

Abstract
We propose a new mechanism to explain autoinhibition of the epidermal growth factor receptor (EGFR/ErbB) family of receptor tyrosine kinases based on a structural model that postulates both their juxtamembrane and protein tyrosine kinase domains bind electrostatically to acidic lipids in the plasma membrane, restricting access of the kinase domain to substrate tyrosines. Ligand-induced dimerization promotes partial trans autophosphorylation of ErbB1, leading to a rapid rise in intracellular [Ca2+] that can activate calmodulin. We postulate the Ca2+/calmodulin complex binds rapidly to residues 645–660 of the juxtamembrane domain, reversing its net charge from +8 to −8 and repelling it from the negatively charged inner leaflet of the membrane. The repulsion has two consequences: it releases electrostatically sequestered phosphatidylinositol 4,5-bisphosphate (PIP2), and it disengages the kinase domain from the membrane, allowing it to become fully active and phosphorylate an adjacent ErbB molecule or other substrate. We tested various aspects of the model by measuring ErbB juxtamembrane peptide binding to phospholipid vesicles using both a centrifugation assay and fluorescence correlation spectroscopy; analyzing the kinetics of interactions between ErbB peptides, membranes, and Ca2+/calmodulin using fluorescence stop flow; assessing ErbB1 activation in Cos1 cells; measuring fluorescence resonance energy transfer between ErbB peptides and PIP2; and making theoretical electrostatic calculations on atomic models of membranes and ErbB juxtamembrane and kinase domains.
INTRODUCTION
The general mechanism by which binding of a ligand to the extracellular domain of a typical receptor tyrosine kinase activates its intracellular protein tyrosine kinase (PTK) domain is well understood: binding produces dimerization (or dimer rearrangement) that leads to trans autophosphorylation of tyrosines in the “activation loop” of the PTK domain (Hubbard and Till, 2000; Schlessinger, 2000, 2003; Huse and Kuriyan, 2002; Jorissen et al., 2003; Hubbard, 2004). The ErbB tyrosine kinase family (ErbB1/HER1/EGFR, ErbB2/HER2, ErbB3/HER3, ErbB4/HER4) appears to be the major exception to this rule because their PTK domains do not require phosphorylation for catalytic competency (Gotoh et al., 1992; Jorissen et al., 2003; Hubbard, 2004). The structure of the ErbB1 PTK domain suggests why this is so: the unphosphorylated activation loop is in an active conformation and the catalytic elements are “primed and ready for phospho-transfer,” suggesting “the regulation of the vital cellular processes influenced by epidermal growth factor receptor (EGFR) signaling must be exerted by control of the delivery of the COOH-terminal substrate tyrosines to the active site” (Stamos et al., 2002). Understanding this control mechanism is important because members of the ErbB family are frequently overactive in solid tumors (Blume-Jensen and Hunter, 2001; Yarden and Sliwkowski, 2001; Gschwind et al., 2004; Paez et al., 2004; Sordella et al., 2004); ErbB2, for example, is overexpressed in 25% of breast cancers, and this overexpression correlates with poor prognosis (Klapper et al., 2000).
Several groups have recently proposed models to explain autoinhibition of the ErbB family. Landau et al. (2004) developed a computational model that suggests direct contact between a positively charged face of the kinase domain and a negatively charged segment of the COOH-terminal tail region of the receptor produces autoinhibition. Alternatively, Aifa et al. (2005) proposed that this negatively charged segment, ErbB1(979–991), interacts with a cluster of basic residues in the juxtamembrane (JM) region of an adjacent ErbB molecule. The ErbB1 basic JM region also plays an important role in the structural model of autoinhibition we propose.
In our model, basic residues in the ErbB JM and PTK domains interact electrostatically with acidic lipids in the membrane, inhibiting catalytic activity in the absence of ligand. This autoinhibitory hypothesis has an obvious corollary: conditions that decrease the electrostatic binding (e.g., an increase in the cytoplasmic salt concentration, exposure to an amphipathic weak base that decreases the negative fixed charge density on the membrane) should release the JM and PTK regions from the membrane, producing ligand-independent trans autophosphorylation. We have tested this corollary by comparing data from our experiments with model membranes and peptides to data in the literature from intact cells.
Our model also suggests a novel positive feedback mechanism by which ligand-induced dimerization may contribute to activation. As discussed below, it is well established that ligand-induced dimerization of ErbB1 leads to a transient (∼10 min) increase in the level of free Ca2+ within a cell. We postulate that calcium/calmodulin (Ca/CaM) binds to the ErbB JM region very rapidly (∼100 ms when [Ca/CaM] = 1 μM), reversing its charge and repelling both it and the PTK domain from the membrane. This implies Ca/CaM binding will increase the initial rate of trans autophosphorylation over and above the rate due to the local concentration effect resulting from ligand-induced dimerization. Our postulated Ca/CaM-mediated activation mechanism will be important only when the [Ca2+] is high enough to produce a significant increase in Ca/CaM.
The model predicts that Ca/CaM can pull peptides corresponding to the ErbB JM region off a membrane rapidly and that CaM inhibitors will inhibit, but not completely block, the initial phase of EGF-mediated ErbB autophosphorylation in cells. We tested these predictions experimentally; while the results are consistent with the predictions, they neither prove that the model is correct nor rule out other potential activation mechanisms that may act in parallel (Jorissen et al., 2003; Schlessinger, 2003). For example, there is much evidence that phosphatases play an important role in controlling the trans autophosphorylation of ErbB (e.g., Reynolds et al., 2003; Tonks, 2003; Ichinos et al., 2004; Matilla et al., 2004); we return to the role of phosphatases in the concluding section of DISCUSSION.
Structural Model
Fig. 1 illustrates our model; the cartoons (Fig. 1, A and B) focus on the ∼40-residue JM domain (residues 645–682 in ErbB1) between the helical transmembrane (TM) and structured PTK domains. Fig. 1 C shows the sequence of the ErbB1 JM domain using color coding to indicate the amino acids that can interact with the bilayer: basic (R and K) residues are blue, acidic residues (E) are red, and hydrophobic residues (F, L, I, and V) are green. The cytoplasmic leaflet of a mammalian cell plasma membrane typically contains 15–30% monovalent acidic phospholipid, mainly phosphatidylserine (PS), which produces a negative surface potential of −30 to −40 mV (McLaughlin, 1989). Hence the membrane bilayer attracts blue and green residues through electrostatic and hydrophobic interactions, respectively, and electrostatically repels red residues.
Figure 1.

Model of ErbB activation. (A) ErbB1 in a quiescent cell (no EGF, low free [Ca2+] in cytosol). The cytoplasmic leaflet of the plasma membrane contains acidic phospholipids (lipids with red minus signs) that strongly attract the reversible membrane anchor region of the JM domain, comprising eight basic (circled blue plus signs) and five hydrophobic residues (green circles). The PTK core (depicted with the COOH-terminal portion of the JM domain as a rectangle of appropriate dimensions, 5 × 6 nm) has a positively charged face (blue plus signs) that also binds electrostatically to the bilayer. We postulate that these membrane interactions inhibit the enzyme. The length of the first two portions of the JM domain, as well as the size of the PTK domain and calcium/calmodulin (Ca/CaM), are drawn approximately to scale with the bilayer (thickness ∼5 nm). (B) Activated ErbB1. After ligand-induced dimerization (not depicted) and partial trans autophosphorylation, we postulate that binding of acidic Ca/CaM (charge = −16) to the basic (charge = +8) reversible membrane anchor forms a high affinity complex with a net charge of –8; this charge reversal repels the complex from the negatively charged bilayer, ripping both the JM and PTK domains off the membrane and fully activating the ErbB1. The PTK core is now free to rotate via the flexible linker, giving the catalytic site access to tyrosines on an adjacent ErbB family member (not depicted), or other substrate. As discussed in the text, Ca/CaM may cycle on and off the JM region at up to a diffusion limited rate (∼102 s−1 if [Ca/CaM] = 10−6 M). (C) Sequence of the JM region of ErbB1. SwissProt accession no. P00533.
