

Abstract
Defects in Brca1 confer susceptibility to breast cancer and genomic instability indicative of aberrant repair of DNA breaks. Brca1 was previously implicated in the homologous recombination pathway via effects on the assembly of recombinase Rad51. Activation-induced cytidine deaminase (AID) deaminates C to U in B lymphocyte immunoglobulin (Ig) DNA to initiate programmed DNA breaks. Subsequent Uracil-glycosylase mediated U removal, and perhaps further processing, leads to four known classes of mutation: Ig class switch recombination that results in a region-specific genomic deletion, Ig somatic hypermutation that introduces point mutations in Ig V-regions, Ig gene conversion in vertebrates that possess Ig pseudo-V genes, and translocations common to B cell lymphomas. We tested the involvement of Brca1 in AID-dependent Ig diversification in chicken DT40 cells. The DT40 cell line diversifies Ig Vλ mainly by gene conversion, and less so by point mutation. Brca1-deficiency caused a shift in Vλ diversification, significantly reducing the proportion of gene conversions relative to point mutations. Thus, Brca1 regulates AID-dependent DNA lesion repair. Interestingly, while Brca1 is required to recruit ubiquitinated FancD2 to DNA damage, the phenotype of Brca1-deficient DT40 differs from the one of FancD2-deficient DT40, in which both gene conversion and non-templated mutations are impaired.
1. Introduction
Fundamental to humoral immunity is the vast diversity and exquisite specificity of antibodies. During early B cell development, germline immunoglobulin (Ig) V, D and J gene segments recombine to generate unique antibody variable region exons (combinatorial diversity). In humans and mice, immense diversity is generated from the possibly limitless combinations of V(D)J segments. Functional, assembled Ig genes are diversified further by three mechanisms: somatic hypermutation introduces point mutations and occasional insertions and deletions into the assembled V(D)J exon and flanks with the purpose to increase the affinity of antigen-specific antibodies. Alternatively, in chickens and a few other vertebrates where a small number of functional V gene segments limits combinatorial diversity (eg., in chickens only single functional V and J genes at L and H chain loci rearrange [1], further diversification is accomplished by gene conversion (GC) between the active V exon and a collection of 5′ pseudo-V gene segments [2,3]. GCs are characterized by mutations in the active V gene derived in sequence tracts of variable length from pseudo-V gene donor sequences during homologous recombination. Third, the Ig constant region isotype is altered by class switch recombination (CSR). CSR targets a genomic deletion that fuses a non-IgM/D constant region to the variable region.
SHM, GC and CSR share in common the requirement for activation-induced deaminase (AID), expression of which is strongly and specifically induced in activated B cells and chicken bursal B cells undergoing GC [4,5]. AID deaminates cytosine bases in Ig genes to produce uracils; untouched, the resulting U-G mismatches lead to transition mutations from G and C (ie., C to T and G to A) [6,7]. Alternatively, the uracil glycosylase Ung and mismatch repair enzymes Msh2/6 initiate further processing of the U-G mismatch to produce DNA breaks that lead to SHM, CSR, or GC by a regulated mechanism that is not understood (reviewed by [8]. Regulation of AID-mediated genomic alterations indeed is crucial because dysregulated AID expression and/or absence of DNA repair proteins involved in regulating the outcome of an AID-induced DNA lesion lead to oncogenic translocations of types frequently observed in lymphomas [9].
The chicken bursal B cell line DT40 constitutively acquires AID-dependent Ig GCs and SHM-like point mutations in the rearranged Vλ allele [10] and has become a popular model system for analyzing the genetic requirements for GC and SHM (as well as other processes) because of the relative ease of generating gene-knockout lines [11]. Moreover, genetic changes at the Ig heavy and λ light chain loci can be monitored phenotypically by gain or loss of surface IgM [12]. For example, the popular CL18 line of DT40 harbors a frameshift mutation in Vλ that abolishes surface IgM expression; the frameshift can be corrected by homologous recombination/GC with one of several Vλ pseudogenes to restore surface IgM. Homologous recombination in vertebrates is catalyzed by Rad51. Like bacterial RecA, Rad51 polymerizes on single-stranded DNA to form nucleoprotein filaments that catalyze pairing and strand exchange with homologous DNA (reviewed by [13]. Both in mammals and DT40, Rad51 is an essential gene [14]. However, DT40 cells deficient for any of the five vertebrate Rad51 paralogs (Rad51B, Rad51C, Rad51D, XRCC2 and XRCC3, which in humans share 20-30% identity with Rad51) are viable though impaired for homologous recombination, and all display a decrease in the frequency of Ig GC, as expected. Impaired Ig GC also is observed in DT40 cells mutated in BRCA2, a breast cancer susceptibility gene whose encoded protein plays a direct role in regulating Rad51 [15]. Surprisingly however, all Rad51 paralog, and BRCA2 mutants also display a concomitant increase in the frequency of point mutations in Vλ [15], a phenotype also achieved by deletion of the λ pseudo-V genes (ie., GC donor sequences) [16]. Together these results suggest that preventing recombination by absence of either the enzymes that catalyze recombination, or the recombination donor DNA, results in accumulation of Vλ point mutations that resemble mammalian SHM.
Ung-deficient DT40 also fail to perform GC but uniquely accumulate G/C transition mutations in Ig genes because they fail to remove AID-deaminated cytosines [6,17]. In contrast, DT40 cells lacking Rev1 (a dCMP transferase, and an adapter protein for error-prone polymerases Polη, Polι, Polκ, and Polζ, [18] generate GCs almost exclusively, indicating that Rev1 is necessary for generating SHM-like point mutations [19,20]. These results imply that a common intermediate – a U/G mismatch generated by AID, processed into a single-strand gap by Ung and subsequent base excision repair enzymes, and perhaps further processing (for instance by the Mre11-Rad50-NbsI endonuclease complex, [21] – initiates SHM or GC depending on the availability of partially homologous V-region donor sequence in cis, and the DNA repair factors involved [22].
Both Brca2 and Brca1 proteins are involved in DNA repair. Defects in either protein confer susceptibility to breast cancer [23], and genomic instability characterized by spontaneous accumulation of chromosomal rearrangements including translocations, deletions and chromosome fusions that indicate aberrant repair of DNA breaks (reviewed by [24]. Unlike Brca2 which binds Rad51 directly, it is not yet clear whether Brca1 interaction with Rad51 is direct or indirect. Nevertheless, Brca1 is involved in homologous recombination (reviewed by [25]; formation of ionizing radiation- and cisplatin-induced Rad51 nuclear foci require Brca1 in mammalian cells and DT40 [26-28], and homologous recombination repair of double-strand breaks is impaired in Brca1-deficient cells [29,30]. However, Brca1 also is reported to function in other DNA repair pathways, and diverse processes including transcription and cell cycle control (reviewed by [13,24]. Moreover, Brca1 is reported to interact with a large number of proteins, of which not all are implicated in DNA repair. In most cases, the functional significance of the Brca1 interaction is unclear, as is the function of Brca1 itself (reviewed by [31]. However, a number of the proteins with which Brca1 interacts are involved in Ig GC and/or SHM, including Brca2 [32], the Mre11/NbsI/Rad50 complex [33], Msh2/6 [34] and FancD2 [35]. Notably, DNA damage-localized Brca1 recruits FancD2, one of 13 proteins defective in Fanconi anemia, and which function in DNA repair (reviewed by [36]. Intriguingly, DT40 cells defective in FancD2 and FancC are impaired for both Ig GC as well as non-templated mutation [35,37].
