

Abstract
Our previous studies demonstrated a significant decline in brain function and behavior in Fischer 344 (F344) rats with age. The present study was designed to test the hypothesis that dysregulation in calcium homeostasis (as assessed through 45Ca flux) may contribute to the increase in age-related vulnerability to oxidative stress in brain regions, and result in a deficit in behavior-mediated signaling. Crude membrane (P-2) and more purified synaptosomal fractions were isolated from the striatum, hippocampus, and frontal cortex of young (6 months) and old (22 months) F344 rats and were assessed for calcium flux and extracellular-regulated kinase activity 1 (ERK) under control and oxidative stress conditions induced by low dose hydrogen peroxide (final concentration 5 μM). The level of oxidative stress responses was monitored by measuring reactive oxygen species (ROS) and glutathione (GSH). The results showed a significant difference in oxidative stress responses between young and old rats in evaluated brain regions. Old rats showed higher sensitivity to oxidative stress than young rats. The present findings show the differential effects of oxidative stress on calcium flux in brain regions with age that are dependent upon the brain areas examined and the fraction assessed. The accumulation of ROS and the decrease in GSH in the frontal cortex were sufficient to decrease ERK activity in old rats. This is the first study, to our knowledge, that demonstrates age-related differential sensitivity to oxidative stress expressed as a function of behavior-mediated signaling and stress levels among different fractions isolated from brain regions controlling behavior.
Keywords: P-2 fraction, Synaptosomes, Calcium, Glutathione, Extracellular signal-regulated kinase
Introduction
Aging is associated with a significant decline in brain function. Although the specific causes of this decline are not known, we have been investigating the hypothesis that alterations in calcium homeostasis (as assessed through Ca45 flux) may contribute to the age-related increase in vulnerability to oxidative stress in brain regions. Substantial evidence shows that multiple brain calcium regulatory mechanisms change with age (Peterson and Gibson 1983; Landfield et al. 1992; Khachaturian 1994; Thibault and Landfield 1996; Thibault et al. 1998, 2001; for review, see Toescu et al. 2004). Declines in calcium regulatory mechanisms with a subsequent increase in calcium entry, and decreases in calcium clearance result in elevated cytosolic calcium in neuronal cells (Verkhratsky et al. 1994; Verkhratsky and Petersen 2002). High cytosolic calcium is associated with substantial decreases in synaptic plasticity, as well as neuronal dysfunction and death (Khachaturian 1994; Verkhratsky and Toescu 1998; Thibault et al. 2001; Verkhratsky and Petersen 2002; Blalock et al. 2003). Our previous results have shown the involvement of calcium dysregulation in increased cellular sensitivity to oxidative stress induced by low dose hydrogen peroxide (5 μM) (Denisova et al. 1997, 1999). Oxidative stress impaired the ability of the cells to extrude or sequester calcium (recovery) and significantly decreased cellular viability (Joseph and Fisher 2003; Joseph et al. 2004).
Prior research suggests that vulnerability to oxidative stress with age results from a greater accumulation of reactive oxygen species (ROS) (Harman 1956; Carrillo et al. 1992; Beckman and Ames 1998; Baek et al. 1999; Yoon et al. 2002; Webber et al. 2005; Liu et al. 2007). ROS have been proposed to act as second messengers in redox-sensitive signal transduction pathways and can damage biomolecules. Reactive oxygen intermediates, represented in the text as ROS, including superoxide radical, hydrogen peroxide and hydroxyl radical, are produced mainly in mitochondria. ROS act as physiological modulators of some mitochondrial functions, but may also damage mitochondria. In this respect, the excess of oxygen-driven radicals have been implicated in neuronal injury and neuronal death.
Antioxidants, such as glutathione (GSH), ascorbic acid, and vitamin E play an important role in neuronal defense against radical-induced damage. In this respect, the pivotal role for GSH in the defense against oxidative stress is well documented (Cooper and Kristal 1997; Rice et al. 2002; Dringen and Hirrlinger 2003; Jenner 2003; Maher 2005; Liu et al. 2007). Earlier studies report a synergistic effect between aging and oxidative stress on GSH levels (Benzi et al. 1989, 1990). Our research also demonstrated an important role of GSH in possibly preventing motor and cognitive behavioral changes in Fischer 344 (F344) rats (Shukitt-Hale et al. 1997, 1998). Shukitt-Hale and colleagues showed that experimentally-induced reductions in brain GSH levels via intracerebroventricular injections of buthionine sulfoximine followed by dopamine produced deficits in motor and cognitive behavior in young animals similar to those seen in old animals (Shukitt-Hale et al. 1997, 1998).
Mechanistically, it appears that depletion of brain GSH has been implicated as important determinant of behavior-mediated signaling, including regulation of extracellular-regulated kinase activity I (ERK). A number of studies have reported abundant expression of ERK in brain tissue and the role of this enzyme in neuronal plasticity and memory formation (Bozon et al. 2003; for review, see Thiels and Klann 2001). A significant decrease in ERK’s activity has been observed in brain tissue with normal aging and age-related neurodegeneration (Zhu et al. 2001, 2002, 2006; Webber et al. 2005). Oxidative stress has been suggested as a possible factor contributing to the deficit in ERK activity (Guyton et al. 1996; Tournier et al. 1997; Gupta et al. 1999; Zhu et al. 2001). The present study was designed to test the hypothesis that dysregulation in calcium homeostasis (as assessed through Ca45 flux) may contribute to the increase in age-related vulnerability to oxidative stress in brain regions, and result in the deficit in behavior-mediated signaling (as assessed through ERK).
Materials and methods
Reagents
Reagents used were of the highest grade available and were obtained from Sigma Chemical (St. Louis, Mo., USA). Fluorescent probes, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) and monochlorobimate (MBCL) to measure reactive oxygen species (ROS) and glutathione (GSH), respectively, were obtained from Molecular Probes (Ore., USA). Radioactive reagents ([32P]-γ -ATP and 45CaCl2) were purchased from NEN Life Science Products (Boston, Mass., USA). ERK1 antibodies were purchased from Santa Cruz Biotechnology (Calif., USA; cat# sc-93).
