

Abstract
Gating of voltage-dependent K+ channels involves movements of membrane-spanning regions that control the opening of the pore. Much less is known, however, about the contributions of large intracellular channel domains to the conformational changes that underlie gating. Here, we investigated the functional role of intracellular regions in Kv4 channels by probing relevant cysteines with thiol-specific reagents. We find that reagent application to the intracellular side of inside-out patches results in time-dependent irreversible inhibition of Kv4.1 and Kv4.3 currents. In the absence or presence of Kv4-specific auxiliary subunits, mutational and electrophysiological analyses showed that none of the 14 intracellular cysteines is essential for channel gating. C110, C131, and C132 in the intersubunit interface of the tetramerization domain (T1) are targets responsible for the irreversible inhibition by a methanethiosulfonate derivative (MTSET). This result is surprising because structural studies of Kv4-T1 crystals predicted protection of the targeted thiolate groups by constitutive high-affinity Zn2+ coordination. Also, added Zn2+ or a potent Zn2+ chelator (TPEN) does not significantly modulate the accessibility of MTSET to C110, C131, or C132; and furthermore, when the three critical cysteines remained as possible targets, the MTSET modification rate of the activated state is ∼200-fold faster than that of the resting state. Biochemical experiments confirmed the chemical modification of the intact α-subunit and the purified tetrameric T1 domain by MTS reagents. These results conclusively demonstrate that the T1–T1 interface of Kv4 channels is functionally active and dynamic, and that critical reactive thiolate groups in this interface may not be protected by Zn2+ binding.
INTRODUCTION
Activation of voltage-gated potassium channels (Kv channels) is directly controlled by the movements of their S4 voltage sensors, and a subsequent concerted conformational change that opens an internal gate (Yellen, 1998; Horn, 2000; Bezanilla and Perozo, 2003). The bundle-crossing of four transmembrane S6 segments constitutes the main activation gate that controls K+ passage at the internal opening of the tetrameric pore structure (Jiang et al., 2002; Webster et al., 2004). Just beneath the main activation gate, the NH2-terminal tetramerization domain (T1) of Kv channels is a fourfold symmetric structure that is responsible for the subfamily-specific coassembly of Kv subunits (Li et al., 1992; Shen et al., 1993). The “side windows” between the T1 domain and the transmembrane core domain provide direct access to the internal mouth of the pore (Kreusch et al., 1998; Gulbis et al., 2000; Kobertz et al., 2000; Sokolova et al., 2001; Kim et al., 2004a). Recent studies have suggested that the T1 domain and other intracellular regions also contribute to the function of Kv channels (Cushman et al., 2000; Gulbis et al., 2000; Minor et al., 2000; Kurata et al., 2002; Hatano et al., 2003; Wray, 2004). However, the underlying molecular mechanisms are not well understood. Here, we demonstrate that internally applied thiol-specific reagents irreversibly inhibit Kv4 channels by chemical modification of specific intracellular locations of the channel protein. Furthermore, by using systematic alanine mutagenesis, kinetic analysis, and coexpression with specific auxiliary subunits, we show that the functional inhibition of Kv4.1 channels by a membrane-impermeable thiol-specific reagent (2-trimethylammonium-ethyl-methanethiosulfonate bromide [MTSET]) is gating state dependent and results from the unexpected modification of thiolate groups that were predicted to coordinate Zn2+ with high affinity in the T1–T1 intersubunit interface. Earlier observations from crystallographic and biochemical studies have demonstrated that the isolated T1 domains of channels in the Kv2, Kv3, and Kv4 subfamilies contain tightly bound Zn2+ in the intersubunit T1–T1 interface, and that Zn2+ binding is necessary for the assembly and stability of the tetrameric structure (Bixby et al., 1999; Jahng et al., 2002; Nanao et al., 2003; Strang et al., 2003). In the crystal structure, this interfacial Zn2+ is coordinated by thiolate groups from two cysteines, the side chain of a histidine and a third thiolate group from a neighboring subunit (a C3H1 motif encoded within the conserved sequence HX 5CX 20CC). Given this structural information, the interfacial T1 thiolate groups should be protected by stably liganded Zn2+, and therefore, could not undergo the observed rapid and dynamic chemical modification that suppresses channel function. Our results strongly suggest that the T1–T1 intersubunit interface is more reactive and dynamic than the original Zn2+-coordinated crystal structure had predicted. We also discuss the functional and structural implications of these findings and possible mechanisms responsible for the inhibition of the Kv4.1 channel by MTSET.
MATERIALS AND METHODS
Chemicals and Reagents
Methanethiosulfonate (MTS) reagents (MTSET and 2-sulfonatoethyl-methanethiosulfonate bromide [MTSES]) were purchased from Toronto Research Chemicals and stored in a desiccator at −20°C. These compounds hydrolyze in water, but are stable for hours at 4°C at concentrations >100 mM. Final working solutions were made up immediately before use (in buffer, MTSET decomposes at pH 7.5 with a half-life of ∼10 min). Primers for mutagenesis and DNA sequencing were obtained from the Nucleic Acid Facility of the Kimmel Cancer Institute (Thomas Jefferson University). MTSEA-biotin and DTT were purchased from Biotium, Inc. and Sigma-Aldrich, respectively. Tetrakis-(2-pyridylmethyl) ethylendiamide (TPEN) was purchased from Molecular Devices. As reported by the manufacturer, TPEN has a KD for Zn2+ of the order of 3 × 10−16 M.
Molecular Biology
Kv4.1 (mouse) and Kv4.3 (rat) were maintained in pBluescript II KS (Stratagene) and pBK/CMV (Invitrogen), respectively. The Kv4.3 cDNA was a gift from J. Nerbonne (Washington University, St. Louis, MO). KChIP-1 was maintained in a modified pBluescript vector, pBJ/KSM (gift from M. Bowlby, Wyeth-Ayerst Research, Princeton, NJ). The cDNA encoding the short splice variant of DPPx (gift from B. Rudy, New York University, New York, NY) was maintained in pSG5 (Stratagene). Capped cRNA for expression in oocytes was made using the Message Machine (Ambion). The QuickChange system (Stratagene) was used for oligonucleotide-directed mutagenesis. All mutations were verified by automated DNA sequencing (Nucleic Acid Facility of the Kimmel Cancer Institute, Thomas Jefferson University).
Heterologous Expression and Oocyte Electrophysiology
Wild-type or mutant Kv4, KChIP-1, and DPPx-s cRNAs were injected into defolliculated Xenopus oocytes using a Nanoject microinjector (Drummond). K+ currents were recorded 1–7 d postinjection. To express ternary Kv4 complexes, the mRNA molar ratio was (α subunit:DPPx-s:KChIP1) 1.5:1:3.7 for wild type, C3xA, C11xA, C12xA; and 5.3:1:3.7 and 7.9:1:3.7 for C13xA and C14xA, respectively. Patch-clamp recording was conducted using an Axopatch 200A (Axon Instruments). Patch pipettes were fabricated from Corning glass 7052 or 7056 (Warner Instrument Corp.). Typically, the tip resistance of the recording pipettes in the bath solution was ∼1 MΩ. The composition of the pipette solution (ND96; mM) was 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES (pH 7.4, adjusted with NaOH). The bath solution (internal solution for inside-out patches) contained (mM) 98 KCl, 0.5 MgCl2, 1 EGTA, 10 HEPES (pH 7.2, adjusted with KOH). Passive leak and capacitive transients from macropatch currents were subtracted online using a P/4 procedure. Currents were recorded at room temperature (22 ± 1°C). The reversibility of the functional effects of MTSET was tested upon washout of the reagent or by applying 6–10 mM DTT to the cystoplasmic side of the inside-out patches. In agreement with the covalent modification of thiolate groups (RESULTS), the inhibition of the Kv4 channels by MTSET was not reversed by washout; but upon application of freshly prepared DTT, the inhibition of the Kv4.1-C11xA mutant was reversible in a pH-dependent manner (not depicted). Partial reversal was observed at pH 7.2 (10–30%) and nearly complete reversal at pH 8.5 (>80%). This difference is consistent with the increased ionization and more aggressive reducing power of DTT at alkaline pH.
