

Abstract
In endocrine cells, plasma membrane (PM)-bound secretory granules must undergo a number of maturation stages (i.e., priming) to become fusion-competent. Despite identification of several molecules involved in binding granules to the PM and priming them, the exact nature of events occurring at the PM still largely remains a mystery. In stimulated BON cells, we used evanescent wave microscopy to study trajectories of granules shortly before their exocytoses, which provided a physical description of vesicle-PM interactions at an unprecedented level of detail, and directly lead to an original mechanistic model. In these cells, tethered (T), nonfusogenic, vesicles are prevented from converting to fusogenic, docked (D) ones in resting conditions. Upon elevation of calcium, T-vesicles perform a 21-nm step toward the PM to become D, and fuse ∼3 s thereafter. Our ability to directly visualize different modes of PM-attachment paves the way for clarifying the exact role of various molecules implicated in attachment and priming of granules in future studies.
INTRODUCTION
Hormones and many other bioactive substances are released into the bloodstream by endocrine cells via Ca2+-regulated exocytosis of large, dense core vesicles, also known as secretory granules. The Ca2+-dependent fusion is preceded by: 1), packaging of the secretory vesicle contents into secretory granules in the trans-Golgi network; 2), translocation to the cell periphery; 3), attachment of the granules to the cell membrane; and 4), maturation stages required for the vesicle to become fusion-competent (1,2). This scheme is based on evidence obtained by different approaches, including biochemical experiments such as cell permeabilization, electrical methods (capacitance and amperometry) and, more recently, optical techniques, especially total internal reflection fluorescence microscopy (TIRFM), also called evanescent wave microscopy (3). In the latter method, a laser beam totally reflected off the glass coverslip-aqueous solution interface creates an evanescent field whose intensity decreases exponentially as a function of distance from the coverslip, with a characteristic decay length of 50–300 nm. Thus, only the first 1–3 layers of secretory granules (typical diameter = 200–300 nm) are visualized, without noise from fluorescently labeled, out-of-focus vesicles, making TIRFM an ideal tool to study attachment of granules to the cell membrane and their fusion. Using this method, secretory vesicle motions close to the plasma membrane (PM) (4–7), the fate of freshly fused granules (8–10), and mechanisms of sequential exocytosis of granules queued-up at the cell membrane (11), have been studied.
The ensemble of maturation stages that a secretory granule must undergo while attached to the PM to become fusion-competent is called priming (2,12). If priming involves a remodeling of the attachment machinery, a change in the distance between a vesicle and the PM may happen. If such a change occurs rapidly compared to the lifetimes of the preceding and succeeding states, and if it spans a sufficient distance, it may be detected by TIRFM, which can have better than ∼10 nm spatial resolution along the z axis, perpendicular to the imaging plane. A number of previous observations indirectly suggested that a remodeling of the vesicle attachment machinery may indeed occur some time before exocytosis. For example, by combining an analysis of the residency-time distribution of vesicles in the evanescent field with other approaches, Toonen et al. (13) proposed the existence of two different modes of granule attachment to the PM. Nofal et al. (14) suggested that primed and not-primed vesicle populations had different lateral mobilities in the imaging (xy) plane, because treatments known to increase the number of rapidly releasable vesicles, such as phorbol myristate acetate, also affected lateral vesicle motions. Finally, in retinal bipolar neurons stimulated by a short depolarization, Zenisek et al. (15) found that some vesicles made a final, sudden 20-nm approach toward the PM shortly before exocytosis. However, all these suggestions were either based on indirect evidence, or were thought not to be generally applicable. In the first study, the lifetime analysis was done in chromaffin cells at rest (not stimulated for secretion) and included all vesicles in the evanescent field, whether or not attached to the cell membrane. As for Nofal et al. (14), motions along the z axis, which could have revealed changes in distances between vesicles and the PM, were not studied. Finally, Zenisek et al. (15) observed only a very limited number of 20-nm approaches and interpreted them as being due to a specific structure called a ribbon, marking active zones of exocytosis, only found in some highly specialized neurons. Interestingly, however, a few 20-nm approaches were also reported by the same authors in nonactive zones, where presumably ribbons were absent, opening the possibility that the stepwise approach may have had a more general origin. In this work, to detect a remodeling of the attachment machinery directly, we focused our attention onto motions that granules underwent shortly before their exocytoses in stimulated endocrine BON cells. This builds upon previous work which had led to a detailed characterization of three-dimensional transient motions of secretory granules in BON cells at rest (16).
The BON cell line, derived from a human pancreatic carcinoid tumor (17), shares many features with enterochromaffin cells of the foregut and is being increasingly used in studies of regulated secretion (11,16,18–21). In vivo, carcinoid tumors secrete serotonin and various proteins and peptides, and are responsible for the carcinoid syndrome characterized by flushing, diarrhea, wheezing and cardiac problems. In vitro, BON cells synthesize and store, in secretory granules, serotonin, chromogranin A, and several regulatory peptides such as pancreastatin and neurotensin. Calcium-regulated secretion is induced by a number of ways, e.g., by secretagogues such as acetylcholine (21,22), second messengers such as phorbol esters or forskolin, or the ionophore ionomycin in the presence of extracellular calcium (17,23–25). Although secretion from these cells is not nearly as well characterized as that from chromaffin or PC-12 cells, they present advantages for TIRFM: they are easy to culture and transfect, possess large secretory vesicles that are easily labeled and visualized, and have good adhesion to bare glass coverslips, eliminating the need to use coating materials which may diffuse light and degrade image quality.