Animals
Fifteen 6-month-old (young) and fifteen 22-month-old (old) male F344 rats were obtained from a colony maintained by the National Institute on Aging. These animals were used in compliance with all applicable laws and regulations and principles expressed in the National Institutes of Health, USPHS, Guide for the Care and Use of Laboratory Animals. This study was approved by the Animal Care and Use Committee of the Jean Mayer, United States Department of Agriculture Human Nutrition Research Center on Aging at Tufts University (Boston, Mass.).
Sample preparation
Rats were decapitated, and striatum, hippocampus, and frontal cortex were dissected on ice. Each distinct brain region from three rats from each age group described above were dissected, combined together, and processed as one experimental sample. There were five samples per each age group. Samples for biochemical measurements were chosen randomly and performed under control and oxidative stress conditions. Oxidative stress was induced by low physiological dose hydrogen peroxide (final concentration 5 μM). Experimental conditions were chosen based on analyses of several graded concentrations of hydrogen peroxide at different time points (Denisova et al. 1998). Calcium flux was measured in crude membrane (P-2) and more purified synaptosomal fractions isolated from two samples as described previously (Joseph et al. 1998). P-2 fractions isolated from the remaining three samples were assessed for GSH, ROS, and ERK (Denisova et al. 1998).
Glutathione and reactive oxygen species
Levels of oxidative stress induced by 5 μM hydrogen peroxide were monitored by measuring total ROS (i.e., superoxide radical, hydrogen peroxide and hydroxyl radical) and GSH concentrations in freshly prepared samples by using fluorescent methods as described previously (Denisova et al. 1998; Joseph et al. 1998). GSH levels were assessed by using MBCL (Denisova et al. 1998). Numerous studies implicate ROS production in biological systems as an oxidative stress marker (Beckman and Ames 1998). The high reactivity and relative instability of ROS make them difficult to detect in biological systems. Therefore, we employed a widely accepted method that allowed us to quantify ROS based on their reaction with a detector molecule (i.e., a molecule that is oxidatively modified to produce a fluorescence signal) (LeBel et al. 1992). The method is based on oxidation of the non-fluorescent substrate 2′,7′- dichlorofluorescin (DCFH) to the fluorescent product 2′,7′-dichlorofluorescein (DCF). The esterified form of 2′,7′-dichlorofluorescin (DCFH), dichlorofluorescin diacetate (DCFH-DA), is capable of crossing cell membranes and then, as a result of deacetylation by intracellular esterases, forming a non-fluorescent DCFH, which in the presence of ROS rapidly oxidizes to highly fluorescent 2′,7′- dichlorofluorescein (DCF). Aliquots of brain samples were incubated with DCFH-DA (final concentration 50 μM) for 30 min., washed, and then exposed to 0 or 5 μM H2O2 (LeBel et al. 1992).
Calcium flux
Calcium activity in P-2 and synaptosomal fractions was assessed based on measurement of 45Ca flux under control and oxidative stress conditions as described previously (Joseph et al. 1998). Briefly, aliquots of freshly isolated P-2 or synaptosomal fractions (1.3 mg protein/ml) were pre-incubated for 5 min at 37°C in the basal medium (BM: 136 mM NaCl; 5 mM KCl; 1.2 mM CaCl2; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65) followed by exposure to 5 μM H2O2. In control samples, H2O2 was equally replaced with basal media. The 45Ca flux was initiated by transferring oxidized or non-oxidized samples into the basal or depolarizing medium (DM: 60 mM KCl; 1.2 mM CaCl2; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65). Both media had previously been supplemented with 45CaCl2 (final concentration, 2 μCi/ml; specific activity, 2.1 mCi/ml). The reaction was stopped by rapid filtration of the samples through Whatman GF/B filters, followed by washing with ice-cold stop medium (SM: 136 mM NaCl; 5 mM KCl; 3 mM EGTA; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65) by using vacuum filtration (BRANDEL ML-48; USA). The radioactivity retained in the filters was measured by a liquid scintillation counter (WALLAC 1409) programmed for automatic quenching correction. The ratio between radioactivities calculated under depolarizing conditions (D-cond.) and basal conditions (B-cond.) was defined as Recovery. Percent of Recovery at 30 s after depolarization was calculated as follows:
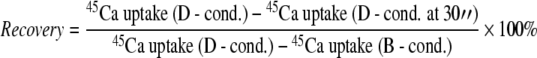 |
Extracellular-regulated kinase activity assay
The lysates from the oxidized and non-oxidized samples were analyzed for ERK activity as described previously (Hassan et al. 2002). Briefly, oxidized and non-oxidized samples were extracted in 5 volumes of buffer: 25 mM HEPES pH 7.5, 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.05% Triton X-100, 20 mM β-glycerophosphate, 0.1 mM orthovanadate, 0.5 mM DTT, 0.4 mM PMSF, 1 mg/ml leupeptin, 1 mg/ml pepstatin, WCE buffer; for 30 min at 4°C. Extracts were then microfuged at 4°C for 15 min and the cleared lysates were frozen at −80°C. Protein A beads (15 ml per assay; Repligen Corporation) were washed with Triton Lysis Buffer (TLB: 20 mM Tris pH 7.4, 137 mM NaCl, 2 mM EDTA pH 7.4, 1% Triton X-100, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 2 mM sodium pyrophosphate, 10% glycerol) and incubated with 10 μl ERK antibodies at room temperature. Beads were then incubated with experimental lysate at 4°C for 2 h. The kinase reaction was carried out by adding the following to the beads: 22 μl purified fusion protein, 1.6 μl 5X KB, 4.4 ml H2O, 1 μl 0.5 mM ATP, 1 μl [32P]-γ -ATP; the beads were then incubated for 20 min at 30°C. This reaction was terminated by adding 20 μl buffer [0.125 M Tris-HCl, pH 6.8; 2% (wt/vol) SDS; 10% vol/vol glycerol; 2% vol/vol β-mercaptoethanol; and 0.01% bromophenol blue]. Samples were then boiled at 100°C for 3 min and analyzed by standardized SDS-PAGE and immunoblotting procedures. Proteins recognized by specific ERK1 antibodies in immunoblots were visualized by standard chemiluminescence methods (Renaissance®, NEN-Life Science Products). Densitometry quantification of immunoblotted membranes was performed with a Molecular Dynamics Densitometer (Amersham Biosciences, Sunnyvale, Calif.).
