

Abstract
Complement activation by targeting ligands is an important issue that governs the fate of targeted colloidal contrast agents for molecular imaging. Here, we extend previous work on a stimulus-responsive microbubble construct, in which the ligand is normally buried by a polymeric overbrush and transiently revealed by ultrasound radiation force, to show reduced complement activation and focused adhesion to cells using a physiological peptide ligand. Attachment of C3/C3b in vitro and production of soluble C3a anaphylotoxin in vitro and in vivo decreased significantly for the buried-ligand architecture versus the conventional exposed-ligand motif and no-ligand control. Additionally, the buried-ligand architecture prevented adhesion of RGD-bearing microbubbles to integrin-expressing HUVEC cells when driven by buoyancy in a static chamber, but it did not affect adhesion efficiency when activated by ultrasound radiation force pulses. These results show, for the first time, the molecular mechanism for reduced immunogenicity for the buried-ligand architecture and feasibility of targeting with this motif using a physiological ligand-receptor pair.
Keywords: microbubble, immunogenicity, targeting, adhesion, RGD, αVβ3 integrin, biotin
Introduction
Ultrasound contrast agents – microbubbles comprising a hydrophobic gas and a stabilizing shell – have enabled clinical contrast echocardiography owing to their enhanced stability in circulation (e.g., see [1-4]). These agents can survive passage through the pulmonary vasculature [5] and exhibit contrast persistence on the order of tens of minutes for a typical human dose. In turn, efforts to engineer contrast-specific ultrasound pulse sequences have led to significant increases in imaging sensitivity and resolution, allowing detection and tracking of microbubbles in vivo. These advances have engendered an interest in ultrasound as a molecular imaging modality, and recent studies have demonstrated ultrasonic imaging of molecular targets associated with angiogenesis [6-9], thrombosis [10] and inflammation [11-13]. Many of the advantages of ultrasound as a molecular imaging tool have been enumerated, such as economy, safety, ease of use and facilitation of single-session, multi-target studies [2, 14-16]. An additional benefit for targeted imaging involves exploitation of microbubble oscillation in the ultrasound field. We and others have shown that oscillation can be used to remotely manipulate the microbubbles by pushing them with primary radiation force to enhance adhesion [17-20] and breaking them by fragmentation to deposit targeted nanoparticles [21].
The typical approach has been to inject ligand-bearing microbubbles, with the ligand maximally exposed to the milieu, and then allow time for the circulating microbubbles to accumulate at the target vascular site. Decorating microbubbles with targeting ligands – such as proteins, peptides or metabolites – could present chemical groups that trigger immune activation. The same holds for other colloidal constructs used as imaging contrast agents. Clearly, avoidance of an immunogenic response is desirable for targeted contrast agents, not just to minimize hypersensitivity reactions, but also to enable long enough circulation persistence for accumulation at the target. Ideally, the targeting ligand would be hidden from the milieu until the contrast agent reaches the target site, at which point it is exposed for binding and, through multiple ligand-receptor interactions, facilitates adhesion.
We recently introduced a new microbubble construct for use with ultrasound radiation force to allow triggered manipulation of the contrast agent adhesiveness [19]. Figure 1 shows a cartoon of the concept. During injection and transit to the target organ, the ligand is hidden by the buried-ligand architecture (BLA), in which the ligand is tethered to the microbubble surface by poly(ethylene glycol) (PEG) and is buried in a PEG overbrush (Fig. 1A). We hypothesized that burying the ligand would conceal it from blood components, such as opsonins and phagocytes. We showed that the buried-biotin architecture (compared to exposed-biotin) on microbubbles reduced adhesion to an avidinated surface during shear flow in a parallel-plate flow chamber, reduced adhesion to resting or activated human neutrophils in vitro, and increased ultrasound contrast persistence in the rat kidney. Panel B shows the concept of revealing the ligand; targeted adhesion is achieved by applying an ultrasound radiation force (USRF) pulse. We hypothesized that ultrasound-induced microbubble oscillation – the forward movement of the bubble’s center of mass coupled with the expansion of the shell – would reveal the ligand to the target epitopes. We showed that, in a hollow fiber with immobilized avidin, adhesion efficiency was completely restored within the acoustic field by application of ultrasound radiation force.