We asked whether Brca1 plays a role in regulating Ig GC and non-templated point mutation in DT40. We hypothesized that Brca1-deficient DT40 might be impaired for Ig GC if Brca1 plays a role in facilitating GC, and further, that Brca1-deficient cells would lack non-templated mutations if Brca1 is epistatic to FancD2.
2. Materials and methods
2.1. DT40 cell lines and culture
The parent of the Brca1-/- cells (Fig.1) was DT40 cre. It and AID-/- derivatives were a gift from J.-M. Buerstedde and H. Arakawa (Institute of Molecular Radiobiology, GSF, Munich, Germany) via M. Nussenzweig and N. Papavasiliou (Rockefeller U., New York). DT40 cre, a derivative of CL18, expresses a v-myb transgene and also is transgenic for tamoxifen-inducible Cre recombinase [38]. Presumably due to v-myb expression, DT40 cre performs GC at a ∼5-fold increased rate in comparison to the DT40 parent line [38]. The parent of the Brca1-/- (clone #8) and the Brca1-/- (“partial” clone #37) lines is an IgM+ derivative of CL18 [39]. This DT40 cell line does not contain a v-myb or Cre transgene. The Brca1-/r lines (clones C1, F10 and L4) are the result of homologous integration of a chicken Brca1 cDNA (with the first two introns retained to promote efficient expression regulated by mRNA splicing) into one allele of the Brca1 -/- “partial” line (O. Yildiz, Y. Li, and D. B., unpublished). CL18 and the XRCC3-/- derivative thereof were generously provided by S. Takeda (Japan). CL18 and derivatives are mostly IgM-, due to a frameshift in Vλ [12]. All cells were cultured in RPMI supplemented with L-glutamine, 10% FBS, 1% chicken serum (Sigma), penicillin/streptomycin, and β-mercaptoethanol. Cells were cultured at 39.5°C, 5% CO2.
Fig. 1.
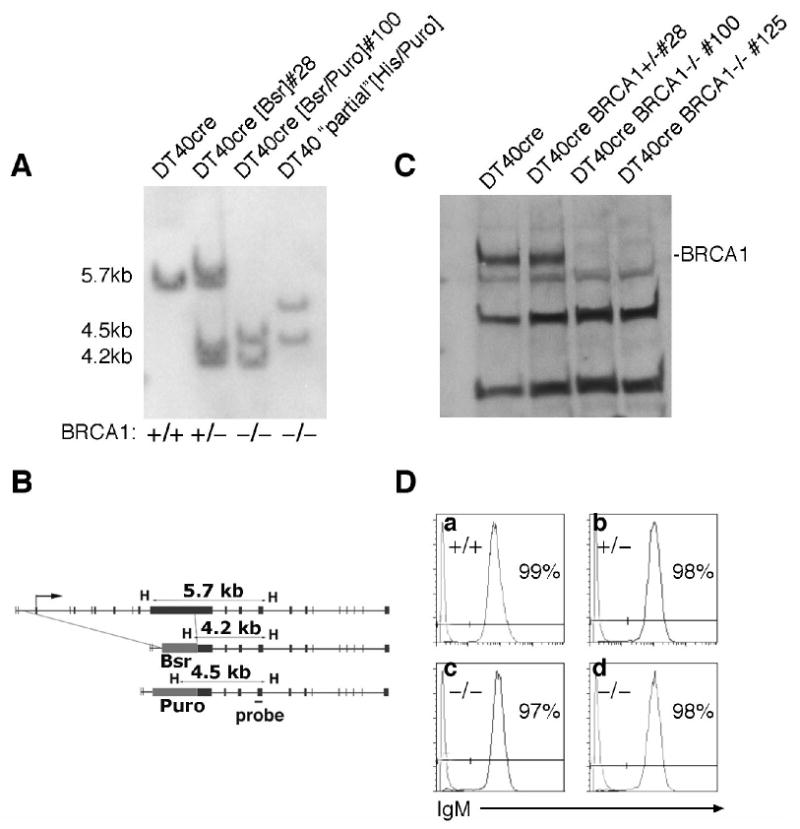
Brca1-deficient derivatives of DT40 cre. (A) Southern blot. (B) Targeting strategy. (C) Western blot of nuclear lysates, blotted and probed with anti-serum to chicken Brca1. The position of Brca1 is indicated. Other bands are non-specific proteins recognized by either the primary antiserum or secondary anti-rabbit IgG used for detection. (D) Flow cytometry of cells stained for surface IgM a – DT40 cre b – DT40 cre Brca1+/- c – DT40 cre Brca1-/- clone #100 d – DT40 cre Brca1-/- clone #125. One to two-week old cultures of each genotype were stained with anti-chicken IgM labeled with FITC, and analyzed by flow cytometry. All cultures are comprised of largely IgM+ cells.
2.2. Transfection and selection of DT40 cre
To obtain the Brca1-targeting vector with the blasticidin-resistance cassette, a histidine-resistance cassette was removed by BamH1 digest of pNRB384, a Brca1-targeting vector (Fig. 1B), and replaced with the blasticidin-resistance cassette removed from pNRB397 by BamH1 restriction. The puromycin-resistance cassette was similarly exchanged using pNRB383. The 5′ targeting flank is homologous with exons 1-2, and the 3′ flank is homologous with exon 11 of chicken Brca1.
For transfecting DT40 cre by electroporation, the targeting vector was linearized with NotI. Drug selection was started 12-16 hrs after electoporation with 25 μg/mL blasticidin (EMD Biosciences) or 0.5 μg/mL puromycin (AG Scientific).
2.3. Screening of drug-resistant DT40 cre clones for targeted integration of the Brca1 locus
One vial of 5-10 x 106 cells of each drug-resistant clone was frozen within 1-2 weeks of transfection to prevent in vitro evolution. Parallel cultures were expanded to obtain 5-10 x 106 cells that were harvested to purify DNA. Southern blots were probed with an 0.4-kb EcoRI fragment homologous with the chicken Brca1 locus just 3′ of the 3′ targeting flank sequence, but just 5′ of a diagnostic HindIII site (Fig, 1B).
2.4. Protein extraction and western blotting to detect Brca1 protein in DT40
The “NE-PER nuclear and cytoplasmic reagents kit” (Pierce), supplemented with a protease inhibitor cocktail (Roche), NaF (1mM) and Na3VO4 (1mM), consistently solubilized Brca1 into the “nuclear,” but not the “cytoplasmic” fraction (Fig. 1C). Cell lysates were resolved on 5-6% denaturing polyacrylamide mini-gels and transferred to PVDF Immobilon-P membranes (Millipore). The membranes were probed with rabbit anti Brca1 (a gift of N. Lowndes, University of Glasgow, Scotland).
