

Abstract
A computational system for the prediction of polymorphic loci directly and efficiently from human genomic sequence was developed and verified. A suite of programs, collectively called pompous (polymorphic marker prediction of ubiquitous simple sequences) detects tandem repeats ranging from dinucleotides up to 250 mers, scores them according to predicted level of polymorphism, and designs appropriate flanking primers for PCR amplification. This approach was validated on an approximately 750-kilobase region of human chromosome 3p21.3, involved in lung and breast carcinoma homozygous deletions. Target DNA from 36 paired B lymphoblastoid and lung cancer lines was amplified and allelotyped for 33 loci predicted by pompous to be variable in repeat size. We found that among those 36 predominately Caucasian individuals 22 of the 33 (67%) predicted loci were polymorphic with an average heterozygosity of 0.42. Allele loss in this region was found in 27/36 (75%) of the tumor lines using these markers. pompous provides the genetic researcher with an additional tool for the rapid and efficient identification of polymorphic markers, and through a World Wide Web site, investigators can use pompous to identify polymorphic markers for their research. A catalog of 13,261 potential polymorphic markers and associated primer sets has been created from the analysis of 141,779,504 base pairs of human genomic sequence in GenBank. This data is available on our Web site (pompous.swmed.edu) and will be updated periodically as GenBank is expanded and algorithm accuracy is improved.
The positional cloning of many disease loci has been facilitated by high-resolution genetic maps. The precise localization of the DNA sequence responsible for a disease usually requires the development of very high-density physical and genetic maps. The availability of multiple polymorphic genetic markers is crucial to this effort (1). Current widely used methods for the identification of new simple sequence repeat polymorphisms involve PCR-based and subcloning strategies (1, 2). Subcloning, once the primary method of isolating new microsatellite sequences (3), largely has been supplanted by PCR-based methods because of the relatively large amount of work and technical difficulties involved in subcloning strategies, including sublibrary construction and screening with oligonucleotide probes (4, 5). PCR-based strategies, although generally faster and more successful than those using subcloning, still suffer from several disadvantages. The researcher is unable to specify with much precision the size, homogeneity, or locations of the repeat loci identified and is forced to use complex or sophisticated methods to complete the process (6). In addition, many of the repeat loci identified are either redundant, difficult to cleanly amplify, or are otherwise unsuitable for use. These limitations restrict the ability of genetic researchers to create local high-density maps in their area of interest and force many to be dependent on large-scale mapping facilities.
The large quantities of human genomic sequence being generated by the Human Genome Project are rapidly making these approaches obsolete. It is anticipated that the Human Genome Project will be more than 95% complete by the year 2002 (7). The knowledge of the frequency and level of polymorphism of the various types and sizes of microsatellites and variable number tandem repeats allows one to predict, a priori, which tandem repeats are likely to be highly polymorphic from a single genomic sequence. For microsatellites, the level of heterozygosity has been observed to be directly proportional to the number of repeated units and inversely proportional to the size of the repeated unit (6, 8–12). Although several software applications for locating some microsatellites or larger tandem repeats currently exist, a comprehensive tool for the identification of, and generation of primer sequences for, those repeats correlated with a high probability of polymorphism has been lacking (13–23). Because of this need, software was written that will take human genomic sequence data as input and will output a list of oligonucleotide sequences that may be used as primers for PCR amplification of those tandem repeat sequences that are predicted to be highly polymorphic based on observations from the literature (Table 1). The 3p21.3 region has been found to be a site of frequent allele loss in lung, breast, and other cancers, as well as being the site of frequent homozygous deletions in lung and breast cancers. As part of a positional cloning effort to identify a putative tumor suppressor gene in this region we constructed a cosmid/P1 contig, which recently has been sequenced by the Washington University Human Genome Center (St. Louis) and the Sanger Centre (Hinxton, Cambridge, U.K.) (31). The closest flanking known polymorphic markers are MFD 93 centromeric and D3S15F2 telomeric. By using standard blastn analysis, various sequence-tagged sites and polymorphic markers previously unmapped can be identified, and the GenBank annotation documents some previously identified tandem repeats. We have discovered in this region on the known genetic linkage map that polymorphic markers would be of great use for further studies of allele loss in tumors and associated preneoplastic lesions as well as for placement of the genes. As a test of pompous (polymorphic marker prediction of ubiquitous simple sequences), we applied this software suite to approximately 750 kilobases of genomic sequence from this human chromosome 3p21.3 region and then determined the utility of the predicted primers in detecting length polymorphisms and allele loss in a panel of human tumor and normal DNAs.
Table 1.
Minimum criteria for polymorphism
| Type of repeat | Threshold number* of repeating units | Minimum homogeneity** |
|---|---|---|
| Dinucleotide | 8† | 0.9 |
| Trinucleotide | 7‡ | 0.9 |
| Tetranucleotide | 6§ | 0.9 |
| 5-mer–9-mer | 6¶ | 0.9 |
| 10-mer–250-mer | 4‖ | <2 consecutive errors |
The threshold number refers to the minimum number of repeated units required for a locus to be scored as a likely polymorphism.
