

Abstract
To examine leptin’s role in human appetite regulation, we studied recombinant methionyl human leptin’s effects on satiation and satiety in a model of leptin insufficiency, lipodystrophy. Eight females with hypoleptinemia and lipodystrophy were given sc injections of A-100 (maximal dose, 200% of that predicted to normalize serum leptin) for 4 months. Satiation and satiety were determined before and again during leptin treatment. Satiation was measured as the time to voluntary cessation of eating from a standardized food array after a 12-h fast. Satiety was determined as the time to hunger sufficient to consume a full meal after consumption of a standardized preload. During leptin treatment, satiation time decreased (41.2 ± 18.2 to 19.5 ± 10.6 min; P = 0.01), satiety time increased (62.9 ± 64.8 to 137.8 ± 91.6 min; P = 0.04), energy consumed to produce satiation decreased (2034 ± 405 to 1135 ± 432 kcal or 8.5 ± 1.7 to 4.7 ± 1.8 MJ; P < 0.01), and the amount of food desired in the postabsorptive state decreased (P < 0.02). Ghrelin concentrations also decreased during leptin administration (284.3 ± 127.9 to 140.6 ± 104.5 pmol/liter; P < 0.002). We conclude that increased leptin in patients with lipodystrophy results in less caloric, shorter, more satiating meals and longer-lived satiety. These data support the hypothesis that leptin plays an important, permissive role in human appetite regulation.
The lipodystrophies are rare disorders that are characterized by selective loss of sc and/or visceral fat due to any of several genetic or acquired conditions that alter the ability to store triglyceride in adipose tissue (1). Consequently, lipodystrophies are frequently associated with hypertriglyceridemia, accumulation of intramyocellular lipid, hepatomegaly and hepatic steatosis, disordered glucose metabolism, hyperphagia, and thermodysregulation.
Patients with lipodystrophy also have abnormally low circulating concentrations of adipocyte-derived leptin, which is decreased in proportion to their reduced sc fat stores. Leptin is a hormone secreted by the adipocyte that is believed to convey information about the status of adipocyte triglyceride content as well as the energy and macronutrient content of recent intake to the hypothalamic centers that control energy intake (2–4). Low concentrations of circulating leptin have been hypothesized to produce the hyperphagia associated with lipodystrophy, because low levels may insufficiently inhibit the hypothalamic appetite-regulating neurons that release orexigenic peptides and insufficiently stimulate neurons that release anorexigenic factors (5). In animal models (6, 7), leptin administration alters the secretion of orexigenic and anorexigenic neuropeptides and significantly decreases energy intake. In humans with homozygous inactivating mutations of the leptin gene (8, 9), leptin administration was reported to decrease energy intake at a single meal, but little is known about how leptin administration affects the temporal dynamics of eating episodes.
Satiation (meal termination) is defined as the point at which an individual becomes full or sated during an isolated eating episode, and satiety (inter-meal interval) is defined as the period during which an individual remains sated after the ingestion of a prescribed amount of food (10, 11). Some data from leptin-sufficient adults suggest a role for leptin in satiety and satiation. For example, obese women subjected to an energy deficit, experience fullness (12) in direct, and hunger (13) in inverse, proportion to circulating leptin concentrations. However, these relationships seem to be less evident for subjects in a eucaloric state (13). Some researchers believe that these findings implicate leptin in the control of food-seeking behavior, rather than with the experience of satiety or satiation (10). However, the role of leptin in modulating either satiation or satiety in humans has not been fully experimentally elucidated. Therefore, we studied the effects of exogenous leptin on satiation and satiety in patients who were hypoleptinemic because of lipodystrophy.
Subjects and Methods
Subjects (Table 1) were eight females (five Caucasian and three African American; mean age, 25.4 ± 12.5 yr) with lipodystrophies, serum leptin levels less than 4 ng/ml (0.32 nmol/ml), and at least one of the following metabolic abnormalities: diabetes mellitus by American Diabetes Association criteria (14), hypertriglyceridemia [fasting triglycerides, >200 mg/dl (2.23 mmol/liter)], or hyperinsulinemia [fasting insulin, >30 µU/ml (215 pmol/liter)] (15). Five of the eight subjects had congenital generalized lipodystrophy (Seip-Berardinelli syndrome), one had Dunnigan’s familial partial lipodystrophy, and the other two had acquired generalized lipodystrophy. Each subject was studied during admission to the NIH Warren Grant Magnuson Clinical Center before and again while they were treated with open-label recombinant methionyl human leptin (A-100, Amgen, Inc., Thousand Oaks, CA) administered sc twice a day for 4 months. The physiological replacement dose was estimated to be 0.03–0.04 mg/kg body weight according to the manufacturer. Subjects received 50% of this dose initially, increasing to 200% of this dose by the end of the second month, and maintained at this level for the third and fourth months of the study. Each subject served as her own control (i.e. the response to leptin at 4 months was compared with baseline response). While admitted, subjects were prescribed a diabetic weight maintenance diet, with adjustments to mimic the subject’s diet at home. Subjects completed a questionnaire of food preferences and underwent studies of satiation and satiety using a food array on successive days. The study was approved by the institutional review board of the NIDDK, NIH. All subjects provided written informed consent. Assent was also obtained for children under the age of 18 yr. Some of the metabolic effects of leptin in six of the study’s subjects have been reported previously (15).
