [PubMed] [Google Scholar]
- Hoenderop, J.G., T. Voets, S. Hoefs, F. Weidema, J. Prenen, B. Nilius, and R.J. Bindels. 2003. Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 and TRPV6. EMBO J. 22:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C.-L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels and its stabilization by Gβγ. Nature. 391:803–808. [DOI] [PubMed] [Google Scholar]
- Kobrinsky, E., T. Mirshahi, H. Zhang, T. Jin, and D.E. Logothetis. 2000. Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat. Cell Biol. 2:507–514. [DOI] [PubMed] [Google Scholar]
- Kozak, J.A., H.H. Kerschbaum, and M.D. Cahalan. 2002. Distinct properties of CRAC and MIC channels in RBL cells. J. Gen. Physiol. 120:221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, Y.-M., W.-Z. Zeng, H.-H. Liou, C.R. Solaro, and C.-L. Huang. 2000. Phosphatidylinositol 4,5-bisphosphate and intracellular pH regulate the ROMK1 potassium channel via separate but interrelated mechanisms. J. Biol. Chem. 275:10182–10189. [DOI] [PubMed] [Google Scholar]
- Liu, B., and F. Qin. 2005. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25:1674–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D., and E.R. Liman. 2003. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. USA. 100:15160–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z., and R. MacKinnon. 1994. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 371:243–246. [DOI] [PubMed] [Google Scholar]
- Mavichak, V., C.M. Coppin, N.L. Wong, J.H. Dirks, V. Walker, and R.A. Sutton. 1988. Renal magnesium wasting and hypocalciuria in chronic cis-platinum nephropathy in man. Clin. Sci. (Lond.). 75:203–207. [DOI] [PubMed] [Google Scholar]
- Montell, C., L. Birnbaumer, V. Flockerzi, R.J. Bindels, E.A. Bruford, M.J. Caterina, D.E. Clapham, C. Harteneck, S. Heller, D. Julius, et al. 2002. A unified nomenclature for the superfamily of TRP cation channels. Mol. Cell. 92:229–231. [DOI] [PubMed] [Google Scholar]
- Nadler, M.J.S., M.C. Hermosura, K. Inabe, A.-L. Perraud, Q. Zhu, J. Stokes, T. Kurosaki, J.P. Kinet, R. Penner, A.M. Scharenberg, and A. Fleig. 2001. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 411:590–595. [DOI] [PubMed] [Google Scholar]
- Nakanishi, S., K.J. Catt, and T. Balla. 1995. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositol phospholipids. Proc. Natl. Acad. Sci. USA. 92:5317–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasuhoglu, C., S. Feng, J. Mao, M. Yamamoto, H.L. Yin, S. Earnest, B. Barylko, J.P. Albanesi, and D.W. Hilgemann. 2002. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal. Biochem. 301:243–254. [DOI] [PubMed] [Google Scholar]
- Nilius, B., R. Vennekens, J. Prenen, J.G. Hoenderop, R.J. Bindels, and G. Droogmans. 2000. Whole-cell and single channel monovalent cation currents through the novel rabbit epithelial Ca2+ channel ECaC. J. Physiol. 527:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., R. Vennekens, J. Prenen, J.G. Hoenderop, G. Droogmans, and R.J. Bindels. 2001. The single pore residue Asp542 determines Ca2+ permeation and Mg2+ block of the epithelial Ca2+ channel. J. Biol. Chem. 276:1020–1025. [DOI] [PubMed] [Google Scholar]
- Oliver, D., C.-C. Lien, M. Soom, T. Baukrowitz, P. Jonas, and B. Fakler. 2004. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 304:265–270. [DOI] [PubMed] [Google Scholar]
- Prakriya, M., and R.S. Lewis. 2002. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J. Gen. Physiol. 119:487–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott, E.D., and D. Julius. 2003. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 300:1284–1288. [DOI] [PubMed] [Google Scholar]