Figure 1.
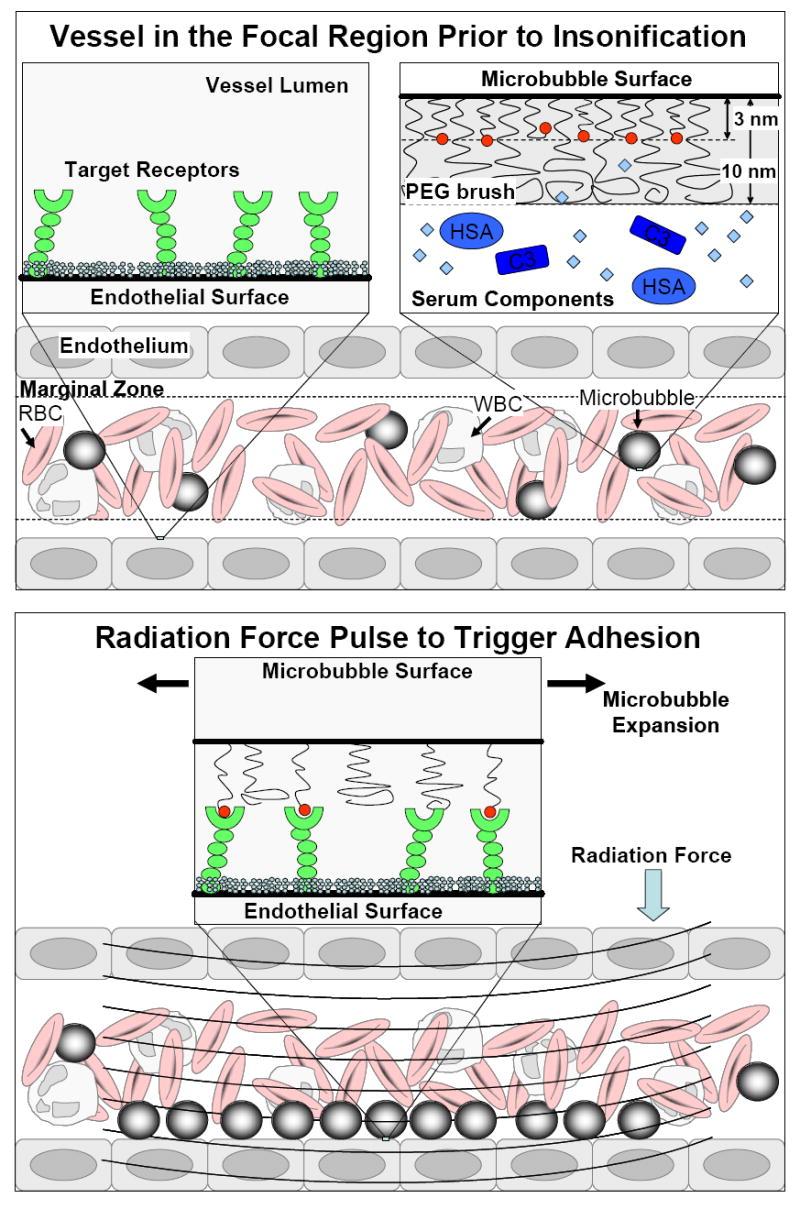
Schematic representation of non-immunogenic, targeted adhesion using ultrasound radiation force (USRF) and microbubbles engineered with the buried ligand architecture (BLA). (Top) Before the acoustic pulse: the microbubble is stealth as it travels to the site of interest. The polymeric overbrush of the BLA shields the ligand from serum components, such as complement proteins, red blood cells (RBCs) and white blood cells (WBCs). (Right) During the acoustic pulse, ultrasound radiation force pushes the microbubble against the endothelium and oscillation reveals the ligand for binding to the target receptor. Multiple ligand-receptor interactions result in targeted adhesion, but only within the beam focus.
However, several questions remained open: Does the BLA actually reduce immunogenicity? Can the BLA conceal a higher molecular weight, peptide ligand? Can targeted adhesion be achieved using the BLA with a physiological ligand-receptor pair? At the heart of these questions lie complex details of the collective molecular interactions at play between the microbubble surface, soluble blood components and the surface of the target endothelial cells.
The purpose of the present study was to experimentally test immunogenicity and ultrasound-targeted adhesion of buried-ligand microbubbles bearing biotin or a common physiological ligand, cyclic Arg-Gly-Tyr (RGD). Biotin represents a class of smaller metabolite ligands, while RGD represents a class of larger peptide ligands. RGD is commonly used to target integrins, such as αVβ3, which is known to be associated with angiogenesis in cancer [22]. Both types of ligands are important for molecular targeting; their shielding and binding capacity – which are based upon molecular mass, chemical structure and polymer dynamics – are expected to be of general interest. Addressing these two issues is key to the elucidation of the molecular mechanisms at play and the assessment of this technology for in vivo molecular imaging.
Materials and Methods
Design of the surface architecture
The lipid shell was engineered in such a way as to achieve the desired vertical segregation to conceal the ligand on the static bubble, and then to reveal the ligand on the oscillating bubble. The analytical expressions governing the microbubble surface architecture were derived from self-consistent field theory [23], and are as follows:
| (1) |
| (2) |
| (3) |
where H0, HS, and HL are the monomodal, short and long chain brush heights; p is the chain stretching parameter (assumed unity for the flexible mPEG); ν is the second virial coefficient (assumed unity for PEG in water at standard pressure and temperature); σ is the normalized surface coverage; NS is the persistence length of the short chains; q is the fraction of long chains and α is the relative difference in chain lengths. The brush heights are equilibrium, time-averaged values. However, the brush is not a static structure; individual polymer chains are always stretching and recoiling under thermal motion, as described by Jeppesen et al. [24]. Importantly, the above analysis does not include intermolecular interactions or mass effects of the ligand moieties attached to a portion of the mPEG chains.
Figure 3 shows a schematic representation of the exposed-ligand and buried-ligand architectures for the RGD and biotin ligands used in this study. The calculated brush heights assume that the lipids are present in their bulk fractions and mix uniformly throughout the monolayer shell. We previously showed that the former assumption is valid [25]. However, we also observed phase separation in the monolayer shell between the PEGylated and lecithin species, and thus the calculated brush heights should be considered as time and spatial averages with local variations due to surface heterogeneities.
Figure 3.
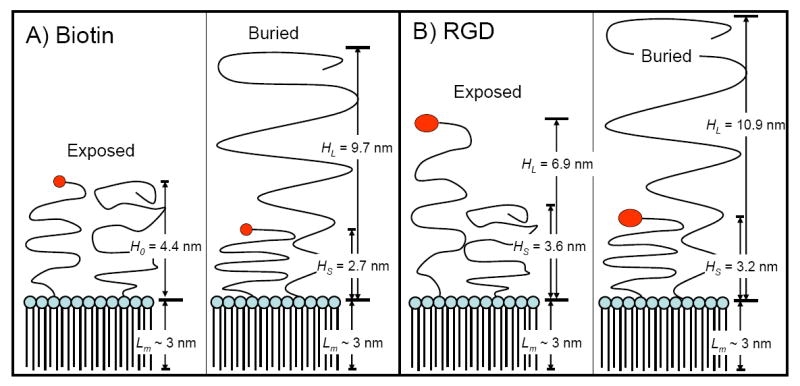
Schematic representation of the surface architectures used in this study for biotin (A) and RGD (B). Brush heights were calculated from Eqns. 1-3 using values of 0.45 nm2 for the average area per lipid molecule and 0.35 nm for the PEG monomer length. The monolayer thickness (Lm) was estimated to be ~ 3 nm from the persistence length of the stearoyl chains [1]. See text for details.