TABLE 1.
Subject characteristics at baseline
| Subject no. | Diagnosis | Age (yr) | Gender | Race | Medications |
|---|---|---|---|---|---|
| 1 | Acquired generalized | 17 | F | W | Orlistat (360 mg/d), acarbose (150 mg/d), metformin (1500 mg/d), fenofibrate, atorvastatin |
| 2 | Congenital generalized | 17 | F | B | Insulin (194 U/d) |
| 3 | Congenital generalized | 17 | F | W | Insulin (1200 U/d), lisinopril |
| 4 | Congenital generalized | 15 | F | W | Insulin (3000 U/d), lisinopril |
| 5 | Congenital generalized | 37 | F | B | Metformin (1500 mg/d) |
| 6 | Congenital generalized | 41 | F | B | Metformin (1500 mg/d), gemfibrozil, rosiglitazone (45 mg/d), enalopril |
| 7 | Acquired generalized | 14 | F | W | Insulin (110 U/d), fenofibrate |
| 8 | Familial partial | 42 | F | W | Insulin (110 U/d), gemfibrozil, pioglitazone, lisinopril |
Food array
Individuals who have lipodystrophy are hyperphagic; therefore, foods in the array were offered in very large quantities (over three times the amount that individuals with lipodystrophy normally would be expected to consume). The array comprised 10,767 kcal of energy in cold luncheon foods, with a macronutrient content of 335.6 g (12.5%) protein, 414.9 g (34%) fat, and 1,473.3 (54.5%) g carbohydrate. The design of the array was based on those used in previous satiation and satiety studies (16, 17). Content purposely departed from the diet order, and foods were chosen to represent a wide range of macronutrient composition so that leptin’s effect on macronutrient intake could be evaluated (Table 2). The array was displayed in exactly the same arrangement and under the same environmental conditions for each patient.
TABLE 2.
Composition of food array
| Food | Amount offered (g) | Energy/100 g (kcal)a | Total calories (kcal)a | Total protein (g) | Total fat (g) | Total carbohydrate (g) |
|---|---|---|---|---|---|---|
| White bread | 180.8 | 267.0 | 482.7 | 14.8 | 6.5 | 89.5 |
| Wheat bread | 156.3 | 260.0 | 406.4 | 14.2 | 6.4 | 73.8 |
| Ham 180.0 | 120.0 | 216.0 | 33.3 | 8.2 | 0 | |
| Turkey | 180.0 | 157.0 | 282.6 | 53.8 | 5.8 | 0 |
| Bologna | 180.0 | 316.0 | 568.8 | 21.0 | 50.9 | 5.0 |
| American cheese | 240.0 | 375.5 | 901.1 | 53.2 | 75.0 | 3.8 |
| Tomatoes | 200.0 | 21.0 | 42.0 | 1.7 | 0.7 | 9.3 |
| Lettuce | 50.0 | 12.0 | 6.0 | 0.5 | 0.1 | 1.0 |
| Banana | 513.7 | 92.0 | 472.6 | 5.3 | 2.5 | 120.4 |
| Apples | 534.4 | 59.0 | 315.3 | 1.0 | 1.9 | 81.5 |
| Grapes | 249.2 | 71.0 | 176.9 | 1.6 | 1.4 | 44.3 |
| Oreos | 137.2 | 472.0 | 647.6 | 6.4 | 28.3 | 96.4 |
| Shortbread | 87.1 | 502.0 | 437.2 | 5.3 | 21.0 | 56.2 |
| Peanut butter | 120.0 | 593.0 | 711.6 | 30.2 | 61.2 | 23.1 |
| Grape jelly | 120.0 | 283.0 | 339.6 | 0.2 | 0.0 | 84.6 |
| Light mayonnaise | 90.0 | 334.0 | 300.6 | 0.5 | 29.6 | 7.6 |
| Light ranch dressing | 90.0 | 266.7 | 240.0 | 3.0 | 21.0 | 9.0 |
| Mustard | 90.0 | 66.0 | 59.4 | 3.6 | 2.8 | 7.0 |
| Baby carrots | 200.0 | 38.0 | 76.0 | 1.7 | 1.1 | 16.3 |
| Potato chips | 120.0 | 536.0 | 643.2 | 8.4 | 41.5 | 63.5 |
| Pretzels | 150.0 | 381.0 | 571.5 | 13.6 | 5.3 | 118.8 |
| Fat-free yogurt | 720.0 | 88.1 | 634.4 | 22.2 | 0 | 133.2 |
| Jelly beans | 120.0 | 367.0 | 440.4 | 0 | 0.6 | 111.7 |
| Candies | 120.0 | 492.0 | 590.4 | 5.2 | 25.4 | 85.4 |
| Water | 850.0 | 0.0 | 0 | 0 | 0 | 0 |
| Milk (2%) | 850.0 | 49.7 | 422.2 | 28.3 | 16.3 | 40.8 |
| Apple juice | 850.0 | 47.0 | 399.5 | 0.5 | 0.9 | 99.3 |
| Orange juice | 850.0 | 45.0 | 382.5 | 5.8 | 0.5 | 91.6 |
To convert kilocalories to megajoules, multiply by 4.184 and divide by 1000.