Ref. 8.
The minimum homogeneity refers to the minimum overall identity score for an alignment of the query sequence against the repeat sequence to which it is most similar.
MATERIALS AND METHODS
Computational Tools.
All codes were written in either ANSI C, HP/C, or Perl and run on an HP/Convex Exemplar with eight processors and 512 MB of shared RAM. Database searches for simple repetitive elements in sequences were performed by using a parallel version of blastn (23). The accession numbers of the cosmid and P1 clone genomic DNA sequences analyzed are listed in the Appendix.
Genomic DNA.
Genomic DNA was extracted by standard methods (32) from 36 human B lymphoblastoid cell lines and paired tumor lines (one mesothelioma, one breast carcinoma, 14 small cell lung carcinoma, and 20 nonsmall cell lung carcinoma).
Primers, PCR Conditions, and Allelotyping.
Genomic DNA was amplified via PCR using the predicted primers (Table 2). Primers for this study were produced by using a MerMade high-throughput oligonucleotide synthesizer (S. Rayner, S. Brignac, R. Bumeister, Y. Belodludtsev, T. Ward, O. Grant, K.M.O., G. A. Evans, and H.R.G., unpublished work). Reactions were run in a Perkin–Elmer 9700 thermocycler with 1.5 mM MgCl2, 200 μM each dNTP (GIBCO), 2 μCi α32P-dCTP, 10 μM each primer, AmpliTaq 0.5 μg/μl (Perkin–Elmer). A touch-down PCR program with a modified hot start was used: initial denaturation at 94°C for 2.5 min followed by 10 cycles with denaturation at 94°C for 30 sec, annealing starting at 68°C and decreasing by 1°C for each cycle for 30 sec, and extension at 72°C for 30 sec followed by 25 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec, and extension at 72°C for 30 sec followed by a final extension at 72°C for 10 min. PCR products were heat-denatured, snap-chilled, and run on a denaturing 6.7% acrylamide gel (acrylamide/bis acrylamide ratio 19:1) with 10 M urea. Results were visualized by autoradiography using BioMax film (Kodak).
Table 2.
PCR-allelotyped primers predicted by pompous for region 3p21.3
| Oligo name* | Forward primer† | Reverse primer† | Size | Sequence |
|---|---|---|---|---|
| LUCA1.1 | CCTCATCCTCTCTGTTGGG | GCAGAATGACGTGAACCC | 266 | (ct)15 |
| LUCA1.2 | TTCCTCCACAGATTCCTCTG | TTGAGAAGGATGTGGATGAC | 277 | (ca)18 |
| LUCA1.3 | GTTCATGATGGCAGACTCTG | CCCAGAACAATCACAAGATG | 259 | (ca)23 |
| LUCA2.1 | GCTCCTCAGGCAGAGTCC | CCACAGCCATCCACTGGAAGG | 371 | (ca)14(ca)14 |
| LUCA2.2 | GGCAGTGTTTGAGCTTACATGGG | CAGGCTCTGGAAACCAAGC | 172 | (ca)29 |
| LUCA4.1 | CTGCCTGCCTCACTACTCC | CAAACTGCCCAGCTTCTG | 182 | (ca)23 |
| LUCA4.2 | CCAGTGAGTACCAAGATATGGG | GAGAATGTGACCCGGGC | 236 | (aaac)6 |
| LUCA4.3 | ACATGGTGGCATGTGCC | AGACCTTTAGCGTGTCATTTAC | 239 | (aaaat)6 |
| LUCA4.4 | GATCATGGAGGCCTTGAATG | TGTTCATTGCCTGCATGG | 407 | (ca)13 |
| LUCA6.3 | AAAGTACAGGGCAGTGTCAG | AGAAGAAGAGTCCAGCAAGC | 480 | (aatg)6 |
| LUCA7.1 | GGAGATCACCTGGACACATC | TAAGTGCTAGGAGGCACTGC | 229 | (agcccc)7 |
| LUCA7.2 | TTGTGGGCACTTCTTGCTAG | ACAACATATGCAAGGCCCAG | 523 | (aatg)6 |
| LUCA8.1 | TAACCCTTGGCTTTGCTG | GGCTACTCTTCAACAAGATGC | 274 | (ca)30 |
| LUCA8.2 | GCTGAGAAATCTCAATTGTGGGTG | GGCTGCTGAGCAGTGTCAGAC | 125 | (ca)11 |
| LUCA8.3 | AATTCAGAACTGCGCCTTG | GCAGAGTGAGTAAAGGTTCACAG | 485 | (aat)25 |
| LUCA11.1 | CACACACATCAGGCCGC | CTGCTCACAACCCCCTACC | 252 | (ca)22 |
| LUCA11.2 | AAGACCAGCAAGGTGGC | TACTAGGGCTCCAGAGAGGG | 238 | (ca)13 |
| LUCA11.4 | CCTGTGCCCACTCCCTATCTC | CCCAGGCTGGGATGCCTTTGG | 177 | (ca)12 |
| LUCA12.1 | TTCTCTGAGGAATGACTCCTGG | AACAGCGCCTTATGATGGAG | 308 | (cca)8 |
| LUCA13.4 | CCGCCTTCCAAGTTTAAGTG | TCAACCTGGGAGGTGGG | 199 | (aat)7 |
| LUCA17.1 | GCTTGATCTTGGCTCACCTC | AGGATCACTTGGGCCTG | 374 | (aaat)8 |
| LUCA17.2 | GTGGGTGGATCATCAGGTC | GTGCCTACAATAGAGAGGGTC | 360 | (ttttg)6 |
| LUCA17.4 | GAACATGGAAGGAGGAGG | CTCGATCTATCTCTTGATCTCAG | 220 | (aaat)8 |
| LUCA19.1 | AGGCAGGCTATATTCAACCC | GAAAAAGCCCCGGAACC | 199 | (ta)15 |
| LUCA19.2 | AATTAGGACCTTCAGGAAACC | CCAGCAGATGGAGGTTGC | 204 | (aaag)6 |
| LUCA20.2 | TGTGGTCCCACCTAATCTGG | CACATGCGTGCACATGC | 304 | (aaag)11, (aagg)11 |
| LUCA22 | GAAGTGGTTCACCCTAAGATC | AAGGGAGCTGGCCTAGGAG | 235 | (ct)12 |
| 3938P1.2 | AAACCTAGTTGATGCCAGGC | AACTTCTGGGATACATGGGC | 413 | (aaac)6 |
| 3938P1.3 | TGCCAGAGCCCCAAAAC | AAATGGGGCCAGGTGTG | 416 | (ttttc)6 |
| 3938P1.4 | TAAACCACTAGGCCCGGC | AGGTGTGGTGTTGTGTGCC | 246 | (ttttg)6 |