A computer-controlled piezoelectric translator (EXFO Burleigh) was used for rapid solution switching experiments involving MTS reagents (see Fig. 9). In this system, a double-barreled pipette simultaneously delivers a control and experimental solutions (gravity driven). The flow was adjusted to produce two sharp streams. The inside-out patch was first placed in the control stream, and the piezoelectric translator controlled the rapid interface crossing of the pipette tip between the two streams. The solution exchange time of this system was assessed by measuring the amplitude of K+ currents when the cytoplasmic side of the membrane is exposed to two distinct concentrations of K+. With inside-out macropatches, these measurements have yielded exchange rates of ∼1–3 ms (Shahidullah et al., 2003).
Figure 9.

State-dependent inhibition of Kv4.1 wild type and Kv4.1-C11xA currents by internally applied MTSET. The Kv4.1 pore-forming subunits were expressed alone and the outward currents were recorded in the inside-out patch configuration. The acquisition program controls the coordinated application of voltage steps and MTSET concentration jumps (MATERIALS AND METHODS). (A) MTSET (200 μM) was applied when the membrane was held at −100 mV (resting channels), and the test current was evoked by the indicated pulse protocol. (B) MTSET (200 μM) was applied during the step depolarization to +80 mV as illustrated in the figure (activated channels). For both resting and activated channels, the duration of the reagent concentration jump was 7 ms. (C) Time courses of the inhibition by MTSET of resting (hollow symbols) and activated (filled symbols) channels. The solid lines are the best-fit exponential decays with the following best-fit parameters: 1/τ = 20 mM−1s−1 and A∞ = 0.4, for activated channels; 1/τ = 0.9 mM−1s−1 and A∞ = 0.2, for resting channels. The observed resting state rate constant is not an accurate estimation of the overall modification rate constant because the brief time frame of the experiment (0–0.2 mM × s) severely limits the observed fraction of the slow decay (see below). However, the current data cannot rule out the possibility of a small fast component (≤10%) in the modification time course of the resting channels. (D) Bar graph summarizing the second-order rate constants of the modification by MTSET (n = 4–6). For a more accurate estimation of the slow rate constant from resting channels, the duration of the MTSET concentration jump was 240 ms and the MTSET concentration was 400 μM. Under these conditions, ≥90% of the decay was exponential.
Data Acquisition and Analysis
Voltage-clamp protocols and data acquisition were controlled by a Pentium-III class desktop computer interfaced to a 12-bit A/D converter (Digidata 1200 using Clampex 8.0; Axon Instruments). Data analysis was conducted using Clampfit (pClamp 8.0; Axon Instruments) and Origin 6.0 (OriginLab Inc.). Current relaxations and other time-dependent processes were described assuming a simple exponential function or the sum of exponential terms. Unless stated otherwise, the results are expressed as mean ± SEM.
Protein Biochemistry
CHO cells in DMEM with 10% serum were transfected with Kv4.2 and KChIP-3 using FuGENE 6 (Roche Diagnostics Corp.) or lipofectamine-2000 (Invitrogen), and then CHO cell membrane fragments were prepared as described previously (Strang et al., 2003; Kunjilwar et al., 2004). The Kv4.2-T1 domain with a poly-His tag at the NH2 terminus was expressed in bacteria and purified by a standard Ni2+ column protocol as described previously (Jahng et al., 2002). For biotinylation, MTSEA-biotin was prepared immediately before use, at 0.2 M in water, and then stored on ice and used within 2 h. Final reaction concentrations were 0.2 mM for membrane preparations and 0.5 mM for Kv4.2-T1 preparations. After reaction with MTSEA-biotin, the protein was prepared for gel electrophoresis and characterized by FPLC (fast protein, peptide, and polynucleotide liquid chromatography) analysis as described previously (Strang et al., 2003). Kv4.2 and T1 proteins that reacted with MTSEA-biotin were detected with streptavidin HRP (Pierce Biotechnology). The streptavidin HRP solution (0.1 mg/ml streptavidin HRP in PBS) was diluted 1/10,000 with PBS containing Tween-20 at 0.05% vol/vol. Western blots for proteins were performed as described previously using the same primary and secondary antisera (Zhou et al., 2004). Blots were preblocked with 1% BSA in the PBS-Tween-20 at 0.05%, instead of the milk solution, to avoid any contamination of milk-based biotin containing peptides and proteins.
RESULTS
Inhibition of Kv4 Channels by Internally Applied Thiol-specific Reagents
To investigate the effect of thiolate group modification on the functional properties of mammalian Kv4 channels heterologously expressed in Xenopus oocytes, we applied thiol-specific reagents to the intracellular side of inside-out macropatches (Fig. 1, A–F). Exposing Kv4.1 and Kv4.3 channels to 200 μM MTSET resulted in a time-dependent and nearly complete inhibition of the outward currents within 2–7 min. The time courses of inhibition were approximately exponential and the second-order rate constants of inhibition were 58 and 248 M−1s−1 for Kv4.1 and Kv4.3, respectively (Fig. 1, legend). At the same concentration, the negatively charged MTSES also inhibited Kv4.1. Although the rate constant of inhibition (37 M−1s−1) was only modestly slower than that of MTSET, ∼40% of the peak current remained at steady state. In addition to methanethiosulfonate reagents, N-ethyl-maleimide (NEM, 2 mM) also induced an approximately exponential and nearly complete inhibition of Kv4.1 currents (Fig. 1, G and H) with a very slow rate constant (8 M−1s−1). In all instances, the inhibition was not reversed upon washout of the reagents, which implies a covalent modification (MATERIALS AND METHODS). The critical thiolate groups targeted by the reagents could only be accessed from the intracellular side. When a membrane-impermeant reagent (MTSET, 0.6–1 mM) was present in the patch pipette solution bathing the external regions of the channel, the Kv4.1 current remained stable over the same period of time that is normally sufficient to observe significant inhibition by internal application of the reagent (Fig. 1, I and J). From these observations, we conclude that thiol-specific compounds react with thiolate groups located in functionally sensitive intracellular domains of Kv4 channels.
Figure 1.
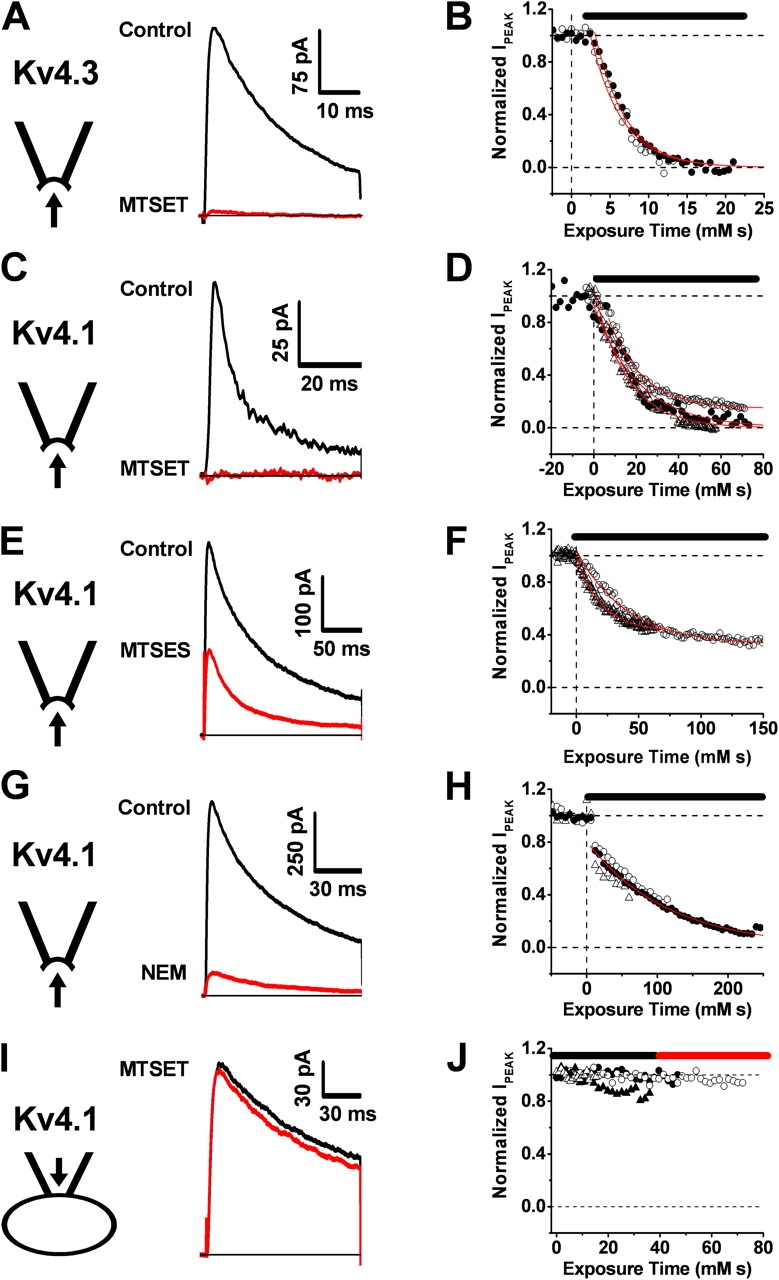
Time-dependent inhibition of Kv4 channels by thiol-specific reagents applied to the intracellular side of the membrane. (A–H) Currents were evoked by a step depolarization from −100 mV to +50 mV at 3-s intervals. The intracellular side of inside-out macropatches from Xenopus oocytes was exposed to the thiol-specific reagents (200–600 μM MTSET, 200 μM MTSES, 2 mM NEM). The current traces are averages of ∼7–20 individual responses recorded before the reagent application (black) and after the inhibition approached steady-state (red). Reagent exposure in the corresponding inhibition time courses (B, D, F, and H) is indicated by a black horizontal bar. Each symbol type represents a separate macropatch (B, n = 2; D, n = 3; F, n = 2; H, n = 3; J, n = 4). Red lines in these graphs are the best fits assuming an exponential decay. The mean values of the derived second-order rate constants (1/(τ × [reagent])) were 248, 58, 37, and 8 M−1s−1, for B, D, F, and H, respectively. (I and J) When MTSET (600 μM) was present in the pipette (external solution), the current remained stable. The black and red traces correspond to the average currents obtained during the first and second half of the experiment, respectively (black and red bars).