Food preference
Taste preferences were examined with a 50-item food questionnaire administered before satiation and satiety testing. The 28 foods on the array were embedded among the 50 items. Foods were rated on Likert scales with the anchor points: 1 (“I strongly dislike the food”) and 10 (“I love the food”). To ensure that subjects would consume a sufficient number of the foods available in the array, at least 50% of the foods in the array had to be rated 6 or more for the baseline satiety and satiation tests to be performed.
Satiety testing
Subjects observed a 12-h fast the night before testing. Immediately before testing, subjects recorded their subjective degree of hunger, fullness, “stomach rumbling,” and the amount of food desired on 100-mm visual analog scales (11, 18). They also rated other factors that might interfere with food intake: sleepiness, nausea, dizziness, indigestion, anxiety, headache, and thirst. Between 0800 and 0900 h, subjects drank a 1000-ml (1 kcal/ml) yogurt shake consisting of 28.8 g protein, 161.7 g carbohydrate, and 23.0 g fat, similar to the formula used in a previous satiety testing study (19). Subjects were encouraged to drink as much of the total volume of the shake within 15 min as possible, but were not allowed to continue to drink the shake after 15 min. The amount of shake consumed was recorded, and the same amount was offered at the 4-month follow-up study. Subjects completed the same scales for hunger and other factors after consumption of the yogurt shake.
After the shake, subjects were instructed not to eat anything or drink any energy-containing beverage until they experienced the sensations of physical hunger strongly enough to consume a full meal. During this time, activity was restricted to those nonathletic pursuits available on the in-patient ward. Subjects then reported the time that they became hungry enough to eat a full meal. The amount of time elapsed between consumption of the yogurt shake and the time of hunger onset constituted the standard satiety time. The scales for hunger and other factors were readministered before the presentation of the test meal (food array).
The test meal was presented to each subject in a specific room containing only chairs and the table holding the food array. The room was devoid of other types of sensory stimuli (e.g. pictures, TV, etc.). Subjects listened to standardized, tape-recorded instructions inviting them to “let yourself go and eat as much as you would like. You may eat as much of anything as you would like to, but you do not have to eat anything that you do not like.” Subjects then self-selected the types and amounts of foods they consumed from the food array. The subject was monitored from outside the room during the entire test meal period. The amount of time spent eating was measured on a stop-watch and constituted the post-preload satiation time. Both the amount of preload and the amount of food consumed were measured by pre- and postweighing the food containers. After consuming as much of the food array as desired, subjects again rated their degree of hunger and the other factors previously monitored. In addition, subjects rated the appearance, smell, taste, and texture of only those foods they actually consumed.
Satiation testing
Satiation testing was carried out within 1 h of 1200 h, after the subjects had observed a minimum 12-h fast. A similar duration fast was observed both at baseline and during leptin treatment for each subject. Before the test meal, subjects completed the same rating scales used in the satiety study. The food array offered to subjects and the test meal procedure were exactly the same as those used for the satiety study. The amount of time spent eating was measured on a stop-watch and constituted the standard satiation time. The amount of food consumed was determined by weighing the food before presentation and reweighing the food returned to the kitchen. After the test meal, subjects again completed the rating scales. Subjects were instructed not to eat anything or drink any energy-containing beverage until they became hungry enough to consume another full meal. Activity during this period was limited to nonathletic pursuits available on the in-patient ward. The time difference between the end of the test meal and the time when the subjects reported the desire to eat another full meal was recorded as the post-test meal satiety time.
Hormones and substrates
Blood specimens were obtained between 0600 and 0700 h for analysis of fasting glucose, triglycerides, insulin, leptin, ghrelin, and glucagonlike peptide 1 (GLP1). Serum glucose and triglyceride levels were measured on a Hitachi 917 analyzer (Tokyo, Japan) using reagents from Roche (Indianapolis, IN). Serum insulin levels were determined by immunoassay using an Immulite 200 machine (Diagnostic Products Corp., Los Angeles, CA) with reagents provided by Abbott Laboratories (Abbott Park, IL). Serum leptin, total ghrelin, and GLP-1 levels were determined by RIA with the use of commercial kits (Linco Research, Inc., St. Charles, MO).