| 3938P1.5 | TGTTCCCTTCCCTATAAACAGAT | GAAAGCAAGGAAGGCACATG | 167 | (ca)24 |
| 3938P1.6 | CATGGTGGCACACAACTG | CCTCACCTGCCTGATTTTTG | 310 | (aaat)6 |
| 3938P1.7 | AGCAGATAGACAGATAAAGAGAGG | TCAGCTTCTTTGTCCCTTTCAC | 211 | (10 mer)5 |
Oligo name represents the LUCA cosmid the primer maps to (see Appendix).
Sequences are listed in a 5′ to 3′ orientation.
RESULTS
Computational Considerations.
Current algorithms and software tools for the identification of simple sequence repeats, commonly performed to remove low-complexity elements from a sequence before computational analysis (filtering or masking), are based on one of two primary methods: database analysis or self-comparison of sequences (13–23) or sophisticated hybrids of both methods (13). Database comparison methods for masking repeat sequences are preferred for microsatellite repeats because they are easily implemented by using available comparison programs such as blastn and are readily optimized to quickly yield meaningful results with a minimum of noise for small repeat sequences (23). These programs typically function by comparing the query sequence with a compiled database of simple sequence repeats and then excluding regions with significant similarity from subsequent genetic database queries. For larger repeating elements, such as minisatellites, self-comparison algorithms are more efficient because repeat databases grow exponentially with repeating element size. Because hypervariable tandem repeats can have repeating units with lengths ranging from two to greater than 50, we chose to use a dual strategy for the large scale, automated analysis of large tracts of genomic sequence, using each type of algorithm where it is most effective. These algorithms are combined with codes that evaluate the likelihood of polymorphism of the identified repeats and then design PCR primers for their amplification to give the researcher a comprehensive tool for length polymorphism identification.
For the strategy we used to detect and evaluate novel microsatellite repeats an exhaustive repeat database was required. A cursory examination of widely used repeat databases found that they are not exhaustive, but contain only those simple sequence repeats that have been previously described and cataloged. For odd repeat unit length, n, the number of nonrepetitive nonredundant sequences is given by:
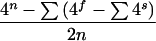 |
for all f = factors of n excluding n, and for all s = factors of f excluding f.
The terms in the summation account for repeats that can be represented as repeats with shorter periods, whereas the n in the denominator accounts for all cyclic permutations and the 2 in the denominator accounts for their complements (for even n the calculation is complicated by the occurrence of sequences that are their own complements because simply dividing by 2 is no longer valid). For example, for repeating unit length n = 9 there are
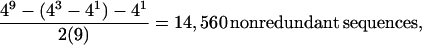 |
whereas the simple sequence repeat databases available contained less than 100. Available repeat sequence generation programs are unsuitable for any large n as they generate all 4n sequences and compare all possible cyclic permutations of each sequence and their complements to all previously selected sequences, compiling a list of unique sequences. This algorithm, though direct, becomes unwieldy quickly [efficiency is O(42n)] as it compares every possible sequence to every other sequence in the exponentially growing list of selected sequences, and this effectively limits its utility to small n. To alleviate this computational obstacle to determining the optimal value of n for the database-centered portion of our approach, we developed an algorithm that uses a branch and bound technique (13) to eliminate the computationally expensive cross-comparison used by other codes. It also has the added advantage of lending itself well to parallel processing. We wrote a program based on this algorithm, called simpleseq, to generate minimal exhaustive simple sequence databases for n ≤13 on an eight-processor HP/Convex Exemplar. These data, once computed, were kept in a series of databases for n ≤ 8, n ≤ 9, … , n ≤ 13 for empirically determining the optimal database size for the database-centered portion of our approach.
