

Summary
Hutchinson-Gilford progeria syndrome (HGPS) is a progeroid syndrome characterized by multiple aging-like disease phenotypes. We recently reported that a protein farnesyltransferase inhibitor (FTI) improved several disease phenotypes in mice with a HGPS mutation (LmnaHG/+). Here, we investigated the impact of an FTI on the survival of LmnaHG/+ mice. The FTI significantly improved the survival of both male and female LmnaHG/+ mice. Treatment with the FTI also improved body weight curves and reduced the number of spontaneous rib fractures. This study provides further evidence for a beneficial effect of an FTI in HGPS.
Keywords: progeria, aging, protein farnesyltransferase inhibitor, knock-in mice
Introduction
Children with Hutchinson-Gilford progeria syndrome (HGPS) appear normal at birth but then exhibit retarded growth, osteolytic lesions in bones, osteoporosis, alopecia, loss of subcutaneous fat, and ultimately occlusive vascular disease and premature death [1]. HGPS is caused by a LMNA point mutation that alters the splicing of the prelamin A pre-mRNA, leading to a 50-amino acid internal deletion within prelamin A [2, 3]. The mutant prelamin A protein in HGPS, often called progerin, is farnesylated and methylated at a carboxyl-terminal CaaX motif [4, 5]. Progerin is targeted to the nuclear rim, where it interferes with the integrity of the nuclear lamina and leads to misshapen nuclei [2]. Several studies have shown that the frequency of misshapen nuclei in HGPS cells is reduced by treating cells with a protein farnesyltransferase inhibitor (FTI) [4, 6-11]. Interestingly, the FTIs do not appear to have significant adverse effects on the growth of wild-type cells [11].
The FTI studies in cultured cells prompted interest in testing FTIs in mouse models of HGPS. Yang et al. [12] created a gene-targeted HGPS allele (LmnaHG) and showed that heterozygous mice (LmnaHG/+) develop a host of progeria-like disease phenotypes (e.g., normality at birth followed by slow growth, osteolytic lesions, spontaneous rib fractures, a reduction in subcutaneous fat, and premature death). Treatment of LmnaHG/+ mice with an FTI (ABT-100) improved a number of bone phenotypes, including the number of spontaneous rib fractures [12].
The results of the initial FTI study in LmnaHG/+ mice were encouraging, but we firmly believed that additional studies in mice were essential before contemplating human clinical trials, in part because the initial FTI study did not assess the impact of the drug on the survival of LmnaHG/+ mice [12]. Also, the initial FTI study involved relatively small numbers of mice [12]. For these reasons, we set up an independent trial of FTI treatment in LmnaHG/+ mice, focusing on whether the drug would improve survival and other hallmark phenotypes of progeria.
Materials and Methods
Male and female LmnaHG/+ and Lmna+/+ mice were bred by mating female C57BL/6 mice with male chimeric mice (generated with LmnaHG/+ mouse embryonic stem cells). Because all of the mice in this study were bred from chimeras, the LmnaHG/+ and Lmna+/+ mice were genetically identical (except for the targeted mutation). Genotyping was performed by PCR with genomic DNA prepared from biopsies of the tail [6]. The mice were fed a chow diet and housed in a virus-free barrier facility with a 12-h light-dark cycle. UCLA's Animal Research Committee approved all procedures.
We administered an FTI, ABT-100 [13], to groups of 8−13 male and female LmnaHG/+ mice and 7−16 male and female Lmna+/+ mice. ABT-100 was mixed in drinking water containing 0.4% hydroxy methyl propyl cellulose and 1.0% ethanol at a concentration of 0.3 mg/ml, so as to deliver an approximate dose of 39 mg/kg/day. The vehicle consisted of drinking water with 0.4% hydroxy methyl propyl cellulose and 1.0% ethanol. The FTI was initiated at four weeks of age and was continued for up to 45 weeks of age (when the last of the LmnaHG/+ mice died). Previous studies have revealed that this dosage of ABT-100 (∼39 mg/kg/day) yields mean plasma ABT-100 concentrations of approximately 0.7 μg/ml [12, 14]. This FTI dosage did not elicit liver pathology, as judged by routine histological studies. In one series of experiments, we administered a higher concentration of ABT-100 to Lmna+/+ (n = 14) and LmnaHG/+ mice (n = 10), so as to deliver 117 mg/kg/d.