We divide the JM domain into three sections (Fig. 1 C) and hypothesize that the 16-residue NH2-terminal region (Arg645 to Gln660 of ErbB1) acts as a reversible membrane anchor. Specifically, we follow the suggestions of Ullrich et al. (1984) and Hunter et al. (1984) and postulate that in a quiescent cell, i.e., when no ligands are present and the free [Ca2+] is low, the eight basic and five hydrophobic amino acids bind this region to the negatively charged inner leaflet of the bilayer. In our model, the 12-residue central region (Glu661 to Gly672 of ErbB1) acts as a flexible linker that joins the reversible membrane anchor to the COOH-terminal section of the JM domain. The flexible linker has a net charge of −2 (3 E, 1R); it should be repelled, albeit weakly, from the membrane bilayer. The 10-residue COOH-terminal region of the JM domain (Glu673 to Ile682) was identified in the structure of the ErbB1 PTK core (Stamos et al., 2002). Fig. 2 illustrates that one face of this COOH-terminal JM region + PTK structure is positively charged (blue), and thus should bind electrostatically to the negatively charged bilayer, as drawn in Fig. 1 A. Fig. 2 also shows that the PTK domain has a strong dipole character (i.e., one face is positive, while the remainder of the molecule surface is mainly negative); thus it will not be free to rotate when bound to the bilayer. A key assumption of our model is that binding of the PTK core to the membrane inhibits its activity; hindered rotation could, for example, limit its contact with, and thus ability to phosphorylate, tyrosines in the COOH-terminal tail of an ErbB family dimer or other substrate.
Figure 2.

Electrostatic potential profile adjacent to the ErbB1 PTK core. The red and blue meshes illustrate the −25 and +25 mV equipotential profiles, respectively. Potentials calculated from the Poisson-Boltzmann equation in 100 mM salt and illustrated using GRASP. (A) ErbB1 PTK (together with residues 673–682 of the JM) domain, as revealed by the crystal structure (Stamos et al., 2002). The orientation is the same as depicted in Fig. 1 A; the membrane is above the basic (blue) face. (B) Structure rotated 90° to show the positively charged face. Residues 673–682 are the extended region at the top of the structure, starting from N.
Under physiological conditions, a ligand such as EGF binds to the ErbB1 extracellular domain and induces dimerization. Recent structural studies of the ErbB1 extracellular domain with (Ogiso et al., 2002) and without (Ferguson et al., 2003) bound EGF indicate that ligand binding triggers a dramatic rearrangement of the four extracellular subdomains in each receptor monomer. A direct intramolecular interaction between cysteine-rich subdomains II and IV in the unactivated receptor is broken upon EGF binding, and loss of contact releases a dimerization arm on domain II that mediates direct ErbB1 monomer–monomer contacts (for reviews see Burgess et al., 2003; Ferguson, 2004).
In our model, the intracellular JM + PTK regions of each dimer pair exist in equilibrium between the membrane-bound (Fig. 1 A) and free (not shown) states, with the autoinhibited membrane-bound state predominating in the absence of Ca/CaM. Upon ErbB dimerization (not depicted in Fig. 1 for simplicity), the small fraction of dimeric receptors with membrane-free JM + PTK regions can trans autophosphorylate. Phosphorylation of tyrosine residues in the ErbB COOH-terminal tail leads to binding and activation of PLC-γ, hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) with concomitant production of inositiol 1,4,5-trisphosphate (IP3), and IP3-mediated release of Ca2+ from internal stores (for review see Jorissen et al., 2003). The transitory release of Ca2+ from intracellular stores mediated by IP3 is followed by a more sustained influx of Ca2+ across the plasma membrane (e.g., Pandiella et al., 1988; Cheyette and Gross, 1991; Hughes et al., 1991; Bezzerides et al., 2004; Li et al., 2004c). Adding 30 nM EGF to A431 cells, for example, increases intracellular [Ca2+] about fivefold, to 600 nM, in ∼5 min; [Ca2+] then declines over ∼10 min to a value only slightly above the basal level (Hughes et al., 1991).
The EGF-mediated transient increase in cytoplasmic [Ca2+] activates CaM, and our data, together with previous work on peptides (Martin-Nieto and Villalobo, 1998) and native ErbB1 (Li et al., 2004a), suggest the Ca/CaM complex can bind rapidly and strongly to residues 645–660 of ErbB1, as shown in Fig. 1 B. This binding reverses the charge on the region from +8 to −8, converting its strong electrostatic attraction to the membrane into a strong electrostatic repulsion. We hypothesize that the binding energy of Ca/CaM for the reversible membrane anchor region and the electrostatic repulsion of the resulting complex from the negatively charged bilayer provide the energy to move the PTK core of an ErbB family member off the bilayer. Thus we refer to this mechanism as an “electrostatic engine” that increases both the frequency at which the JM + PTK domain moves from its autoinhibitory membrane-bound conformation (Fig. 1 A) to a freely rotating active state (Fig. 1 B) and the duration of time it spends in this active state. We discuss below experiments that suggest this putative engine may cycle rapidly (∼10–100 s−1).
Our fluorescence resonance energy transfer (FRET) and PLC activity measurements also show that the cluster of basic residues on the JM domain can, when bound to the membrane, electrostatically sequester PIP2. Thus ErbB may function as a scaffolding protein with its JM domain rapidly concentrating and releasing PIP2 in the vicinity of PLC-γ and phophoinositide 3-kinase (PI3K), enzymes that are bound to the ErbB COOH-terminal region and use PIP2 as a substrate.
MATERIALS AND METHODS
Materials
1-palmitoyl-2-oleoyl-sn-PS, 1-palmitoyl-2-oleoyl-sn-phosphatidylcholine (PC), 1-palmitoyl-2-oleoyl-sn-phosphatidylglycerol (PG), and the ammonium salt of l-α-phosphatidyl-d-myo-inositol 4,5-bisphosphate (PIP2) were purchased from Avanti Polar Lipids. Labeled [dioleoyl-1-14C]L-α-dioleoylphosphatidylcholine and [ethyl-1,2-3H]N-ethylmaleimide (NEM) were from PerkinElmer Life Sciences. 6-Acryloyl-2-dimethylaminonaphthalene (acrylodan), Alexa488, and Texas red were from Molecular Probes, Inc. Bodipy-TMR-PIP2 was purchased from Echelon. Sphingosine was from Sigma-Aldrich.
All peptides were obtained from American Peptide Co., Inc. Each peptide was blocked with an acetyl group at its NH2 terminus and an amide group at its COOH terminus. We performed binding measurements with peptides corresponding to the regions of ErbB receptors shown in Fig. S6 (available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1); the peptides had an extra Cys group at the NH2 terminus, which permitted covalent attachment of either a radioactive (NEM) or fluorescent (acrylodan for stop flow, Texas red for FRET, Alexa488 for FCS measurements) probes as described elsewhere (Wang et al., 2002, and references therein). We used peptides without Cys for the zeta potential and surface pressure measurements. Labeled peptides were purified by high pressure liquid chromatography and MALDI-time-of-flight mass spectroscopy. We obtained similar results (see Fig. 4; Table I) with bovine brain (Sigma-Aldrich and Calbiochem) and human brain (Calbiochem) calmodulin, although different samples varied approximately twofold in their affinity for a given peptide.
Figure 4.

Ca/CaM binds with high affinity to ErbB1(645–660) and prevents its association with lipid vesicles. The percent bound peptide, present only at a trace concentration (∼5 nM), is plotted as a function of the concentration of CaM in the presence (open circles, [Ca2+]free ∼20 μM) or absence (filled circles) of Ca2+. The total lipid concentration is 2 × 10−5 M and the solutions contain 100 mM KCl, 1 mM MOPS, pH 7.0, 100 μM EGTA, ±120 μM CaCl2. The solid curve illustrates the prediction of Eq. 2, taking the association constant of the peptide with the membrane, K = 106 M−1 (Fig. 3), and deducing the association constant of the peptide with Ca/CaM, KCaM = 108 M−1 from the fit of Eq. 2 to the data. The fraction of radioactively labeled peptide bound to the 100 nm 2:1 PC/PS large unilamellar vesicles (LUVs) was determined by a centrifugation technique.
TABLE I.
Affinity of Peptides for Membranes and Ca/CaM
| Peptide | Molar Partition Coefficient (K) | Ca/CaM Association Constant (KCaM) |
|---|---|---|
| ErbB1(645–660) | 1 × 106 M−1 | 1 × 108 M−1 |
| ErbB2(676–692) | 5 × 105 M−1 | 6 × 107 M−1 |
| ErbB3(667–683) | 2 × 104 M−1 | 3 × 106 M−1 |
| ErbB4(676–692) | 2 × 105 M−1 | 2 × 107 M−1 |
The centrifugation assay was used to measure the binding of ErbB peptides to 2:1 PC/PS LUVs (numbers correspond to position in sequence of native protein; see Fig. S6, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1, for sequences). The values of K and KCaM were determined from Eqs. 1 and 2, respectively. The solutions contained 100 mM KCl, 1 mM MOPS, pH 7; solutions used in Ca/CaM binding experiments also contained 100 μM EGTA + 120 μM Ca2+.