- Putney, J.W., Jr. 2003. Capacitance calcium entry in the nervous system. Cell Calcium. 34:339–344. [DOI] [PubMed] [Google Scholar]
- Rohács, T., C.M. Lopes, I. Michailidis, and D.E. Logothetis. 2005. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8:626–634. [DOI] [PubMed] [Google Scholar]
- Romani, A.M., and A. Scarpa. 2000. Regulation of cellular magnesium. Front. Biosci. 5:D720–D734. [DOI] [PubMed] [Google Scholar]
- Runnels, L.W., L. Yue, and D.E. Clapham. 2001. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 291:1043–1047. [DOI] [PubMed] [Google Scholar]
- Runnels, L.W., L. Yue, and D.E. Clapham. 2002. The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat. Cell Biol. 4:329–336. [DOI] [PubMed] [Google Scholar]
- Schlingmann, K.P., S. Weber, M. Peters, L. Niemann-Nejsum, H. Vitzthum, K. Klingel, M. Kratz, E. Haddad, E. Ristoff, D. Dinour, et al. 2002. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat. Genet. 31:166–170. [DOI] [PubMed] [Google Scholar]
- Schmitz, C., A.L. Perraud, C.O. Johnson, K. Inabe, M.K. Smith, R. Penner, T. Kurosaki, A. Fleig, and A.M. Scharenberg. 2003. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 114:191–200. [DOI] [PubMed] [Google Scholar]
- Shyng, S.-L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Sui, J.L., J. Petit-Jacques, and D.E. Logothetis. 1998. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc. Natl. Acad. Sci. USA. 95:1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suki, W.N., E.D. Lederer, and D. Rouse. 2000. Renal transport of calcium, magnesium, and phosphate. Brenner & Rector's The Kidney. Sixth edition. B.M. Brenner, editor. Saunders, Philadelphia. 520–574.
- Vennekens, R., J. Prenen, J.G.J. Hoenderop, R.J.M. Bindels, G. Droogmans, and B. Nilius. 2001. Pore properties and ionic block of the rabbit epithelial calcium channel expressed in HEK 293 cells. J. Physiol. 530:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets, T., A. Janssens, J. Prenen, G. Droogmans, and B. Nilius. 2003. Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. J. Gen. Physiol. 121:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets, T., B. Nilius, S. Hoefs, A.W. van der Kemp, G. Droogmans, R.J. Bindels, and J.G.J. Hoenderop. 2004. a. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J. Biol. Chem. 279:19–25. [DOI] [PubMed] [Google Scholar]
- Voets, T., A. Janssens, G. Droogmans, and B. Nilius. 2004. b. Outer pore architecture of a Ca2+-selective TRP channel. J. Biol. Chem. 279:15223–15230. [DOI] [PubMed] [Google Scholar]
- Walder, R.Y., D. Landau, P. Meyer, H. Shalev, M. Tsolia, Z. Borochowitz, M.B. Boettger, G.E. Beck, R.K. Englehardt, R. Carmi, and V.C. Sheffield. 2002. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 31:171–174. [DOI] [PubMed] [Google Scholar]
- Warnock, D.G. 2002. Renal genetic disorders related to K+ and Mg2+. Annu. Rev. Physiol. 64:845–876. [DOI] [PubMed] [Google Scholar]
- Yeh, B.-I., T.-J. Sun, J.Z. Lee, H.-H. Chen, and C.-L. Huang. 2003. Mechanism and molecular determinant for regulation of rabbit transient receptor potential type 5 (TRPV5) channel by extracellular pH. J. Biol. Chem. 278:51044–51052. [DOI] [PubMed] [Google Scholar]
- Yeh, B.-I., Y.K. Kim, W. Jabbar, and C.-L. Huang. 2005. Conformational changes of pore helix coupled to gating of TRPV5 by protons. EMBO J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., and R. MacKinnon. 2003. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol. 333:965–975. [DOI] [PubMed] [Google Scholar]