Functional Profiling of Intracellular Cysteine Residues
To pinpoint the location of the reactive internal thiol group(s), we identified all candidate cysteines of the Kv4 subunit that might be exposed to the intracellular milieu (Fig. 2). In Kv4.1, there are four thiol groups in the intracellular NH2-terminal region (C105, C110, C131, and C132); one in the S2–S3 loop (C257); one in the S4–S5 loop (C322); and eight in the COOH-terminal region (C392, C467, C484, C490, C532, C533, C589, and C642). Even though C392 is located in the S6 transmembrane segment, we considered it as a candidate because the reagents may gain access to the C392 thiol group from the ion permeation pathway. Only 5 out of the 14 thiol groups listed above are Kv4.1 specific (C105, C257, C467, C490, and C642). The rest are highly conserved in both vertebrate and invertebrate variants of the Kv4 subfamily. According to the most recent structural models of Kv channels (Cuello et al., 2004; Durell et al., 2004), other highly conserved Kv4.1 cysteines are found in the extracellular S1–S2 loop (C209 and C223) and the NH2-terminal external half of S2 (C233 and C338). These extracellular thiol groups are not accessible or do not play a functional role because externally applied MTSET had no effect on Kv4.1 currents (Fig. 1, I and J).
Figure 2.

Mutagenesis of intracellular cysteines in Kv4.1 channels. (A) Schematic topology of the Kv4.1 pore-forming subunit. Filled red circles mark the approximate positions of the 14 cysteine residues likely exposed to the intracellular milieu. (B) Nomenclature of the Kv4.1 mutant channels. Alanine was substituted for cysteine at the indicated positions. CZn indicates that the marked cysteine contributes to Zn2+ binding in the crystal structure of the isolated T1 tetramer.
The 14 intracellular cysteines were mutated to alanines to test their functional impact. Fig. 2 illustrates the specific sites and substitutions, and defines the corresponding nomenclature of the mutants. Extensive cysteine to alanine mutations did not significantly affect channel function. Kv4.1-C11xA mutant (with only the three Zn2+ site cysteines) exhibited peak conductance–voltage relationship and macroscopic inactivation kinetics comparable to those of Kv4.1 wild-type channel, as previously described (Beck and Covarrubias, 2001) (Fig. 3). Thus, most intracellular cysteines are not functionally critical. In contrast, Kv4.1 mutations of the three remaining cysteines (C110A, C131A, C132A, and C3xA) and complete mutation of all 14 intracellular cysteines to alanines (Kv4.1-C14xA) abolished functional expression. These results are in agreement with the contribution of C110, C131, and C132 to the interfacial T1 Zn2+ binding site, which is critical for the formation and stability of the tetramer.
Figure 3.

Functional properties of Cys-depleted Kv4.1 channels. The Kv4.1 wild type and the mutant C11xA (Fig. 2) were functionally expressed in the absence of auxiliary subunits. (A and B) In the cell-attached configuration, macropatch outward currents were evoked by step depolarizations from −100 mV to command voltages ranging between −80 and +60 mV in 20-mV increments. (C) Voltage dependence of the peak conductance. Filled and hollow symbols correspond to wild-type and C11xA channels, respectively. The solid lines are the corresponding best-fit fourth-order Boltzmann functions with the following best-fit parameters for wild type: Va = −53.6 ± 1.8 mV, V1/2 = −10.9 ± 2.9, k = 25.6 ± 1.2 mV (n = 5); and for C11xA: Va = −63.9 ± 4.5 mV, V1/2 = −8.1 ± 6.4 mV, k = 33.6 ± 1.3 mV (n = 5). (D) The time constants of inactivation were determined from the best biexponential fits describing the decay of the currents at the examined membrane potentials (Beck and Covarrubias, 2001; Shahidullah et al., 2003). (E) The ratio of the amplitudes (AFAST/ASLOW) of the biexponential fits plotted against the examined membrane potentials. Note that the kinetic parameters of current decay are only modestly affected by the C11xA mutation.
To characterize the function of the Zn2+ site mutants, we took advantage of the surprising ability of KChIP1 or KChIP3 to repair the apparently lethal phenotype induced by the disruption of the Zn2+ binding site (Kunjilwar et al., 2004). KChIPs are soluble cytosolic proteins related to the neuronal Ca2+ sensor that specifically interact with Kv4 channels (An et al., 2000). KChIP1 facilitates trafficking of the Kv4 tetramer from the endoplasmic reticulum to the plasma membrane and remodels Kv4 gating (An et al., 2000; Beck et al., 2002). Thus, KChIP1 conceivably compensates for the loss of Zn2+ binding by promoting the assembly of the Kv4 tetramer, enhancing its stability, and ensuring the formation of a functional channel complex (Kunjilwar et al., 2004). Accordingly, the function of the Kv4.1-C14xA mutant and C3xA was rescued by coexpression with KChIP1; however, the functional expression levels were insufficient to observe reliable macroscopic currents in Xenopus oocyte inside-out macropatches. In light of this important observation, we decided to investigate the ternary Kv4 complexes composed of the pore-forming Kv4.1 α-subunit and the auxiliary subunits KChIP1 and DPPx-s. DPPx-s is a membrane-bound auxiliary subunit related to the CD26 surface antigen that also remodels gating and up-regulates surface expression of Kv4 channels (Nadal et al., 2003). The Kv4 ternary complex is likely to underlie the somatodendritic A-type K+ current in the brain (Nadal et al., 2003). The similarity between the functional profile of the ternary complexes including either Kv4.1-wild type, Kv4.1-C11xA, Kv4.1-C12xA, Kv4.1-C13xA, Kv4.1-C14xA, or Kv4.1-C3xA suggests that the basic biophysical properties of the Kv4 channel and their remodeling by auxiliary subunits are independent of intracellular thiol groups, including those that may contribute to Zn2+ binding in the T1 domain (Fig. 4, A–C; Table I). The peak conductance–voltage relations of the mutants and the wild-type channels were similar, and the time constants of inactivation were only modestly changed by the most extensive mutations (Fig. 4, D and E; Table I). The time constants of inactivation of wild-type and mutant channels increased with membrane depolarization in a similar fashion, a property that is characteristic of certain neuronal A-type K+ channels (Hoffman et al., 1997). From the results obtained in the presence of auxiliary subunits, we confirmed that the Zn2+ binding site in the T1 domain of Kv4 channels is not essential for surface expression and qualitatively normal activation and inactivation gating.
Figure 4.