A Perl script, tandmin, was written to take as input the results of a blastn of the sequence of interest against this simple repeat database and parse the high scoring hits to determine the repeat unit length, level of homogeneity, type of repeat, and the number of repeating units and evaluate these parameters against the criteria listed in Table 1. These minimum thresholds were drawn from the extensive literature on microsatellite and minisatellite repeat polymorphisms and represent a conservative extrapolation of the data found therein to reliably identify repeat sequence loci with a greater than 50% probability of having a heterozygosity (H) of at least 0.50 (see Table 1 for references). We ran tandmin on several cosmid and PAC genomic sequences by using each of the simple sequence repeat databases containing all repeats with length n less than or equal to 13 and found that tandmin run times increased dramatically when we went from a database representing all repeats with n ≤ 9 to one with n ≤ 10 (from ≈4 sec to several minutes), we therefore selected n ≤ 9 as the optimal database size for use (Table 1). A large increase was expected because of details of the implementation, primarily because of our minimum required homogeneity threshold of 90% identity. To be scored as repeats, adjacent sequences must be at least 90% identical to database repeat core sequence. For repeat units of length 10 or greater, a single base mismatch in every repeat no longer drops the identity score below 90%.
For the detection of larger repeats the database approach is clearly not feasible (for n = 37 the minimal sequence set is 1.9 × 1022 sequences). To identify these longer repeats a second program, tandmax, was written that compares the query sequence to itself at offsets of 10–250 bases, looking for stretches of near identity (no more than two consecutive mismatches or indels) that are longer than the offset. This approach is analogous to graphic matrix analysis wherein the presence of diagonals close to the main diagonal in a binary comparison matrix is indicative of tandem repeats (14, 34). Those sequences satisfying these criteria then are analyzed further to determine the number of repeating units. Because large tandem repeats frequently have units deleted in the course of cloning and sequencing, the threshold number of repeats is reduced to four for minisatellites of repeating unit length from 10 to 250 nt. In addition, Jeffreys et al. (12) report that minisatellites appear to have a different mechanism of mutation than microsatellites that may not be dependent on copy number (see also ref. 30).
pompous is a Perl script that automates the entire process (Fig. 1). It runs the parallel blastn (sppblastn) on the query sequence, sends the output to tandmin and then invokes tandmax. The outputs of tandmin and tandmax are integrated, consolidating any redundant or adjacent hits. pompous then takes these results and translates them into the proper input format of the primer prediction software, primo (35). pompous executes primo, which designs multiple flanking oligonucleotides for PCR amplification of all selected repeat regions. Each oligonucleotide is checked for optimal length, melting temperature, GC content, self-complementary, GC clamps, base quality (if available), and complementary to common human repeats (e.g., Alu, LINE1, THE). The final output is a file containing a list of repetitive sequences, statistics such as length and identity, and predicted primers and conditions that can be used for amplification of the repeat loci.
Figure 1.

Program flow. pompous takes as input a sequence of length up to 250,000 bases in fasta format and processes it first by a parallel sequence homology search (sppblastn), which then is parsed by tandmin for hits with the properties listed in Table 1. The sequence then is scanned for larger tandem repeats by tandmax, and the outputs are consolidated. pompous then converts the consolidated data into a format that is usable by primo (regions file), and primo uses this file to predict flanking oligonucleotide primers for subsequent PCR amplification.
pompous Identifies Polymorphic Loci.