Statistical analysis
The results of biochemical measurements were analyzed by analysis of variance (ANOVA) and Fisher Least-Significant-Difference (LSD) tests. All statistical analyses were performed using Systat version 10.0; (SPSS. Chicago, Ill.). All tests were two-sided. Results were considered statistically significant if the observed significance value was no greater than 0.05.
Results and discussion
Glutathione and reactive oxygen species
The level of oxidative stress was assessed by monitoring the levels of ROS and GSH in freshly isolated P-2 fractions exposed to 5 μM hydrogen peroxide for 15 min. The results show a significant (P < 0.05) increase in the concentration of the ROS in the striatal (Fig. 1a), hippocampal (Fig. 1b) and cortical (Fig. 1c) fractions. There was a significant difference in the accumulation in the ROS between the two age groups [F(2,8) = 12.44; P = 0.044]. The levels of the ROS were significantly higher in the old rats.
Fig. 1.
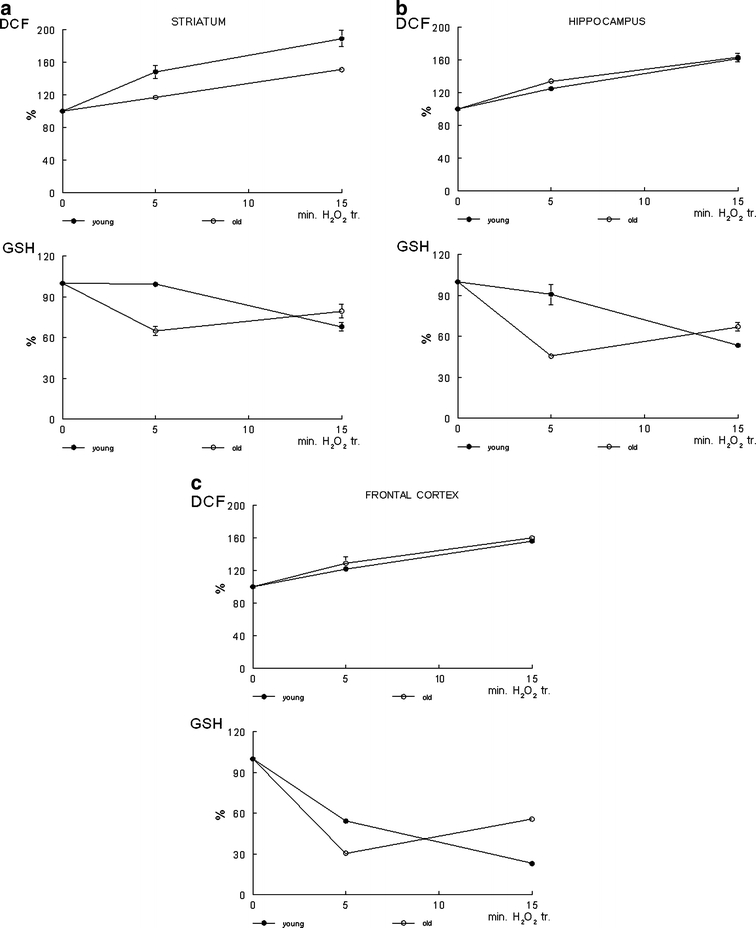
Glutathione and reactive oxygen species. Glutathione (GSH) and reactive oxygen species (ROS) were assessed in P-2 fractions freshly isolated from the striatum (a), hippocampus (b), and frontal cortex (c) from young (-•-) and old (-○-) groups of F344 rats. The effect of oxidative stress, induced by hydrogen peroxide (final concentrations, 5 μM; 0–15 min) on GSH and ROS were assessed by using fluorescent methods for 96 well plates as described in Materials and Methods. Fluoresence was monitored on a Cyto Fluor Multi Well Plate Reader with λEx/Em at 360/460 nm (for GSH determination) and λEx/Em 485/530 nm (for ROS determination) (PerSeptive Biosystem, Framingham, Mass., USA) for 0–15 min. Data are means from three individual experiments ± SEM
A number of studies demonstrated the crucial role of GSH in protecting cells from oxidative stress (D’Alessio et al. 2003; Liuzzi et al. 2003; Pillai et al. 2007). Our data show that the accumulation of the ROS in the brain fractions exposed to oxidative stress were associated with decreases in GSH levels in evaluated fractions (Fig. 1). Importantly, there were significant differences in GSH levels between young and old rats in the hippocampus [F(1,4) = 11.23; P = 0.029; Fig. 1b] and frontal cortex [F(1,4) = 19.43; P = 0.012; Fig. 1c], and marginal significance in the striatum (P = 0.074; Fig. 1a). The decreases in GSH levels in response to oxidative stress were significantly higher in the hippocampal and cortical fractions isolated from the old rats. Our previous data implicated GSH as an important determinant of calcium activity in PC12 cells (Denisova et al. 1999). It appeared from that study that low levels of intracellular GSH in PC12 cells resulted in a decline in the ability of the cells to restore calcium recovery under oxidative stress conditions induced by 5 μM hydrogen peroxide. Repletion of GSH in PC12 cells with 20 mM N-acetylcysteine was sufficient to attenuate the damaging effect of oxidative stress on calcium recovery (Denisova et al. 1999). Calcium recovery in the present study was assessed through 45Ca flux as a function of age and oxidative stress.