Functional properties of Cys-depleted Kv4.1 channels coexpressed with auxiliary subunits. All mutants examined here were expressed as ternary complex including the Kv4.1 pore-forming subunit, DPPx-s and KChIP-1. (A–C) In the cell-attached configuration, currents were evoked by step depolarizations from −100 mV to command voltages ranging between −90 and +110 mV in 20-mV increments. (D) Kv4.1-C11xA, Kv4.1-C12xA, Kv4.1-C13xA, and Kv4.1-C14xA exhibit modestly accelerated inactivation but maintain preferential closed-state inactivation as evident from the voltage dependence of the time constant of inactivation (Jerng et al., 2004b). The solid lines are the best-fit exponential growth functions (Table I). (E) Kv4.1-C11xA, Kv4.1-C12xA, Kv4.1-C13xA, and Kv4.1-C14xA do not exhibit significantly altered voltage dependence of the peak conductance. The solid lines are the corresponding best-fit fourth-order Boltzmann functions (Table I).
TABLE I.
Biophysical Properties of Kv4.1 Cys→Ala Mutants
| Peak Conductance–Voltage Relation
|
Inactivation
|
||||
|---|---|---|---|---|---|
| Va | V1/2 | k | τV = +90 | z | |
| mV | mV | mV/e-fold | ms | e0 | |
| Kv4.1-wild type | −54 ± 1.5 | −9.6 ± 1.5 | 26.8 ± 0.5 | 67 ± 2.1 | 0.38 ± 0.02 |
| n = 16 | n = 13 | ||||
| Kv4.1-C3xA | −61.9 ± 3.1 | −17.8 ± 1.9 | 26.5 ± 1.9 | 63.6 ± 6.9 | 0.21 ± 0.02 |
| n = 3 | n = 3 | ||||
| Kv4.1-C11xA | −44.9 ± 1.7 | −2.1 ± 2.4 | 28.3 ± 1.2 | 23.7 ± 0.8 | 0.41 ± 0.02 |
| n = 16 | n = 16 | ||||
| Kv4.1-C12xA | −47.4 ± 3.5 | −4 ± 4.6 | 30.9 ± 1 | 25.4 ± 0.7 | 0.37 ± 0.02 |
| n = 17 | n = 16 | ||||
| Kv4.1-C13xA | −54.9 ± 3.8 | −9.1 ± 3 | 27.5 ± 2 | 35.3 ± 2.3 | 0.30 ± 0.02 |
| n = 8 | n = 8 | ||||
| Kv4.1-C14xA | −57.7 ± 1.3 | −11.2 ± 1.9 | 28 ± 0.9 | 31.9 ± 1.4 | 0.34 ± 0.01 |
| n = 16 | n = 17 | ||||
The Kv4.1 channels were expressed as ternary complex including the pore-forming subunit, DPPx-s, and KChIP1 (MATERIALS AND METHODS). Va, activation voltage derived from a fourth-order Boltzmann fit; k, slope factor derived from a fourth-order Boltzmann fit; V1/2, voltage at G = 50%; τV = +90, time constant of inactivation at +90 mV; z, apparent valence of macroscopic inactivation derived from the best fit exponential growth function (Fig. 4 D).
To examine the possibility of dynamic Zn2+-dependent modulation involving the thiolate groups in the high-affinity interfacial T1 Zn2+ site, we exposed the intracellular side of inside-out patches expressing the ternary complex of Kv4.1-C11xA or Kv4.1-C14xA to 10 μM ZnCl2. Neither the amplitude nor the kinetics of the currents was significantly affected by this treatment (Fig. 5, A, C, and D). We also investigated the modulatory role of intracellular Zn2+ by applying 20 μM TPEN (a high-affinity Zn2+-specific chelator) to the intracellular side of inside-out patches expressing the ternary complexes of Kv4.1-C11xA and Kv4.1-C14xA. This treatment reduced both the peak current and the time constant of inactivation (Fig. 5, B, C, and D). These effects are, however, not caused by the removal of Zn2+ from the T1 Zn2+ binding site because the mutant lacking the critical Zn2+ site cysteines (Kv4.1-C14xA) yielded similar results (Fig. 5, C and D). Because there is no evidence of additional Zn2+ sites in other regions of the Kv4 channel complex, TPEN appears to exert collateral effects on Kv4 channel function that are independent of its Zn2+ chelating activity. These TPEN effects (reduced peak current and accelerated inactivation) are reminiscent of an open-channel occlusion mechanism. Overall, the Zn2+ and TPEN experiments suggest that the T1 Zn2+ site of Kv4 channels does not contribute to dynamic Zn2+-dependent modulation of channel function.
Figure 5.

Effects of internal Zn2+ or TPEN on Kv4 channels with intact (C11xA) or disrupted (C14xA) T1 Zn2+ sites. Mutant channels were expressed as described in Fig. 4 legend. (A and B) Outward Kv4.1-C11xA currents evoked by a step depolarization to +80 mV from a holding potential of −100 mV. These currents were recorded from inside-out patches before (black) and after (red) the intracellular application of Zn2+ or TPEN at the indicated concentrations. (C) Bar graph comparing the normalized peak current (at +80 mV) before and after the application of Zn2+ or TPEN. (D) Bar graph comparing the effects of Zn2+ and TPEN on the time constant of inactivation at +80 mV (Fig. 4). The Zn2+ and TPEN experiments were conducted separately; therefore, the data are presented in two groups. Note that independently of the integrity of the T1 Zn2+ site, the peak current and time constant of inactivation decreased upon TPEN application (see text).
The Zn2+ Site Thiolate Groups in the T1 Domain Are Responsible for the Inhibition of Kv4 Channels by Thiol Reagents
If Zn2+ is tightly bound in the T1–T1 intersubunit interface of the intact Kv4 ternary complex, as found in the crystal structure of the isolated Kv4-T1 domain (Bixby et al., 1999; Jahng et al., 2002; Nanao et al., 2003) (see Fig. 10), the critical Zn2+ site cysteines (C110, C131, and C132) would be protected and therefore are inaccessible to chemical modification by MTSET or any other thiol-specific reagent (Smith et al., 2005). To further investigate this possibility, we examined the effect of internally applied MTSET (200 μM) on ternary complexes that included Kv4.1 wild type (14 intracellular thiol groups) or Kv4.1 mutants with three, two, and one remaining thiolate group in the Zn2+ site (C11xA, C12xA, and C13xA, respectively; Fig. 2). Note that the remaining cysteines in Kv4.1-C12xA are C131 and C132 and the remaining cysteine in Kv4.1-C13xA is C110. Because all other intracellular cysteines have been mutated to alanines in these mutants, we can be assured that the membrane-impermeant MTSET is targeting the remaining thiolate group(s) only. Like the Kv4.1 wild type, all mutants with an intact (C11xA) or partially disrupted Zn2+ site (C12xA and C13xA) exhibited an exponential and nearly complete (≥77%; Fig. 6) inhibition by MTSET with rate constants not differing by >2.4-fold (28–67 M−1s−1; Figs. 6 and 7). In sharp contrast, as mentioned earlier, the Kv4.1 mutant channel that has no intracellular thiolate groups (Kv4.1-C14xA) was not inhibited by MTSET (Fig. 6, I and J). The contribution of the auxiliary subunits in the ternary complex is also ruled out because the Kv4.1-C11xA mutant expressed alone exhibits inhibition by MTSET similar to that of the ternary complex (unpublished data). Altogether, these results provide compelling evidence demonstrating that at least one out of three thiolate groups in the Zn2+ site of Kv4.1 wild type and Kv4.1-C11xA is always accessible to chemical modification by MTSET, which results in a potent inhibition of the channel. Although the results from mutants with the impaired Zn2+ site (C12xA and C13xA) suggest modest changes in the accessibility of the targeted thiolate group, it is clear that even with an intact Zn2+ site, metal coordination cannot simultaneously protect all thiolate groups in the T1 Zn2+ site. The exponential time dependence of the inhibition suggests that reaction with a single thiolate group in the T1–T1 intersubunit interface would be sufficient to suppress channel function.
Figure 10.