The accumulation of prelamin A in liver was assessed by western blots. Liver samples were collected and frozen in liquid nitrogen, and urea-soluble extracts were prepared and analyzed by SDS-polyacrylamide gel electrophoresis and western blotting. The antibody dilutions were 1:6000 for a rabbit anti-prelamin A antiserum [6, 7], 1:400 for a goat anti-lamin A/C antibody (Santa Cruz Biotechnology), 1:500 for a mouse anti-HDJ-2 antibody (NeoMarkers), 1:1000 for a goat anti-actin IgG (Santa Cruz Biotechnology), 1:6000 for anti-goat IgG-HRP antibody (Santa Cruz Biotechnology), 1:6000 for anti-rabbit IgG-HRP antibody (Santa Cruz Biotechnology), and 1:6000 for HRP-labeled anti-mouse IgG (Amersham Biosciences). Antibody binding was detected with the ECL Plus chemiluminescence system (Amersham) and exposure to X-ray film.
Body weights of Lmna+/+ and LmnaHG/+ mice were assessed weekly. Body weight curves were compared with repeated-measures ANOVA and the log rank test. The number of surviving male and female mice was recorded weekly and expressed as a percentage of the total number of mice. The significance of differences was determined by the Kaplan-Meier method.
At the time of death, the number of spontaneous rib fractures in LmnaHG/+ mice was documented. After opening the thoracic cavity and removing the heart and lungs, the interior of the thorax was photographed with a digital camera, and rib fractures were counted. The number of rib fractures in FTI- and vehicle-treated LmnaHG/+ mice was compared with a two-tailed Student's t test.
Results
We administered an FTI, ABT-100 (39 mg/kg/day) [13], to groups of 8−13 male and female LmnaHG/+ mice and 7−16 male and female Lmna+/+ mice. As expected, the FTI treatment interfered with the biogenesis of mature lamin A and resulted in an increased amount of nonfarnesylated prelamin A, as judged by western blots on liver extracts from FTI-treated mice (Fig. 1). The FTI treatment also blocked the farnesylation of HDJ-2, resulting in the appearance of nonfarnesylated HDJ-2 (Fig. 1).
Fig. 1.

Western blot analysis of liver extracts. FTI treatment of Lmna+/+ mice leads to the appearance of nonfarnesylated HDJ-2 (with an antibody against HDJ-2) in liver extracts. FTI treatment also leads to the appearance of nonfarnesylated prelamin A (as judged by a western blot with a prelamin A–specific antibody). However, the amount of prelamin A accumulation was small, as judged by a western blot with an antibody against lamin A/C (prelamin A is a faint shadow above a large lamin A band). The FTI did not reduce the levels of mature lamin A.
Kaplan-Meier survival curves revealed that FTI treatment significantly improved survival in both male (P = 0.05) and female LmnaHG/+ mice (P = 0.0027) (Fig. 2), extending the median survival by 6−8 weeks.
Fig. 2.

Kaplan-Meier survival plots for LmnaHG/+ mice treated with an FTI or vehicle alone. Male LmnaHG/+ mice on vehicle (n = 8); male LmnaHG/+ mice on FTI (n = 10); female LmnaHG/+ mice on vehicle (n = 8); female LmnaHG/+ mice on FTI (n = 13).
FTI treatment improved body weight curves in male (P < 0.0001) and female LmnaHG/+ mice (P < 0.0001) (Figs. 3A and 3B). The FTI treatment was well tolerated in both Lmna+/+ and LmnaHG/+ mice. In previous studies [12, 14], we found a small but significant reduction in body weight in the Lmna+/+ mice treated with an FTI. We observed a slight trend towards reduced weight gain in FTI-treated male and female Lmna+/+ mice in this study (Figs. 3A and 3B), but these differences did not achieve statistical significance.
Fig. 3.