2.5. RT-PCR
RNA was extracted using the RNAqueous kit (Ambion). First-strand cDNA was produced using the Superscript II cDNA synthesis kit (Gibco-BRL) according to the manufacturer's directions. Negative control templates were generated by identical first-strand synthesis reactions, but without addition of reverse transcriptase. Serial dilutions of first-strand cDNA were used as template for gene-specific PCR. Gene-specific primers and PCR conditions were as follows: ggRad6F: 5′ tgatgcgggacttcaagagattgc, ggRad6R 5′ ctgtggcacttgaaattcttggtagc; ggPCNAF 5′ agggctcggtgctcaagc; ggPCNAR 5′ cttcaatctttggagccaggtag; ggActinF 5′ ccccaagcttactcccacagccagccatgg; ggActinR 5′ ggctctagatagtccgtcaggtcacggcca. PCR conditions were 95°C, 4 min (1 cycle), 95°C, 30 seconds-56°C, 30 seconds-72°C, 1 minute (30 cycles) and 72°C, 7 minutes (1 cycle), using 3-fold dilutions of input first-strand cDNA.
2.6. Fluctuation analyses of IgM loss and gain in DT40
For each experiment, 12-24 independent cultures (derived by limiting dilution, manually or “automated” by flow sorting single cells into 96-well round-bottom plates) were first expanded to 1 mL in 24-well plates, then passaged daily for 2-6 weeks as indicated for each experiment (and determined by the doubling times for different DT40 lines; on average, 90-100 cell generations occurred in one experiment), maintaining cells at approximately 0.3-1.2 x 106 cells/mL. Cells rinsed in PBS and transferred to 96-well plates were then stained (or not, as a control) for surface IgM (with anti-chicken IgM, FITC-labeled, Bethyl Labs) in PBS 0.3%BSA, and analyzed on a FACScanII flow cytometer. 10,000-30,000 events were collected for each independent culture.
The percent of surface IgM loss in each culture was determined as described [12]; for each sample, live cells were gated on forward- and side-scatter plots, then displayed as histograms to detect FITC on the FL1 channel. For each sample, the IgM- (FITC-negative) gate boundaries were set to 8-fold below the median FL1 channel of the whole live population. The percent of cells falling within the entire IgM- gate represent the frequency of IgM loss in one culture. In a set of cultures of one genotype, the median of the IgM loss frequency was also calculated.
Genomic DNA was collected using the DNEasy kit (Qiagen) from a few clones of each set of cultures at the end of each experiment, from cells either sorted, or not, on the basis of their changed surface IgM phenotype.
2.7. Cloning, sequencing and mutation analysis of the DT40 Vλ locus
Analysis of events occurring at the Vλ locus in DT40 over time were performed essentially as described [12]. Briefly, genomic DNA from ∼10,000 cell-equivalents was used as a template for PCR-amplification with Pfu polymerase using CVLF6 (5′ CAGGAGCTCGCGGGGCCGTCACTGATTGCCG 3′) and CVLR3 (5′ GCGCAAGCTTCCCCAGCCTGCCGCCAAGTCCAAG 3′) with the following PCR cycling: 1 cycle at 94°C for 4 minutes, 9 cycles of 94°C for 1 minute, 68°C for 1 minute (decreased by 1°C every cycle and 72°C for 1.5 minutes, followed by 21-22 cycles of 94°C for 30 seconds, 60°C for 1 minute, and 72°C for 1,5 minutes, lastly extending for 7 minutes at 72°C. PCR products were cloned into pTopoII Blunt (Invitrogen) according to the manufacturer's instructions and used for transformation of E. coli Top10 cells. Bacterial colonies were sent for semi-automated culturing, plasmid preparation, and sequencing to the DNA core facility, University of Chicago, using M13F and M13R primers (Invitrogen).
Sequence changes in each clone were compared against a template sequence (Figure S2). Sequence changes that occurred in clones derived from the same culture were scored only once since all cells in one culture are clonally related, but if the same change occurred in multiple (but not all cultures of the same genotype – these were considered polymorphisms) cultures, the change was scored once for each independent culture. This is evident in Figure S2, where the same mutation occurred multiple times at the same nucleotide position in different subclones.
Classification of sequence changes as GCs, non-templated point mutations, or ambiguous was performed as described [12]. We used as references the sequences of the 25 known pseudo-Vλ genes [2] with modifications specific for the λG4 rearranged allele (DT40 is heterozygous for the Igλ locus, the unrearranged allele being λS3) as described [40]. Mutation frequencies for DT40 cre and Brca1-deficient derivatives were calculated as the number of mutational events (where one GC or one point mutation is one event) per number of total bps sequenced of clones not sorted for surface IgM phenotype.
2.6. Growth and clonogenic survival assays
Doubling times were determined by trypan blue exclusion using a Levy Brightline hemacytometer. Methylcellulose clonogenic assays were performed in 6-well plates containing DMEM-F12 (Gibco) supplemented with 7.5% FBS (Atlas), 1% chicken serum (Sigma) and 6% methyl-cellulose (Sigma). Cells were irradiated using a Maxitron generator calibrated for this purpose (250 KVp photons, 15 mA, 0.5 mm Cu filtration, dose rate=1.55 Gy/min).
3. Results
3.1. Generation of Brca1-deficient DT40
Brca1-deficient derivatives of the DT40 cre cell line were generated by targeted integration. This derivative of DT40 was chosen because it undergoes a high rate of Ig GC relative to the “parental” DT40 line [41]. Targeting vectors for deleting chicken Brca1 were designed to remove a major portion of the Brca1 coding sequence between exons two and eleven, as depicted in Fig. 1B. As assayed by Southern blotting (Fig. 1A), targeted integration efficiency for the Brca1 locus in DT40 cre was fairly low (4/41 clones targeted to obtain the heterozygous line) in comparison to reported homologous integration frequencies in DT40 for many loci (eg., [11], but similar low frequencies have been reported for some loci (eg., [17]. Targeting at the second allele occurred with a significantly lower frequency (3/139; P=0.048), perhaps because heterozygosity for Brca1 reduces the efficiency of homologous integration. Homologous integration frequencies for the Brca1 locus are summarized in Table 1. Two lines (#100 and #125) homozygous for Brca1 disruption were analyzed for Brca1 protein by western blotting in parallel with DT40 cre and the Brca1-heterozygous intermediate (clone #28). Neither homozygous Brca1-disrupted line produces detectable amounts of Brca1 protein (Fig. 1C).