Statistics
Parametric data were analyzed on a Macintosh G3 using StatView 5.0.1 and SuperAnova 1.11 software (Abacus Concepts, Inc., Berkeley, CA). Data were log- or arcsine, square root-transformed, and geometric means and sds are reported where appropriate. Paired, two-tailed t tests were conducted to determine differences between groups for weight, laboratory measurements, satiety time, satiation time, and intake at baseline vs. 4 months of leptin therapy. Multiple regression was used to test for associations between intake, leptin concentrations, and variables that are potential moderators of leptin: insulin, glucose, and triglycerides. Post hoc tests were corrected for multiple comparisons using the Bonferroni-Holm procedure (20).
Results
After 4 months of leptin therapy, serum leptin concentrations increased significantly, from 1.3 ± 1.9 to 12.3 ± 2.1 ng/ml (P<0.0001) to a point within normal range for weight. Weight decreased 5% on the average (from 54.5 ± 15.1 to 51.5 ± 14.1 kg; P < 0.01). Serum insulin concentrations decreased significantly from 63.6 ± 2.9 to 28.6 ± 2.7 µIU/ml (P < 0.01) as did glucose (from 14.5 ± 6.0 to 7.5 ± 1.8 mmol/liter; P < 0.01), triglyceride (from 15.9 ± 12.8 to 4.2 ± 3.3 mmol/liter; P = 0.025), and ghrelin (from 284.3 ± 127.9 to 140.6 ± 104.5 pmol/liter; P < 0.002) concentrations. The GLP1 concentration did not change significantly (from 57 ± 51 to 26 ± 25 pmol/liter; P = 0.28).
All subjects were taking glucose-lowering medications before leptin therapy (Table 1). After 4 months of leptin treatment, six patients no longer had fasting hyperglycemia. Two subjects were able to discontinue insulin injections, three had insulin dosages lowered 40–80%, and the remaining three discontinued their oral agents.
Leptin therapy markedly affected both the temporal characteristics of meals and food consumption (Table 3 and Fig. 1). The standard satiation time (the amount of time the subject consumed food during an isolated eating episode before feeling sated) decreased significantly from 41.3 ± 18.2 to 19.5 ± 10.6 min (Fig. 1A; P = 0.01). Intake during the satiation test also decreased significantly, from 2034 ± 405 to 1135 ± 432 kcal (8.5 ± 1.7 to 4.7 ± 1.8 MJ; Fig. 1B; P = 0.007). In addition, the standard satiety time (the amount of time the subject remained sated after ingestion of a standardized preload) increased from 62.8 ± 64.8 to 137.8 ± 91.6 min (Fig. 1C; P = 0.04).
TABLE 3.
Changes in standard satiation, satiety, and intake before and after leptin therapy
| Subject no. | Satiation (min) |
Satiety (min) |
Intake (kcal)a |
|||
|---|---|---|---|---|---|---|
| Baseline | Leptin | Baseline | Leptin | Baseline | Leptin | |
| 1 | 52.4 | 16.2 | 32.8 | 202.8 | 2588 | 553 |
| 2 | 35.4 | 7.7 | 33.9 | 19.5 | 1845 | NA |
| 3 | 67.3 | 30.6 | 13.7 | 206.8 | 2063 | 1171 |
| 4 | 54.8 | 29.0 | 12.1 | 58.3 | 2528 | 1582 |
| 5 | 48.0 | 14.6 | 2251 | 1232 | ||
| 6 | 15.5 | 14.8 | 197.0 | 270.2 | 1398 | 1111 |
| 7 | 17.2 | 7.6 | 85.2 | 128.0 | 1782 | 616 |
| 8 | 39.7 | 35.7 | 65.6 | 79.6 | 1820 | 1682 |
To convert kilocalories to megajoules, multiply by 4.184 and divide by 1000.
NA, Not applicable.
FIG. 1.