We formed multilamellar vesicles (MLVs) for zeta potential measurements, 100-nm-diameter large unilamellar vesicles (LUVs) for FRET, FCS, and stop flow fluorescence measurements, and 100-nm sucrose-loaded LUVs for centrifugation binding measurements, as described previously (Wang et al., 2002).
Methods
Measurements of Peptide Binding to LUVs.
We measured the binding of [3H]NEM-labeled peptides to sucrose-loaded PC/PS LUVs using a centrifugation technique described previously (Wang et al., 2002; Gambhir et al., 2004). In brief, we mixed sucrose-loaded LUVs and a trace concentration (∼5 nM) of [3H]NEM-labeled peptide, and then centrifuged the mixture at 100,000 g for 1 h. We calculated the percentage of peptide bound from counts of the radioactive peptide in the supernatant and in the pellet.
Zeta Potential of MLVs.
We measured the electrophoretic mobility (velocity/field) of single MLVs and calculated the zeta potential, the electrostatic potential at the hydrodynamic plane of shear (∼0.2 nm from the surface), using the Helmholtz-Smoluchowski equation (Wang et al., 2002, and references therein).
Surface Pressure Measurements.
We deposited a stock lipid–chloroform solution onto the surface of a 15-ml aqueous solution in a 5-cm-diameter Teflon trough with a magnetic stirrer at the bottom. Once the chloroform had evaporated, we measured the surface pressure of the monolayer using a square piece of filter paper and a balance from Nima Technology Ltd. We then added the peptide to the subphase and measured the change in surface pressure as described previously (Wang et al., 2002).
FRET.
We monitored FRET between a Bodipy TMR label on PIP2 and a Texas red label attached to membrane-bound ErbB1(645–660) as described previously (Gambhir et al., 2004); the membrane contained 69.7% PC, 30% PS, and 0.3% PIP2.
Circular Dichroism (CD) Spectroscopy.
We obtained the CD spectrum of ErbB1(645–660) bound to isotropic bicelles on an Olis DSM CD spectrometer (Olis Instruments) with a 0.2-mm path-length cell. The bicelles were composed of a mixture of DMPC, DMPG, and DHPC in a 10:3:13 molar ratio. The buffered bicelle solution (20 mM sodium phosphate, pH 7.0) was 10% (wt/vol) lipid and the peptide:lipid ratio was 1:100. Measurements of a bicelle solution without peptide prepared in parallel were subtracted as background. The corrected CD spectrum exhibits the strong negative ellipticity at 200 nm characteristic of random-coil structures.
Fluorescence Correlation Spectroscopy (FCS).
We used a Carl Zeiss MicroImaging, Inc. Confocor II microscope to monitor the binding of Alexa488-labeled ErbB1(645–660) to 2:1 PC/PS 100-nm LUVs and to study the ability of Ca/CaM to remove the peptide from the membrane. The experimental techniques were similar to those described in detail in Rusu et al. (2004). In brief, the correlation times of the peptide bound to 100-nm LUVs and to Ca/CaM are 1,700 μs and 100 μs, respectively; hence we could distinguish the two correlation times easily. We determined the affinity of Ca/CaM for the peptide by plotting the fraction of membrane-bound peptide against the concentration of Ca/CaM in the solution, as shown for experiments using the centrifugation technique (see Fig. 4).
Stop Flow Kinetics.
We made fluorescence stop flow kinetic measurements to determine the rate at which Ca/CaM removes membrane-bound acrylodan-labeled ErbB1(645–660) from PC/PS vesicles; adding Ca/CaM increased the fluorescence approximately fourfold as the peptide moved from vesicle to Ca/CaM. Specifically, one solution contained 200 or 400 nM acrylodan-labeled ErbB1(645–660) bound to 100-nm 85:14:1 PC/PS/NBD-PS vesicles (100 μM accessible lipid; the 1% NBD-PS in these membranes quenches the acrylodan fluorescence), and the other solution contained 0.5, 1, 2, 4, or 7 μM CaM, and 50 μM CaCl2. Both solutions contained 100 mM KCl buffered to pH 7.0 with 1 mM MOPS. We measured the time constants of the exponential increase in fluorescence, τ, and determined the slope of 1/τ vs [CaM]. This slope is equal to the transfer rate constant. The two peptide concentrations produced identical time constants, as expected. We repeated the stop flow measurements with vesicles containing 10, 12, and 18% PS. The results and experimental details are similar to those shown in Fig. 7 of Arbuzova et al. (1997) for a different basic/hydrophobic acrylodan-labeled peptide.
Calculation of Electrostatic Potentials.
We built atomic models of the 2:1 PC/PS bilayer (Wang et al., 2002) and ErbB1(645–660) in an extended conformation using the Insight Biopolymer and Discover modules (Accelrys); the atomic radii and partial charges assigned to the peptide were taken from the CHARMM forcefield. We solved the nonlinear Poisson-Boltzmann equation for atomic models of these systems in 100 mM KCl. The resulting potential maps, as well as the atomic coordinates for the peptide/membrane/CaM models, were displayed using GRASP.
Online Supplemental Material
The supplemental material for this paper comprises eight figures (available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1). Fig. S1 shows how the binding of the ErbB1 JM peptide depends on the mole fraction of acidic lipid in the membrane. Fig. S2 shows FRET between the ErbB1 JM peptide and PIP2. Fig. S3 shows the effect of the ErbB1 JM peptide on PLC-catalyzed PIP2 hydrolysis. Fig. S4 shows an atomic model of membrane, adsorbed ErbB1 JM peptide and calcium/calmodulin, and illustrates the predicted electrostatic potentials of the membrane and molecules. Fig. S5 shows the electrostatic potential adjacent to a complex of calmodulin and a peptide similar to the ErbB1 JM domain. Fig. S6 shows ErbB family members share a common basic/hydrophobic JM region. Fig. S7 shows the kinase domains of ErbB family members have a positively charged face in common. Fig. S8 shows the patterns of ErbB1 phosphorylation predicted by the model under different conditions.
RESULTS
ErbB1(645–660) Binds Strongly to Phospholipid Vesicles through Nonspecific Electrostatic Interactions
We first tested the postulate that the ErbB1 reversible membrane anchor region binds to the plasma membrane by determining if a peptide corresponding to this region, ErbB1(645–660), binds to phospholipid vesicles. The data in Fig. 3 show this is indeed the case: ErbB1(645–660) binds strongly to vesicles containing physiological (15–30%) mole fractions of the monovalent acidic lipid PS. We describe the binding using Eq. 1 (Arbuzova et al., 2000):
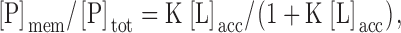 |
(1) |
where [P]mem/[P]tot is fraction of peptide bound, [L]acc is the accessible lipid concentration (1/2 the total lipid concentration because we add the peptide to preformed vesicles), and K is the molar partition coefficient. Three characteristics of the binding indicate it is due mainly to nonspecific electrostatic interactions: the binding energy is independent of the chemical nature of the monovalent acidic lipid, decreases markedly if the salt concentration increases, and increases linearly (K increases exponentially) with the mole fraction of acidic lipid; see Fig S1 (available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1).
Figure 3.

Binding of ErbB1(645–660) to phospholipid vesicles. Vesicles were formed from a mixture of the zwitterionic lipid PC and the acidic lipid PS. 100 nm LUVS were formed from 2:1 PC/PS (filled circles) or 5:1 PC/PS (open circles); the external aqueous solutions contained 100 mM KCl, 1 mM MOPS, pH 7.0. The binding measurements were made with a low (∼4 nM) concentration of radiolabeled (NEM) peptide using a centrifugation assay. The solid lines through the points illustrate the fit of Eq. 1 to the data. The molar partition coefficient, K (or apparent association constant of the peptide with a lipid), is the reciprocal of the lipid concentration that binds 50% of the peptide; the dotted line indicates K ≅ 2 × 104 M−1 for 5:1 PC/PS vesicles. The bars through the data points illustrate the SD of n = 4 independent experiments.