1. Israelachvili J (1992). Intermolecular and Surface Forces. 2nd ed: Academic Press.
Materials
The lipids 1,2-Distearoyl-sn-Glycero-3-Phosphocholine (DSPC), 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-2000] (DSPE-mPEG2000), 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-5000] (DSPE-mPEG5000) and 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Biotinyl(Polyethylene Glycol)2000] (DSPE-PEG2000-biotin) were purchased from Avanti Polar Lipids (Alabaster, AL). Figure 2 shows the lipopolypeptide (LPP), which was synthesized in house by solid phase peptide synthesis. The chemical structure of the bioconjugate peptide was cyclo(Arg-Gly-Tyr(Me)-Lys-Glu) (i.e., c(RGDY(Me)KE)). Final product was purified with HPLC (Beckman Coulter) and molecular weights were confirmed with MALDI-TOF (Applied Biosystems).
Figure 2.
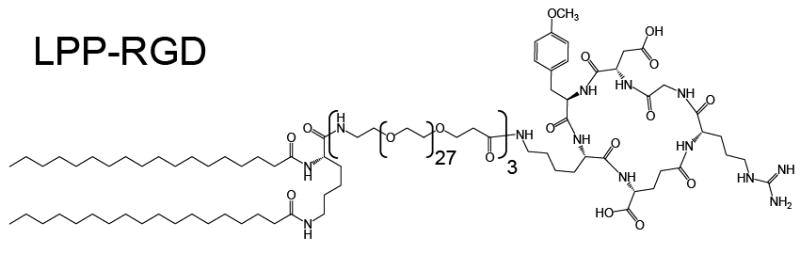
Molecular structure of the lipopolypeptide (LPP) with the cyclic RGD bioconjugate ligand.
Human serum was obtained from two sources. Fresh serum was isolated from healthy human volunteers according to the University of California, Davis, approved human subjects protocol (protocol identification 993120). Complement-preserved human serum was purchased from Valley Biomedical (catalog no. HS1004HC, Winchester, VA). Purchased serum was thawed once to aliquot into 10-mL vials and then refrozen and stored at -80 °C. Serum was thawed and/or stored on ice prior to experiments.
Microbubble preparation
Microbubbles were prepared as previously described [26]. All lipid compositions contained 90 mol% DSPC and 10 mol% PEGylated species. The compositions were 10 mol% DSPE-mPEG2000 for control; 5 mol% DSPE-PEG2000-biotin and either 5 mol% DSPE-mPEG5000 or 5 mol% DSPE-mPEG2000 for the buried-biotin or exposed-biotin architecture, respectively; or 2 mol% LPP-RGD and either 8 mol% DSPE-mPEG5000 or 8 mol% DSPE-mPEG2000. To make the precursor solution, the lipids were mixed in chloroform, dried and suspended in phosphate buffered saline (PBS, Dubelco) containing Mg2+ and Ca2+ to 1.0 mg-lipid/mL. The precursor solution was then transferred to 3-mL serum vials and sealed, and the headspace was exchanged with perfluorobutane. Microbubbles were formed by shaking with a VialMix (ImaRx Therapeutics, Tucson, AZ) for 45 sec. Microbubbles were then diluted to 10 mL PBS and washed three times by centrifugation-flotation in a bucket-rotor centrifuge at 300×g for 3 minutes. Sizing and measuring concentration was performed using an Accusizer optical particle counter (Particle Sizing Systems, Santa Barbara, CA).
Human complement C3 binding to the microbubble surface
Microbubbles were diluted in a syringe to ~107 per mL in 3 mL PBS and mixed with 2 mL serum. The mixture was incubated on a benchtop rotator at room temperature for 30 min. After incubation, the mixture was centrifuged at 300×g for 3 minutes to isolate microbubbles. The infranatant was saved for analysis of C3a content (see below). The microbubble cake was re-suspended into PBS and washed three times. Microbubbles were then mixed with a 2-mL solution (25 μg/mL) of fluorescently tagged antibody to human C3/C3b (CL2103F, Cedarlane, Hornby, Ontario), incubated for 30 min, and washed three times. The presence of bound antibody to the microbubble surface was visually confirmed by epi-fluorescence microscopy (Mikron Instruments, San Marcos, CA) and imaged using a digital camera (Cascade 512B, Roper Scientific) controlled by Simple PCI software (Compix). Median fluorescence intensity per microbubble was determined by FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ).
Human anaphalatoxin C3a production in vitro
The infranatant obtained after incubation in human serum and centrifugation-flotation (see above) was diluted 1:3 in 20 mM EDTA and stored at -80 °C. The amount of C3a per sample was assayed using an ELISA kit purchased from BD Biosciences (Cat# 550499) following the manufacturer’s instructions.
Mouse anaphylatoxin C3a production in vivo
Sixteen adult C3H mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed at facilities provided by the Experimental Mouse Shared Services at the Arizona Cancer Center, Tucson. Mice were divided into four groups (n=4 per group), including blank (PBS only), control (no-ligand), exposed-biotin or buried-biotin. Each mouse received a 0.150-mL tail vein injection of the indicated solution. Microbubbles were freshly prepared and injected without further dilution or washing for each experiment. Five minutes after injection, blood was taken by a retro-orbital bleed (~0.1 mL blood) and immediately chilled on ice. Serum was isolated by centrifugation and stored at -80 °C. Experiments were repeated once each week for a total of five weeks. Animals were routinely observed for hypersensitivity reactions.
Mouse serum samples were warmed to room temperature and pooled for each indication and for each week (n=5 per group). Anaphylatoxin C3a was assayed by ELISA using primary and secondary antibodies (cat. #’s 558250 and 558251) and standard reagents from BD Biosciences.
Targeted adhesion of RGD-microbubbles to plated HUVEC
Human umbilical vein endothelial cells (HUVEC) were received from Cascade Biologics as proliferating cultures. Cells were grown in HUVEC media (Medium 200, cat. # M-200-500) with 8% Low Serum Growth Supplement (cat. # S-003-10, 2% FBS) to ~50% confluence in 5% CO2/95% air, at 37°C. Once cells reached the desired confluence, they were subcultured using 0.25% Trypsin/EDTA (Invitrogen, cat. # 25200-056). Trypsin was neutralized with Trypsin Neutralizing Solution (Cascade Biologics, cat. # R-002-100). Cells were collected in a 15 mL conical tube and centrifuged at 180×g for 7 minutes. Once pelleted, supernatant is removed and cells were frozen down in a 10% DMSO/20% FBS/70% M-200 media solution. Cells are plated from frozen stocks in HUVEC media at a density of 2,500 cells/cm2 onto glass gelatin-coated coverslips. HUVEC cells have been shown to express the cell adhesion integrin αVβ3, which binds with high affinity to disintegrins containing cyclic RGD [27].