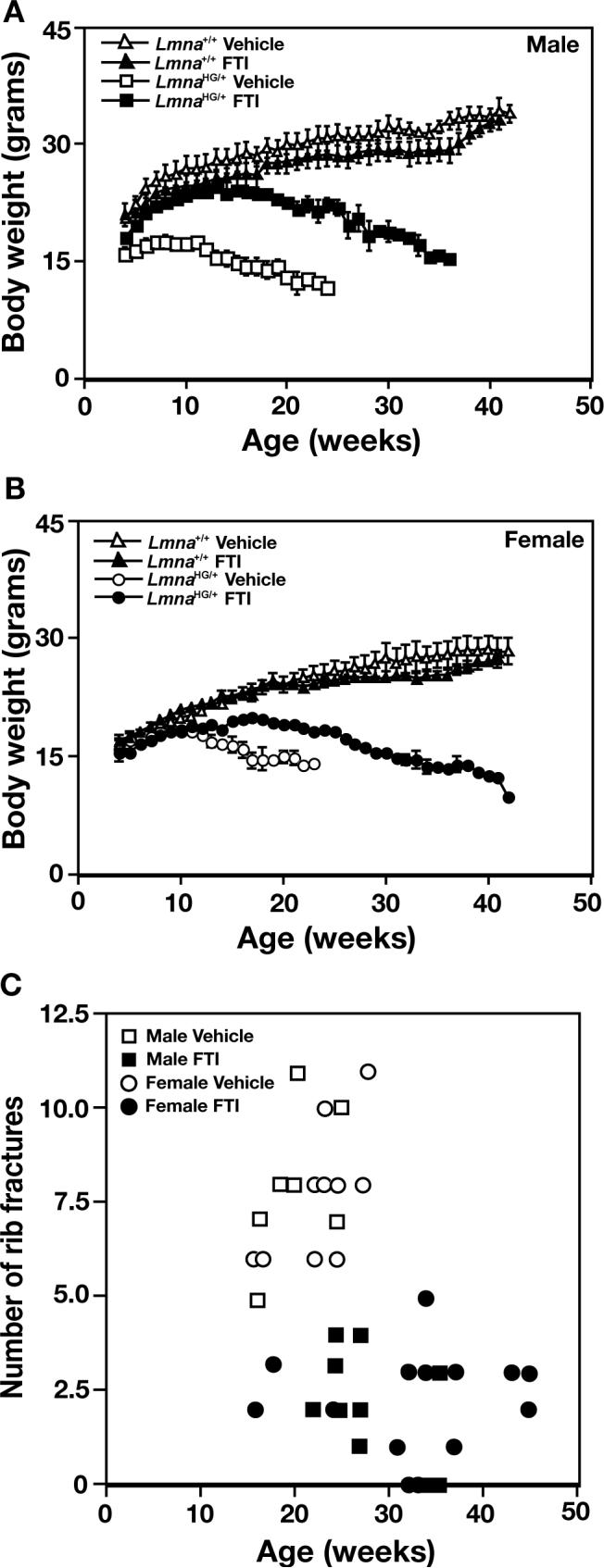
Disease phenotypes in LmnaHG/+ mice with FTI treatment. (a and b) Body weight curves for male (a) and female (b) LmnaHG/+ and littermate Lmna+/+ mice treated with an FTI or vehicle alone, beginning at four weeks of age. The body weight curves for FTI-treated LmnaHG/+ mice were significantly different than those of the vehicle-treated LmnaHG/+ mice, both in males and in females (P < 0.0001, as judged by the repeated-measure analysis of variance method). (c) Spontaneous rib fractures in male (squares) and female (circles) FTI- and vehicle-treated LmnaHG/+ mice at the time that they died. The FTI-treated mice had fewer fractures (P < 0.0001 for both males and females).
The FTI treatment significantly reduced the number of spontaneous rib fractures in LmnaHG/+ mice (P < 0.0001) (Fig. 3C). This was the case in both male and female LmnaHG/+ mice (P < 0.0001 in both groups).
We attempted to deliver higher doses of the FTI to both Lmna+/+ (n = 14) and LmnaHG/+ mice (n = 10), with the hope of achieving greater improvements in disease phenotypes in LmnaHG/+ mice. However, increasing the concentration of ABT-100 in the drinking water (so as to deliver 117 mg/kg/d) was toxic, even in wild-type mice. During the first week on the higher FTI dosage, 58% of the FTI-treated mice lost >10% of their body weight; by the end of the second week, 74% of the mice had lost >10% of their body weight. During the first two weeks of the higher dose of FTI, five mice died (three Lmna+/+ mice and two LmnaHG/+ mice). The mice that died had necrosis of the liver. [Finding liver toxicity with the higher dose of ABT-100 was not surprising; liver toxicity with high doses of the drug had been reported by the manufacturer in the MSDS (Material Safety Data Sheet).]
Discussion
This study provides additional support for a favorable effect of FTI therapy in LmnaHG/+ mice. Importantly, we found that the FTI improved disease phenotypes. Kaplan-Meier survival curves revealed that FTI treatment significantly improved survival in both male and female LmnaHG/+ mice. In addition, body weight curves were improved by the FTI, and the number of spontaneous bone fractures was reduced.