Calcium flux and extracellular-regulated kinase activity
Figure 2 represents the effect of aging and oxidative stress, induced by 5 μM H2O2, on 45Ca flux in P-2 and synaptosomal fractions isolated from the striatum, hippocampus and frontal cortex of young and old rats. Our data support previous findings (Peterson and Gibson 1983; Michaelis et al. 1984; Martinez-Serrano et al. 1992; Foster and Norris 1997; Tanaka et al. 1996; Verkhratsky and Toescu 1998) and demonstrate a significant deficit in calcium activity in the brain with age. Age-related declines in 45Ca flux were observed in both P-2 and synaptosomal fractions across brain regions in the absence of oxidative stress (Fig. 2; see 0 μM cond.; striatum — P-2 fraction, P < 0.01; synaptosomal fraction, P < 0.03; hippocampus — P-2 fraction, P < 0.01; synaptosomal fraction, P < 0.001; frontal cortex — P-2 fraction, P < 0.03; synaptosomal fraction, P < 0.02). There was a significant dysregulation in calcium flux under oxidative stress conditions in both P-2 and synaptosomal fractions across evaluated brain regions (striatum — P-2 fraction, P < 0.001; synaptosomal fraction, P < 0.02; hippocampus — P-2 fraction, P < 0.01; synaptosomal fraction, P < 0.02; frontal cortex — P-2 fraction, P < 0.01; synaptosomal fraction, P < 0.01) (Fig. 2; see 0 μM vs 5 μM H2O2). P-2 fractions isolated from the young rats showed brain region-specific changes in calcium flux as a function of oxidative stress. Specifically, striatal and cortical P-2 fractions isolated from the young rats showed higher oxidative stress-induced dysregulation in calcium flux as compared to hippocampal fractions. However, P-2 fractions isolated from the old rats showed a significant dysregulation in calcium flux as a function of oxidative stress across all brain regions (Fig. 2).
Fig. 2.
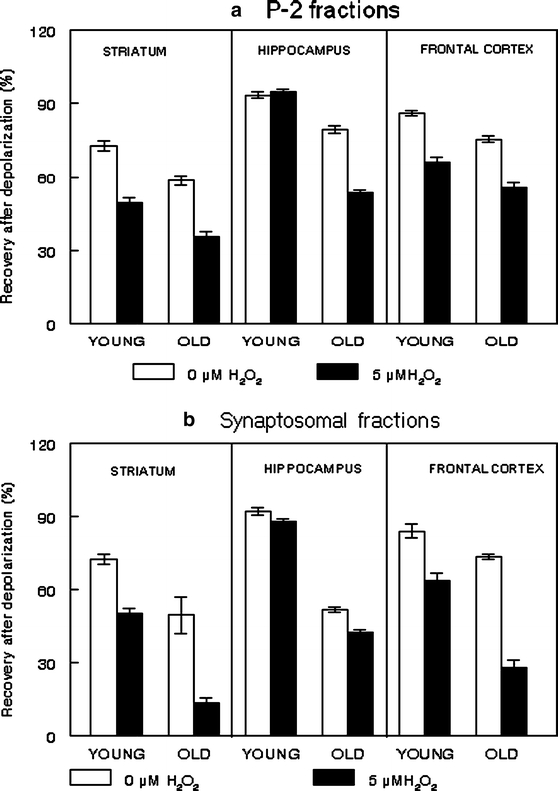
Calcium flux. 45Ca flux were assessed in P-2 (a) and synaptosomal (b) fractions freshly isolated from the striatum, hippocampus, and frontal cortex of young and old F344 rats as described in Materials and Methods. Briefly, aliquots of P-2 or synaptosomal fractions (50 μl; at the protein concentration 1.3 mg protein/ml) were pre-incubated for 5 min at 37°C in the basal medium (BM: 136 mM NaCl; 5 mM KCl; 1.2 mM CaCl2; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65) followed by exposure to 5 μM hydrogen peroxide. The 45Ca flux was initiated by transferring oxidized or non-oxidized samples into the basal or depolarizing medium (DM: 60 mM KCl; 1.2 mM CaCl2; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65). Both media had previously been supplemented with 45CaCl2 (final concentration, 2 μCi/ml; specific activity, 2.1 mCi/ml). The reaction was stopped by rapid filtration samples through Whatman GF/B filters, followed by washing filters with ice-cold stop medium (SM: 136 mM NaCl; 5 mM KCl; 3 mM EGTA; 1.3 mM MgCl2; 10 mM glucose; 20 mM Tris; pH 7.65) by using vacuum filtration (BRANDEL ML-48, U.S.A.). The radioactivity retained in the filters was measured by a liquid scintillation counter (WALLAC 1409) programmed for automatic quenching correction. Percent of Recovery at 30 s after depolarization was calculated as described in Materials and Methods. Data are means from two individual experiments ± SEM
More purified synaptosomal fractions show higher oxidative stress-induced dysregulation in calcium flux with age as compared to P-2 fractions (P < 0.04). The differences in calcium flux between P-2 and synaptosomal fractions may be partially attributed to the difference between these fractions in calcium sequestration mechanisms controlled by mitochondria, calcium binding proteins, and microsomes. Numerous morphological and biochemical data showed enrichment of P-2 fractions with mitochondria and microsomes, whereas synaptosomal fractions were enriched with neuronal membranes and synaptosomal mitochondria (Sun and Sun 1972; Rao et al. 1993; Gylys et al. 2000; Battino et al. 2001). The essential role of the sequestration mechanisms for age-related calcium activity in synaptosomal membranes was reported in earlier studies (Michaelis et al. 1984, 1992, 1996; Satrustegui et al. 1996; Zaidi et al. 1998; Zaidi and Michaelis 1999). By using modern gene microarray technology, Landfield and co-authors demonstrated a significant association between functional brain aging and gene regulation as a function of oxidative stress, inflammation, and calcium activity (Blalock et al. 2003). They demonstrated that elevated intracellular calcium concentration reduced neuronal activity by activating inhibitory calcium-dependent conductances, and calcium-regulated genes (Blalock et al. 2003). Moreover, this group showed that the viability and functions of the hippocampal neurons are mediated by the amplification of calcium influx and release with age (Thibault and Landfield 1996; Blalock et al. 2003). The present findings show the differential effects of oxidative stress on calcium flux in brain regions with age that are dependent upon the brain areas examined and the fraction assessed. Fractions isolated from the hippocampus and frontal cortex of old rats showed a significantly higher sensitivity to oxidative stress as reflected through the dysregulation of calcium flux than fractions isolated from the young rats (P < 0.01). Our present data showed that old rats expressed a significantly higher sensitivity to experimental oxidative stress than young rats.