Prior to microbubble production, 0.001 mL of 1-mM DiI(C18) solution (Vybrant, Invitrogen) was added per mL of lipid precursor solution to fluorescently tag the microbubble shells. Following washing, microbubbles were diluted to ~107 per mL and added to an acoustic chamber as previously described [28]. In static adhesion experiments, the chamber was inverted to allow the microbubbles to rise by buoyancy to the cell plate for 5 min. In acoustic experiments, the chamber was placed upright in a custom water bath that allowed application of ultrasound radiation force pulses at the focus of a single piston transducer. The setup was previously described [29]. Two sequential 5-sec bursts were applied at 2.5 MHz and 40 kPa peak negative pressure (PNP). The pulse was generated by an arbitrary waveform generator (AWG2021, Tecktronics, Beaverton, OR) attached to a radio-frequency amplifier (325LA, ENI, Rochester, NY) and 2.25-MHz, single-element transducer with a 2-inch focus and 0.75-inch diameter (V305, Panametrics, Waltham, MA). Following treatment, the acoustic chambers were dissembled, and the cells were washed by gently flowing PBS over the plate surface. Adhesion of microbubbles was observed and imaged by epi-fluorescence and bright-field microscopy. Images were analyzed for adhesion density using ImageJ (http://rsb.info.nih.gov/ij) by drawing a region of interest within the transducer focal zone and measuring the area fraction of bright pixels from the dark mode slice.
Statistics
Paired, two-tailed Student’s t-tests were performed to test the significance of difference in the means between the buried-ligand, exposed-ligand and control microbubbles.
Results
Microbubble generation
For each set of lipid components, vial activation by shaking produced a milky, white suspension of microbubbles that was stable over the experimental timeframe. Figure 4 shows that, following three washing steps, the size distribution for each shell composition was centered at ~1.8 μm diameter with secondary peaks at ~4.5 and ~7.5 μm diameter and a small sub-micron bubble population. For both biotin and RGD, the exposed-ligand architecture resulted in a significant decrease in microbubble concentration in comparison to the buried-ligand or the control. The buried-ligand composition, however, yielded concentrations similar to the control. Finally, shaking yielded more biotin-microbubbles than RGD-microbubbles.
Figure 4.
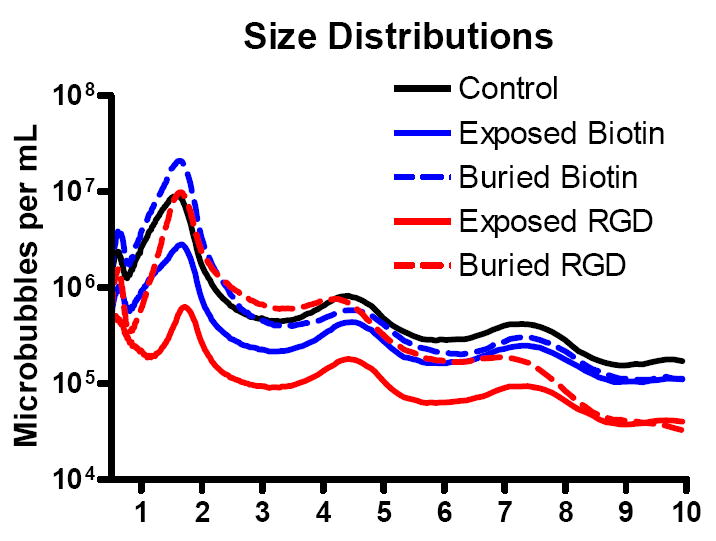
Average production yields of microbubbles with different surface architectures. Measurements were taken after three washing steps and are normalized to 1-mL of the precursor solution.
Binding of human C3/C3b to the microbubble shell
Microbubbles of each composition were stable during incubation in human serum in vitro and could be rapidly isolated and washed by centrifugation-flotation to allow observation by microscopy and flow cytometry. Detection of the fluorescent antibody allowed an assessment of complement protein C3/C3b binding to the microbubble shells. Figure 5 shows a typical exposed-biotin microbubble exhibiting a heterogeneous distribution of fluorescent antibody over the surface. Similar patchy fluorescence was observed on all such microbubbles that were large enough (>3 μm diameter) to examine the surface microstructure by optical microscopy.
Figure 5.
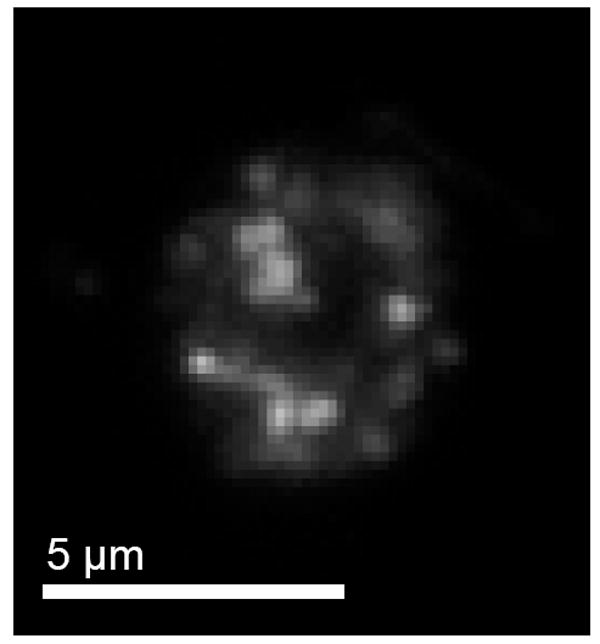
Micrograph showing the distribution of fluorescent antibody bound to C3/C3b on the surface of a typical microbubble with the exposed-biotin surface architecture.