A structural model of the Kv4-T1 domain and the contribution of T1–T1 intersubunit interface to the gating state–dependent inhibition of the Kv4 channel by MTSET. (A) Model of the Kv4 tetramer depicting a theoretical model of the transmembrane α-core (Durell et al., 2004) and the likely location of the intracellular T1 domain (red). The depicted 3-D model of the T1 domain is based on the crystal structure of the isolated Kv4-T1 domain (Nanao et al., 2003). (B) Magnified views of the T1 domain model. In both, the lateral (left) and top (right) views, the blue spheres represent the location of Zn2+ atoms in the T1–T1 intersubunit interface as found in the crystal structure. The four layers of the T1 scaffolding are indicated next to the lateral view as L1–L4. Note that the Zn2+ site is located between L3 and L4. In the intact channel, L4 is directly connected to the NH2-terminal end of the transmembrane S1 segment, which links T1 with the voltage-sensing regions of the channel. (C) Magnified view of a single interfacial Zn2+ site in T1. The coordinating amino acid side chains are explicitly shown in a scaled ball and stick representation, and the outlined circle (dashed line) represents the Zn2+ atom. H104, C131, and C132 are from the same subunit, and C110 is from the neighboring subunit. H104 is behind the backbone of C131 and C132. A standard color scheme is used to represent the relevant atoms (sulfur atoms in yellow). Note that the peptide bond between C131 and C132 is also shown. (D) Working hypothesis explaining the gating state dependence of the inhibition of Kv4 channels by MTSET. In the resting state (at hyperpolarized voltages), at least one thiolate group (e.g., C131) exhibits low accessibility to MTSET. Upon activation by a strong depolarization, a concerted clockwise displacement of L4 exposes the Zn2+ site thiolate groups to MTSET. The data suggest that modification of a single thiolate group in the T1–T1 interface would be sufficient to explain the inhibition of the channel (see text). A possible location of Zn2+ in these models is indicated by the dashed perimeter of a circle in the T1–T1 interface. The exact alternative architecture of the T1 Zn2+ site that leaves at least one free thiolate in the T1–T1 interface is not known (see text).
Figure 6.
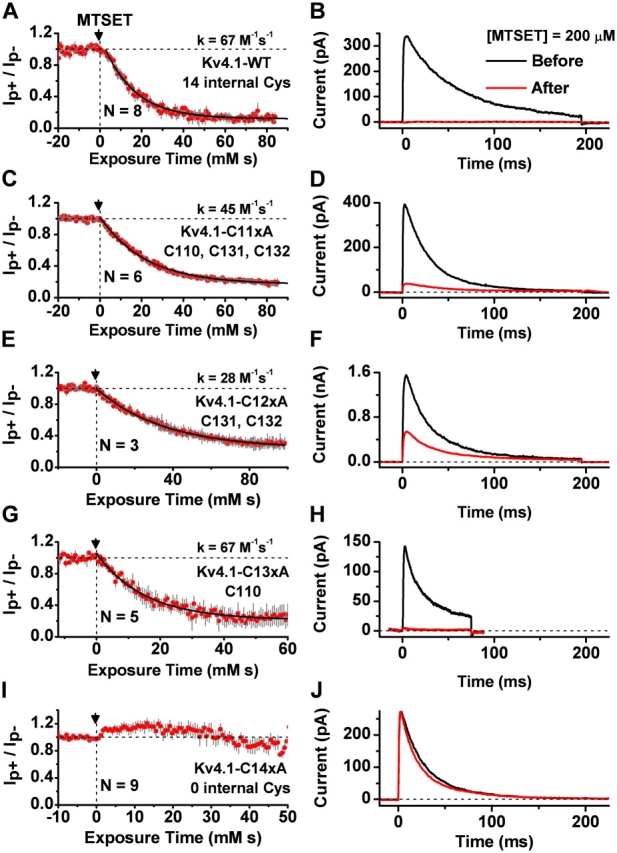
Inhibition of Kv4.1-wild type and Zn2+ site mutants by internal MTSET. Mutant channels were expressed as described in Fig. 4 legend. (A, C, E, and G) Time-dependent inhibition of wild type, C11xA, C12xA, C13xA, and C14xA (Fig. 2 for mutant nomenclature) by internally applied MTSET (arrow, 200 μM). The y axis is the normalized peak current (peak current after MTSET/peak current before MTSET). These experiments were conducted as explained in Fig. 1 legend. The solid black line through the data points corresponds to the best-fit exponential. The second-order rate constants are indicated within the corresponding panels. (I) When all internal cysteines were mutated to alanines (C14xA), there was no inhibition by MTSET. Red symbols and gray bars represent the mean ± SEM from the number of independent measurements indicated in the corresponding panels. (B, D, F, H, and J) Representative current traces (corresponding to the left panels in each row) evoked by a step depolarization from −100 to +80 mV (inside-out patch configuration). The traces are averages (∼7–20 traces) taken before (black) and after (red) approaching the steady-state level of the inhibition.
Figure 7.

Inhibition of Kv4.1 Zn2+ site mutants by intracellular MTSET in the presence of Zn2+ or a Zn2+-specific chelator. The experiments were conducted as explained in Fig. 6 legend. (A) MTSET-induced inhibition in the presence of 10 μM ZnCl2 in the internal solution (no EGTA added). The solid line across the symbols corresponds to the best-fit exponential function (1/τ = 0.067 mM−1s−1; A∞ = 0.2; A∞ is the estimated level of the remaining current). (B) MTSET-induced inhibition in the presence of 20 μM TPEN in the internal solution. The solid line across the symbols corresponds to the best-fit exponential function (1/τ = 0.050 mM−1s−1; A∞ = 0.16). The insets in A and B display the currents before (thick line) and after (thin line) application of MTSET to the internal side of the inside-out patch. The dashed line marks the zero current level. (C) Bar graph summarizing the MTSET second-order inhibition rate constants (1/(τ × [MTSET])) for various Kv4.1 mutants (Fig. 2) examined under control conditions or in the presence of ZnCl2 or TPEN.
Does the Occupancy of the T1 Zn2+ Site Modulate the Inhibition of Kv4 Channels by MTSET?
If the Zn2+ coordination site is not protecting the T1 interfacial thiolate groups against chemical modification by MTSET, it is conceivable that the native T1 domain of the Kv4 channel adopts a conformation that has lower affinity for Zn2+, or does not coordinate Zn2+ in the manner observed in the crystals of the isolated T1 oligomers (see Fig. 10 C). Thus, the metal ion could dissociate from the intact channel upon patch excision, exposing the site; or dynamically expose one or more thiolate coordination groups depending upon the specific structural constraints in the intact Kv4 channel. To test whether differential Zn2+ occupancy affects the reactivity of the relevant thiolate groups when the site is intact and functional, we exposed the intracellular side of inside-out patches expressing the Kv4.1-C11xA mutant to 1–10 μM Zn2+ or 20 μM TPEN during the application of 200 μM MTSET (Fig. 7, A and B). If some Zn2+ sites were free and occupancy plays a significant role in regulating thiolate group accessibility and reactivity, we would expect a slower rate of inhibition in the presence of added Zn2+ (Zn2+ binding would protect critical thiolate groups against MTSET); and if the sites were still Zn2+ bound, we would expect an accelerated rate of inhibition in the presence of TPEN (the chelator would deplete the Zn2+ site, leaving the thiolate groups unprotected). Our results showed that both Zn2+ and TPEN modestly increased the rate constant of the inhibition of Kv4.1-C11xA by ∼50% (Fig. 7 C). Similarly, the inhibition of Kv4.1-C12xA, with a partially disrupted Zn2+ site, was also increased by TPEN (<50%; Fig. 7 C). Although the effect of TPEN on these mutant channels is qualitatively consistent with the simple Zn2+ occupancy hypothesis involving a relatively weak interaction, the effect of Zn2+ is not. Diverging from the crystal structure, the results suggest that Zn2+ could be preferentially liganded to two or three coordinating groups (e.g., H104 and C132 in C12xA or H104, C131, and C132 in C11xA) in a way that always leaves at least one thiolate group exposed and poorly protected against the MTS reagent (e.g., C131 in C12xA, and C110 in C11xA) (see Fig. 10 C). Weakly liganded Zn2+ could exert a modest acceleration of the inhibition by MTSET through local rearrangements and steric interactions.
As a negative control, we also investigated the effects of MTSET, TPEN, and Zn2+ on Kv4.1-C3xA, a mutant lacking Zn2+ site thiolate groups in the T1 domain but maintaining 11 intracellular cysteines, which are not likely to contribute to metal coordination (Fig. 2). Kv4.1-C3xA coexpressed with KChIP1 and DPPx-s was partially inhibited by 200 μM MTSET (∼60%), with a modification rate constant that was only modestly decreased by Zn2+ or TPEN (Fig. 7 C). Therefore, there is at least another thiol group located within a functionally critical intracellular region outside of the T1 Zn2+ site. This additional reactive thiolate group is not in the auxiliary proteins because the ternary complex of Kv4.1-C14xA, which lacks all intracellular thiolate groups in the pore-forming subunit (Fig. 2), exhibited no response to internally applied MTSET (Fig. 6, I and J). From these results, we concluded that there are two loci for MTSET reagents in the cytoplasmic domains of Kv4.1, one involving thiolate groups in the T1 Zn2+ site and the other at an as yet unidentified intracellular cysteine. The relatively small and variable effects of Zn2+ and TPEN on the modification rate constants of various Kv4.1 mutants (including Kv4.1-C3xA with no Zn2+ site thiolate groups; Fig. 7) suggest that these reagents may exert weak and possibly nonspecific effects on the modification rate constant of MTSET. More significantly, however, the main evidence argues strongly for the presence of one or more reactive thiolate groups in the functionally active T1 interfacial Zn2+ site (Figs. 6 and 7). Further research beyond the scope of this study is necessary to solve the exact configuration and occupancy of the Zn2+ site in the native channel and the interactions of thiol-specific reagents with other internal Kv4 thiol groups outside of the T1 domain.