The application of these findings in relation to aging and behavior clearly requires additional research. Numerous studies have demonstrated a significant enrichment of hippocampus and frontal cortex with cholinergic neurons (Wurtman 1992; Perry et al. 2005; Sahin et al. 2006). The dysfunction of the cholinergic neurons has been strongly associated with the behavioral deficits observed during normal aging and age-related neurodegeneration (Wurtman 1992; Sahin et al. 2006; Chwang et al. 2006; Fodale et al. 2006; Mowla et al. 2007). Our recent study also suggests that differential sensitivity to oxidative stress among various muscarinic receptor subtypes (Joseph and Fisher 2003) might also contribute to the previously observed behavioral and neuronal deficits. Our present data suggest that oxidative stress-induced dysregulation of calcium flux in the hippocampus and frontal cortex with age may be an important contributor to the deficit in functions of cholinergic neurons. In this respect, there is evidence that the viability and functions of cholinergic neurons are strongly associated with ERK activity. ERK is a member of a large family of serine/threonine protein kinases that function in signaling cascades that primarily mediate a variety of functions that include cellular growth and differentiation as well as survival in mammalian cells (Volmat and Pouyssegur 2001). Extra cellular signal related kinases isoforms (ERK1/2) have been shown in numerous experiments to be important for neuroprotection, such as oxidative stress/inflammation which plays an important role in neurodegeneration (Cavanaugh et al. 2006). The up-regulation of ERK-mediated signaling has been implicated as a pro-survival neuronal mechanism against oxidative stress (Lee et al. 2006; Luo and DeFranco 2006; Abdul and Butterfield 2007; Boulos et al. 2007). Additional studies have provided evidence indicating that activation of ERK and the antiapoptotic B-cell CLL/lymphoma 2 (bcl-2) play a role in growth factor-mediated neuroprotection from 6-hydroxydopamine (6-OHDA) toxicity in a dopaminergic cells (Zigmond 2006), hypoxia in cortical neurons (Ma and Quirion 2005), protection from stroke (Mehta et al. 2007), and a variety of oxidative stressors (McCubrey et al. 2006). Similar findings have been reported regarding inflammation (Malemud 2006).
The protein kinase B (Akt)-dependent signaling pathway is part of the MAPK/ERK induction system that is important in the regulation of cell growth and differentiation and is also important in mediating oxidative stress and inflammation. As with ERK, AKT, in addition to the oxidant-inducing conditions cited above, has also been shown to be effective along with ERK in stress conditions involving glaucoma (Tatton et al. 2003), telomerase activity (Dong et al. 2007), acute LPS-induced inflammatory responses (Zhang et al. 2007), endothelial cell protection by HDL (Norata and Catapano 2005), the rescue of neurons from MPTP-induced cell death (Weinreb et al. 2006), N-methyl-D-aspartate receptors (NMDAR)-induced neuronal survival (Hetman and Kharebava 2006), and inhibition of glycogen synthase kinase-3 (GSK3) in the prevention of cell death in neurodegenerative diseases (Takashima 2006). The essential role of this enzyme for brain functions and behavior has been extensively studied (Blum et al. 1999; Sweatt 2004; Webber et al. 2005; Sahin et al. 2006; Lee et al. 2006).
Our results show a significant difference in ERK activity between the two age groups in the different brain regions [F(1,4) = 7.61; P = 0.05; Fig. 3]. The age-related difference in enzyme activity was particularly expressed in the frontal cortex (P = 0.04). The exposure of cortical fractions to oxidative stress resulted in increased ERK in the young group of rats, and decreased enzyme activity in old rats [F(1,4) = 9.53; P = 0.037; Fig. 3c]. Our previous data showed that decreases in brain GSH levels produced deficits in motor and cognitive behavior in young Fischer 344 rats similar to those seen in old animals (Shukitt-Hale et al. 1997, 1998). Present data suggests that oxidative stress-induced decreases in GSH followed by decreases in ERK activity in the cortex in the old rats may contribute to the previously reported behavioral deficit observed in the Fisher rats with age.
Fig. 3.
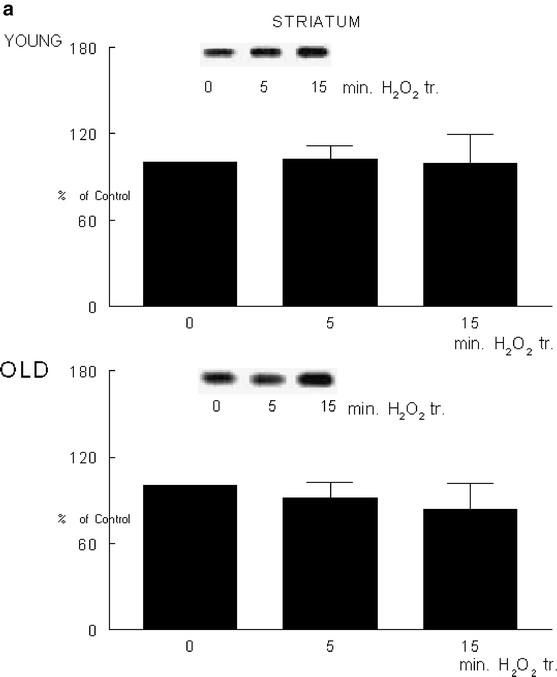
Extracellular signal-regulated kinase activity. Extracellular signal-regulated kinase (ERK) activity was assessed in an aliquot of P-2 fractions from striatum (a), hippocampus (b), and frontal cortex (c) from young and old F344 rats. The enzyme activity was determined as described in Materials and Methods under control and oxidative stress conditions. The blots on the top of the figure represent the typical blot from samples exposed to 0 or 5 μM hydrogen peroxide. Data are means from three individual experiments ± SEM
The increase in enzyme activity by high dose hydrogen peroxide has been previously reported (Abe et al. 1998). Our results showed that low physiological dose of hydrogen peroxide were not sufficient to alter enzyme activity in the striatal (Fig. 3a) and hippocampal (Fig. 3b) fractions. Oxidative stress-induced changes in the ROS and GSH are important mediators of the age-related differences in the calcium flux and ERK activity in the fractions isolated from the frontal cortex. This is the first study, to our knowledge, that demonstrates age-related differential sensitivity to oxidative stress as a function of behavior-mediated signaling and stress levels among different fractions isolated from the brain regions controlling behavior.