Table 1. Targeted integration frequencies for the Brca1 locus in DT40 cre.
| Host cell | Selection-marker | Screened | Targeted |
|---|---|---|---|
| DT40 cre | blasticidin (Bsr) | 41 | 4
(clone numbers 28, 29, 41 and 46) |
| DT40 cre Brca1+/- [Bsr] clone #41 | puromycin | 30 | 0 |
| DT40 cre Brca1+/- [Bsr] clone #29 | puromycin | 75 | 0 |
| DT40 cre Brca1+/- [Bsr] clone #46 | puromycin | 6 | 0 |
| DT40 cre Brca1+/- [Bsr] clone #28 | puromycin | 139 | 3
(clone numbers 3, 100 and 125) |
Compared with the DT40 cre parent, Brca1-deficient lines display a dose-dependent increase in cell cycle time: doubling times were 8.3, 8.5, 10.3 and 9.7 hours for DT40 cre, DT40 cre Brca1+/- clone 28, and DT40 cre Brca1-/- clones 100 and 125, respectively (Fig.S1A). In addition, both Brca1-/- lines are significantly more sensitive to X-ray-induced cell death (ie, at 1 Gray exposure, slightly less than 50% of cells of both Brca1-deficient lines survive, whereas almost 80% of the DT40 cre parent survive this dose (Fig. S1B). Both the reduced growth rate and the X-ray sensitivity phenotypes are consistent with phenotypes of Brca1-deficient lines of a different DT40 heritage [28].
3.2. Frequency of surface IgM-loss and Vλ sequence changes in DT40 cre and Brca1-deficient derivatives
To assess the role of Brca1 in Ig GC, fluctuation analysis of surface IgM-loss frequency was performed. Multiple independent cultures (12-24) of the DT40 cre Brca1-deficient lines were cultured in parallel with the Brca1-proficient parent line for 2 to 5 weeks. All DT40 cre and DT40 cre-derived lines described here were surface IgM-positive at the start of culture (Fig. 1D). Over time in culture, surface IgM-loss mutants accumulate as assayed by flow cytometry of cells stained with anti-chicken IgM antibody. This change in surface IgM is an indicator of genetic changes occurring at Ig loci. The percentage of cells that lost surface IgM in each independent culture is plotted in Figure 2, in addition to the median IgM-loss frequency of each population. At two and four-week time points, the median IgM-loss frequency of the DT40 cre population was 0.4% and 0.9%, respectively. In comparison, Brca1-deficient cultures accumulated an increased frequency of IgM-loss cells (2.4% at two weeks and 5.7-6.5% at four weeks), with an intermediate frequency exhibited by the Brca1-heterozygous line (1.2% at two weeks and 3.2% at four weeks, Fig. 2A).
Fig. 2.


Frequency of IgM loss and patterns of non-templated point mutations in Vλ in DT40 cre and derivatives deficient for Brca1. (A) Frequency of IgM loss. Each symbol represents the percentage of IgM-loss variants in an individual subclone after two (filled symbols) or four (open symbols) weeks of culture. The median percentage of IgM-loss of all subclones of a particular genotype is plotted as a large dash, with the value indicated above each set. (B) Patterns in Vλ of non-templated point mutations in cells of DT40 cre or Brca1- /- derivatives either selected, or not, for lack of surface IgM. Numbers are raw numbers of each class of base change.
At the end of the culture period, three to five clones that displayed significant, but variable (ie., median to high) frequency of IgM-loss were chosen to analyze for DNA sequence changes in Vλ. DNA was isolated either from the total pool of cells of a particular clone, or from IgM- cells of a particular clone isolated by fluorescence-activated cell sorting (FACS) in order to analyze mutations in a 427-bp region of the rearranged Vλ gene of DT40. Sequence changes in Vλ were classified either as GCs derived from one (or more) of 25 available pseudo-Vλ donors }} or non-templated point mutations, as described [12]. Strikingly, whereas there were 5 times as many GCs as non-templated events in the Brca1+/+ line, this disparity was reversed in the Brca1-/- line which showed 1 GC to about 4 non-templated events (Tables 2A and 2B). Despite the difference in type of mutation, mutations appear to be distributed similarly in Brca1-proficient and –deficient cells within the region of Vλ analyzed (Fig. 3). Although only very few non-templated mutations were found in DT40 cre, their point mutation pattern resembled that found in the Brca1-deficient derivatives, which were predominantly mutations from G and C (Fig. 2B). Mutation frequencies were 0.43 x 10-3 bp for DT40 cre and 1.6 x 10-3 bp for Brca1-/- derivatives.
Table 2.
| Table 2A. Sequence changes in Vλ of IgM-loss variants from four-week cultures | ||||||
|---|---|---|---|---|---|---|
| Genotype | Gene conversionb (% of total events) |
Non-templated point mutationc (% of total events) |
Gene conversion:point mutation ratiod | Insertion/deletion/duplication
(% of total events) |
Ambiguouse (% of total events) |
Total events
(# of sequenced clones) |
| DT40 cre | 20
(53) |
4
(11) |
83:17 |
4
(11) |
10
(27) |
38
(46) |
| DT40 cre Brca1+/-(clone #28) | 20
(48) |
8
(19) |
71:29 |
7
(17) |
7
(17) |
42
(36) |
| DT40 cre Brca1-/- (clones #100 and 125, pooled) a | 11
(14) |
46
(60) |
19:81 |
14
(17) |
6
(7) |
77
(81) |
| Table 2B. Sequence changes in Vλ of four-week cultures, without selection for surface IgM phenotype | ||||||
| Genotype | Gene conversion (% of total events) | Non-templated point mutation (% of total events) | Gene conversion:point mutation ratio | Insertion/deletion/duplication (% of total events) | Ambiguous (% of total events) | Total events (# of sequenced clones) |
| DT40 cre | 19
(26) |
15
(21) |
55:45 |
5
(7) |
33
(46) |
72
(187) |
| DT40 cre Brca1-/- (clones #100 and 125, pooled) | 25
(12) |
118
(59) |
17:83 |
15
(7) |
43
(21) |
201
(286) |
Because the frequencies of IgM loss, gene conversion to point mutation ratios, point mutation pattern, and frequency of mutations was similar in the two independent Brca1-/- DT40 cre lines #100 and #125, data from the two clones were pooled.
Sequence changes/events of two or more mutations were classified as a single gene conversion if they together matched a contiguous stretch of pseudo-Vλ gene sequence of ≥ 9 basepairs.
Events were classified as non-templated point mutations if no matching stretch of ≥ 9 basepairs could be found in any pseudo-Vλ sequence.
Expressed as a ratio of percents when total events are comprised only of gene conversions plus non-templated mutations.
Single basepair mutations with no nearby mutations were classified as ambiguous if a stretch of ≥ 9 basepairs containing the change was present in pseudo-Vλ sequence.
Fig. 3.

Mutation distributions in randomly selected DT40 cre and Brca1-deficient derivatives. Each horizontal line is a map of events in one individual clone plotted with respect to the location in a 427-basepair region of Vλ. Nucleotide positions are indicated on the X-axis, and the three gray rectangles indicate the location of the three CDRs. Mutations from cells sorted for IgM loss are shown in plots (A) and (B), and from unsorted populations in plots (C) and (D). Filled circles represent non-templated point mutations, black lines represent gene conversions, filled trapezoids and triangles represent insertions and deletions, and gray squares are ambiguous events.