Chemical Modification of the Kv4 Channel Protein by a Thiol-specific Reagent
To directly show the chemical modification of the Kv4 protein by the MTS reagent, we performed biochemical labeling experiments using MTSEA-biotin. For full-length channels, we used intact Kv4.2 channel expressed in CHO cells (Fig. 8 A), and for specific labeling of the Zn2+ site cysteines, we used the Kv4.2-T1 domain expressed and purified from bacteria (Fig. 8 B). The Kv4.2 subunit was preferred for these experiments because we have good antisera against this subunit, and the only cysteines in its T1 domain are three cysteines involved in Zn2+ coordination. For both, the full-length Kv4 channel as well as the Kv4-T1 domain, Western blot analysis revealed strong specific signals upon streptavidin-HRP detection. Thus, thiolate groups in the intact Kv4.2 channel and the isolated Kv4.2-T1 domain are reacting with MTSEA-biotin. The modification of the isolated Kv4-T1 protein with the MTS reagent is especially significant because it shows that at least one out of three cysteines equivalent to those remaining in the Kv4.1-C11xA mutant is reacting with MTS reagents on its own. We also examined the effect of MTSEA-biotin on the T1 tetramer stability by using size exclusion chromatography FPLC. The FPLC profile showed that before and after the treatment with MTSEA-biotin, the T1 oligomer remained fully tetrameric (Fig. 8 C). Thus, the MTSEA-biotin may react with one or more thiolate groups in the Kv4-T1 interface, but this reaction does not destabilize the tetramer. Therefore, tetramer dissociation does not seem to be the cause of the rapid inhibition of Kv4 channel function by the thiol-specific reagents. More likely, MTSET inhibits the Kv4 currents by altering gating, which results from the chemical modification of free critical thiolate groups in the T1–T1 interfacial Zn2+ binding sites. Even though previous studies have shown that Zn2+ is bound to the isolated Kv4.2-T1 oligomeric protein (Jahng et al., 2002; Nanao et al., 2003; Strang et al., 2003), the biochemical data demonstrate that at least one out of the three relevant thiolate groups must be free to react with MTSEA-biotin when the protein is in solution.
Figure 8.

Biochemical evidence of the chemical modification of Kv4.2 and Kv4.2-T1 by MTSEA-biotin. (A) Membrane fragments containing Kv4.2 and KChIP3 (Kv4.2:KChIP3, 1:3) were reacted with MTSEA-biotin for 20 min at room temperature, and then electrophoresed and blotted with either anti-Kv4.2 or streptavidin-HRP (MATERIALS AND METHODS). (B) Likewise, the purified T1 domain of Kv4.2 was also reacted with the biotinylated MTSEA reagent and screened with streptavidin-HRP. The indicated molecular weights correspond to those of Kv4.2 a-monomer (67 kD) and the monomeric Kv4.2-T1 protein (14 kD). (C) FPLC profile of the Kv4.2-T1 protein before (black) and after (red) treatment with MTSEA-biotin (0.5 mM). AU280, normalized absorbance units at 280 nM. The expected elution times of the T1 tetramer and the T1 monomer are schematically marked above the abscissa.
Inhibition of Kv4.1 Channels by Internal MTSET Is Gating State Dependent
Does the inhibition of Kv4.1 channels by a thiol-specific reagent depend on conformational changes associated with gating? This is a relevant question because thiolate groups are found in regions that are believed to play a role in gating of Kv channels (Fig. 2), including the T1 domain (Cushman et al., 2000; Minor et al., 2000). Also, it is conceivable that conformational changes initiated by the movement of the voltage sensor propagate into the T1 domain and other intracellular regions. To examine this question, the intracellular side of inside-out patches expressing Kv4.1 wild type and Kv4.1-C11xA (in the absence of auxiliary subunits) were exposed to 200 μM MTSET using controlled concentration jumps combined with two distinct voltage-clamp protocols (Fig. 9, A and B): (1) to test the inhibition of resting channels, a 7-ms pulse of the reagent was applied when the membrane potential was held at −100 mV and the available current was tested by a subsequent 5-ms step depolarization to +80 mV; and (2) to test the inhibition of activated channels, a 7-ms pulse of the reagent was applied during a short 12-ms step depolarization to +80 mV to mainly encompass the peak of the current. Significant current inhibition by MTSET was observed with both protocols (Fig. 9, A and B). However, the time course of the inhibition was very different when comparing the modification of resting and activated channels, even though the application time of MTSET was the same (Fig. 9 C). With both, Kv4.1 wild type and Kv4.1-C11xA, the observed second-order inhibition rate constant of the activated state (∼2 × 104 M−1s−1) was ∼200-fold faster than that of the resting state (∼100 M−1s−1) (Fig. 9 D). This result strongly suggests that gating rearrangements change the accessibility of critical thiolate groups in the interfacial T1 Zn2+ site; and therefore, we conclude that the T1–T1 interface of the Kv4.1 channel is functionally active.
DISCUSSION
This study found that, regardless of their chemical structure or polarity, thiol-specific reagents applied to the intracellular side of the membrane cause irreversible inhibition of Kv4 channels. This result suggests that the steric effect of the resulting adduct at the modified site(s) inhibits gating or blocks permeation. Since the inhibition was still robust with only three thiolate groups in the Zn2+ site of the T1 domain remaining intact, and the T1 domain does not contribute to the permeation pathway, we conclude that the intracellular T1–T1 intersubunit interface near the Zn2+ site is functionally active and that the inhibition may result from a packing perturbation that interferes with channel gating. Biochemical experiments confirmed that tetramer dissociation is not the likely cause of the inhibition because the Kv4-T1 protein remained tetrameric upon chemical modification by MTSET.
Mechanistic and Physiological Implications
The solute accessibility or reactivity of the targeted thiolate group(s) is dramatically enhanced when the channel activates. This result may be interpreted as the propagation of a conformational change that starts with movements in the voltage sensing domain (transmembrane segments S1–S4) followed by rearrangements in the T1–T1 intersubunit interface (where the reactive thiolate groups are found; Fig. 10, A and B) and the opening of an internal pore gate. From electron microscopy studies of Kv4 channels (Kim et al., 2004b), the four T1–T1 interfacial Zn2+ sites are predicted to sit just below the “side windows” that provide aqueous access to the pore from the cytoplasmic side (Fig. 10 A) (Bixby et al., 1999; Nanao et al., 2003). Conceivably, the T1–T1 intersubunit interfaces may rearrange during channel gating as part of a conformational change that widens the side windows to enhance solute access to the open pore. Supporting this model, the Zn2+ site is located between T1 layers 3 and 4 (L3 and L4) in a region that is probably close to the core of the channel (Fig. 10, B and C). L4 is a short α-helix directly connected to a short linker that joins T1 to the NH2-terminal end of the S1 transmembrane segment; therefore, it may serve as a physical link between T1 and the voltage-dependent activation machinery of the channel. We propose a clockwise displacement of L4 (viewed from the membrane side of T1) that results from voltage-dependent activation gating (Fig. 10 D). This movement would effectively expose the thiolate groups in the Zn2+ site and account for the dramatic 200-fold increase in the modification rate constant upon channel activation (Fig. 9). The exponential time course of the inhibition suggests that perturbation of one site would be sufficient to cause inhibition because the T1 tetramer may act as a functional unit that undergoes a concerted conformational change. The proposed rearrangement of the T1–T1 interfaces (Fig. 10 D) may be a prerequisite for a subsequent expansion of the lateral windows and opening of the inner mouth of the pore. Chemical modification of a single thiolate group in the Zn2+ site may thus obliterate this conformational change and favor a nonconducting conformation of the channel. A previous study found that NH2-terminal thiolate groups of Kv2.1 (including those that contribute to Zn2+ binding) are modified by MTS reagents, which primarily delayed the single channel latency to the first opening (Pascual et al., 1997). Although this study did not determine the exact location of the targeted thiolate groups, it suggested a contribution of the intracellular NH2-terminal region to activation gating of Kv channels.