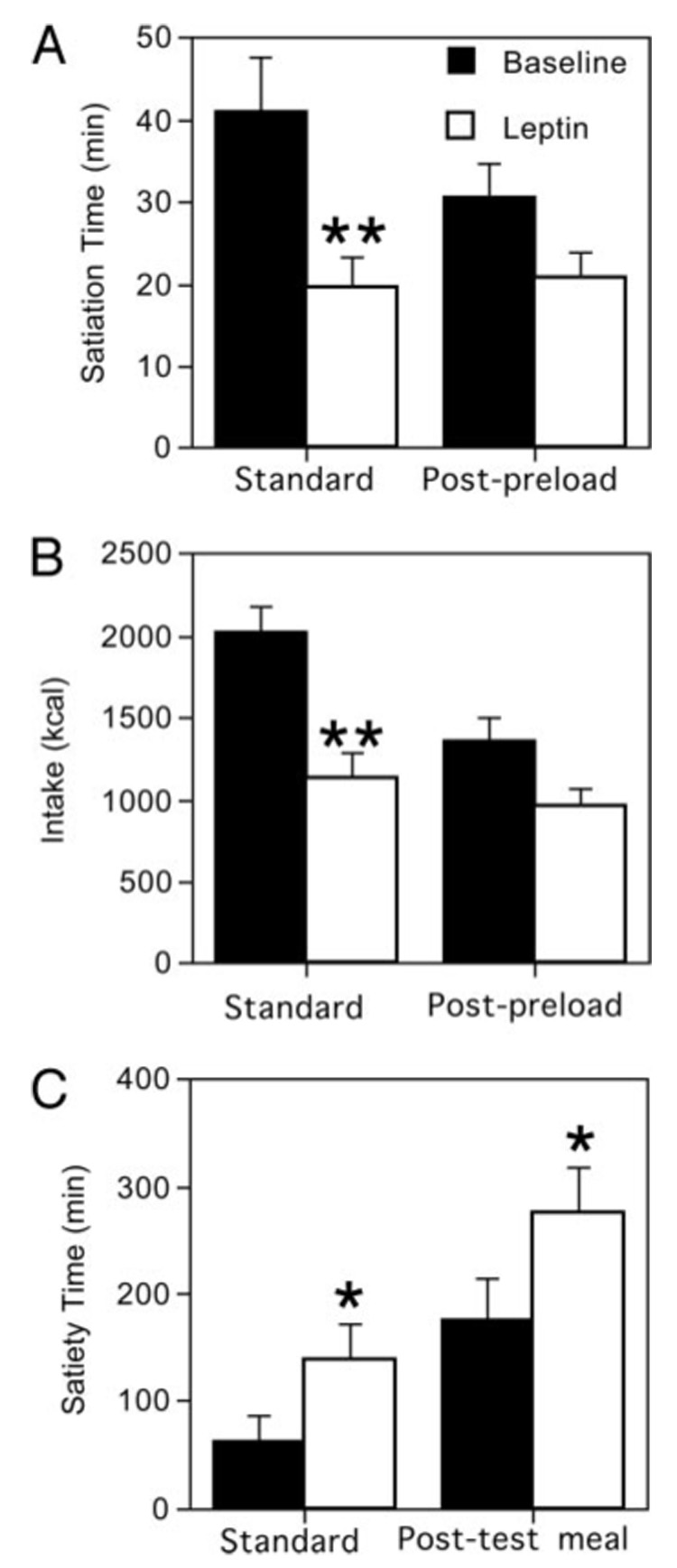
Changes in indicators of appetite before and after leptin therapy. A, Satiation time; B, intake; C, satiety. *, P ≤ 0.05; **, P ≤ 0.01.
Similar results were obtained for the less standardized measurements of post-preload satiation and post-test meal satiety. The post-preload satiation time trended downward from 30.6 ± 10.8 to 20.7 ± 8.8 min (Fig. 1A; P = 0.07). Post-preload intake decreased from 1360 ± 391 to 963 ± 300 kcal (5.7 ± 1.6 to 4.0 ± 1.2 MJ; Fig. 1B; P = 0.07). The post-test meal satiety time increased significantly from 176.5 ± 101.5 to 275.7 ± 103.2 min (Fig. 1C; P = 0.05). Simple and multiple regression analyses examining the relationships between changes in insulin, leptin, glucose, ghrelin, GLP1, and triglycerides, and intake did not reveal any significant associations (all P > 0.08). There were also no significant correlations between any of these variables either at baseline or after 4 months of leptin therapy after correction for multiple comparisons.
The combination of foods subjects ate during the test meal had a macronutrient composition similar to that recommended by the American Dietetic Association (15–20% protein, 25–30% fat, and 55–60% carbohydrate) (21) both before and after leptin therapy. The percentage of energy from fat and carbohydrate did not change significantly (P > 0.4); however, the percentage of energy from protein in the post-preload test meal decreased slightly (from 15.7 ± 4.3% to 11.0 ± 7.6%; P = 0.04).
The number of foods that subjects rated less than 6 on a 10-point scale for appearance, smell, taste, and texture increased significantly during leptin treatment from 9.2 ± 3.3 to 13.0 ± 4.4 (P < 0.01). Food preference ratings for individual foods consumed before and after leptin therapy changed in only a few foods, decreasing for milkshakes, mayonnaise, and orange juice. After correction for multiple comparisons, these changes were no longer statistically significant.