3.3. Comparison of non-templated mutation patterns in Brca1- and XRCC3-deficient DT40
Fluctuation analysis of surface IgM-loss and sequence analysis of Vλ from 4-week cultured clones also was performed with Brca1-deficient lines derived independently from a DT40 parent (an IgM+ clone derived from the CL18 line, [39] for which an XRCC3-deficient derivative was described previously [42]. As shown in Fig. 4A (Expt. #1), XRCC3-deficient DT40 accumulated IgM-loss variants at a significantly higher frequency than the XRCC3-proficient cells, as reported [12], whereas the median frequency of IgM-loss variants accumulating in the Brca1-deficient lines was only slightly elevated. XRCC3-deficient cells also were tested in parallel with two independent Brca1-deficient derivatives of DT40 (both of which express no detectable Brca1 protein, but which differ in that Brca1-/- “partial” was generated with the targeting vector depicted in Fig. 1, whereas the Brca1-/- “complete” used for most studies in this section, harbors a more extensive deletion of the Brca1 locus, [39]. In comparison with the XRCC3-/- cultures, both Brca1-/- lines accumulated a significantly lower frequency of IgM-loss variants (Fig. 4A, Expt. #2). Sequence analysis of Vλ revealed that these Brca1-deficient DT40 accumulated mostly point mutations (albeit with a lower frequency despite a similar number of cell generations in culture), and very few GCs, like the XRCC3-deficient cells (Table 3A and 3B). The fact that both DT40 and DT40 cre-derived Brca1-/- cells accumulate non-templated point mutations largely in favor over GCs implicates Brca1 specifically in Ig GC. The pattern of non-templated mutations in the XRCC3- and Brca1-deficient cells is similar (Fig. 4B), as is their distribution (Fig. S2).
Fig. 4.


Frequency of IgM loss and mutation pattern in DT40 and derivatives deficient for XRCC3 or Brca1. (A) Frequency: Two independent experiments are shown. Each symbol represents the percentage of IgM-loss variants in an individual subclone after five weeks of culture. The median percentage of IgM-loss of all subclones of a particular genotype is plotted as a large dash, with the value indicated above each set. (B) Patterns of non-templated point mutations in Vλ, with or without selection for surface IgM phenotype. Raw numbers (% of total mutations).
Table 3. Patterns of mutation in Vλ of Brca1-/-, XRCC3-/- and Brca1-rescued DT40.
| A Sequence changes in Vλ, without and without selection for surface IgM phenotype (all events pooled) | ||||||
|---|---|---|---|---|---|---|
| Genotypea | Gene conversion
(% of total events) |
Non-templated point mutation
(% of total events) |
Gene conversion: point mutation ratio | Insertion/deletion/duplication
(% of total events) |
Ambiguous
(% of total events) |
Total events
(# of sequenced clones) |
| Brca1-/- “complete” clone 8 | 11
(5) |
166
(71) |
6:94 |
28
(12) |
29
(12) |
234
(548) |
| XRCC3-/- | 1
(12) |
139
(59) |
1:99 |
15
(7) |
21
(21) |
176
(277) |
| Brca1-/- “partial” | 0 | 11
(58) |
0:100 | 3
(16) |
5
(26) |
19
(68) |
| Brca1-/r (Brca1 knock-in rescue, clone F10) | 6
(22) |
12
(44) |
33:67 | 3
(11) |
6
(22) |
27
(147) |
| B Non-templated point mutation frequencies | ||||||
| Genotype | IgM- sorted | Not sorted | ||||
| Point mutations/total sequenced | Mutation frequencyb | Point mutations/total sequenced | Mutation frequencyb | |||
| Brca1-/- “complete” clone 8 | 132/337 | 0.92 x 10-3 | 32/211 | 0.36 x 10-3 | ||
| XRCC3-/- | 110/136 | 1.9 x 10-3 | 41/141 | 0.68 x 10-3 | ||
Brca1-/- “partial” is an independent clone with homozygous deficiency for Brca1, generated in the DT40 parent, with the same targeting vectors used to generate the Brca1-deficient DT40 cre derivatives. No Brca1 protein is apparent in this line. The Brca1-/- “complete,” clone 8, line has a more extensive deletion of the Brca1 locus, and also makes no detectable Brca1 protein. Numbers in table are raw numbers of each class (with all duplicated mutations from the same clone excluded to avoid resampling of the same mutations in a clonal lineage) followed in brackets by the percentage of the total in that class.
Calculated as the number of point mutations per total basepairs sequenced (total clones sequenced times 427 – the length of Vλ analyzed)
Targeted hemizygous integration of Brca1 partially reconstituted Brca1 expression in DT40 Brca1-/- cells (Fig. S3). Sequence analysis of a small set of Brca1-rescued clones revealed a rescue of GC ability at the expense of untemplated mutations (Table 3A), suggesting restoration of Brca1 function.
3.4. Expression of AID, Rad6 and PCNA is not affected in DT40 cells lacking Brca1
To begin to address whether Brca1 plays a direct or indirect role in Ig GC and the efficiency of non-templated point mutations, candidate genes encoding proteins with known or possible effects on Ig gene diversification were analyzed for expression in DT40 Brca1-deficient cells and controls. In microarray analyses, overexpression of Brca1 in mammalian cells induced expression of Rad6, involved in regulating the error-free versus error-prone branches of DNA repair [43] and PCNA, the clamp that loads and holds DNA polymerases onto DNA. Ubiqutination of PCNA by the Rad6/Rad18 Ub ligase regulates the frequency of non-templated mutations in DT40 Vλ [44]. However, no change in transcript levels was detected by semi-quantitative RT-PCR for either PCNA or Rad6 in Brca1-deficient DT40 cells (Figure 5).
Figure 5.

RT-PCR analysis of Rad6 and PCNA in DT40 cells deficient for Brca1. As compared with the control gene, β-actin, no difference in expression of Rad6 and PCNA was observed in DT40 Brca1-/- cells. AID-/- cells are in the DT40 cre background and were used as an additional control.
3.5. Aberrant genomic rearrangements involving the Vλ locus were not detectable either in Brca1-deficient, or XRCC3-deficent DT40
The increased ratio of non-templated point mutations to GCs in the Brca1- and XRCC3-deficient DT40 indicates that the favored outcome of AID-induced lesions toward GC in Vλ is perturbed. Absence of some DNA repair proteins (eg., ATM, NbsI, p53) results in AID-dependent chromosomal translocations involving Ig V- and Ig S-region DNA [45]. To investigate whether loss of Brca1 could lead to Ig V-region translocations in DT40 too, we analyzed by Southern blotting the gross integrity of the Vλ locus in DT40 and derivatives lacking Brca1 or XRCC3. As reported previously, individual CL18 and XRCC3-deficient clones after 5 weeks of passage all retained the expected genomic fragments following digestion with BamHI/SalI and a Vλ- promoter probe (Fig. S4). Similarly, in DT40cre and DT40cre Brca1-/- clones, no aberrant banding pattern was observed in DNA derived from 3-week old cultures (Fig. S5).
Genomic translocations might cause a growth disadvantage in long-term culture. Therefore, we also analyzed Vλ DNA in larger, multiply-passaged cultures from which individual clones were obtained by limiting dilution and passed for only one week (long enough to accumulate enough cells for Southern blotting). Again, no aberrant banding patterns associated with the Vλ locus were observed (Fig. S6). Therefore, genomic translocations involving the Vλ locus in DT40 and Brca1-/- derivatives may not be an outcome of aberrant processing of AID-induced lesions, or if they occur, they occur at a frequency of <2.13%.