Is the binding strong enough to anchor this region of ErbB1 to a plasma membrane, which typically contains 15–30% acidic phospholipid (Holthuis and Levine, 2005)? Our measurements (see online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1) show that the peptide binds strongly to membranes containing either monovalent PS (104 < K < 106 M−1 for PC/PS vesicles with 15–30% PS) or multivalent PIP2 (K = 6 × 104 M−1 for 99:1 PC/PIP2 vesicles). Moreover, the adjacent transmembrane helix of ErbB molecules tethers the basic region of the JM domain to the bilayer (Fig. 1), so it experiences a high effective lipid concentration, [lipid]eff > 10−2 M. Extrapolating our peptide binding results to the tethered 645–660 region of ErbB1 suggests this region is bound to the plasma membrane >99% of the time (ratio of bound/free = K[lipid]eff > 100). We used a peptide comprising both the TM and the basic–hydrophobic cluster of the JM domains, ErbB1(622–660), which has four additional amino acids (RRRS) added at the NH2 terminus, to obtain more direct evidence to support this conclusion. Reconstitution of ErbB1(622–660) with a COOH-terminal Texas red label into vesicles containing ∼1% PIP2 with a Bodipy-TMR label produces strong FRET between the fluorophores (Sato, T., personal communication). The simplest interpretation of this result, together with complementary NMR data showing the TM helix breaks close to the membrane interface, is that residues 645–660 in the ErbB1(622–660) peptide are bound to the bilayer.
ErbB1(645–660) Laterally Sequesters PIP2
When the 645–660 region of ErbB1 is bound to the bilayer component of the plasma membrane, it produces a local positive electrostatic potential that will attract multivalent acidic lipids, even when monovalent acidic lipids are present in excess (see Fig. S4, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1). FRET and PLC activity measurements demonstrate directly that membrane-bound ErbB1(645–660) laterally sequesters PIP2, even in membranes comprising physiological levels of both PS and PIP2 (Figs. S2 and S3).
Biological Experiments Consistent with our Autoinhibition Hypothesis
A quantitative comparison of three results from earlier experiments on cell membranes and data from peptide/phospholipid vesicle studies indicate that electrostatic interactions can explain ErbB autoinhibition. First, hyperosmotic shock stimulates tyrosine phosphorylation of ErbB1 and ErbB2 in the absence of ligand (King et al., 1989; Rodriguez et al., 2002). The model predicts this effect because high salt should reduce the electrostatic attraction of the JM + PTK domains for the membrane; binding measurements show that increasing the salt concentration threefold reduces ErbB1 (645–660) binding ∼500-fold (online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1). Second, 1 mM Mn2+ or 10 mM Mg2+ activates ErbB1 in a broken cell preparation (Carpenter et al., 1979). Our model also predicts this effect because these divalent cations bind to membranes containing acidic lipids, reducing the magnitude of the negative electrostatic potential; binding measurements show adding 1 mM Mn2+ or 10 mM Mg2+ reduces binding of ErbB1(645–660) to 2:1 PC/PS vesicles by ∼100-fold or ∼1,000-fold, respectively. Third, 2–5 μM sphingosine, an amphipathic, membrane-permeable weak base, activates ErbB1 in WI-38 fibroblasts, provided the receptor is in an intact membrane (Davis et al., 1988); in contrast, EGF can activate ErbB1 both in membranes and in solubilized form. The model predicts that amphipathic weak bases should reduce the negative charge on the inner leaflet of the bilayer and thus its electrostatic attraction for the basic JM region; our data demonstrate that 2 μM sphingosine reverses the charge (sign of the zeta potential, direction of the electrophoretic movement) of a 2:1 PC/PS vesicle and causes 75% of the ErbB1(645–660) peptide to desorb from PC/PS vesicles (online supplemental material).
Ca/CaM Removes ErbB JM Peptides from Membranes Rapidly
The mechanism shown in Fig. 1 B, i.e., Ca/CaM binds to the membrane anchor region and removes it and the PTK domain to facilitate EGF-mediated activation, is admittedly speculative, but peptide experiments provide evidence that it is feasible. We first tested whether Ca/CaM competes with phospholipid membranes for binding of ErbB1(645–660); Fig. 4 shows peptide binding to 2:1 PC/PS vesicles in the presence of increasing [Ca/CaM]. In the absence of Ca/CaM, 90% of the peptide binds to the vesicles, as expected from both theoretical calculations (unpublished data; see Murray et al., 2002, for methods) and the binding results in Fig. 3. The open circles in Fig. 4 illustrate the effect of increasing [Ca/CaM]: adding 0.1 μM Ca/CaM reduces binding by ∼50% (total [CaM] ∼ 10–100 μM in a mammalian cell). Adding CaM in the absence of free Ca2+ does not affect ErbB1(645–660) binding to the vesicles (filled circles). These data indicate that Ca/CaM binds to ErbB1(645–660) with high affinity (Kd = 10 nM), preventing the peptide from binding to a phospholipid vesicle. We used a modified version of Eq. 1 to describe the effect of Ca/CaM on ErbB1(645–660) membrane binding, incorporating the assumption that Ca/CaM and the membrane compete for the peptide:
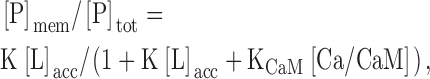 |
(2) |
where K is the molar partition coefficient of the peptide onto the 2:1 PC/PS vesicles (106 M−1 from Fig. 3) and KCaM is the association constant of the peptide with Ca/CaM (KCaM = 108 M−1 from the fit of Eq. 2 to data in Fig. 4).
We repeated the measurements shown in Fig. 4 using 3:1 rather than 2:1 PC/PS vesicles and obtained a similar value for KCaM. We also used an independent method, fluorescence correlation spectroscopy (FCS), to validate the centrifugation technique used to obtain the measurements shown in Fig. 4. FCS measures the correlation time (inversely proportional to diffusion constant) of a fluorescent label, in this case Alexa488 attached to ErbB1(645–660). The labeled peptide diffuses slowly when bound to the large (100 nm) vesicles and rapidly when bound to the smaller Ca/CaM; thus we can determine the fraction of membrane-bound peptide as a function of the concentration of Ca/CaM. Both centrifugation and FCS measurements with Alexa488-labeled ErbB1(645–660) indicate KCaM = 107 M−1 (U. Golebiewska, personal communication; unpublished data). Thus, we conclude that the FCS measurements validate the centrifugation technique. As expected from electrostatics, the negatively charged Alexa probe decreases the affinity of the peptide for Ca/CaM (and vesicles) ∼10-fold.
Phosphorylation of Thr654 also decreases the affinity of ErbB1(645–660) for Ca/CaM. Experiments similar to those shown in Fig. 4, but conducted with an ErbB1(645–660) peptide with a phosphorylated Thr654, show that phosphorylation reduces KCaM 20-fold: removal of 50% of the phosphopeptide from the vesicles requires 20-fold more Ca/CaM than shown in Fig. 4 under similar conditions, i.e., when 90% of the phosphopeptide is bound initially. Phosphorylation also decreases the membrane binding of the peptide ∼10-fold. These results agree well with previous measurements showing that phosphorylation “drastically hampers” (Martin-Nieto and Villalobo, 1998) or “totally inhibits” (Aifa et al., 2002) the ability of Ca/CaM to bind to this region of ErbB1. The model shown in Fig. 1 thus predicts that phosphorylation of Thr654 by PKC should significantly attenuate at least the early phase of ErbB1 autophosphorylation, a prediction consistent with experiment (Couchet et al., 1984; Hunter et al., 1984; Countaway et al., 1990; Welsh et al., 1991). However, phosphorylation of Thr654 also produces a less robust inhibition of ErbB1 autophosphorylation in model systems, such as A431 cell membranes (e.g., Couchet et al., 1984), which must be due to some other phenomenon.
How rapidly can Ca/CaM remove the ErbB1 645–660 region from a plasma membrane? We approached this question by making fluorescence stop flow measurements to determine how rapidly Ca/CaM can remove acrylodan-labeled ErbB1(645–660) from model membranes that have a physiologically relevant fraction of acidic lipid, i.e., 85:15 PC/PS vesicles. The vesicles had 1% NBD-PS to quench the fluorescence. If τ is the measured time constant for moving a peptide from a vesicle to Ca/CaM, 1/τ increases linearly from 3 s−1 to 30 s−1 as [Ca/CaM] increases to 5 μM. Put another way, adding 5 μM Ca/CaM reduces the lifetime of the peptide on the vesicles 10-fold, from ∼0.3 to 0.03 s. The transfer rate constant, defined as the slope of the (1/τ) vs. [CaM] data, is 5 × 106 M−1s−1 for 85:15 PC/PS vesicles and 5 × 107 M−1s−1 for 90:10 PC/PS vesicles, a value close to the diffusion limited rate (∼108 M−1s−1) at which Ca/CaM combines with other proteins in solution (online supplemental material, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1).