The only significant difference associated with leptin treatment in the subjective ratings of hunger, etc., was the score for the amount of food desired before the standard meal test (72 ± 17 vs. 51 ± 21; P = 0.02). After consumption of satiation test meals, leptin treatment was associated with trends toward decreases in hunger, the amount of food desired, and “stomach rumbling,” and also with slight increases in fullness (Table 4). The subjective ratings taken before and after the satiety tests also changed in the expected directions, but did not reach statistical significance (Table 4).
TABLE 4.
Subjective ratings associated with satiety and satiation tests
| Baseline | Leptin | P value | |
|---|---|---|---|
| Standard satiety test | |||
| Before yogurt shake | |||
| Degree of hunger | 55.6 ± 30.8 | 48.0± 29.9 | 0.69 |
| Degree of fullness | 7.0 ± 5.0 | 24.7 ± 19.3 | 0.06 |
| Desire to eat | 63.3 ± 17.7 | 46.7 ± 20.9 | 0.26 |
| Stomach rumbling | 34.6 ± 29.7 | 21.3 ± 24.9 | 0.24 |
| After yogurt shake | |||
| Degree of hunger | 42.8 ± 30.2 | 17.6 ± 25.0 | 0.07 |
| Degree of fullness | 48.3 ± 30.8 | 72.4 ± 20.9 | 0.06 |
| Desire to eat | 38.4 ± 25.9 | 16.8 ± 16.2 | 0.06 |
| Stomach rumbling | 19.1 ± 27.8 | 11.3 ± 9.8 | 0.48 |
| Standard satiation test | |||
| Before test meal | |||
| Degree of hunger | 66.1 ± 25.5 | 53.2 ± 17.6 | 0.20 |
| Degree of fullness | 10.3 ± 8.3 | 12.0 ± 10.9 | 0.29 |
| Desire to eat | 72.0 ± 16.7 | 51.1 ± 21.3 | 0.01 |
| Stomach rumbling | 42.0 ± 33.6 | 23.3 ± 21.0 | 0.14 |
| After test meal | |||
| Degree of hunger | 17.0 ± 17.9 | 4.8 ± 2.8 | 0.21 |
| Degree of fullness | 68.7 ± 27.9 | 80.7 ± 16.2 | 0.24 |
| Desire to eat | 21.6 ± 25.3 | 8.3 ± 8.6 | 0.24 |
| Stomach rumbling | 4.7 ± 7.1 | 3.3 ± 1.0 | 0.19 |
Discussion
We found that leptin administration markedly affected both satiation and satiety in lipodystrophic women with hypoleptinemia. When serum leptin concentrations were increased to values seen in adults with normal amounts of body fat, the time for induction of satiety decreased by 53%, energy intake at a meal decreased by 44%, and the length of time subjects remained sated after a standardized preload increased by 219% These alterations in meal characteristics occurred in conjunction with improvements in glucose tolerance and triglyceride concentrations (15) and significant decreases in fasting ghrelin concentrations.
In this study satiation was examined twice, during the standard satiation test meal (after a 12 h fast) and at the post-preload test meal. The energy consumed at these two test meals were significantly different at baseline [2034 ± 405 kcal (8.5 ± 1.7 MJ) after the 12-h fast vs. 1360 ± 391 kcal (5.7 ± 1.6 MJ) post-preload], but quite comparable to one another during leptin administration in both macronutrient composition and energy content [1135 ± 432 kcal (4.7 ± 1.8 MJ) vs. 963 ± 300 kcal (4.0 ± 1.2 MJ)]. The observation that a similar amount of food was able to terminate an eating episode in both fasted and post-preload states when plasma leptin levels were increased suggests that the role of leptin in satiation may be permissive, allowing some of the gastrointestinal factors that normally regulate meal dynamics to act. After an overnight fast, in the absence of leptin, the acute, gastrointestinal indicators of satiation and hunger, e.g. cholecystokinin (CCK), peptide YY, and ghrelin, may be overridden by stronger signals released in response to low leptin, e.g. neuropeptide Y and agouti-related peptide. However, after a preload, more chronic indicators of metabolic status, such as insulin, would exert their effects also, moderating intake at the second meal array and partially compensating for the lack of leptin. Conversely, with leptin replacement, gut signals are able to regulate intake in both instances.
Satiety was also studied twice, once after a standard quantity of preload with known composition and once after a standardized test meal array. In both cases the amount of time that the patients remained sated improved dramatically, increasing 74.9 ± 78.0 and 66.1 ± 128.5 min (P = 0.79) during the two studies. The trend (P = 0.06) in subjective ratings toward an increased degree of fullness before and after the standardized satiety preload also support the suggestion that adequate leptin allows the hypothalamus to register gastrointestinal satiety signals released in response to moderate, controlled energy loads. Leptin may also influence satiety by moderating the onset of hunger. Ghrelin, a peptide produced by the stomach, is implicated in the short-term control of hunger onset through its activation of neuropeptide Y/agouti-related peptide neurons (22). Barazzoni et al. (23) assert that leptin negatively regulates ghrelin. We found that humans with lipodystrophy and leptin deficiency have high circulating ghrelin. Greater ghrelin may conceivably contribute to the hyperphagia observed in patients with lipodystrophy. Leptin replacement was associated with decreases in fasting ghrelin, which may have contributed to the increased satiety observed.