4. Discussion
Brca1 is a DNA repair protein that participates in many cellular processes [24]. The precise role of Brca1 in these processes is undefined, but irreplaceable. At least one mechanism that might explain the cancer predisposition is the genomic instability conferred by loss of Brca1 function. Brca1 is reported to be involved in several pathways of DNA repair, particularly homologous recombination [28,29,46]. The results presented here implicate Brca1 also in Ig GC. Brca1-deficiency in three independent lines of DT40 results in a shift in Vλ diversification from GC to point mutation, substantiating the role of Brca1 in homologous recombination paths of DNA repair, and indicating a role for Brca1 in AID-initiated DNA mutation. This Brca1-/- phenotype is similar to cells mutant for Brca2, or deficient for one of the Rad51 paralogs [12,15]. Whether the role of Brca1 in Ig GC is direct or indirect is unknown. Expression of two genes (ie., Rad6 and PCNA) involved in DT40 Ig gene diversification is not affected by loss of Brca1. Brca1 is known to recruit Rad51 to DNA damage-induced nuclear foci [26,28,47] a function likely to be important for facilitating GC at Ig loci. Interestingly, Brca1 fails to localize significantly at AID-initiated S-region DNA breaks in murine B cells induced for switch recombination [48]. However, whether Brca1 is involved in CSR, a homology-independent, region-specific recombination mechanism, is unknown; homozygous Brca1 deletion is lethal in mice.
Repair of an I-SceI site-specific DSB by homologous recombination is impaired significantly in DT40 cells lacking Brca1 [30]. DSBs are thought to be repaired mainly by either of two pathways: faithfully by homologous recombination with an intact DNA sequence donor (eg., a sister chromatid in S and G2 phases of the cell cycle), or by NHEJ that ligates double-stranded DNA ends with little or no regard for sequence homology of the participating DNAs [49], reviewed by [50]. Brca1 is reported also to regulate the fidelity of NHEJ by preventing DNA end resection that might otherwise result in extensive deletions [51,52]. We did not observe more, or larger, Vλ deletions in subclones of Brca1-/- DT40 as compared with controls, suggesting that in DT40 Ig gene diversification, Brca1 plays no role in NHEJ and/or that a DSB is not an intermediate.
That Ig GC involving Brca1 is initiated by a single-strand gap is an attractive possiblity (see [53] and references therein), especially considering that the frequency of AID deamination is estimated to be too low in Vλ to initiate simultaneous deamination, Ung U-glycosylation, and abasic site nicking within a few bps on opposite strands to generate a DSB, as estimated to be too low in Vλ in DT40 cells lacking Ung or the pseudo Vλ genes [16,17,54]. Single-strand nicks could be converted to DSBs by a passing replication fork, but the fact that Ku70-deficiency has little [55,56] to no (our unpublished findings) apparent effect on the frequency of Ig GC in DT40 encourages the idea that single-strand gaps may be candidates for initiating Ig GC.
The shift toward favoring non-templated point mutations over GCs due to Brca1-deficiency is similar to the effect of deficiency of the Rad51 paralogs and Brca2. However, although the frequency of Vλ point mutations in Brca1-/- cells is elevated with respect to the Brca1-proficient control, they are not elevated to the level observed in XRCC3- deficient cells (Fig. 4, Table 3 and [12]). The pattern and distribution of non-templated mutations is similar in Brca1-/- and XRCC3-/- cells, suggesting that the fundamental mechanisms by which non-templated mutations are generated in Ig genes of DT40 cells are similar in the absence of Brca1 or other homologous recombination factors, as well as in the absence of pseudo-gene donor sequence. Non-templated point mutations in DT40 Ig genes appear to be generated by a mechanism(s) analogous to that of SHM in mammals (though perhaps using different translesion polymerases); at the very least, these mechanisms are linked by their dependence on AID, Ung and Rev1 [19, 20, 57]. This study and others in DT40 reveal no defect in non-templated mutation in DT40 lacking Brca1, Brca2, nor any of the Rad51 paralogs. Therefore these proteins probably are not required for bona fide mammalian SHM, but may regulate (i) the occurrence of SHM rather than genomic translocation or recombination, and (ii) SHM frequency (based on the difference in frequency of non-templated mutation accumulation in Brca1 versus XRCC3; see further below).
Ung-deficient DT40 produce only C to T (G to A) point mutations in Vλ at a frequency that likely represents the approximate frequency of AID deaminations in Vλ e.g., Ref. [17]. This mutation frequency is lower than that of the XRCC2-deficient cells [54]. One interpretation of this observation is that some AID-induced damage is restored to the original Vλ sequence via XRCC2-mediated homologous recombination, for instance with a sister chromatid [54], or with pseudo-Vλ sequence that is identical with the active Vλ locus sequence. The latter is supported by the report that deletion of the pseudo-Vλ genes in the DT40cre line causes a 2.5-fold higher mutation rate than XRCC3-deficiency in the same DT40 cell line [16]. Thus, since Brca1 deficiency increases non-templated mutations, Brca1 also would be implicated in repairing some AID-induced lesions by error-free recombination with pseudo-Vλ sequences. In addition, processing of the deaminated C to an abasic site by Ung, and perhaps further to a DNA strand break by enzymes able to nick at abasic sites, might facilitate an increase in the mutation rate by creating a substrate(s) for translesion polymerases when recombination is unavailable, or prevented. Translesion polymerases then would have the potential to generate additional point mutations beyond those introduced by AID alone. The degree to which translesion polymerases gain access might therefore regulate the frequency of mutation. For instance, XRCC2- and XRCC3-deficiency might stall homologous recombination early enough (though later than deletion of the pseudo-V recombination donor sequence) to promote frequent translesion polymerase access; in contrast, Brca1-deficiency might stall replication intermediates later, at a stage less favorable for access by translesion polymerases. In support of this idea, DT40 lacking Rad54, which has a number of activities thought to contribute to homologous recombination including late-acting functions in filament disassembly [58] and Holliday junction migration [59], are defective for GC, but do not accumulate non-templated point mutations [60]. A similar defect occurs in DT40 cells lacking either FancC or FancD2, whose functions in DNA repair are unknown [35,37].
A key event in activating the Fanconi anemia (FA) DNA repair pathway is ubiquitination of FancD2, which results in relocalization FancD2 to chromatin (reviewed by [36]). Although ubiquitination of FancD2 occurs independently of Brca1 [61], Brca1 is required to recruit ubiquitinated FancD2 to DNA damage [61,62]. Because FancD2- and FancC-deficient DT40 are impaired for both Ig GC and non-templated mutations, one hypothesis for the role of these proteins would be that they promote both homologous recombination and translesion synthesis pathways (eg, [63]. According to this idea, absence of Brca1 would lead to failure of FancD2 recruitment, and therefore a reduced frequency relative to the Brca1-proficient control of both GC and point mutations, similar to FancD2-/- and FancC-/- DT40. This we did not observe. Perhaps the role of Brca1 in Ig gene diversification is independent of FancD2/C, or possibly, the absence of Brca1 still allows hampered, or transient recruitment, of FancD2 to AID-induced lesions, allowing some repair. This would explain the lower frequency of non-templated mutations in Brca1- versus XRCC2-, XRCC3-deficient cells.