Acknowledgments
This work was supported by USDA Cooperative Agreement 58-1950-7-707. The authors acknowledge Beverly Dobson and Sherley Casseus for valuable help in preparation of this manuscript.
References
- Abdul HM, Butterfield DA (2007) Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by cotreatment of acetyl-L-carnitine and alpha-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Free Radic Biol Med 42:371–384 [DOI] [PMC free article] [PubMed]
- Abe MK, Kartha S, Karpova AY, Li J, Liu PT, Kuo WL, Hershenson MB (1998) Hydrogen peroxide activates extracellular signal-regulated kinase via protein kinase C, Raf-1, and MEK1. Am J Respir Cell Mol Biol 18:562–569 [DOI] [PubMed]
- Baek BS, Kwon HJ, Lee KH, Yoo MA, Kim KW, Ikeno Y, Yu BP, Chung HY (1999) Regional difference of ROS generation, lipid peroxidation, and antioxidant enzyme activity in rat brain and their dietary modulation. Arch Pharm Res 22:361–366 [DOI] [PubMed]
- Battino M, Bompadre S, Leone L, Villa RF, Gorini A (2001) Coenzymes Q9 and Q10, vitamin E and peroxidation in rat synaptic and non-synaptic occipital cerebral cortex mitochondria during ageing. J Biol Chem 382:925–931 [DOI] [PubMed]
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78:547–581 [DOI] [PubMed]
- Benzi G, Marzatico F, Pastoris O, Villa RF (1989) Relationship between aging, drug treatment and the cerebral enzymatic antioxidant system. Exp Gerontol 24:137–148 [DOI] [PubMed]
- Benzi G, Marzatico F, Pastoris O, Villa RF (1990) Influence of oxidative stress on the age-linked alterations of the cerebral glutathione system. J Neurosci Res 26:120–128 [DOI] [PubMed]
- Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW (2003) Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci 23:3807–3819 [DOI] [PMC free article] [PubMed]
- Blum S, Moore AN, Adams F, Dash PK (1999) A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci 19:3535–3544 [DOI] [PMC free article] [PubMed]
- Boulos S, Meloni BP, Arthur PG, Majda B, Bojarski C, Knuckey NW (2007) Evidence that intracellular cyclophilin A and cyclophilin A/CD147 receptor-mediated ERK1/2 signalling can protect neurons against in vitro oxidative and ischemic injury. Neurobiol Dis 25:54–64 [DOI] [PubMed]
- Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S (2003) MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B 358:805–814 [DOI] [PMC free article] [PubMed]
- Carrillo MC, Kanai S, Sato Y, Kitani K (1992) Age-related changes in antioxidant enzyme activities are region and organ, as well as sex, selective in the rat. Mech Ageing Dev 65:187–198 [DOI] [PubMed]
- Cavanaugh JE, Jaumotte JD, Lakoski JM, Zigmond MJ (2006) Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J Neurosci Res 84:1367–1375 [DOI] [PubMed]
- Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD (2006) ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem 13:322–328 [DOI] [PMC free article] [PubMed]
- Cooper AJ, Kristal BS (1997) Multiple roles of glutathione in the central nervous system. Biol Chem 378:793–802 [PubMed]
- D’Alessio M, Cerella C, De Nicola M, Bergamaschi A, Magrini A, Gualandi G, Alfonsi AM, Ghibelli L (2003) Apoptotic GSH extrusion is associated with free radical generation. Ann NY Acad Sci 1010:449–452 [DOI] [PubMed]
- Denisova NA, Strain J, Joseph JA (1997) Oxidant injury in PC12 cells: a possible model of calcium “dysregulation” in aging II. Interactions with membrane lipids. J Neurochem 69:1259–1266 [DOI] [PubMed]
- Denisova NA, Tsaioun K, Bielinski D, Palmer H, Paulson E, Joseph JA (1998) Age-related and area-specific regulation of mitogen-activated protein kinases by oxidative stress in synaptosomes. FASEB J 12:A1844
- Denisova NA, Fisher D, Provost M, Joseph JA (1999) The role of glutathione, membrane sphingomyelin, and its metabolites in oxidative stress-induced calcium “dysregulation” in PC12 cells. Free Radic Biol Med 27:1292–1301 [DOI] [PubMed]
- Dong XX, Hui ZJ, Xiang WX, Rong ZF, Jian S, Zhu CJ (2007) Ginkgo biloba extract reduces endothelial progenitor-cell senescence through augmentation of telomerase activity. J Cardiovasc Pharmacol 49:111–115 [DOI] [PubMed]
- Dringen R, Hirrlinger J (2003) Glutathione pathways in the brain. Biol Chem 384:505–516 [DOI] [PubMed]
- Fodale V, Quattrone D, Trecroci C, Caminiti V, Santamaria LB (2006) Alzheimer’s disease and anaesthesia: implications for the central cholinergic system. Br J Anaesth 97:445–452 [DOI] [PubMed]
- Foster TC, Norris CM (1997) Age-associated changes in Ca(2+)-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus 7:602–612 [DOI] [PubMed]
- Gupta A, Rosenberger SF, Bowden GT (1999) Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progress mouse keratinocyte cell lines. Carcinogenesis 20:2063–2073 [DOI] [PubMed]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ (1996) Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 271:4138–4142 [DOI] [PubMed]
- Gylys KH, Fein JA, Cole GM (2000) Quantitative characterization of crude synaptosomal fraction (P-2) components by flow cytometry. J Neurosci Res 61:186–192 [DOI] [PubMed]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Ger 11:298–300 [DOI] [PubMed]