To test the effectiveness of pompous in identifying polymorphic repeat loci that are useful in genetic studies, we ran the analysis on an approximately 750-kilobase region of human chromosome 3p21.3 (28). This region is involved in homozygous deletions in small cell lung carcinoma and breast cancer and is an area of intense search for a tumor suppressor gene (31, 36–39). pompous predicted primer sets for 40 tandem repeat loci among the 19 cosmid and P1 clones. For this study we tested all 40 pompous-predicted primer sets. Table 2 lists all primer sets yielding specific products. To determine the frequency of heterozygosity and utility of the pompous predicted primer set we tested it for amplification, specificity, and heterozygosity in a panel of 36 predominately Caucasian patient DNAs, including matched B lymphoblastoid and tumor cell lines. In this way we were able to determine not only the heterozygosity of the selected loci, but also their utility in studies of allele loss. In addition to the usual controls (normal human DNA and a water blank), DNAs from two small cell lung cancer lines, NCI-H740 and H1450, were included that carry ≈1.5 Mbp and ≈.75 Mbp homozygous deletions within the 3p21.3 region, respectively. H1450 has a paired B lymphoblastoid line that also was included in these studies. These homozygous deletion DNAs provide a specificity check, as the predicted primers sets should yield no product in reactions with these templates. The results are summarized in Table 3. Thirty-three reactions yielded no product from the homozygously deleted tumor line DNAs (Fig. 2A, DEL) while giving clean product of the approximate expected size in matched B lymphoblastoid lines (Fig. 2). Two previously discovered polymorphic markers [K1.1CA and D3S1568(Z23748)] were identified and predicted by pompous (Table 3). Primer sets yielding product from the homozygously deleted lines (five sets) or giving no product from normal lines (two sets) were excluded from further analysis (Table 3). Of the 33 loci yielding clean amplification patterns 22 were found to be polymorphic with heterozygosities ranging from 0.03 to 0.86 (mean H = 0.42) (Table 3). This gives an estimate of one marker for every 31 kilobases on average. All of the patients were heterozygous for at least one of the 22 informative markers (average of nine heterozygous loci per patient), and as predicted from many previous studies of 3p21.3, allele loss was found to be common, occurring in 75% of the tumor line DNAs.
Table 3.
Heterozygosity rate for pompous-predicted polymorphic markers
| Marker | Repeat type* | Repeat homogeneity | No. of alleles | Rate of heterozygosity† (n = 36 individuals) |
|---|---|---|---|---|
| LUCA1.1 | (ct)15 | 0.97 | 2 | 0.42 |
| LUCA1.2 | (ca)18 | 0.94 | 1 | <0.03 |
| LUCA1.3 | (ca)23 | 1.00 | 4 | 0.64 |
| LUCA2.1 | (ca)14(ca)14 | 0.97 | 4 | 0.42 |
| LUCA2.2‡ | (ca)29 | 0.98 | 7 | 0.69 |
| LUCA4.1 | (ca)23 | 1.00 | 4 | 0.64 |
| LUCA4.2 | (aaac)6 | 1.00 | 1 | <0.03 |
| LUCA4.3 | (aaaat)6 | 0.94 | 1 | <0.03 |
| LUCA4.4 | (ca)13 | 0.92 | 1 | <0.03 |
| LUCA6.2 | (15mer)4 | 0.97 | NA | No product |
| LUCA6.3 | (aatg)6 | 0.97 | 2 | 0.03 |
| LUCA7.1 | (agcccc)7 | 0.95 | 3 | 0.08 |
| LUCA7.2 | (aatg)6 | 0.96 | 2 | 0.03 |
| LUCA7.3 | (10 mer)6 | 0.85 | NA | No product |
| LUCA8.1§ | (ca)30 | 1.00 | 7 | 0.72 |
| LUCA8.2 | (ca)11 | 1.00 | 3 | 0.53 |
| LUCA8.3 | (aat)25 | 0.96 | 3 | 0.64 |
| LUCA11.1 | (ca)22 | 1.00 | 4 | 0.50 |
| LUCA11.2 | (ca)13 | 1.00 | 2 | 0.03 |
| LUCA11.4 | (ca)12 | 1.00 | 2 | 0.22 |
| LUCA12.1 | (cca)g | 0.96 | 1 | <0.03 |
| LUCA13.1 | (10 mer)4 | 0.73 | NA | Nonspecific product |
| LUCA13.4 | (aat)7 | 0.95 | 2 | 0.17 |
| LUCA13.5 | (ttg)10 | 0.96 | NA | Nonspecific product |
| LUCA17.1 | (aaat)g | 0.91 | 1 | <0.03 |
| LUCA17.2 | (ttttg)6 | 1.00 | 3 | 0.31 |
| LUCA17.3 | (13 mer)4 | 0.85 | NA | Nonspecific product |
| LUCA17.4 | (aaat)8 | 1.00 | 1 | <0.03 |
| LUCA19.1 | (ta)15 | 0.93 | 6 | 0.86 |
| LUCA19.2 | (aaag)6 | 0.92 | 1 | <0.03 |
| LUCA20.1 | (aaag)12, (aaag)12, (10 mer)6 | 1.00 | NA | Nonspecific product |
| LUCA20.2 | (aaag)11, (aagg)11 | 0.94 | 4 | 0.83 |
| LUCA22 | (ct)12 | 0.96 | 1 | <0.03 |
| P1.1 | (aat)13 | 0.97 | NA | Nonspecific product |
| P1.2 | (aaac)6 | 0.92 | 2 | 0.03 |
| P1.3 | (ttttc)6 | 0.90 | 1 | <0.03 |
| P1.4 | (ttttg)6 | 0.96 | 2 | 0.58 |
| P1.5 | (ca)24 | 1.00 | 5 | 0.83 |
| P1.6 | (aaat)6 | 0.96 | 1 | <0.03 |
| P1.7 | (10 mer)5 | 0.82 | 2 | 0.03 |
NA, not available.