Flow cytometry histograms typically gave log-normal distributions in fluorescence intensity, although bimodal distributions were occasionally observed (e.g., see buried-biotin plot in Fig. 6A). Shifting of the distribution to the right indicated an increase in fluorescent antibody binding to C3/C3b on the surface. For both ligands, the median fluorescence intensity (MFI) increased significantly for the exposed-ligand microbubbles versus the buried-ligand or the control. The MFI increased only slightly, although significantly, for the buried-ligand compared to the control.
Figure 6.
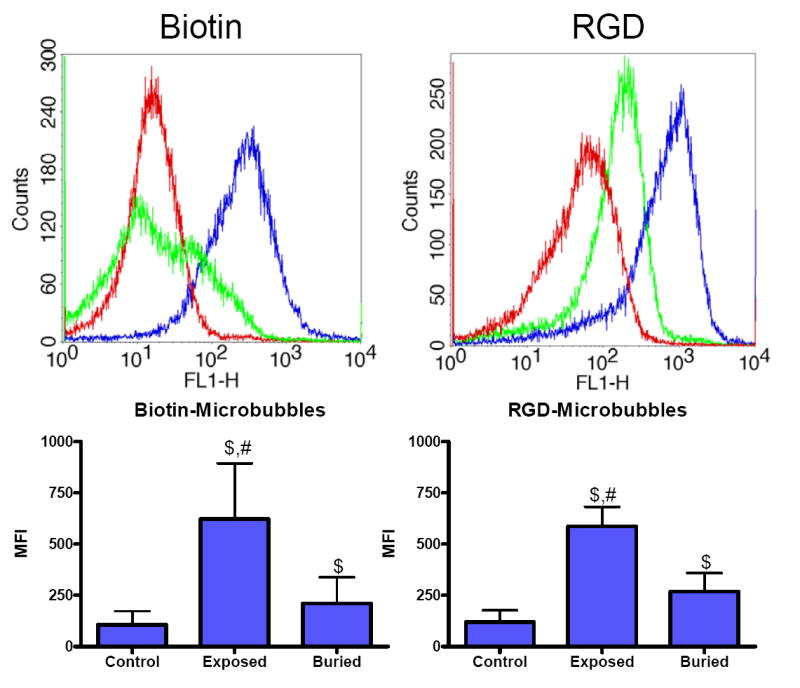
Flow cytometric analysis of human C3/C3b bound to microbubbles with different surface architectures. (Top) Selected histograms showing control (red), exposed-ligand (blue) and buried-ligand (green) architectures. (Bottom) Median fluorescent intensities (±SD), where “$” denotes a significant increase versus control and “#” indicates an increase over buried-ligand (p<0.05).
Anaphylatoxin C3a production
ELISA analysis of the infranatant taken after centrifuging the microbubbles in serum allowed determination of C3a content. Figure 7 shows measurements of C3a in human and mouse sera after exposure to biotin-bearing microbubbles (vs. blank and control) for experiments in vitro and in vivo. Experiments in freshly isolated human serum showed a significant increase in C3a production for the exposed-biotin microbubbles versus buried-biotin or control microbubbles (Fig. 7A). A similar trend was observed for C3a in mouse blood 5 minutes after intravenous injection (Fig. 7B); anaphylatoxin production increased significantly for exposed-biotin microbubbles compared to control or buried-biotin microbubbles. Figure 7 also shows that the presence of control microbubbles did not significantly increase C3a present in isolated human serum (versus blank), although it did result in a significant increase in C3a present in the mouse circulation. Similarly, a significant increase in C3a was measured for buried-biotin versus control in mice, but not in isolated human serum. It is important to note that, during the tail vein injections, the microbubbles were often destroyed owing to high pressure exerted on the syringe plunger, as observed by a sharp decrease in the optical turbidity. In general, the mice did not display adverse hypersensitivity reactions to any of the treatments, although one mouse was removed from the study due to infection from the retro-orbital bleed.
Figure 7.
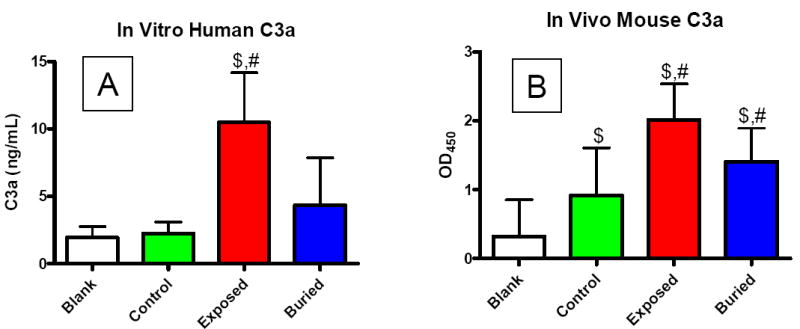
ELISA results of C3a production (mean ±SD) for the different surface architectures and biotin as the ligand. (A) Anaphalatoxin production in human serum following 30 min incubation in vitro. (B) Anaphalatoxin production in mouse serum measured 5 min after intravenous injection in vivo. “$” denotes statistical significance over blank, and “#” denotes statistical significance over control (p<0.05).
RGD-microbubble adhesion to Huvec cells
The acoustic chambers allowed direct visualization and quantification of microbubble adhesion to plated HUVEC cells. We examined adhesion due to buoyancy and ultrasound radiation force (USRF). Figure 8 shows a typical micrograph of the acoustic focus during USRF-assisted targeting. Panel A shows a punctate fluorescent spot where buried-RGD microbubbles have adhered. The circle denotes the predicted area of the main lobe (out to zero pressure) of the acoustic field. The focal region was clearly defined by the presence of the adherent microbubbles, with very sparse adhesion in the outlying areas. Panels B and C are magnified regions of the same image showing that the fluorescence regions correlated with the presence of microbubbles, as clearly defined in the bright field. Microbubbles remained intact after insonification and often were found in clusters on the surfaces of individual cells. Microbubbles were absent on the cell-free regions.
Figure 8.