- Hassan W, Cantuti-Castelvetri I, Denisova NA, Joseph JA, Paulson EK (2002) The nitrone-spin trap PBN alters the cellular response to H2O2: calcium-dependent activation of the EGF receptor/ERK pathway. Free Radic Biol Med 32:551–561 [DOI] [PubMed]
- Hetman M, Kharebava G (2006) Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem 6:787–799 [DOI] [PubMed]
- Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53:26–36 [DOI] [PubMed]
- Joseph JA, Fisher DR (2003) Muscarinic receptor subtype determines vulnerability to amyloid beta in transfected COS-7 cells. J Alzheimer’s Dis 4:1–12 [DOI] [PubMed]
- Joseph JA, Shukitt-Hale B, Denisova NA, Prior RL, Cao G, Martin A, Taglialatela G, Bickford PC (1998) Long-term dietary strawberry, spinach, or vitamin E supplementation retards the onset of age-related neuronal signal-transduction and cognitive behavioral deficits. J Neurosci 18:8047–8055 [DOI] [PMC free article] [PubMed]
- Joseph JA, Fisher DR, Carey A (2004) Fruit extracts antagonize Abeta- or DA-induced deficits in CA2+ flux in M1-transfected COS-7 cells. J Alzheimer’s Dis 6:403–411 [DOI] [PubMed]
- Khachaturian ZS (1994) Calcium hypothesis of Alzheimer’s disease and brain aging. Ann NY Acad Sci 747:1–11 [DOI] [PubMed]
- Landfield PW, Thibault O, Mazzanti ML, Porter NM, Kerr D (1992) Mechanisms of neuronal death in brain aging and Alzheimer’s disease: role of endocrine-mediated Ca2+ dyshomeostasis. J Neurobiol 23:1247–1260 [DOI] [PubMed]
- LeBel CP, Ischiropolos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescein as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5:227–231 [DOI] [PubMed]
- Lee SY, Kang JS, Song GY, Myung CS (2006) Stress induces the expression of heterotrimeric G protein beta subunits and the phosphorylation of PKB/Akt and ERK1/2 in rat brain. Neurosci Res 56:180–192 [DOI] [PubMed]
- Liu Q, Xie F, Rolston R, Moreira PI, Nunomura A, Zhu X, Smith MA, Perry G (2007) Prevention and treatment of Alzheimer disease and aging: antioxidants. Mini Rev Med Chem 7:171–180 [DOI] [PubMed]
- Liuzzi F, Fanelli C, Ciriolo MR, Cerella C, D’Alessio M, Denicola M, Magrini A, Bergamaschi A, Ghibelli L (2003) Rescue of cells from apoptosis by antioxidants occurs downstream from GSH extrusion. Ann NY Acad Sci 1010:441–445 [DOI] [PubMed]
- Luo Y, DeFranco DB (2006) Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem 281:16436–16442 [DOI] [PubMed]
- Ma W, Quirion R (2005) The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin Ther Targets 9:699–713 [DOI] [PubMed]
- Maher P (2005) The effects of stress and aging on glutathione metabolism. Ageing Res Rev 4:288–314 [DOI] [PubMed]
- Malemud CJ (2006) Small molecular weight inhibitors of stress-activated and mitogen-activated protein kinases. Mini Rev Med Chem 6:689–698 [DOI] [PubMed]
- Martinez-Serrano A, Blanco P, Satrustegui J (1992) Calcium binding to the cytosol and calcium extrusion mechanisms in intact synaptosomes and their alterations with aging. J Biol Chem 267:4672–4679 [PubMed]
- McCubrey JA, Lahair MM, Franklin RA (2006) Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 8:1775–1789 [DOI] [PubMed]
- Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66 [DOI] [PubMed]
- Michaelis ML, Johe K, Kitos TE (1984) Age-dependent alterations in synaptic membrane systems for Ca2+ regulation. Mech Ageing Dev 25:215–225 [DOI] [PubMed]
- Michaelis ML, Foster CT, Jayawickreme C (1992) Regulation of calcium levels in brain tissue from adult and aged rats. Mech Ageing Dev 62:291–306 [DOI] [PubMed]
- Michaelis ML, Bigelow DJ, Schoneich C, Williams TD, Ramonda L, Yin D, Huhmer AF, Yao Y, Gao J, Squier TC (1996) Decreased plasma membrane calcium transport activity in aging brain. Life Sci 59:405–412 [DOI] [PubMed]
- Mowla A, Mosavinasab M, Pani A (2007) Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? A double-blind, placebo-controlled, clinical trial. J Clin Psychopharmacol 27:67–70 [DOI] [PubMed]
- Norata GD, Catapano AL (2005) Molecular mechanisms responsible for the antiinflammatory and protective effect of HDL on the endothelium. Vasc Health Risk Manag 1:119–129 [DOI] [PMC free article] [PubMed]
- Perry E, Ziabreva I, Perry R, Aarsland D, Ballard C (2005) Absence of cholinergic deficits in “pure” vascular dementia. Neurology 64:132–133 [DOI] [PubMed]
- Peterson C, Gibson GE (1983) Aging and 3,4-diaminopyridine alter synaptosomal calcium uptake. J Biol Chem 258:11482–11486 [PubMed]
- Pillai A, Parikh V, Terry AV Jr, Mahadik SP (2007) Long-term antipsychotic treatments and crossover studies in rats: differential effects of typical and atypical agents on the expression of antioxidant enzymes and membrane lipid peroxidation in rat brain. J Psychiatr Res 41:372–386 [DOI] [PubMed]
- Rao MA, Igbavboa U, Semotuk M, Schroeder F, Wood G (1993) Kinetics size of cholesterol lateral domains in synaptosomal membranes: modification by sphingomyelinase and effects on membrane enzyme activity. Neurochem Int 23:45–52 [DOI] [PubMed]
- Rice ME, Forman RE, Chen BT, Avshalumov MV, Cragg SJ, Drew K (2002) Brain antioxidant regulation in mammals and anoxia-tolerant reptiles: balanced for neuroprotection and neuromodulation. Comp Biochem Physiol C Toxicol Pharmacol 133:515–525 [DOI] [PubMed]