The kinetics results are perhaps surprising: the simplest interpretation of our equilibrium measurements (Fig. 4) is that Ca/CaM acts as a passive peptide buffer. That is, adding it to a solution containing peptides bound to vesicles should merely decrease the equilibrium concentration of free peptide in the bulk aqueous phase, allowing peptide to desorb from the vesicles at its spontaneous rate until a new equilibrium is attained. The stop flow measurements, however, reveal that Ca/CaM increases the rate at which ErbB1(645–660) desorbs from the vesicles, presumably by ripping the peptide directly from the surface; we have discussed this mechanism elsewhere (Arbuzova et al., 1997, 1998).
Figs. S4 and S5 (available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1) show, respectively, calculations of the electrostatic potential for an atomic model of Ca/CaM approaching a membrane-bound ErbB1(645–660) and calculations of the electrostatic potential adjacent to Ca/CaM bound to a basic peptide. The atomic models illustrate how electrostatic attraction may guide Ca/CaM (net charge z = −16) to the membrane-associated JM region (z = +8), how electrostatic interactions may help rip the peptide from the surface (Arbuzova et al., 1998), and how the Ca/CaM-JM region complex (z = −8) may then be repelled from the membrane surface.
We performed binding measurements similar to those shown in Figs. 3 and 4 with peptides corresponding to the reversible membrane anchor region of other members of the ErbB family. The basic/hydrophobic nature of this region is moderately conserved (Fig. S6, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1), and Table I shows they all bind to both membranes and Ca/CaM, suggesting that the electrostatic engine model may apply to activation of all four molecules. Moreover, the PTK core regions of all ErbB family members share a positively charged face (Fig. S7).
CaM Inhibitors Reduce the Initial Rate of EGF-mediated Autophosphorylation in Cos1 Cells
A key prediction of the mechanism shown in Fig. 1 B is that exposing cells to membrane-permeable CaM inhibitors such as W-7 should inhibit the initial rate of EGF-mediated ErbB1 autophosphorylation. According to our model, CaM inhibitors should affect autophosphorylation only for a short time (<15 min) after EGF stimulation because the concomitant increase in cytoplasmic Ca2+ to values that activate CaM significantly is only transient. For example, Hughes et al. (1991) reported the intracellular [Ca2+] in A431 cells increases to 600 nM in 5 min, but falls to 150 nM by 15 min. Nojiri and Hoek (2000) show the intracellular [Ca2+] in hepatocytes increases to 450 nM in 1–2 min, and then falls to 150 nM in the next 2–3 min. Fig. 5 shows treating Cos1 cells with W-7 produces dose-dependent inhibition of ErbB1 tyrosine phosphorylation measured 10 min after addition of EGF; the concentration range used, 20–50 μM, was selected on the basis of the inhibitor's affinity for Ca/CaM (Osawa et al., 1998). EGF-stimulated ErbB1 autophosphorylation is maximal ∼10 min after adding EGF to Cos1 cells under our conditions (unpublished data). Recent detailed studies in two other cell types showed that the CaM inhibitors W-7, W-12, and W-13 inhibit the initial peak of ErbB1 autophosphorylation, but not the steady-state value observed for times >20 min (Li et al., 2004a), as expected from our model. Li et al. (2004a) report an important control experiment: W-7, W-12, and W-13 do not inhibit the tyrosine kinase activity of a purified ErbB1 preparation.
Figure 5.

The calmodulin inhibitor W7 inhibits the EGF-mediated autophosphorylation of the ErbB1 receptor. Cos1 cells were treated with 20–50 μM W-7 for 30 min, and then exposed to 100 ng/ml (∼20 nM) EGF for 10 min. The cells were lysed and treated with rabbit anti-ErbB1 to immune precipitate the receptor. Western blots were performed on the immunoprecipitates using the murine antiphosphotyrosine monoclonal antibody 4G10, (top panel). Reprobing of the same blot with a rat anti-ErbB1 monoclonal antibody (bottom panel) demonstrated that W-7 treatment did not affect receptor levels during the time frame of the experiment.
DISCUSSION
Membrane Binding of ErbB Intracellular Regions and Postulated Role of Ca/CaM
Our peptide experiments and/or theoretical calculations support the hypothesis that both the JM region and PTK domain of ErbB family members bind electrostatically to the inner leaflet of the plasma membrane. If our postulate that this binding produces autoinhibition is correct, factors that reduce these electrostatic interactions should stimulate cellular ErbB autophosphorylation in the absence of ligand. Specifically, we postulate that autophosphorylation depends on both the rate at which ErbB monomers collide in the plasma membrane and the probability the JM + PTK domain is in a membrane-dissociated active conformation. Our model membrane experiments allowed us to measure quantitatively the levels of salt, divalent cations, or the amphipathic weak base sphingosine required to disengage a peptide corresponding to the membrane anchor region from a lipid bilayer; when we compared the values to those reported for ligand-independent activation of ErbB1 in cells or broken membrane preparations (Carpenter et al., 1979; Davis et al., 1988; King et al., 1989), there was quantitative agreement in each case. Thus the mechanism illustrated in Fig. 1 A provides a plausible and experimentally testable explanation for autoinhibition, but it does not address the specific mechanism by which membrane binding inhibits tyrosine kinase activity. Possible mechanisms could include restricted rotation of the PTK domain that prevents interaction of the catalytic site and substrate, or structural effects on the activation loop within the PTK domain (Stamos et al., 2002; Wood et al., 2004); several recent reviews discuss autoinhibitory mechanisms for receptor tyrosine kinases in some detail (Jorissen et al., 2003; Schlessinger, 2003; Hubbard, 2004).
Our data show that Ca/CaM binds strongly to peptides corresponding to the membrane anchor region of ErbB1 (Fig. 4) and the corresponding JM regions in other ErbB family members (Table I). The results agree qualitatively with previous reports that Ca/CaM binds strongly to these regions of ErbB1 (Martin-Nieto and Villalobo, 1998; Li et al., 2004a) and ErbB2 (Li et al., 2004b). We note that Martin-Nieto and Villalobo (1998) reported KCaM = 3 × 106 M−1, but their Ca/CaM binding measurements employed 0.3 × 10−6 M of a GST-ErbB1(645–660) construct; thus we interpret their measurements to mean KCaM > 3 × 106 M−1, which agrees with our estimate. These data suggest that the activation mechanism illustrated in Fig. 1 B is feasible and could enhance the activation produced by ligand-induced dimerization, which presumably acts through a local concentration effect (Schlessinger, 2000).
The Electrostatic Engine Mechanism in ErbB Activation
Our model suggests the following process for EGF-mediated trans autophosphorylation. When EGF-stimulated dimerization occurs, the level of intracellular [Ca2+], and thus Ca/CaM, is initially low. The ErbB JM + PTK regions, however, move off the membrane spontaneously, albeit at a low rate (e.g., stop flow measurements demonstrate acrylodan-labeled ErbB1(645–660) moves off a 85:15 PC/PS vesicle at a rate of 3 s−1). The small fraction of receptors with dissociated JM + PTK domains will produce a low level of trans autophosphorylation even in the absence of Ca/CaM. When PLC-γ binds to a phosphorylated ErbB and hydrolyzes its substrate PIP2, it produces IP3, which in turn releases Ca2+ from internal stores, increasing the concentration of Ca/CaM (for review see Jorissen et al., 2003). Our measurements show that Ca/CaM increases both the rate at which peptides corresponding to the ErbB1 membrane anchor region leave the membrane and the fraction of membrane-dissociated peptide (Fig. 4). As the JM and kinase domains of an ErbB move off the membrane, the latter becomes catalytically active in our model, as illustrated for one member of the ErbB1 dimer in Fig. 1 B. Thus we postulate that Ca/CaM drives a positive feedback mechanism that produces maximal activation of ErbB. The mechanism functions only when the local [Ca2+] is sufficiently elevated to provide a source of Ca/CaM; intracellular [Ca2+] measurements indicate this occurs only for times <15 min after exposure to EGF (Hughes et al., 1991; Nojiri and Hoek, 2000). This electrostatic engine model predicts that CaM inhibitors (or agents that increase the intracellular Ca2+ buffering capacity) will diminish the transient increase in trans autophosphorylation observed within 15 min of EGF stimulation, but have little effect on the steady-state level of activity measured at later times. CaM inhibitors do indeed inhibit peak level of EGF-mediated ErbB1 autophosphorylation in Cos1 (Fig. 5) and two other cell types (Li et al., 2004a), but have no effect on the steady-state level of autophosphorylation (Li et al., 2004a). Agents that deplete intracellular stores of Ca2+, such as thapsigargin, should and do inhibit receptor autophosphorylation in A431 cells measured 5 min after addition of EGF (Friedman et al., 1989).