Leptin sufficiency may also be required for the satiation signals from gut-derived hormones, such as CCK, enterostatin, and peptide YY, and/or the baroreceptors to be recognized by hypothalamic feeding centers. This hypothesis is supported by the work of Barrachina et al. (24), who found that CCK exhibited a synergistic interaction with leptin when administered peripherally in mice. Although CCK’s action is immediate, and leptin’s is somewhat delayed when the hormones act alone, the combination of the two hormones decreased food intake evenly across the entire 7-h postprandial time period measured. In addition, McMinn et al. (25) found that leptin replacement prevented the attenuation of the CCK-induced satiety response in fasted rats. Leptin has been suggested to induce hypersensitization of neurons in the nucleus tractus solitarius/dorso-motor nucleus of the vagus nerve to the satiety-inducing effects of CCK and perhaps other meal-related signals, thereby resulting in increased satiety and smaller meals (26). Future studies should examine changes in these meal-related peptides. It is also possible that leptin’s actions on meal characteristics are at least in part mediated through its effects on insulin action; by improving glucose homeostasis, leptin may also have decreased total daily energy requirements. This hypothesis is supported by the decreases in resting energy expenditure observed with leptin administration in patients with lipodystrophies (15).
The macronutrient composition of the test meals changed little before and during leptin therapy. This was not unexpected, because leptin is believed to convey the status of adipocyte energy stores to the hypothalamus without regard to the original source of triglyceride. Data from animal models (27) suggest that serum leptin concentrations are not influenced by the gut hormones that sense the composition of a meal, such as CCK, which is released in proportion to meal fat content (28). In addition, there were no significant changes in the preference ratings for any particular food or type of food, supporting the hypothesis that leptin is a general, rather than a specific, satiety signal.
This study is limited by its small sample size, the lack of a placebo-treated control group, and the fact that not all of the subjects had the same type of lipodystrophy. However, all subjects were similar in their low endogenous leptin production, high baseline energy intake, and their marked response to leptin treatment. Thus, although this paper must be considered a preliminary investigation of the effects of leptin on satiation and satiety, the uniformity of response suggests that these data accurately represent leptin’s ability to alter meal dynamics.
In conclusion, although leptin is not considered a primary satiety factor in humans because changes in food intake do not induce short-term increases in blood leptin concentrations (29), we found that normalization of serum leptin concentration decreased fasting ghrelin concentrations, decreased satiation time, decreased caloric intake, increased satiety time, and influenced subjective perceptions of hunger, fullness, and desire to eat in patients with hypoleptinemia and lipodystrophy. We hypothesize that leptin’s effects on satiety is permissive; leptin may affect satiety by sufficiently inhibiting central nervous system orexigens so as to allow satiety signals from gut hormones and baroreceptors to affect eating behavior.
Acknowledgments
This work was supported in part by grants from the NIH (ZO1-HD-00641 and RO1-DK-54387). Additional support was provided through a materials transfer agreement from Amgen, Inc.