Kv4 channels exhibit preferential closed-state inactivation (Beck and Covarrubias, 2001; Bähring et al., 2001; Shahidullah and Covarrubias, 2003), which may involve channels that fail to open because they become effectively desensitized to changes in membrane potential, as proposed for HCN channels (Shin et al., 2004). Previous studies of Kv4 channels have suggested that the opening step is not strongly forward biased (Bähring et al., 2001; Beck and Covarrubias, 2001; Beck et al., 2002; Shahidullah et al., 2003). Through the mechanism proposed above, MTSET modification of intracellular thiolate groups in the T1–T1 interface could induce further shifting of the opening equilibrium toward the inactivation permissive preopened closed state, and thereby promote inactivation that would result in a partial or complete suppression of current, depending on the magnitude of the shift. Additionally, MTSET could directly promote closed-state inactivation, either by stabilizing the closed-inactivated state or by enhancing the rate of entry into the closed-inactivated state.
The presence of free reactive thiolate groups in a functionally active T1–T1 intersubunit interface of Kv4 channels can have broad physiological implications because these groups are subject to oxidation under normal or pathological conditions. Kv4 channels underlie the somatodendritic A-type K+ current (ISA) (Johnston et al., 2003; Jerng et al., 2004a) in brain and the fast transient K+ current in the heart and smooth muscle (ITO) (Nerbonne et al., 2001; Amberg et al., 2003). ISA is a key neuronal moderator of electrical excitability that, among various functions, regulates spike firing frequency and dampens backpropagating action potentials (Johnston et al., 2003; Jerng et al., 2004a). These functions contribute to signal coding, dendritic integration, and plasticity (Cai et al., 2004). ITO in the heart regulates cardiac excitability by shaping phase-1 of the cardiac action potential. Suppression of the ITO is arrhythmogenic. Mammalian Kv4 channels (Kv4.1, Kv4.2, and Kv4.3) are highly conserved and vertebrate and invertebrate orthologues share the determinants of the Zn2+ binding site (Jerng et al., 2004a). In light of our observations, future studies need to examine whether oxidative stress (hypoxia and ischemia-reperfusion) and physiological modulators (e.g., NO and its reactive metabolites) in brain and heart have an impact on Kv4 channel function through the chemical modification of the thiol groups in the intracellular T1–T1 intersubunit interface. Our results may explain the irreversible inhibition of native Kv4 channel in rat ventricular myocytes by thiol-specific reagents (Rozanski and Xu, 2002).
Structural Implications
Earlier studies of native or engineered Zn2+ binding sites in nonchannel proteins have shown that high-affinity Zn2+ binding protects the coordinating thiolate groups against chemical modification by thiol-specific reagents, including fluorescent maleimide derivatives and NEM (Fu et al., 1996; Smith et al., 2005). Thus, what can be said about the configuration and chemical state of the T1–T1 thiolate groups in the Kv4 channel? Although originally identified as part of a high-affinity Zn2+ coordination site in isolated Kv4-T1 crystals (Fig. 10, A–C), the relevant thiolate groups are more reactive and dynamic than expected from this original model. In solution, even the isolated T1 domain containing the three Zn2+ site thiolate groups only exhibits chemical modification by MTSEA (Fig. 8), which suggests that at least one thiolate group is not protected by Zn2+ binding. Also, as the number of thiolate groups in the Zn2+ site of the full-length channel is progressively reduced by mutagenesis (Figs. 6 and 7), the accessibility of the remaining thiolate(s) is similar (within a twofold range) and only modestly affected by a potent Zn2+ chelator (Fig. 7). Thus, the T1 Zn2+ site of these mutants seems to be unoccupied under the conditions of our inside-out patch recordings; or Zn2+ is only weakly liganded in the T1 site. This conclusion is tempered by the absence of an inhibitory effect of adding Zn2+ on thiolate group accessibility (Fig. 7), which suggests that either (a) the site is unable to bind Zn2+, or (b) that the architecture of the Zn2+ site differs from that in the T1 crystal structure in such a way that at least one free thiolate group (e.g., from the interfacial C131; Fig. 10 C) is accessible to the MTS reagents in a gating state–dependent manner (Fig. 9). High-affinity Zn2+ binding per se may help to assemble and stabilize the tetramer in the early biogenesis of the Kv4 protein. However, once the Kv4 protein and auxiliary subunits form the native channel complex, the functional configuration of the oligomer may no longer support high-affinity Zn2+ binding involving all four coordinating groups from the C3H1 motif in the T1–T1 interface. In agreement with this hypothesis, KChIP1 or KChIP3 can override the essential need of Zn2+ for tetramer formation and stability; and the Kv4-KChIP channel complex without the T1 Zn2+ site is fully functional and exhibits qualitatively normal biophysical properties (Fig. 4) (Kunjilwar et al., 2004). Future studies need to consider these observations and possibilities to fully understand the function of Zn2+ in Kv channels.
Conclusion
It has been shown previously that the intersubunit T1 Zn2+ site plays an important structural role in Kv4 channels (Jahng et al., 2002; Nanao et al., 2003; Strang et al., 2003). Other studies have also suggested that the Kv channel T1 domain may play a more direct functional role (Pascual et al., 1997; Cushman et al., 2000; Minor et al., 2000; Kurata et al., 2002). Our data suggest strongly that the interfacial T1 Zn2+ sites are functionally active because MTSET suppresses channel function through the chemical modification of the relevant thiolate groups that are not protected by Zn2+. The preferential inhibition in the activated state of the channel by MTSET is especially attractive because it suggests significant structural rearrangements involving the T1–T1 intersubunit interface and potential modulation of Kv4 gating by the redox state of the cell. Similar interactions may take place in Kv2 and Kv3 channels, which share the interfacial Zn2+ site in their T1 domain (in contrast to Kv1 channels) (Kreusch et al., 1998; Bixby et al., 1999).
Acknowledgments
We thank Dr. Mark Bowlby for providing KChIP-1 cDNA and Dr. Bernardo Rudy for providing DPPx-s cDNA. We also thank Drs. Senyon Choe, Carol Deutsch, and Richard Horn for their critical reading of the manuscript; Dr. Arthur Karlin for insightful suggestions; and Dr. Robert Guy (National Cancer Institute, Bethesda, MD) for providing the coordinates to create the theoretical model of the Kv α-core in Fig. 10 A.
This work was supported by U.S. Public Health Service (USPHS) research grants R01 NS32337 (M. Covarrubias); R01 NS31583 and P01 NS37444 (P.J. Pfaffinger); and USPHS training grant AA07463 (M. Shahidullah).
Olaf S. Andersen served as editor.
M. Shahidullah's present address is Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, PA.
Abbreviations used in this paper: FPLC, fast protein, peptide, and polynucleotide liquid chromatography; MTS, methanethiosulfonate; MTSES, 2-sulfonatoethyl-methanethiosulfonate bromide; MTSET, 2-trimethylammonium-ethyl-methanethiosulfonate bromide; NEM, N-ethyl-maleimide; TPEN, tetrakis-(2-pyridylmethyl) ethylendiamide.