Less direct evidence in support of this hypothesis comes from studies of Ca2+-induced transactivation of ErbB1 (i.e., activation that occurs without addition of a ligand that binds directly to ErbB1). Transactivation can occur in response to activation of Gq-coupled receptors, opening of ion channels selective for Ca2+, or addition of Ca2+ ionophores (for review see Zwick et al., 1999). Much recent work on the transactivation of ErbB1 has focused on the unique triple-membrane-passing signal (Prenzel et al., 1999; for reviews see Gschwind et al., 2001; Blobel, 2005), but our model provides a clue as to how an increase in [Ca2+] and Ca/CaM could initiate this interesting phenomenon. If Ca2+ helps initiate transactivation by the mechanism shown in Fig. 1 B, both CaM inhibitors and Ca2+ buffers should inhibit transactivation. Murasawa et al. (1998) reported both the CaM inhibitor W-7 and the Ca2+ buffer BAPTA-AM inhibit angiotensin II–stimulated ErbB1 transactivation in cardiac fibroblasts.
Rigorous testing of the model will require extensive molecular and cell biological experiments as well as biophysical measurements on larger peptides corresponding to the ErbB TM + JM domains reconstituted into vesicles, which are in progress. If additional work supports our postulate that Ca/CaM may act in concert with dimerization to stimulate ErbB1 activation, the model shown in Fig. 1 can probably be extrapolated to the ErbB2 and ErbB4 (Carpenter, 2003) family members: peptides corresponding to their JM regions also bind with high affinity to both membranes and Ca/CaM (Table I).
ErbB as a Scaffolding Protein
Our model also suggests a new and potentially important function for ErbB in signal transduction. Binding of the reversible membrane anchor region to the negatively charged membrane should produce a local positive potential (see Fig. S4, available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1) that acts as a basin of attraction for multivalent acidic lipids such as PIP2. FRET and PLC hydrolysis experiments show that ErbB1(645–660) laterally sequesters PIP2, even when the membrane includes a physiologically relevant 100-fold excess of monovalent acidic lipids (Figs. S2 and S3). PIP2 is the substrate of PLC-γ and PI3 kinase, enzymes that bind to the phosphorylated COOH-terminal regions of ErbB family members (Schlessinger, 2000). Thus a corollary of our hypothesis is that ErbB family members act as scaffolding proteins (Wong and Scott, 2004), binding both enzymes and their substrate. PLC cannot hydrolyze PIP2 sequestered by peptides corresponding to the ErbB JM basic cluster, however (Fig. S3); PIP2 must first be released from the basic cluster.
How rapidly can Ca/CaM bind to the JM region and release the electrostatically sequestered PIP2? Our stop flow experiments reveal that 2 μM Ca/CaM can remove acrylodan-labeled ErbB1(645–660) peptides from PC/PS vesicles at rates of 10 and 100 s−1 for vesicles containing 15 and 10% PS, respectively. It is difficult to extrapolate these results using a model system to a living cell for several reasons. For example, ErbB1 may be in noncaveolar cholesterol- and PIP2-enriched “rafts” that have a different lipid composition than the bulk plasma membrane (e.g., Chen and Resh, 2002; Roepstorff et al., 2002; Westhover et al., 2003; Simons and Vaz, 2004, and references therein). We can state that if the Ca/CaM level rises to ∼1 μM in a cell, the maximum (diffusion limited) rate at which Ca/CaM could bind to the 645–660 JM region of ErbB1 and rip it off the plasma membrane is ∼100 s−1.
How long will the Ca/CaM remain bound to the JM region? The lifetime of Ca/CaM bound to a solubilized ErbB1 molecule is probably ∼1 s (KCaM/kon = 108 M−1/108 M−1s−1), but theoretical considerations suggest the lifetime of Ca/CaM bound to ErbB1 in a membrane will decrease significantly (possibly to 0.01 s) as the mole fraction of acidic lipid in the membrane increases. This is because the acidic lipids repel the negatively charged Ca/CaM bound to the JM region.
Thus the electrostatic engine shown in Fig. 1 (A and B) could cycle 10–100 times a second when the [Ca/CaM] increases to ∼1 μM. Even if the JM region remains bound to the bilayer for only ∼0.01 s, this provides more than sufficient time, t, for PIP2 to equilibrate with the basic cluster through diffusion (from the Einstein relation, t = x2/4D, where x is the distance PIP2 must diffuse in the plasma membrane and D is its diffusion constant). FCS measurements show that the diffusion constant of Bodipy-PIP2 in a fluid phase PC phospholipid membrane has the expected value of D = 3 × 10−8 cm2s−1 (Golebiewska, U., personal communication). In a plasma membrane, D could be 10-fold lower because cholesterol increases the viscosity, and as much as 90% of the PIP2 could be sequestered such that x = distance between PIP2 free to diffuse = 30 nm; even under these conditions the diffusion time is only ∼0.001 s. The maximal rate at which PLCs can hydrolyze PIP2 is ∼102 s−1, so the JM region of ErbB can potentially cycle on and off the membrane at a frequency that could facilitate the hydrolysis of PIP2 by an adjacent PLCγ.
One caveat concerning this electrostatic engine mechanism: the free [Ca/CaM] in cytoplasm may be significantly lower than the total cellular [CaM] of 10–100 μM. Recent measurements suggest that much of the Ca/CaM in cells may be bound to target proteins (Persechini and Stemmer, 2002; Black et al., 2004; Kim et al., 2004; Rakhilin et al., 2004).
Predictions of the Model
Fig. S8 (available at http://www.jgp.org/cgi/content/full/jgp.200509274/DC1) illustrates how the model shown in Fig. 1 can be used to predict the time course of ErbB1 trans autophosphorylation after stimulation by EGF. For simplicity, we assume that only three factors affect phosphorylation: ligand-induced dimerization, which increases phosphorylation by a local concentration effect; phosphatases, which remove phosphates from ErbB1; and the calmodulin-dependent positive feedback mechanism shown in Fig. 1, which operates only when intracellular [Ca2+] is elevated. The plots show the predicted percent trans autophosphorylation as a function of time after addition of EGF in three different cases: permeabilized cells lacking both CaM and phosphatases, cells exposed to CaM inhibitors, and normal cells. Recent experimental results appear to agree well with the predictions. We stress, however, that the calculations in Fig. S8 represent a highly oversimplified scheme; for example, we make no attempt to incorporate the well documented endocytosis of activated ErbB1 and PLC-γ1 (Matsuda et al., 2001; Wang et al., 2001; Wang and Wang, 2003). Endocytosis has been considered quantitatively in models for ErbB family activation by Lauffenburger and others (e.g., Wiley et al., 2003; Hendriks et al., 2005). We also acknowledge that other, more complex, quantitative models for short term signaling by ErbB1 (Kholodenko et al., 1999; Moehren et al., 2002) can account for the maximum in ErbB1 autophosphorylation by a different mechanism than the one we invoke in Fig. 1. The advantages of formulating quantitative models of signal transduction phenomena are discussed in a recent commentary entitled “Why biophysicists make models” (Shapiro, 2004) and in Papin et al. (2005). In our view, the main advantage of these models (e.g., our Fig. 1; Kholodenko et al., 1999; Moehren et al., 2002; Wiley et al., 2003; Landau et al., 2004; Hendriks et al., 2005), is that they make quantitative predictions and can thus be easily falsified or modified by future experiments.
Supplemental Material
Acknowledgments
We thank B. Kholodenko, T. Miller, J. Pessin, and J. Staros for helpful discussions and G. Hangyás-Mihályné, I. Zaitseva, and I. Ischenko for excellent technical assistance.
This work was supported by National Institutes of Health grants GM24971 (to S. McLaughlin), GM69651 (to S.O. Smith), CA28146 (to M. Hayman), National Science Foundation/Molecular and Cellular Biosciences 0212362 (to D. Murray), and a Carol M. Baldwin Foundation Breast Cancer Research Award (to S. McLaughlin).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: Ca/CaM, calcium/calmodulin; CD, circular dichroism; EGFR, epidermal growth factor receptor; FCS, fluorescence correlation spectroscopy; FRET, fluorescence resonance energy transfer; IP3, inositiol 1,4,5-trisphosphate; JM, juxtamembrane; LUV, large unilamellar vesicle; MLV, multilamellar vesicle; NEM, [ethyl-1,2-3H]N-ethylmaleimide; PC, phosphatidylcholine; PIP2, phosphatidylinositol 4,5-bisphosphate; PS, phosphatidylserine; PTK, protein tyrosine kinase; TM, transmembrane.