Finally, because Brca1-deficiency affects the type (ie., point mutation versus GC) of mutation in Vλ, mutations that all are initiated by AID, Brca1 regulation of AID/Ung-generated DNA lesion repair is implied. In humans and mice, AID-initiated lesions have the potential to generate translocations. In human lymphomas with cell surface markers characteristic of germinal center activated B cells (eg., Hodgkin's lymphoma), translocations between c-myc and IgH S-regions, and occasionally in V-regions, are hallmarks [64]. Similarly, some translocations in murine B cell lymphomas between IgH loci and c-myc are AID- and Ung-dependent, and their frequency is elevated in murine activated B cells that lack DNA repair factors including ATM, NbsI and p53 [45]. As such, AID-dependent DNA breaks, and perhaps downstream DNA intermediates of SHM, CSR and Ig GC, seem to be substrates for at least four pathways (i) somatic point mutation, generally thought to be achieved by error-prone DNA synthesis; (ii) CSR resulting from AID-dependent breaks that are joined to accomplish deletion of DNA between activated S-regions, and fusion of the VH-region to the switched C isotype; (iii) Ig GC that uses AID-dependent DNA breaks to initiate Ig GC; and (iv) inappropriate translocation of Ig gene sequence to another chromosome. The first three paths are developmentally programmed, and somehow regulated to achieve co-opting of proteins that normally would repair DNA, into processes that generate mutations. The fourth represents the canonical view of DNA repair -- to prevent genomic instability. Although Brca1 is involved in Ig GC, in mammals Brca1 may be crucial to the fourth pathway to prevent aberrant, non-regulated processing of AID-induced DNA breaks in activated B cells or other cells, a likely possibility given that many of the DNA repair proteins involved in CSR like Nbs1, Msh2, and ATM also regulate and/or interact with Brca1, and in fact may be regulated by Brca1 [51]. We did not observe an increase in translocations involving Vλ in Brca1-/- DT40, but Vλ translocations may be too rare. Checking for popular known translocations in mammalian cells deficient for Brca1 awaits a model system for depleting Brca1 in activated B cells.
Supplementary Material
Fig. S1- Growth and clonogenic survival of DT40cre and Brca1-/- derivatives. (A) Cells were diluted to a density of 5 X 104/ml and samples taken at the indicated times, stained with trypan blue and live cells counted by hemacytometry. Data represent mean and standard deviation of 3 independent replicates. Calculated doubling times are presented in tabular form at right. (B) Cells were plated in triplicate into 6 well dishes in 6% methylcellulose medium, exposed to the indicated dose of X-rays and counted 7-10 days following treatment. Data are presented as percent survival relative to untreated controls with mean and standard deviation of triplicate data.
Fig. S2- Distribution of non-templated mutations in a 427-basepair region of Vλ in Brca1-/- and XRCC3 -/- DT40 clones (mutation sets pooled from cells sorted and not for IgM-loss). The template sequence against which mutated sequences were compared is shown in bold, with the three CDR regions indicated as gray boxes. Mutations in Brca1-/- clones are indicated above the template; mutations in XRCC3-/- clones are shown below the template.
Fig. S3- Rescue of Brca1-/- derivatives of DT40 by targeted integration of one allele with chicken Brca1 coding sequence rescues Brca1 protein production. Western blot of nuclear extracts of cell lines indicated above each lane probed with an anti-serum raised against chicken Brca1. The position of Brca1 is indicated on the right.
Fig. S4- Southern blots to examine Vλ locus integrity of 4-week old subclones of CL18 and XRCC3-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic in Fig.S6. The genotype of the clones is indicated along the top (three Ku70-/- and a Brca1+/- subclone are included), size markers are shown on the right, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.
Fig. S5- Southern blots to examine Vλ locus integrity of 3-week old subclones of DT40 cre and Brca1-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic in Fig. S6. The genotype of the clones is indicated along the top, size markers are shown on the left, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.
Fig. S6- Southern blots to examine Vλ locus integrity of 1-week old subclones of DT40 cre and Brca1-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic at the bottom. The genotype of the clones is indicated along the side, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.
Acknowledgments
We thank S.Takeda for CL18 and XRCC3-/- derivative thereof; H. Arakawa and J.-M. Buerstedde (via M. Nussenzweig and N. Papavasiliou) for DT40cre and the AID-/- derivative; N. Lowndes for anti Brca1 antibodies. We are grateful to O. Yildiz for help in vector construction and B. Buikema and C. Hall of the University of Chicago Sequencing Facility for excellent technical assistance. We gratefully acknowledge support by the University of Chicago Cancer Research Center for the use of the DNA Sequencing and Flow Cytometry Facilities. This work was supported by NIH grants AI47380 and AI053130 to U.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reynaud CA, Anquez V, Dahan A, Weill JC. A single rearrangement event generates most of the chicken immunoglobulin light chain diversity. Cell. 1985;40:283–291. doi: 10.1016/0092-8674(85)90142-4. [DOI] [PubMed] [Google Scholar]
- 2.Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- 3.Reynaud CA, Dahan A, Anquez V, Weill JC. Somatic hyperconversion diversifies the single Vh gene of the chicken with a high incidence in the D region. Cell. 1989;59:171–183. doi: 10.1016/0092-8674(89)90879-9. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 5.Withers DR, Davison TF, Young JR. Developmentally programmed expression of AID in chicken B cells. Dev Comp Immunol. 2005;29:651–662. doi: 10.1016/j.dci.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 7.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 8.Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Ramiro A, San-Martin BR, McBride K, Jankovic M, Barreto V, Nussenzweig A, Nussenzweig MC. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol. 2007;94:75–107. doi: 10.1016/S0065-2776(06)94003-6. [DOI] [PubMed] [Google Scholar]
- 10.Buerstedde JM, Reynaud CA, Humphries EH, Olson W, Ewert DL, Weill JC. Light chain gene conversion continues at high rate in an ALV-induced cell line. Embo J. 1990;9:921–927. doi: 10.1002/j.1460-2075.1990.tb08190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buerstedde JM, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 12.Sale JE, Calandrini DM, Takata M, Takeda S, Neuberger MS. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature. 2001;412:921–926. doi: 10.1038/35091100. [DOI] [PubMed] [Google Scholar]
- 13.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatanaka A, Yamazoe M, Sale JE, Takata M, Yamamoto K, Kitao H, Sonoda E, Kikuchi K, Yonetani Y, Takeda S. Similar effects of Brca2 truncation and Rad51 paralog deficiency on immunoglobulin V gene diversification in DT40 cells support an early role for Rad51 paralogs in homologous recombination. Mol Cell Biol. 2005;25:1124–1134. doi: 10.1128/MCB.25.3.1124-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arakawa H, Saribasak H, Buerstedde JM. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004;2:E179. doi: 10.1371/journal.pbio.0020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saribasak H, Saribasak NN, Ipek FM, Ellwart JW, Arakawa H, Buerstedde JM. Uracil DNA glycosylase disruption blocks Ig gene conversion and induces transition mutations. J Immunol. 2006;176:365–371. doi: 10.4049/jimmunol.176.1.365. [DOI] [PubMed] [Google Scholar]