While the FTI-induced improvements were statistically significant, we emphasize that the beneficial effects of the drug fell well short of a complete cure. The FTI-treated LmnaHG/+ mice still had abnormal body weight curves, developed rib fractures, and died prematurely. One possibility for the less-than-complete response to FTI therapy is that the maximum tolerated doses do not fully block the farnesylation of progerin. Western blots revealed the presence of nonfarnesylated prelamin A and nonfarnesylated HDJ-2 in tissue extracts from the FTI-treated mice, indicating that the drug is active and that it inhibits prelamin A farnesylation (Fig. 1). However, the current studies as well as previously published studies [12] have suggested that the inhibition of lamin A biogenesis in vivo with ABT-100 treatment is far from complete. For example, in Fig. 1, there was little effect of the FTI on the biogenesis of mature lamin A, and the amount of nonfarnesylated prelamin A, relative to mature lamin A, was small.
A second possible explanation for the less-than-complete response to FTI therapy is that the nonfarnesylated prelamin A and nonfarnesylated progerin (which accumulate during FTI treatment) have toxic properties. In cell culture experiments, the accumulation of these nonfarnesylated lamin proteins with FTIs does not appear to be associated with significant toxicity [4, 6-11]. However, at this point, we have limited data regarding the actual toxicity of nonfarnesylated prelamin A at the “whole animal” level. In the future, it would be desirable to define the toxicity of these molecules by creating and characterizing gene-targeted mice that synthesize the nonfarnesylated forms of prelamin A and progerin.
Even though the impact of the FTI fell short of a complete cure, the results of the current study provide some hope that FTIs could help humans with HGPS. There are, however, significant differences between the mouse experiments described here and the ongoing trial of FTIs in humans with HGPS (http://www.progeriaresearch.org/progeria_ clinical_drug_trial_has_begun.html). In the mouse studies, the FTI was started at four weeks of age—before the onset of significant disease phenotypes. In humans, the FTI is given to patients who already manifest severe disease phenotypes. To be effective in humans, the FTI will need to reverse established disease and block the progression of disease. Our best guess, based on the mouse studies, is that the FTI will ameliorate disease in human HGPS patients, but that this treatment will fall well short of a complete cure. If this turns out to be the case, it will be important to investigate other strategies for treating HGPS. Fortunately, there are other potential therapeutic strategies. For example, several groups have proposed that antisense or RNAi strategies might be useful for reducing the synthesis of progerin in HGPS patients [15-17]. These alternative therapeutic strategies have been discussed in recent reviews [4, 18].
Acknowledgments
The authors thank Abbott Laboratories for supplying ABT-100 and Dr. David Frost and Ms. Joy Bauch (Abbott Laboratories) for their advice and technical expertise. Supported by NIH grants AR050200, HL86683 and HL76839, grants from the Progeria Research Foundation, and The Ellison Medical Foundation. S.H.Y was supported by a postdoctoral fellowship grant from the American Heart Association, Western States Affiliate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Debusk FL. The Hutchinson-Gilford progeria syndrome. J. Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N. Lamin A truncation in Hutchinson–Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 4.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—New evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Dechat T, Shimi T, Adam S, Rusinol A, Andres D, Spielmann H, Sinensky M, Goldman R. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci U S A. 2007;104:4955–4960. doi: 10.1073/pnas.0700854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 11.Pan Y, Garg A, Agarwal AK. Mislocalization of prelamin A Tyr646Phe mutant to the nuclear pore complex in human embryonic kidney 293 cells. Biochem Biophys Res Commun. 2007;355:78–84. doi: 10.1016/j.bbrc.2007.01.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. Treatment with a protein farnesyltransferase inhibitor improves disease phenotypes in mice with a targeted Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferguson D, Rodriguez LE, Palma JP, Refici M, Jarvis K, O'Connor J, Sullivan GM, Frost D, Marsh K, Bauch J, Zhang H, Lin NH, Rosenberg S, Sham HL, Joseph IB. Antitumor activity of orally bioavailable farnesyltransferase inhibitor, ABT-100, is mediated by antiproliferative, proapoptotic, and antiangiogenic effects in xenograft models. Clin Cancer Res. 2005;11:3045–3054. doi: 10.1158/1078-0432.CCR-04-2041. [DOI] [PubMed] [Google Scholar]
- 14.Fong L, Frost D, Meta M, Qiao X, Yang S, Coffinier C, Young S. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 15.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang S, Chen L, Libina N, Janes J, Martin GM, Campisi J, Oshima J. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum Genet. 2005;118:444–450. doi: 10.1007/s00439-005-0051-7. [DOI] [PubMed] [Google Scholar]
- 17.Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116:743–752. doi: 10.1172/JCI27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meta M, Yang SH, Bergo MO, Fong LG, Young SG. Protein farnesyltransferase inhibitors and progeria. Trends Mol Med. 2006 doi: 10.1016/j.molmed.2006.08.006. [DOI] [PubMed] [Google Scholar]