Micrographs of fluorescent, RGD-coated microbubbles with the buried-ligand architecture adherent to Huvec cells after two 5-sec bursts at 2.5 MHz, 40 kPa PNP. Huvec cells were approximately 50% confluent. (A) Mosaic image showing entire transducer focal area; circle denotes the entire main lobe of the acoustic field. (B) Magnified epi-fluorescent image corresponding to the box in A. (C) Magnified bright field image corresponding to box in B. Dark spheres are individual microbubbles and clusters.
Figure 9 shows quantitative results from the adhesion experiments, in which the area fraction of fluorescence was determined within a region of interest near the center of the transducer focal region (or near the center of the plate in static experiments). For exposed-RGD, the amount of microbubbles that were driven to adhesion by either buoyancy or USRF was approximately equal. In contrast, a large difference in adhesion was observed for the buried-RGD microbubbles when driven by buoyancy versus acoustic radiation force. In the static experiments, very few buried-RGD microbubbles were observed to adhere. In the acoustic experiments, however, buried-RGD microbubbles were found to adhere at a high density, similar to the exposed-RGD case.
Figure 9.
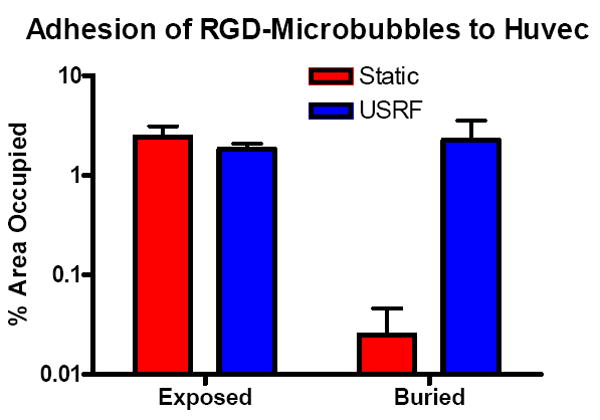
Area fractions (mean ±SD) of fluorescent microbubbles on plated Huvec cells (~50% confluence) within a circular region of interest near the center of the plate (~2-mm diameter). Shown are static adhesion results, where microbubbles were allowed to adhere owing to buoyancy force during a 5-min incubation. Also shown are USRF-assisted adhesion results, where microbubbles were pushed within the transducer focus as shown in Figure 7.
Discussion
The Effect of Ligand Exposure on Immunogenicity
The immunity pathways converge on the complement protein C3, which is converted to the fragments C3b and C3a by C3-convertase [30]. The complement system is activated when C3b binds to the surface of a foreign particle. Immobilized C3b can be recognized by phagocytic cells, and it can interact with the other complement proteins to stimulate humoral immunity and form the membrane attack complex. This poses a significant challenge for engineering therapeutic devices for injection or transplantation [31-33]. Particle surfaces with exposed nucleophilic groups (hydroxyl, amine, thiol etc.) are vulnerable to C3b attachment, which in turn leads to rapid particle clearance by the reticuloendothelial system (RES). Limiting C3b attachment, therefore, is crucial to reducing immunogenicity and avoiding premature clearance by the RES.
The exposed-ligand architecture led to an increase in complement activity compared to the no-ligand control, for both biotin and RGD. This is evidenced by the increase in antibody binding to C3/C3b on the microbubble surface (Fig. 6) and the increase in soluble C3a anaphylatoxin (Fig. 7). We suggest that this effect is due to chemical groups on the ligands that interact directly with C3 molecules, or an intermediate opsonin [34], which leads to C3 immobilization and complement activation. C3 contains an unstable thioester bond [31] and has been shown to bind well to surfaces with exposed hydroxylated and other nucleophilic groups [30, 35]. Our ligands contained such groups, such as the sulfur on the tetrahydrothiophene ring of biotin, or the carbonyl group on the aspartate of RGD (Fig. 2).
Buried Ligand Architecture
The buried-ligand architecture renders the targeted microbubble less immunogenic, as evidenced by the reduction in C3/C3b binding to the microbubble shell (Fig. 6), for both biotin and RGD, as well as the decrease in anaphylatoxin C3a production for biotin (Fig. 7). The latter result for the in vivo study is particularly telling (Fig. 7B), because the buried-ligand microbubbles were approximately an order of magnitude more concentrated than their exposed-ligand counterparts (Fig. 4). These results support our hypothesis that the ligand itself is immunogenic, and that shielding it from serum factors by an intervening mPEG brush renders the particle as stealth (as depicted in Fig. 1A). The shielding is brought about by vertical segregation in the bimodal brush architecture (Fig. 3), which is the entropically favored configuration [23, 36]. In order to penetrate the brush and bind to the ligand, the C3 macromolecule (~185 kDa) must overcome osmotic and steric repulsive forces [37]. Shielding of the immunogenic moiety also explains our previous results of decreased neutrophil adhesion and increased circulation persistence compared to the exposed-ligand architecture [19].
The buried-ligand architecture was not completely stealth. Transient extensions of the ligand tethers past the overbrush layer were expected [24], which could explain the slight increase in C3/C3b binding and C3a production in comparison to the no-ligand control (Fig. 6). Also, microstructural defects in the shell could have rendered the ligand accessible at certain locations (e.g., domain edges). This latter scenario is supported by the heterogeneous fluorescence observed on the shell (Fig. 5).
Ligand-Receptor Pairs by Insonation
Microbubbles bearing the physiological ligand, cyclic RGD, in the buried-ligand architecture were able to firmly adhere to cells displaying the target receptor, αVβ3. Remarkably, adhesion was only observed when the microbubbles were pushed by ultrasound radiation force for 10 seconds, but not when driven by buoyancy for 5 minutes (Fig. 9). Moreover, adherent microbubbles were at approximately the same density as for the exposed-RGD case. This supports our hypothesis that the ligand could be transiently revealed to achieve firm adhesion by the application of ultrasound radiation force (as depicted in Fig. 1B). Thus, we have a molecular probe that can be reversibly switched from stealth to sticky, which should prove useful in applications where it is important that the tracer remain inert to tangential physiological processes, such as avoiding inflammation in angiogenesis detection.