Abbreviations
- CCK
Cholecystokinin
- GLP2
glucagon-like peptide 1
References
- 1.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Havel PJ. Control of energy homeostasis and insulin action by adipocyte hormones: leptin, acylation stimulating protein, and adiponectin. Curr Opin Lipidol. 2002;13:51–59. doi: 10.1097/00041433-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Havel PJ, Townsend R, Chaump L, Teff K. High-fat meals reduce 24-h circulating leptin concentrations in women. Diabetes. 1999;48:334–341. doi: 10.2337/diabetes.48.2.334. [DOI] [PubMed] [Google Scholar]
- 4.Weigle DS, Cummings DE, Newby PD, Breen PA, Frayo RS, Matthys CC, Callahan HS, Purnell JQ. Roles of leptin and ghrelin in the loss of body weight caused by a low fat, high carbohydrate diet. J Clin Endocrinol Metab. 2003;88:1577–1586. doi: 10.1210/jc.2002-021262. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz MW. Brain pathways controlling food intake and body weight. Exp Biol Med. 2001;226:978–981. doi: 10.1177/153537020122601103. [DOI] [PubMed] [Google Scholar]
- 6.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 7.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 8.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 9.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O’Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blundell JE, Goodson S, Halford JC. Regulation of appetite: role of leptin in signalling systems for drive and satiety. Int J Obes Relat Metab Disord. 2001;25 Suppl 1:S29–S34. doi: 10.1038/sj.ijo.0801693. [DOI] [PubMed] [Google Scholar]
- 11.Rolls BJ, Hetherington M, Burley VJ. The specificity of satiety: the influence of foods of different macronutrient content on the development of satiety. Physiol Behav. 1988;43:145–153. doi: 10.1016/0031-9384(88)90230-2. [DOI] [PubMed] [Google Scholar]
- 12.Heini AF, Lara-Castro C, Kirk KA, Considine RV, Caro JF, Weinsier RL. Association of leptin and hunger-satiety ratings in obese women. Int J Obes Relat Metab Disord. 1998;22:1084–1087. doi: 10.1038/sj.ijo.0800731. [DOI] [PubMed] [Google Scholar]
- 13.Keim NL, Stern JS, Havel PJ. Relation between circulating leptin concentrations and appetite during a prolonged, moderate energy deficit in women. Am J Clin Nutr. 1998;68:794–801. doi: 10.1093/ajcn/68.4.794. [DOI] [PubMed] [Google Scholar]
- 14.Peters AL, Schriger DL. The new diagnostic criteria for diabetes: the impact on management of diabetes and macrovascular risk factors. Am J Med. 1998;105:15S–19S. doi: 10.1016/s0002-9343(98)00206-x. [DOI] [PubMed] [Google Scholar]
- 15.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 16.Hadigan CM, Kissileff HR, Walsh BT. Patterns of food selection during meals in women with bulimia. Am J Clin Nutr. 1989;50:759–766. doi: 10.1093/ajcn/50.4.759. [DOI] [PubMed] [Google Scholar]
- 17.Hetherington MM, Altemus M, Nelson ML, Bernat AS, Gold PW. Eating behavior in bulimia nervosa: multiple meal analyses. Am J Clin Nutr. 1994;60:864–873. doi: 10.1093/ajcn/60.6.864. [DOI] [PubMed] [Google Scholar]
- 18.Sepple CP, Read NW. Gastrointestinal correlates of the development of hunger in man. Appetite. 1989;13:183–191. doi: 10.1016/0195-6663(89)90011-1. [DOI] [PubMed] [Google Scholar]
- 19.Kissileff HR, Walsh BT, Kral JG, Cassidy SM. Laboratory studies of eating behavior in women with bulimia. Physiol Behav. 1986;38:563–570. doi: 10.1016/0031-9384(86)90426-9. [DOI] [PubMed] [Google Scholar]
- 20.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–803. [Google Scholar]
- 21.Position of the American Dietetic Association, Dietitians of Canada, and the American College of Sports Medicine: Nutrition and athletic performance. J Am Diet Assoc. 2000;100:1543–1556. doi: 10.1016/S0002-8223(00)00428-4. [DOI] [PubMed] [Google Scholar]
- 22.Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 23.Barazzoni R, Zanetti M, Stebel M, Biolo G, Cattin L, Guarnieri G. Hyperleptinemia prevents increased plasma ghrelin concentration during short-term moderate caloric restriction in rats. Gastroenterology. 2003;124:1188–1192. doi: 10.1016/s0016-5085(03)00281-6. [DOI] [PubMed] [Google Scholar]
- 24.Barrachina MD, Martinez V, Wang L, Wei JY, Tache Y. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proc Natl Acad Sci USA. 1997;94:10455–10460. doi: 10.1073/pnas.94.19.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMinn JE, Sindelar DK, Havel PJ, Schwartz MW. Leptin deficiency induced by fasting impairs the satiety response to cholecystokinin. Endocrinology. 2000;141:4442–4448. doi: 10.1210/endo.141.12.7815. [DOI] [PubMed] [Google Scholar]
- 26.Baskin DG, Blevins JE, Schwartz MW. How the brain regulates food intake and body weight: the role of leptin. J Pediatr Endocrinol Metab. 2001;14 Suppl 6:1417–1429. [PubMed] [Google Scholar]
- 27.Romon M, Lebel P, Velly C, Marecaux N, Fruchart JC, Dallongeville J. Leptin response to carbohydrate or fat meal and association with subsequent satiety and energy intake. Am J Physiol. 1999;277:E855–E861. doi: 10.1152/ajpendo.1999.277.5.E855. [DOI] [PubMed] [Google Scholar]
- 28.Matzinger D, Degen L, Drewe J, Meuli J, Duebendorfer R, Ruckstuhl N, D’Amato M, Rovati L, Beglinger C. The role of long chain fatty acids in regulating food intake and cholecystokinin release in humans. Gut. 2000;46:688–693. doi: 10.1136/gut.46.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jequier E. Leptin signaling, adiposity, and energy balance. Ann NY Acad Sci. 2002;967:379–388. doi: 10.1111/j.1749-6632.2002.tb04293.x. [DOI] [PubMed] [Google Scholar]