References
- Amberg, G.C., S.D. Koh, Y. Imaizumi, S. Ohya, and K.M. Sanders. 2003. A-type potassium currents in smooth muscle. Am. J. Physiol. Cell Physiol. 284:C583–C595. [DOI] [PubMed] [Google Scholar]
- An, W.F., M.R. Bowlby, M. Betty, J. Cao, H.P. Ling, G. Mendoza, J.W. Hinson, K.I. Mattsson, B.W. Strassle, J.S. Trimmer, and K.J. Rhodes. 2000. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 403:553–556. [DOI] [PubMed] [Google Scholar]
- Bähring, R., L.M. Boland, A. Varghese, M. Gebauer, and O. Pongs. 2001. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E.J., and M. Covarrubias. 2001. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys. J. 81:867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E.J., M. Bowlby, W.F. An, K.J. Rhodes, and M. Covarrubias. 2002. Remodelling inactivation gating of Kv4 channels by KChIP-1, a small-molecular-weight calcium binding protein. J. Physiol. 538:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla, F., and E. Perozo. 2003. The voltage sensor and the gate in ion channels. Adv. Protein Chem. 63:211–241. [DOI] [PubMed] [Google Scholar]
- Bixby, K.A., M.H. Nanao, N.V. Shen, A. Kreusch, H. Bellamy, P.J. Pfaffinger, and S. Choe. 1999. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat. Struct. Biol. 6:38–43. [DOI] [PubMed] [Google Scholar]
- Cai, X., C.W. Liang, S. Muralidharan, J.P. Kao, C.M. Tang, and S.M. Thompson. 2004. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 44:351–364. [DOI] [PubMed] [Google Scholar]
- Cuello, L.G., D.M. Cortes, and E. Perozo. 2004. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 306:491–495. [DOI] [PubMed] [Google Scholar]
- Cushman, S.J., M.H. Nanao, A.W. Jahng, D. DeRubeis, S. Choe, and P.J. Pfaffinger. 2000. Voltage dependent activation of potassium channels is coupled to T1 domain structure. Nat. Struct. Biol. 7:403–407. [DOI] [PubMed] [Google Scholar]
- Durell, S.R., I.H. Shrivastava, and H.R. Guy. 2004. Models of the structure and voltage-gating mechanism of the shaker K+ channel. Biophys. J. 87:2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H.W., J.F. Moomaw, C.R. Moomaw, and P.J. Casey. 1996. Identification of a cysteine residue essential for activity of protein farnesyltransferase. Cys299 is exposed only upon removal of zinc from the enzyme. J. Biol. Chem. 271:28541–28548. [DOI] [PubMed] [Google Scholar]
- Gulbis, J.M., M. Zhou, S. Mann, and R. MacKinnon. 2000. Structure of the cytoplasmic β subunit-T1 assembly of voltage-dependent K+ channels. Science. 289:123–127. [DOI] [PubMed] [Google Scholar]
- Hatano, N., S. Ohya, K. Muraki, R.B. Clark, W.R. Giles, and Y. Imaizumi. 2003. Two arginines in the C-terminus domain are essential for voltage-dependent regulation of A-type K+ current in the Kv4 channel subfamily. J. Biol. Chem. 279:5450–5459. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.A., J.C. Magee, C.M. Colbert, and D. Johnston. 1997. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 387:869–875. [DOI] [PubMed] [Google Scholar]
- Horn, R. 2000. Conversation between voltage sensors and gates of ion channels. Biochemistry. 39:15653–15658. [DOI] [PubMed] [Google Scholar]
- Jahng, A.W., C. Strang, D. Kaiser, T. Pollard, P. Pfaffinger, and S. Choe. 2002. Zinc mediates assembly of the T1 domain of the voltage-gated K+ channel 4.2. J. Biol. Chem. 277:47885–47890. [DOI] [PubMed] [Google Scholar]
- Jerng, H.H., P.J. Pfaffinger, and M. Covarrubias. 2004. a. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell. Neurosci. 27:343–369. [DOI] [PubMed] [Google Scholar]
- Jerng, H.H., Y. Qian, and P.J. Pfaffinger. 2004. b. Modulation of Kv4.2 channel expression and gating by dipeptidyl peptidase 10 (DPP10). Biophys. J. 87:2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Johnston, D., B.R. Christie, A. Frick, R. Gray, D.A. Hoffman, L.K. Schexnayder, S. Watanabe, and L.L. Yuan. 2003. Active dendrites, potassium channels and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L.A., J. Furst, M.H. Butler, S. Xu, N. Grigorieff, and S.A. Goldstein. 2004. a. Ito channels are octomeric complexes with four subunits of each Kv4.2 and K+ channel-interacting protein 2. J. Biol. Chem. 279:5549–5554. [DOI] [PubMed] [Google Scholar]
- Kim, L.A., J. Furst, D. Gutierrez, M.H. Butler, S. Xu, S.A. Goldstein, and N. Grigorieff. 2004. b. Three-dimensional structure of I(to); Kv4.2-KChIP2 ion channels by electron microscopy at 21 Å resolution. Neuron. 41:513–519. [DOI] [PubMed] [Google Scholar]
- Kobertz, W.R., C. Williams, and C. Miller. 2000. Hanging gondola structure of the T1 domain in a voltage-gated K+ channel. Biochemistry. 39:10347–10352. [DOI] [PubMed] [Google Scholar]
- Kreusch, A., P.J. Pfaffinger, C.F. Stevens, and S. Choe. 1998. Crystal structure of the tetramerization domain of the Shaker potassium channel. Nature. 392:945–948. [DOI] [PubMed] [Google Scholar]
- Kunjilwar, K., C. Strang, D. DeRubeis, and P.J. Pfaffinger. 2004. KChIP3 rescues the functional expression of Shal channel tetramerization mutants. J. Biol. Chem. 279:54542–54551. [DOI] [PubMed] [Google Scholar]
- Kurata, H.T., G.S. Soon, J.R. Eldstrom, G.W. Lu, D.F. Steele, and D. Fedida. 2002. Amino-terminal determinants of U-type inactivation of voltage-gated K+ channels. J. Biol. Chem. 277:29045–29053. [DOI] [PubMed] [Google Scholar]
- Li, M., Y.N. Jan, and L.Y. Jan. 1992. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 257:1225–1230. [DOI] [PubMed] [Google Scholar]
- Minor, D.L., Y.F. Lin, B.C. Mobley, A. Avelar, Y.N. Jan, L.Y. Jan, and J.M. Berger. 2000. The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell. 102:657–670. [DOI] [PubMed] [Google Scholar]
- Nadal, M.S., A. Ozaita, Y. Amarillo, E.V. de Miera, Y. Ma, W. Mo, E.M. Goldberg, Y. Misumi, Y. Ikehara, T.A. Neubert, and B. Rudy. 2003. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 37:449–461. [DOI] [PubMed] [Google Scholar]
- Nanao, M.H., W. Zhou, P.J. Pfaffinger, and S. Choe. 2003. Determining the basis of channel-tetramerization specificity by x-ray crystallography and a sequence-comparison algorithm: family values (FamVal). Proc. Natl. Acad. Sci. USA. 100:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne, J.M., C.G. Nichols, T.L. Schwarz, and D. Escande. 2001. Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ. Res. 89:944–956. [DOI] [PubMed] [Google Scholar]
- Pascual, J.M., C.C. Shieh, G.E. Kirsch, and A.M. Brown. 1997. Contribution of the NH2-terminus of Kv2.1 to channel activation. Am. J. Physiol. 273:C1849–C1858. [DOI] [PubMed] [Google Scholar]
- Rozanski, G.J., and Z. Xu. 2002. Sulfhydryl modulation of K+ channels in rat ventricular myocytes. J. Mol. Cell. Cardiol. 34:1623–1632. [DOI] [PubMed] [Google Scholar]
- Shahidullah, M., and M. Covarrubias. 2003. The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys. J. 84:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah, M., T. Harris, M.W. Germann, and M. Covarrubias. 2003. Molecular features of an alcohol binding site in a neuronal potassium channel. Biochemistry. 42:11243–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, N.V., X. Chen, M.M. Boyer, and P.J. Pfaffinger. 1993. Deletion analysis of K+ channel assembly. Neuron. 11:67–76. [DOI] [PubMed] [Google Scholar]
- Shin, K.S., C. Maertens, C. Proenza, B.S. Rothberg, and G. Yellen. 2004. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron. 41:737–744. [DOI] [PubMed] [Google Scholar]
- Smith, J.J., D.W. Conrad, M.J. Cuneo, and H.W. Hellinga. 2005. Orthogonal site-specific protein modification by engineering reversible thiol protection mechanisms. Protein Sci. 14:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova, O., L. Kolmakova-Partensky, and N. Grigorieff. 2001. Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure (Camb.). 9:215–220. [DOI] [PubMed] [Google Scholar]
- Strang, C., K. Kunjilwar, D. DeRubeis, D. Peterson, and P.J. Pfaffinger. 2003. The role of Zn2+ in Shal voltage-gated potassium channel formation. J. Biol. Chem. 278:31361–31371. [DOI] [PubMed] [Google Scholar]
- Webster, S.M., D. Del Camino, J.P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- Wray, D. 2004. The roles of intracellular regions in the activation of voltage-dependent potassium channels. Eur. Biophys. J. 33:194–200. [DOI] [PubMed] [Google Scholar]
- Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- Zhou, W., Y. Qian, K. Kunjilwar, P.J. Pfaffinger, and S. Choe. 2004. Structural insights into the functional interaction of KChIP1 with Shal-type K+ channels. Neuron. 41:573–586. [DOI] [PubMed] [Google Scholar]