Another interesting property is that the adhesion only occurred within the transducer focal zone (Fig. 8). This yields the possibility of enhancing specificity through focusing of the ultrasound beam. Thus, contrast agents bearing more generic (less specific) ligands could be used. This architecture may also enhance molecular imaging techniques employing pulse sequences engineered to delineate free from adherent agents in real time as the tissue is being scanned [38].
Conclusions
We showed that the buried-ligand architecture reduces ligand-mediated immunogenicity of microbubbles, as observed by a decrease in C3/C3b binding and anaphylatoxin C3a production. On the other hand, application of ultrasound radiation force reveals the ligand and facilitates targeted adhesion within the focal region. Importantly, these effects have been shown here, for the first time, for microbubbles bearing the physiological ligand (RGD) binding to cells expressing the target epitope, αVβ3 integrin. These results prove the concept of a stimulus-responsive contrast agent that should prove useful in future ultrasound molecular imaging studies.
Molecular imaging is then performed by detecting adherent microbubbles in the beam focus. The advantage of this technique is that it gave nearly instantaneous adhesion only within the beam focus (i.e., focused adhesion), with the contrast agent remaining unbound in all regions outside the beam. The buried-ligand (uninsonified) form of the contrast agent was non-immunogenic.
Acknowledgments
We thank Bethany Skovan for mouse handling and Kate Watson for cell culture. We acknowledge funding for this work from NIH R01 CA097360 to RJG and NIH R01 CA103828 to KWF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindner JR. Microbubbles in medical imaging: current applications and future directions. Nat Rev Drug Discovery. 2004;3:527–532. doi: 10.1038/nrd1417. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara KW, Pollard RE, Borden MA. Ultrasound microbubble contrast agents: fundamentals and application to drug and gene delivery. An Rev Biomed Engr. 2007;9:415–447. doi: 10.1146/annurev.bioeng.8.061505.095852. [DOI] [PubMed] [Google Scholar]
- 3.Feinstein SB. The powerful microbubble: from bench to bedside, from intravascular indicator to therapeutic delivery system, and beyond. Am J Physiol. 2004;287:H450–H457. doi: 10.1152/ajpheart.00134.2004. [DOI] [PubMed] [Google Scholar]
- 4.Tsutsui JM, Kusler M, Porter TR. Intravenous myocardial contrast echocardiography for the diagnosis of coronary artery disease. Curr Opin Cardiol. 2005;20:381–385. doi: 10.1097/01.hco.0000176404.48496.a2. [DOI] [PubMed] [Google Scholar]
- 5.Lindner JR, Song J, Jayaweera AR, Sklenar J, Kaul S. Microvascular rheology of definity microbubbles after intra-arterial and intravenous administration. J Am Soc Echocardiography. 2002;15:396–403. doi: 10.1067/mje.2002.117290. [DOI] [PubMed] [Google Scholar]
- 6.Ellegala DB, Leong-Poi H, Carpenter JE, Klibanov AL, Kaul S, Shaffrey ME, Sklenar J, Lindner JR. Imaging tumor angiogenesis with contrast ultrasound and microbubbles targeted to alpha(v)beta3. Circulation. 2003;108:336–41. doi: 10.1161/01.CIR.0000080326.15367.0C. [DOI] [PubMed] [Google Scholar]
- 7.Leong-Poi H, Christiansen J, Klibanov AL, Kaul S, Lindner JR. Noninvasive assessment of angiogenesis by ultrasound and microbubbles targeted to alpha(v)-integrins. Circulation. 2003;107:455–60. doi: 10.1161/01.cir.0000044916.05919.8b. [DOI] [PubMed] [Google Scholar]
- 8.Weller GER, Wong MKK, Modzelewski RA, Lu EX, Klibanov AL, Wagner WR, Villanueva FS. Ultrasonic imaging of tumor angiogenesis using contrast microbubbles targeted via the tumor-binding peptide arginine-arginine-leucine. Cancer Res. 2005;65:533–539. [PubMed] [Google Scholar]
- 9.Stieger SM, Dayton PA, Borden MA, Caskey CF, Griffey SM, Wisner ER, Ferrara KW. Imaging of Angiogenesis Using Cadence Contrast Pulse Sequencing and Targeted Contrast Agents. 2007 doi: 10.1002/cmmi.224. Submitted. [DOI] [PubMed] [Google Scholar]
- 10.Unger E, Metzger P, 3rd, Krupinski E, Baker M, Hulett R, Gabaeff D, Mills J, Ihnat D, McCreery T. The use of a thrombus-specific ultrasound contrast agent to detect thrombus in arteriovenous fistulae. Invest Radiol. 2000;35:86–9. doi: 10.1097/00004424-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Lindner JR, Song J, Christiansen J, Klibanov AL, Xu F, Ley K. Ultrasound assessment of inflammation and renal tissue injury with microbubbles targeted to P-selectin. Circulation. 2001;104:2107–2112. doi: 10.1161/hc4201.097061. [DOI] [PubMed] [Google Scholar]
- 12.Song J, Christiansen J, Klibanov AL, Ley K, Kaul S, Lindner JR. Adhesion of P-selectin-targeted microbubbles to venules: Implications for imaging inflammation. J Am Coll Cardiol. 2001;37:383A–383A. [Google Scholar]
- 13.Linker RA, Reinhardt M, Bendszus M, Ladewig G, Briel A, Schirner M, Maurer M, Hauff P. In vivo molecular imaging of adhesion molecules in experimental autoimmune encephalomyelitis (EAE) J Autoimmun. 2005;25:199–205. doi: 10.1016/j.jaut.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 14.Kaufmann BA, Lindner JR. Molecular imaging with targeted contrast ultrasound. Curr Opin Biotech. 2007;18:11–16. doi: 10.1016/j.copbio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Dayton PA, Ferrara KW. Targeted imaging using ultrasound. J Magn Reson Imaging. 2002;16:362–377. doi: 10.1002/jmri.10173. [DOI] [PubMed] [Google Scholar]