- 18.Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. Embo J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross AL, Sale JE. The catalytic activity of REV1 is employed during immunoglobulin gene diversification in DT40. Mol Immunol. 2006;43:1587–1594. doi: 10.1016/j.molimm.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. Embo J. 2003;22:1654–1664. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yabuki M, Fujii MM, Maizels N. The MRE11-RAD50-NBS1 complex accelerates somatic hypermutation and gene conversion of immunoglobulin variable regions. Nat Immunol. 2005;6:730–736. doi: 10.1038/ni1215. [DOI] [PubMed] [Google Scholar]
- 22.Neuberger MS, Harris RS, Di Noia J, Petersen-Mahrt SK. Immunity through DNA deamination. Trends Biochem Sci. 2003;28:305–312. doi: 10.1016/S0968-0004(03)00111-7. [DOI] [PubMed] [Google Scholar]
- 23.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 25.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21:8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 27.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1 deficient breast tumors. Cancer Research. 2007 doi: 10.1158/0008-5472.CAN-07-0290. In press. [DOI] [PubMed] [Google Scholar]
- 29.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 30.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 31.Venkitaraman AR. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer. 2004;4:266–276. doi: 10.1038/nrc1321. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- 33.Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Zhang H, Guerrette S, Chen J, Mazurek A, Wilson T, Slupianek A, Skorski T, Fishel R, Greene MI. Adenosine nucleotide modulates the physical interaction between hMSH2 and BRCA1. Oncogene. 2001;20:4640–4649. doi: 10.1038/sj.onc.1204625. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao H, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, Komatsu K, Thompson LH, Takata M. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol Cell Biol. 2005;25:34–43. doi: 10.1128/MCB.25.1.34-43.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orelli BJ. The Division of the Biological Sciences and the Pritzker School of Medicine. Committee on Cancer Biology. University of Chicago; Chicago: 2004. DT40 cells as a model for the role of Brca1 in DNA repair. [Google Scholar]
- 40.McCormack WT, Hurley EA, Thompson CB. Germ line maintenance of the pseudogene donor pool for somatic immunoglobulin gene conversion in chickens. Mol Cell Biol. 1993;13:821–830. doi: 10.1128/mcb.13.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 42.Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 44.Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, Buerstedde JM. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, Nussenzweig A, Nussenzweig MC. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440:105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, Koller BH. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. Embo J. 2007;26:3506–3507. doi: 10.1038/sj.emboj.7601559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Direct DNA binding by Brca1. Proc Natl Acad Sci U S A. 2001;98:6086–6091. doi: 10.1073/pnas.111125998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006;66:1401–1408. doi: 10.1158/0008-5472.CAN-05-3278. [DOI] [PubMed] [Google Scholar]
- 53.Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst) 2007;6:1018–1031. doi: 10.1016/j.dnarep.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Noia JM, Neuberger MS. Immunoglobulin gene conversion in chicken DT40 cells largely proceeds through an abasic site intermediate generated by excision of the uracil produced by AID-mediated deoxycytidine deamination. Eur J Immunol. 2004;34:504–508. doi: 10.1002/eji.200324631. [DOI] [PubMed] [Google Scholar]
- 55.Cook AJ, Raftery JM, Lau KK, Jessup A, Harris RS, Takeda S, Jolly CJ. DNA-dependent protein kinase inhibits AID-induced antibody gene conversion. PLoS Biol. 2007;5:e80. doi: 10.1371/journal.pbio.0050080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang ES, Martin A. NHEJ-deficient DT40 cells have increased levels of immunoglobulin gene conversion: evidence for a double strand break intermediate. Nucleic Acids Res. 2006;34:6345–6351. doi: 10.1093/nar/gkl830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solinger JA, Kiianitsa K, Heyer WD. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 59.Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006;442:590–593. doi: 10.1038/nature04889. [DOI] [PubMed] [Google Scholar]
- 60.Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, Buerstedde JM. Reduced X-ray resistance and homologous recombination frequencies in a RAD54-/- mutant of the chicken DT40 cell line. Cell. 1997;89:185–193. doi: 10.1016/s0092-8674(00)80198-1. [DOI] [PubMed] [Google Scholar]
- 61.Vandenberg CJ, Gergely F, Ong CY, Pace P, Mallery DL, Hiom K, Patel KJ. BRCA1-independent ubiquitination of FANCD2. Mol Cell. 2003;12:247–254. doi: 10.1016/s1097-2765(03)00281-8. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 63.Hinz JM, Nham PB, Salazar EP, Thompson LH. The Fanconi anemia pathway limits the severity of mutagenesis. DNA Repair (Amst) 2006;5:875–884. doi: 10.1016/j.dnarep.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 64.Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–5594. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1- Growth and clonogenic survival of DT40cre and Brca1-/- derivatives. (A) Cells were diluted to a density of 5 X 104/ml and samples taken at the indicated times, stained with trypan blue and live cells counted by hemacytometry. Data represent mean and standard deviation of 3 independent replicates. Calculated doubling times are presented in tabular form at right. (B) Cells were plated in triplicate into 6 well dishes in 6% methylcellulose medium, exposed to the indicated dose of X-rays and counted 7-10 days following treatment. Data are presented as percent survival relative to untreated controls with mean and standard deviation of triplicate data.
Fig. S2- Distribution of non-templated mutations in a 427-basepair region of Vλ in Brca1-/- and XRCC3 -/- DT40 clones (mutation sets pooled from cells sorted and not for IgM-loss). The template sequence against which mutated sequences were compared is shown in bold, with the three CDR regions indicated as gray boxes. Mutations in Brca1-/- clones are indicated above the template; mutations in XRCC3-/- clones are shown below the template.
Fig. S3- Rescue of Brca1-/- derivatives of DT40 by targeted integration of one allele with chicken Brca1 coding sequence rescues Brca1 protein production. Western blot of nuclear extracts of cell lines indicated above each lane probed with an anti-serum raised against chicken Brca1. The position of Brca1 is indicated on the right.
Fig. S4- Southern blots to examine Vλ locus integrity of 4-week old subclones of CL18 and XRCC3-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic in Fig.S6. The genotype of the clones is indicated along the top (three Ku70-/- and a Brca1+/- subclone are included), size markers are shown on the right, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.
Fig. S5- Southern blots to examine Vλ locus integrity of 3-week old subclones of DT40 cre and Brca1-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic in Fig. S6. The genotype of the clones is indicated along the top, size markers are shown on the left, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.
Fig. S6- Southern blots to examine Vλ locus integrity of 1-week old subclones of DT40 cre and Brca1-/- derivatives. Each lane is DNA from a single subclone, digested with BamHI and SalI, and probed with a sequence that hybridizes to the Vλ promoter region as shown in the schematic at the bottom. The genotype of the clones is indicated along the side, and the positions of the rearranged (R) and unrearranged (UR) alleles are marked.