(Repeat sequence)number of repeats.
Rate of heterozygosity = heterozygous cell lines/36.
Previously identified as K1.1CA.
Previously identified as D3S1568(Z23748).
Figure 2.

Representative allelotyping results. Polyacrylamide allelotyping gels for paired B lymphoblastoid and tumor DNAs visualized by autoradiography. (A) Marker LUCA2.2, (ca)n repeat. (B) LUCA20.2, (aaag)n(aagg)n repeat. The bracket above each lane contains DNA pairs from the same individual, first normal DNA and second tumor DNA. Arrows denote different alleles for each marker. DEL, H1450 deletion line; HET, retention of heterozygosity; LOH, loss of heterozygosity; NI, noninformative.
We then went on to analyze 55,402 entries in GenBank comprising 141,779,504 bp of human genomic sequence selected to include the words “human” or “Sapiens” and exclude the word “mRNA” within the annotation of GenBank gbpri1 and gbpri2 databases. We downloaded these sequences from GenBank on March 16, 1998. pompous identified 13,261 probable polymorphic markers in 4,669 of the GenBank entries. If 67% of the predicted markers are polymorphic as was reported in this study that would mean ≈8,885 potential additional markers have been identified, giving an average marker density of 1 marker per 16 kilobases. The actual number of markers undoubtedly will be less than this as many clones in the database contain overlapping or redundant sequences, and pompous analyzes each sequence independently. Additionally, for efficiency reasons, primo is not set to check primers for complementarity to genomic sequences other than common repeats, and this likely contributed to our observed PCR amplification failure rate of 17.5% (7/40). This catalog of putative polymorphic markers, primer sequences, and repeat characteristics, which is now available on our Web site (pompous.swmed.edu), is anticipated to be of value to the postgenomics community of researchers involved in gene identification and localization. Given that this sequence represents ≈4% of the human genome, and coding sequences are overrepresented in the accumulated GenBank database, at least 200,000 new polymorphic markers could be identified from the complete sequence now being generated by the Human Genome Project. Repeats with unit sizes from 2 to 229 were identified, for a total of 76 different unit sizes in the range of 2 to 250 checked by this code. As a percentage of the total repetitive DNA analyzed, the repeat unit sizes were: 50.0% di-, 10.2% tri-, 24.4% tetra-, 5.7% penta-, 2.2% 6 mer-9 mer, and 7.5% 10 mer-229 mer. This represents a total of 710,825 bp, or 0.5% of the GenBank human sequence being part of a repetitive unit that is potentially polymorphic.
DISCUSSION
The identification of polymorphic genetic markers from the huge quantities of genomic sequence generated by the Human Genome Project is necessary for the genetic researcher to localize specific sequences associated with phenotypes and disease. Although massively parallel single nucleotide polymorphism genotyping technologies hold great promise for the future of genetic mapping, they currently cannot be predicted from a single genomic sequence, and their identification relies on large-scale resequencing of genomic DNA from multiple individuals. Length polymorphisms therefore are still the preferred markers for genetic linkage studies. We have developed a suite of programs, called pompous, to predict putative length polymorphisms directly from a single copy of genomic DNA sequence. pompous is designed to detect tandem repeats with repeating unit length ranging from 2 to 250, select those that are likely to be highly polymorphic, and design flanking oligonucleotide primers for use in PCR amplification. To test the accuracy of pompous we allelotyped 33 of these predicted loci in B lymphoblastoid cell DNAs from 36 individuals (72 chromosomes) to determine their levels of polymorphism.
We found 22 to be polymorphic with heterozygosity rates ranging from 0.03 to 0.86, with 11 having heterozygosities of at least 0.5. With no consanguinity and modest mutation rates, the probability that any one individual would be homozygous for these 11 loci is 3.4 × 10−6 and for all 22 polymorphic loci is 1.9 × 10−7. Thus, this panel of polymorphic markers will be useful for tumor allelotyping studies as well as for accurately placing genes in this region on the human recombination map. As a first test of their utility as such we allelotyped the 36 matched tumor cell lines and found very frequent loss of heterozygosity of this region, consistent with the presence of a 3p21.3 tumor suppressor gene.
There was a difference in utility of loci identified by tandmin (repeat unit less than 10 bases) and those identified by tandmax (repeat unit of length 10 or longer). Longer repeats were more difficult to cleanly amplify, largely because of weak homology to the repeats in the flanking sequences from which primers were designed by primo. Some degenerate highly repeated sequences were identified by pompous (including a minisatellite with a 49-bp core sequence repeated approximately 35 times), but not scored as likely polymorphisms because no more than three consecutive repeats were highly conserved. We currently are developing a more sophisticated algorithm for the identification of longer repeats and modifying the primer selection process to minimize these effects. Additionally, because of the fundamentally different algorithm and criteria used in their detection and selection, longer repeats also had identities well below those of repeats scored by tandmin (Table 3). As a more statistically significant quantity of data becomes available, the threshold criteria will be refined to make pompous more efficient at selecting only the most useful loci.