- 16.Bloch SH, Dayton PA, Ferrara KW. Targeted imaging using ultrasound contrast agents: progress and opportunities for clinical and research applications. IEEE Eng Med Biol Mag. 2004;23:18–29. doi: 10.1109/memb.2004.1360405. [DOI] [PubMed] [Google Scholar]
- 17.Dayton PA, Morgan KE, Klibanov ALS, Brandenburger G, Nightingale KR, Ferrara KW. A preliminary evaluation of the effects of primary and secondary radiation forces on acoustic contrast agents. IEEE Trans Ultrason, Ferroelect, Freq Contr. 1997;44:1264–1277. [Google Scholar]
- 18.Zhao S, Borden MA, Bloch S, Kruse D, Ferrara KW, Dayton PA. Radiation-force assisted targeting facilitates ultrasonic molecular imaging. Molecular Imaging. 2004;3:135–148. doi: 10.1162/1535350042380317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borden MA, Sarantos MR, Stieger SM, Simon SI, Ferrara KW, Dayton PA. Ultrasound Radiation Force Modulates Ligand Availability on Targeted Contrast Agents. Molecular Imaging. 2006;5:139–147. [PMC free article] [PubMed] [Google Scholar]
- 20.Rychak JJ, Klibanov AL, Hossack JA. Acoustic radiation force enhances targeted delivery of ultrasound contrast microbubbles: in vitro verification. IEEE Trans Ultrason, Ferroelect, Freq Contr. 2005;52:421–433. doi: 10.1109/tuffc.2005.1417264. [DOI] [PubMed] [Google Scholar]
- 21.Lum AFH, Borden MA, Dayton PA, Kruse DE, Simon SI, Ferrara KW. Ultrasound radiation force enables targeted deposition of model drug carriers loaded on microbubbles. J Control Release. 2006;111:128–134. doi: 10.1016/j.jconrel.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks PC, Clark RAF, Cheresh DA. Requirement of Vascular Integrin Alpha(V)Beta(3) for Angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 23.Lai PY, Zhulina EB. Structure of a Bidisperse Polymer Brush - Monte-Carlo Simulation and Self-Consistent Field Results. Macromolecules. 1992;25:5201–5207. [Google Scholar]
- 24.Jeppesen C, Wong JY, Kuhl TL, Israelachvili JN, Mullah N, Zalipsky S, Marques CM. Impact of polymer tether length on multiple ligand-receptor bond formation. Science. 2001;293:465–468. doi: 10.1126/science.293.5529.465. [DOI] [PubMed] [Google Scholar]
- 25.Borden MA, Martinez GV, Ricker J, Tsvetkova N, Longo M, Gillies RJ, Dayton PA, Ferrara KW. Lateral phase separation in lipid-coated microbubbles. Langmuir. 2006;22:4291–4297. doi: 10.1021/la052841v. [DOI] [PubMed] [Google Scholar]
- 26.Borden MA, Kruse D, Caskey C, Zhao S, Dayton P, Ferrara K. Influence of lipid shell physicochemical properties on ultrasound-induced microbubble destruction. IEEE Trans Ultrason, Ferroelect, Freq Contr. 2005;52:1992–2002. doi: 10.1109/tuffc.2005.1561668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juliano D, Wang YQ, Marcinkiewicz C, Rosenthal LA, Stewart GJ, Niewiarowski S. Disintegrin interaction with alpha(v)beta(3) integrin on human umbilical vein endothelial cells: expression of ligand-induced binding site on beta(3) subunit. Experimental Cell Research. 1996;225:132–142. doi: 10.1006/excr.1996.0164. [DOI] [PubMed] [Google Scholar]
- 28.Dayton PA, Zhao SK, Bloch SH, Schumann P, Penrose K, Matsunaga TO, Zutshi R, Doinikov A, Ferrara KW. Application of ultrasound to selectively localize nanodroplets for targeted imaging and therapy. Molecular Imaging. 2006;5:160–174. [PMC free article] [PubMed] [Google Scholar]
- 29.Shortencarier MJ, Dayton PA, Bloch SH, Schumann PA, Matsunaga TO, Ferrara KW. A method for radiation-force localized drug delivery using gas-filled lipospheres. IEEE Trans Ultrason, Ferroelect, Freq Contr. 2004;51:822–831. doi: 10.1109/tuffc.2004.1320741. [DOI] [PubMed] [Google Scholar]
- 30.Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 31.Janssen BJC, Huizinga EG, Raaijmakers HCA, Roos A, Daha MR, Nilsson-Ekdahl K, Nilsson B, Gros P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437:505–511. doi: 10.1038/nature04005. [DOI] [PubMed] [Google Scholar]
- 32.Kazatchkine MD, Carreno MP. Activation of the Complement-System at the Interface between Blood and Artificial Surfaces. Biomaterials. 1988;9:30–35. doi: 10.1016/0142-9612(88)90066-x. [DOI] [PubMed] [Google Scholar]
- 33.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27:4356–4373. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 34.Andersson J, Ekdahl KN, Lambris JD, Nilsson B. Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials. 2005;26:1477–1485. doi: 10.1016/j.biomaterials.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Sperling C, Maitz MF, Talkengerger S, Gouzy M, Groth T, Werner C. In vitro blood reactivity to hydroxylated and non-hydroxylated polymer surfaces. Biomaterials. 2007;28:3617–3625. doi: 10.1016/j.biomaterials.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 36.Dan N, Tirrell M. Effect of Bimodal Molecular-Weight Distribution on the Polymer Brush. Macromolecules. 1993;26:6467–6473. [Google Scholar]
- 37.Kuhl TL, Leckband DE, Lasic DD, Israelachvili JN. Modulation Of Interaction Forces Between Bilayers Exposing Short-Chained Ethylene-Oxide Headgroups. Biophys J. 1994;66:1479–1488. doi: 10.1016/S0006-3495(94)80938-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao S, Kruse DE, Ferrara KW, Dayton PA. Selective imaging of adherent targeted ultrasound contrast agents. Phys Med Biol. 2007;52:2055–2072. doi: 10.1088/0031-9155/52/8/002. [DOI] [PMC free article] [PubMed] [Google Scholar]