References
- Aifa, S., K. Johansen, U.K. Nilsson, B. Liedberg, I. Lundstrom, and S.P.S. Svensson. 2002. Interactions between the juxtamembrane domain of the EGFR and calmodulin measured by surface plasmon resonance. Cell. Signal. 14:1005–1013. [DOI] [PubMed] [Google Scholar]
- Aifa, S., J. Aydin, G. Nordvall, I. Lundstrom, S.P.S. Svensson, and O. Hermanson. 2005. A basic peptide within the juxtamembrane region is required for EGF receptor dimerization. Exp. Cell Res. 302:108–114. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., J. Wang, D. Murray, J. Jacob, D.S. Cafiso, and S. McLaughlin. 1997. Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J. Biol. Chem. 272:27167–27177. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., D. Murray, and S. McLaughlin. 1998. MARCKS, membranes and calmodulin: kinetics of their interaction. Biochim. Biophys. Acta. 1376:369–379. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., L. Wang, J. Wang, G. Hangyàs-Mihàlynè, D. Murray, B. Honig, and S. McLaughlin. 2000. Membrane binding of peptides containing both basic and aromatic residues. Biochemistry. 39:10330–10339. [DOI] [PubMed] [Google Scholar]
- Bezzerides, V.J., I.S. Ramsey, S. Kotecha, A. Greka, and D.E. Clapham. 2004. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 6:709–720. [DOI] [PubMed] [Google Scholar]
- Black, D.J., Q.-K. Tran, and A. Persechini. 2004. Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+. Cell Calcium. 35:415–425. [DOI] [PubMed] [Google Scholar]
- Blobel, C.P. 2005. Adams: key components in EGFR signaling and development. Nat. Rev. Mol. Cell Biol. 6:32–43. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signalling. Nature. 411:355–365. [DOI] [PubMed] [Google Scholar]
- Burgess, A.W., H.-S. Cho, C. Eigenbrot, K.M. Ferguson, T.P.J. Garrett, D.J. Leahy, M.A. Lemmon, M.X. Sliwkowski, C.W. Ward, and S. Yokoyama. 2003. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 12:541–552. [DOI] [PubMed] [Google Scholar]
- Carpenter, G. 2003. ErbB-4: mechanism of action and biology. Exp. Cell Res. 284:66–77. [DOI] [PubMed] [Google Scholar]
- Carpenter, G., L. King Jr., and S. Cohen. 1979. Rapid enhancement of protein phosphorylation in A-431 cell membrane preparations by epidermal growth factor. J. Biol. Chem. 254:4884–4891. [PubMed] [Google Scholar]
- Chen, X., and M.D. Resh. 2002. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J. Biol. Chem. 277:49631–49637. [DOI] [PubMed] [Google Scholar]
- Cheyette, T.E., and D.J. Gross. 1991. Epidermal growth factor-stimulated calcium ion transients in individual A431 cells: initiation kinetics and ligand concentration dependence. Cell Regul. 2:827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couchet, C., G.N. Gill, J. Meisenhelder, J.A. Cooper, and T. Hunter. 1984. C-kinase phosphorylates the epidermal growth factor receptor and reduces its epidermal growth factor-stimulated tyrosine protein kinase activity. J. Biol. Chem. 259:2553–2558. [PubMed] [Google Scholar]
- Countaway, J.L., P. McQuilkin, N. Girones, and R.J. Davis. 1990. Multisite phosphorylation of the epidermal growth factor receptor. J. Biol. Chem. 265:3407–3416. [PubMed] [Google Scholar]
- Davis, R.J., N. Gironès, and M. Faucher. 1988. Two alternative mechanisms control the interconversion of functional states of the epidermal growth factor receptor. J. Biol. Chem. 263:5373–5379. [PubMed] [Google Scholar]
- Ferguson, K.M. 2004. Active and inactive conformations of the epidermal growth factor receptor. Biochem. Soc. Trans. 32:742–745. [DOI] [PubMed] [Google Scholar]
- Ferguson, K.M., M.B. Berger, J.M. Mendrola, H.S. Cho, D.J. Leahy, and M.A. Lemmon. 2003. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell. 11:507–517. [DOI] [PubMed] [Google Scholar]
- Friedman, B., J. van Amsterdam, H. Fujiki, and M.R. Rosner. 1989. Phosphorylation at threonine-654 is not required for negative regulation of the epidermal growth factor receptor by non-phorbol tumor promoters. Proc. Natl. Acad. Sci. USA. 86:812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir, A., G. Hangyás-Mihályné, I. Zaitseva, D.S. Cafiso, J. Wang, D. Murray, S. Pentyala, S.O. Smith, and S. McLaughlin. 2004. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86:2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh, N., A. Tojo, M. Hino, Y. Yazaki, and M. Shibuya. 1992. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 186:768–774. [DOI] [PubMed] [Google Scholar]
- Gschwind, A., E. Zwick, N. Prenzel, M. Leserer, and A. Ullrich. 2001. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 20:1594–1600. [DOI] [PubMed] [Google Scholar]
- Gschwind, A., O.M. Fischer, and A. Ullrich. 2004. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 4:361–370. [DOI] [PubMed] [Google Scholar]
- Hendriks, B.S., G. Orr, A. Well, H.S. Wiley, and D.A. Lauffenburger. 2005. Parsing ERK activation reveals quantitatively equivalent contributions from epidermal growth factor receptor and HER2 in human mammary epithelial cells. J. Biol. Chem. 280:6157–6169. [DOI] [PubMed] [Google Scholar]
- Holthuis, J.C.M., and T.P. Levine. 2005. Lipid traffic: floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol. 6:209–220. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.R. 2004. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5:464–470. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.R., and J.H. Till. 2000. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 69:373–398. [DOI] [PubMed] [Google Scholar]
- Hughes, A.R., G.S. Bird, J.F. Obie, O. Thastrup, and J.W. Putney Jr. 1991. Role of inositol (1,4,5)trisphosphate in epidermal growth factor-induced Ca2+ signaling in A431 cells. Mol. Pharmacol. 40:254–262. [PubMed] [Google Scholar]
- Hunter, T., N. Ling, and J.A. Cooper. 1984. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature. 311:480–483. [DOI] [PubMed] [Google Scholar]
- Huse, M., and J. Kuriyan. 2002. The conformational plasticity of protein kinases. Cell. 109:275–282. [DOI] [PubMed] [Google Scholar]
- Ichinos, J., M. Murata, T. Yanagida, and Y. Sako. 2004. EGF signaling amplification induced by dynamic clustering of EGFR. Biochem. Biophys. Res. Commun. 324:1143–1149. [DOI] [PubMed] [Google Scholar]
- Jorissen, R.N., F. Walker, N. Pouliot, T.P.J. Garrett, C.W. Ward, and A.W. Burgess. 2003. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 284:31–53. [DOI] [PubMed] [Google Scholar]
- Kholodenko, B.N., O.V. Demin, G. Moehren, and J.B. Hoek. 1999. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 274:30169–30181. [DOI] [PubMed] [Google Scholar]
- Kim, S.A., K.G. Heinze, M.N. Waxham, and P. Schwille. 2004. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. USA. 101:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, C.R., I. Borrello, L. Porter, P. Comoglio, and J. Schlessinger. 1989. Ligand-independent tyrosine phosphorylation of EGF receptor and the erbB-2/neu proto-oncogene product is induced by hyperosmotic shock. Oncogene. 4:13–18. [PubMed] [Google Scholar]
- Klapper, L.N., M.H. Kirschbaum, M. Sela, and Y. Yarden. 2000. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv. Cancer Res. 77:25–79. [PubMed] [Google Scholar]
- Landau, M., S.J. Fleishman, and N. Ben-Tal. 2004. A putative mechanism for downregulation of the catalytic activity of the EGF receptor via direct contact between its kinase and C-terminal domains. Structure (Camb.). 12:2265–2275. [DOI] [PubMed] [Google Scholar]
- Li, H., M.J. Ruano, and A. Villalobo. 2004. a. Endogenous calmodulin interacts with the epidermal growth factor receptor in living cells. FEBS Lett. 559:175–180. [DOI] [PubMed] [Google Scholar]
- Li, H., J. Sanchez-Torres, A. Del Carpio, V. Salas, and A. Villalobo. 2004. b. The ErebB2/Neu/HER2 receptor is a new calmodulin-binding protein. Biochem. J. 381:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., L. Tsiokas, S.C. Sansom, and R. Ma. 2004. c. Epidermal growth factor activates store-operated Ca2+ channels through an inositol 1,4,5-trisphosphate-independent pathway in human glomerular mesangial cells. J. Biol. Chem. 279:4570–4577. [DOI] [PubMed] [Google Scholar]