Future studies will allow precise mapping of very small areas of allele loss as have been found for preneoplastic bronchial epithelial lesions (33). Likewise, by comparing the particular alleles at multiple loci in different tumors with normal tissues and individuals, it will be possible to define allelotype sets that are associated with pathogenicity. This information will be useful for future recombination analysis, genetic epidemiology studies, and predisposition diagnosis.
Acknowledgments
We thank Hewlett Packard, specifically David Valenta, David Field, and Don North, for use of the Exemplar computer on which this work was performed. We thank Dr. Glen Evans for providing space for the Exemplar computer and Dr. Evans and Ron Bumeister for providing some of the oligonucleotide primers used in this study. We also thank Dr. Chris Davies, Peter Schilling, Dr. Adi Gazdar, Tina Chen, Dr. Carmen Behrens, Dr. Ivan Wistuba, Dr. Arvind Virmani, and Victor Stastny for many useful discussions and reagents. This work was supported by the Whitaker Foundation, the P. O’Brien Montgomery Distinguished Chair in Developmental Biology, National Cancer Institute Grants CA 71618 and SPORE P50 CA70907, Texas Advanced Research Projects Grant 003660–080, and the G. Harold and Leila T. Mathers Charitable Foundation.
ABBREVIATION
- pompous
polymorphic marker prediction of ubiquitous simple sequences
Appendix
Database Availability.
pompous has been run for all human genomic sequences in GenBank as of March 16, 1998 (GenBank release v105.0). The results are downloadable as a database and will be expanded to include all of GenBank and will be updated monthly as GenBank expands. All results are available for FTP downloading from our Web site (pompous.swmed.edu). In addition, a pompous server will be available at that site for the analysis of any submitted sequences, with the submission and results conducted through the World Wide Web.
Accession/Contig Numbers.
Analyses were performed on the following sequences: Luca1, Z74618; Luca2, Z77852; Luca3, Z74023; Luca4, Z74019; Luca5, Z74582; Luca6, Luca06.00286, Luca06.01215, Luca06.01317, Luca06.01351, Luca06.01396 (all Luca6 contigs have accession no. Z84493); Luca7, LUCA7.00598, LUCA7.00720 (all Luca7 contigs have accession no. Z84494); Luca8, Z84495; Luca9, Z75743; Luca10, Z75742; Luca11, LUCA11.00789, LUCA11.00039, LUCA11.01287 (all Luca11 contigs have accession no. Z84492); Luca12, AC002481; Luca13, AC002455; Luca14, U73167; Luca15, U73166; Luca16,U73169; Luca17, AC002077; Luca19, AC000063; Luca20.Contig17, Luca20.Contig18 (St. Louis Genome Center); Luca22, U73168; 3938P1.Contig16 (St. Louis Genome Center). Marker LUCA11.1 is in contig LUCA11.00789, and markers LUCA11.2–11.4 are located in contig LUCA11.00039.
References
- 1.Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. Nature (London) 1996;38:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Pulido J C, Duyk G M. Genomics. 1995;25:1–8. doi: 10.1016/0888-7543(95)80103-s. [DOI] [PubMed] [Google Scholar]
- 3.Malo D, Vidal S M, Hu J, Skamene E, Gros P. Genomics. 1993;16:655–663. doi: 10.1006/geno.1993.1244. [DOI] [PubMed] [Google Scholar]
- 4.Cornelis F, Hashimoto L, Loveridge J, MacCarthy A, Buckle V, Julier C, Bell J. Genomics. 1992;13:820–825. doi: 10.1016/0888-7543(92)90159-p. [DOI] [PubMed] [Google Scholar]
- 5.Ostrander E A, Jong P M, Rine J, Duyk G. Proc Natl Acad Sci USA. 1992;89:3419–3423. doi: 10.1073/pnas.89.8.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lathrop M. Nature (London) 1992;359:794–801. doi: 10.1038/359794a0. [DOI] [PubMed] [Google Scholar]
- 7.Marshall E, Pennisi E. Science. 1996;272:188–189. doi: 10.1126/science.272.5259.188. [DOI] [PubMed] [Google Scholar]
- 8.Charmley P, Concannon P, Hood L, Rowen L. Genomics. 1995;29:760–765. doi: 10.1006/geno.1995.9940. [DOI] [PubMed] [Google Scholar]
- 9.Iizuka M, Makino R, Sekiya T, Hayashi K. Genet Anal Tech Appl. 1993;10:2–5. doi: 10.1016/1050-3862(93)90017-d. [DOI] [PubMed] [Google Scholar]