- Sahin HA, Emre M, Ziabreva I, Perry E, Celasun B, Perry R (2006) The distribution pattern of pathology and cholinergic deficits in amygdaloid complex in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol (Berl) 111:115–125 [DOI] [PubMed]
- Satrustegui J, Villalba M, Pereira R, Bogonez E, Martinez-Serrano A (1996) Cytosolic and mitochondrial calcium in synaptosomes during aging. Life Sci 59:429–434 [DOI] [PubMed]
- Shukitt-Hale B, Denisova NA, Strain JG, Joseph JA (1997) Psychomotor effects of dopamine infusion under decreased glutathione conditions. Free Radic Biol Med 23:412–418 [DOI] [PubMed]
- Shukitt-Hale B, Erat SA, Joseph JA (1998) Spatial learning and memory deficits induced by dopamine administration with decreased glutathione. Free Radic Biol Med 24:1149–1158 [DOI] [PubMed]
- Sun GY, Sun AY (1972) Phospholipids and acyl groups of synaptosomal and myelin membranes isolated from the cerebral cortex of squirrel monkey (Saimiri sciureus). Biochem Biophys Acta 280:306–315 [DOI] [PubMed]
- Sweatt JD (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317 [DOI] [PubMed]
- Takashima A (2006) GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J Alzheimer’s Dis 9:309–317 [DOI] [PubMed]
- Tanaka Y, Hasegawa A, Ando S (1996) Impaired synaptic functions with aging as characterized by decreased calcium influx and acetylcholine release. J Neurosci Res 43:63–76 [DOI] [PubMed]
- Tatton W, Chen D, Chalmers-Redman R, Wheeler L, Nixon R, Tatton N (2003) Hypothesis for a common basis for neuroprotection in glaucoma and Alzheimer’s disease: anti-apoptosis by alpha-2-adrenergic receptor activation. Surv Ophthalmol 48(Suppl 1):S25–S37 [DOI] [PubMed]
- Thibault O, Landfield PW (1996) Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272:1017–1020 [DOI] [PubMed]
- Thibault O, Porter NM, Chen KC, Blalock EM, Kaminker PG, Clodfelter GV, Brewer LD, Landfield PW (1998) Calcium dysregulation in neuronal aging and Alzheimer’s disease: history and new directions. Cell Calcium 24:417–433 [DOI] [PubMed]
- Thibault O, Hadley R, Landfield PW (2001) Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci 21:9744–9756 [DOI] [PMC free article] [PubMed]
- Thiels E, Klann E (2001) Extracellular signal-regulated kinase, synaptic plasticity, and memory. Rev Neurosci 12:327–345 [DOI] [PubMed]
- Toescu EC, Verkhratsky A, Landfield PW (2004) Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci 10:614–620 [DOI] [PubMed]
- Tournier C, Thomas G, Pierre J, Jacquemin C, Pierre M, Saunier B (1997) Mediation by arachidonic acid metabolites of the H2O2-induced stimulation of mitogen-activated protein kinases (extracellular-signal-regulated kinase and c-Jun NH2-terminal kinase). Eur J Biochem 244:587–595 [DOI] [PubMed]
- Verkhratsky A, Petersen OH (2002) The endoplasmic reticulum as an integrating signaling organelle: from neuronal signaling to neuronal death. Eur J Pharmacol 447:141–154 [DOI] [PubMed]
- Verkhratsky A, Toescu EC (1998) Calcium and neuronal ageing. Trends Neurosci 21:2–7 [DOI] [PubMed]
- Verkhratsky A, Shmigol A, Kirischuk S, Pronchuk N, Kostyuk P (1994) Age-dependent changes in calcium currents and calcium homeostasis in mammalian neurons. Ann NY Acad Sci 747:365–381 [DOI] [PubMed]
- Volmat V, Pouyssegur J (2001) Spatiotemporal regulation of the p42/p44 MAPK pathway. Biol Cell 93:71–79 [DOI] [PubMed]
- Webber KM, Smith MA, Lee HG, Harris PL, Moreira P, Perry G, Zhu X (2005) Mitogen- and stress-activated protein kinase 1: convergence of the ERK and p38 pathways in Alzheimer’s disease. J Neurosci Res 79:554–560 [DOI] [PubMed]
- Weinreb O, Amit T, Bar-Am O, Sagi Y, Mandel S, Youdim MB (2006) Involvement of multiple survival signal transduction pathways in the neuroprotective, neurorescue and APP processing activity of rasagiline and its propargyl moiety. J Neural Transm 70 (Suppl):457–465 [DOI] [PubMed]
- Wurtman RJ (1992) Choline metabolism as a basis for the selective vulnerability of cholinergic neurons. Trends Neurosci 15:117–122 [DOI] [PubMed]
- Yoon SO, Yun CH, Chung AS (2002) Dose effect of oxidative stress on signal transduction in aging. Mech Ageing Dev 123:597–604 [DOI] [PubMed]
- Zaidi A, Michaelis ML (1999) Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free Radic Biol Med 27:810–821 [DOI] [PubMed]
- Zaidi A, Gao J, Squier TC, Michaelis ML (1998) Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol Aging 19:487–495 [DOI] [PubMed]
- Zhang WJ, Wei H, Hagen T, Frei B (2007) Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA 104:4077–4082 [DOI] [PMC free article] [PubMed]
- Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA (2001) Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev 123:39–46 [DOI] [PubMed]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA (2002) The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 11:270–281 [DOI] [PubMed]
- Zhu Y, Carvey PM, Ling Z (2006) Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res 1090:35–44 [DOI] [PMC free article] [PubMed]
- Zigmond MJ (2006) Triggering endogenous neuroprotective mechanisms in Parkinson’s disease: studies with a cellular model. J Neural Transm 70 (Suppl):439–442 [DOI] [PubMed]