- Martin-Nieto, J., and A. Villalobo. 1998. The human epidermal growth factor receptor contains a juxtamembrane calmodulin-binding site. Biochemistry. 37:227–236. [DOI] [PubMed] [Google Scholar]
- Matilla, E., T. Pellinen, J. Nevo, K. Vuoriluoto, A. Arjonen, and J. Evaska. 2004. Negative regulation of EGFR signaling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7:78–85. [DOI] [PubMed] [Google Scholar]
- Matsuda, M., H.F. Paterson, R. Rodriguez, A.C. Fensome, M.V. Ellis, K. Swann, and M. Katan. 2001. Real time fluorescence imaging of PLCγ translocation and its interaction with the epidermal growth factor receptor. J. Cell Biol. 153:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin, S. 1989. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 18:113–136. [DOI] [PubMed] [Google Scholar]
- Moehren, G., N. Markevich, O. Demin, A. Kiyatkin, I. Goryanin, J.B. Hoek, and B.N. Kholodenko. 2002. Temperature dependence of the epidermal growth factor receptor signaling network can be accounted for by a kinetic model. Biochemistry. 41:306–320. [DOI] [PubMed] [Google Scholar]
- Murasawa, S., Y. Mori, Y. Nozana, N. Gotoh, M. Shibuya, H. Masaki, K. Maruyama, Y. Tsutsumi, Y. Moriguchi, Y. Shibazaki, et al. 1998. Angiotensin II type 1 receptor-induced extracellular signal-regulated protein kinase activation is mediated by Ca2+/calmodulin-dependent transactivation of epidermal growth factor receptor. Circ. Res. 82:1338–1348. [DOI] [PubMed] [Google Scholar]
- Murray, D., A. Arbuzova, B. Honig, and S. McLaughlin. 2002. The role of electrostatic and nonpolar interactions in the associations of peripheral proteins with membranes. Curr. Top. Membranes. 52:277–307. [Google Scholar]
- Nojiri, S., and J.B. Hoek. 2000. Suppression of epidermal growth factor-induced phospholipase C activation associated with actin rearrangement in rat hepatocytes in primary culture. Hepatology. 32:947–957. [DOI] [PubMed] [Google Scholar]
- Ogiso, H., R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J.H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu, and S. Yokoyama. 2002. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 110:775–787. [DOI] [PubMed] [Google Scholar]
- Osawa, M., M.B. Swindells, J. Tanikawa, T. Tanaka, T. Mase, T. Furuya, and M. Ikura. 1998. Solution structure of calmodulin-W-7 complex: the basis of diversity in molecular recognition. J. Mol. Biol. 276:165–176. [DOI] [PubMed] [Google Scholar]
- Paez, J.G., A.J. Pasi, J.C. Lee, S. Tracy, H. Grenlich, S. Gabriel, P. Herman, F.J. Kaye, N. Lindeman, T.J. Boggon, et al. 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304:1497–1500. [DOI] [PubMed] [Google Scholar]
- Papin, J.A., T. Hunter, B.O. Palsson, and S. Subramaniam. 2005. Reconstruction of cellular signaling networks and analysis of their properties. Nat. Rev. Mol. Cell Biol. 6:99–111. [DOI] [PubMed] [Google Scholar]
- Pandiella, A., L. Beguinot, T.J. Velu, and J. Meldolesi. 1988. Transmembrane signalling at epidermal growth factor receptors overexpressed in NIH 3T3 cells. Phosphoinositide hydrolysis, cytosolic Ca2+ increase and alkalinization correlate with epidermal-growth-factor-induced cell proliferation. Biochem. J. 254:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persechini, A., and P.M. Stemmer. 2002. Calmodulin is a limiting factor in the cell. Trends Cardiovasc. Med. 12:32–37. [DOI] [PubMed] [Google Scholar]
- Prenzel, N., E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, and A. Ullrich. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 402:884–888. [DOI] [PubMed] [Google Scholar]
- Rakhilin, S.V., P.A. Olson, A. Nishi, N.N. Starkova, A.A. Fienberg, A.C. Nairn, D.J. Surmeier, and P. Greengard. 2004. A network of control mediated by regulator of calcium/calmodulin-dependent signaling. Science. 306:698–701. [DOI] [PubMed] [Google Scholar]
- Reynolds, A.R., C. Tischeer, P.J. Verveer, O. Rocks, and P.I.H. Bastiaens. 2003. EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat. Cell Biol. 5:447–453. [DOI] [PubMed] [Google Scholar]
- Rodriguez, I., M. Kaszkin, A. Holloschi, K. Kabsch, M.M. Marques, X. Mao, and A. Alonso. 2002. Hyperosmotic stress induces phosphorylation of cytosolic phospholipase A2 in HaCaT cells by an epidermal growth factor receptor-mediated process. Cell. Signal. 14:839–848. [DOI] [PubMed] [Google Scholar]
- Roepstorff, K., P. Thomsen, K. Samndvig, and B. van Deurs. 2002. Sequestration of epidermal growth factor receptors in non-caveolar lipid rafts inhibits ligand binding. J. Biol. Chem. 277:18954–18960. [DOI] [PubMed] [Google Scholar]
- Rusu, L., A. Gambhir, S. McLaughlin, and J. Radler. 2004. Fluorescence correlation spectroscopy studies of peptide and protein binding to phospholipid vesicles. Biophys. J. 87:1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 103:211–225. [DOI] [PubMed] [Google Scholar]
- Schlessinger, J. 2003. Signal transduction. Autoinhibition control. Science. 300:750–752. [DOI] [PubMed] [Google Scholar]
- Shapiro, M.S. 2004. Why biophysicists make models: quantifying modulation of the M current. J. Gen. Physiol. 123:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, K., and W.L.C. Vaz. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33:269–295. [DOI] [PubMed] [Google Scholar]
- Stamos, J., M.X. Sliwkowski, and C. Eigenbrot. 2002. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277:46265–46272. [DOI] [PubMed] [Google Scholar]
- Sordella, R., D.W. Bell, D.A. Haber, and J. Settleman. 2004. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 305:1163–1167. [DOI] [PubMed] [Google Scholar]
- Tonks, N. 2003. PTP1B: from the sidelines to the front lines! FEBS Lett. 546:140–148. [DOI] [PubMed] [Google Scholar]
- Ullrich, A., L. Coussens, J.S. Hayflick, T.S. Dull, A. Gray, A.W. Tam, J. Lee, Y. Yarden, T.A. Liberman, J. Schlessinger, et al. 1984. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 309:418–425. [DOI] [PubMed] [Google Scholar]
- Yarden, Y., and M.X. Sliwkowski. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Gambhir, G. Hangyás-Mihályné, D. Murray, U. Golebiewska, and S. McLaughlin. 2002. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277:34401–34412. [DOI] [PubMed] [Google Scholar]
- Wang, X.-J., H.-J. Liao, A. Chattopadhyay, and G. Carpenter. 2001. EGF-dependent translocation of green fluorescent protein-tagged PLC-γ1 to the plasma membrane and endosomes. Exp. Cell Res. 267:28–36. [DOI] [PubMed] [Google Scholar]
- Wang, Y., and Z. Wang. 2003. Regulation of EGF-induced phospholipase C-γ1 translocation and activation by its SH2 and PH domains. Traffic. 4:618–630. [DOI] [PubMed] [Google Scholar]
- Welsh, J.G., G.N. Gill, M.G. Rosenfeld, and A. Wells. 1991. A negative feedback loop attenuates EGF-induced morphological changes. J. Cell Biol. 114:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhover, E.J., D.F. Cover, H.L. Brockman, R.E. Brown, and L.J. Pike. 2003. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. J. Biol. Chem. 278:51125–51133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H.S., S.Y. Shvartsman, and D.A. Lauffenburger. 2003. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 13:43–50. [DOI] [PubMed] [Google Scholar]
- Wong, W., and J.D. Scott. 2004. AKAP signaling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5:959–970. [DOI] [PubMed] [Google Scholar]
- Wood, E.R., A.T. Truesdale, O.B. McDonald, D. Yuan, A. Hassell, S.H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, et al. 2004. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib). Cancer Res. 64:6652–6659. [DOI] [PubMed] [Google Scholar]
- Zwick, E., P.O. Hackel, N. Prenzel, and A. Ullrich. 1999. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol. Sci. 20:408–412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.