- 10.Weber J L. Genomics. 1990;7:524–530. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 11.Gastier J M, Pulido J C, Sunden S, Brody T, Buetow K H, Murray J C, Weber J L, Hudson T J, Sheffield V C, Duyk G M. Hum Mol Genet. 1995;4:1829–1836. doi: 10.1093/hmg/4.10.1829. [DOI] [PubMed] [Google Scholar]
- 12.Jeffreys A J, Wilson V, Thein S L. Nature (London) 1985;314:67–73. doi: 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- 13.Benson G, Waterman M S. Nucleic Acids Res. 1994;22:4828–4836. doi: 10.1093/nar/22.22.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claverie J-M, States D J. Comput Chem. 1993;17:191–201. [Google Scholar]
- 15.Wooton J C, Federhen S. Comput Chem. 1993;17:149–163. [Google Scholar]
- 16.Brutlag D L, Dautricourt J-P, Diaz R, Fier J, Moxon B, Stamm R. Comput Chem. 1993;17:203–207. [Google Scholar]
- 17.Pearson W R, Lipman D J. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipman D J, Pearson W R. Science. 1985;227:1435–1441. doi: 10.1126/science.2983426. [DOI] [PubMed] [Google Scholar]
- 19.Brendel V, Bucher P, Nourbakhsh I R, Blaisdell B E, Karlin S. Proc Natl Acad Sci USA. 1992;89:2002–2006. doi: 10.1073/pnas.89.6.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shpaer E. In: Methods in Molecular Biology Sequence Data Analysis Guidebook. Swindell S, editor. Totowa, NJ: Humana; 1996. [Google Scholar]
- 21.Hancock J M, Armstrong J S. Comput Appl Biosci. 1994;10:67–70. doi: 10.1093/bioinformatics/10.1.67. [DOI] [PubMed] [Google Scholar]
- 22.Staden R. Mol Biotechnol. 1996;5:233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Watkins W S, Bamshad M, Jorde L B. Hum Mol Genet. 1995;4:1485–1491. doi: 10.1093/hmg/4.9.1485. [DOI] [PubMed] [Google Scholar]
- 25.Hammond H A, Jin L, Zhong Y, Caskey C T, Chakraborty R. Am J Hum Genet. 1994;55:175–189. [PMC free article] [PubMed] [Google Scholar]
- 26.Sheffield V C, Weber J L, Buetow K H, Murray J C, Even D A, Wiles K, Gastier J M, Pulido J C, Yandava C, Sunden S L, et al. Hum Mol Genet. 1995;4:1837–1843. doi: 10.1093/hmg/4.10.1837. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Rasool O, Grander D, Lindblom A, Einhorn S. Hum Mol Genet. 1995;4:727–729. doi: 10.1093/hmg/4.4.727. [DOI] [PubMed] [Google Scholar]
- 28.Armour J A L, Jeffreys A J. Curr Opin Genet Dev. 1992;2:850–856. doi: 10.1016/s0959-437x(05)80106-6. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura Y, Leppert M, O’Connell P, Wolff R, Holm T, Culver M, Martin C, Fujimoto E, Hoff M, Kumlin E, White R. Science. 1987;235:1616–1622. doi: 10.1126/science.3029872. [DOI] [PubMed] [Google Scholar]
- 30.Andreassen R, Egeland T, Olaisen B. Am J Hum Genet. 1996;59:360–367. [PMC free article] [PubMed] [Google Scholar]
- 31.Wei M-H, Latif F, Bader S, Kashuba V, Chen J-Y, Duh F-M, Sekido Y, Lee C C, Geil L, Kuzmin I, et al. Cancer Res. 1996;56:1487–1492. [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 33.Chung G T, Sundaresan V, Hasleton P, Rudd R, Taylor R, Rabbitts P H. Oncogene. 1995;11:2591–2598. [PubMed] [Google Scholar]
- 34.Maizel J V, Jr, Lenk R P. Proc Natl Acad Sci USA. 1981;78:7665–7669. doi: 10.1073/pnas.78.12.7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P, Kupfer K C, Davies C J, Burbee D, Evans G A, Garner H R. Genomics. 1997;40:476–485. doi: 10.1006/geno.1996.4560. [DOI] [PubMed] [Google Scholar]
- 36.Daly M C, Xiang R-H, Buchhagen D, Hensel C H, Garcia D K, Killary A M, Minna J D, Naylor S L. Oncogene. 1993;8:1721–1729. [PubMed] [Google Scholar]
- 37.Todd S, Franklin W A, Varella-Garcia M, Kennedy T, Hilliker C E, Jr, Hahner L, Anderson M, Wiest J S, Drabkin H A, Gemmill R M. Cancer Res. 1997;57:1344–1352. [PubMed] [Google Scholar]
- 38.Kok K, Naylor S L, Buys C H C M. In: Advances in Cancer Research. Klein G, Vande Woude G F, editors. Vol. 71. San Diego: Academic; 1997. pp. 27–92. [DOI] [PubMed] [Google Scholar]
- 39.Sekido, Y., Ahmadian, M., Wistuba, I. I., Latif, F., Bader, S., Wei, M.-H., Duh, F.-H., Gazdar, A. F., Lerman, M. I. & Minna, J. D. (1998) Oncogene 15, in press. [DOI] [PubMed]