We stimulated BON cells for secretion either by digitonin permeabilization in a Ca2+-containing medium (18,26) or by flash photolysis of an intracellular caged-calcium compound (11,27). Single exocytotic events of secretory granules labeled with neuropeptide Y-green fluorescent protein (NPY-GFP) were detected at the basal membrane using TIRFM and the behavior of fusing and nonfusing vesicles was investigated by determining their trajectories both in the xy plane and along the z axis. Detailed analyses of >2000 exocytoses revealed that a significant fraction of vesicles performed a rapid, well-defined 20-nm stepwise approach toward the cell membrane shortly before fusing with it. We propose that such a step originates when a cell-membrane-attached vesicle undergoes a transition from the tethered to the docked state in preparation for exocytosis.
MATERIALS AND METHODS
Cell culture and vesicle labeling
Cells were cultured and labeled as described in Huet et al. (16). The human carcinoid BON cell line was kindly provided by C. M. Townsend (University of Texas Medical Branch, Galveston, TX). For fluorescence observations, cells were transfected by a plasmid coding for the chimera between the human pro-neuropeptide Y and the enhanced green fluorescent protein (28), as described (16). The plasmid was kindly provided by W. Almers (Oregon Health Sciences University, Portland, OR). The processed form of the expressed protein, NPY-GFP, specifically labels the lumens of large, dense core vesicles (28). Either the original cell line (wild-type, or WT) or a subclone named N13 were used in the experiments. The BON N13 clone was generated in our laboratory for another study (29) as a negative control, stably transfected with an empty pcDNA3 vector (Invitrogen, Carlsbad, CA). After transfection, cells were plated onto uncoated glass-bottom dishes (P50G-1.5-14-F, MatTek Cultureware, Ashland, MA). Observations were performed at 31–33°C, between 48 and 72 h after plating, in Locke solution (5.6 mM Glucose, 3.6 mM  159.6 mM Cl−, 157.6 mM Na+, 5.6 mM K+, 5 mM HEPES-NaOH, 2.5 mM CaCl2, 1.2 mM MgCl2).
159.6 mM Cl−, 157.6 mM Na+, 5.6 mM K+, 5 mM HEPES-NaOH, 2.5 mM CaCl2, 1.2 mM MgCl2).
TIRFM observation and single-vesicle tracking
The setup and calibration are described in detail in Huet et al. (16). Briefly, an upright microscope (BX50WI; Olympus, Tokyo, Japan) was adapted to TIRFM by use of a hemisphere, allowing convenient variation of the incidence angle and hence the evanescence depth, δ. An evanescent wave was generated using the 488-nm line of an argon laser (177-G02; Spectra Physics, Newport, Irvine, CA) radially entering a BK7 glass hemisphere from its curved side. The beam totally reflected off the planar face of the hemisphere which was optically coupled to a glass-bottom culture dish using immersion oil (518 C; Carl Zeiss MicroImaging, Oberkochen, Germany). During stream acquisition, laser power was attenuated to ∼1 mW and illumination was restricted to image acquisition by a shutter coupled to the camera to minimize photobleaching. Cells were observed through a water immersion objective (LUMPlanFL/IR 60×/0.9 numerical aperture; Olympus), and a 515–550 nm band-pass or a >515 nm high-pass filter (both from Olympus). Frames were captured with a charge-coupled device camera (CoolSnap HQ; Photometrics, Roper Scientific, Tucson, AZ) after choosing a region of interest typically encompassing a single cell. Under these conditions, one pixel corresponded to 107.5 nm. Frames were acquired for 60–120 s at 2–40 Hz using MetaVue software (Universal Imaging, Molecular Devices, Downingtown, PA).
Most vesicles appeared as diffraction-limited fluorescent spots, since their sizes were similar to those obtained with 200-nm diameter fluorescent beads. Single exocytic events were detected as described in Tran et al. (11), and the position and the frame number of each detected exocytosis event was marked.
Single vesicles were tracked as described in detail in Huet et al. (16). Briefly, two-dimensional (xy) fluorescent spot trajectories in the imaging plane were obtained from stacks of images by single-particle tracking using the Metamorph software (Universal Imaging, Molecular Devices, Sunnyvale, CA). We did single-particle tracking one spot at a time. The position of a given spot (xy coordinates) on every frame was detected as the centroid of the corresponding pixels (30). To minimize positioning error, it was important to exclude as much of the image background as possible from the centroid calculation. This was achieved by setting a threshold brightness. Pixel values below the threshold were taken to be zero whereas those above remained unchanged.
Two-dimensional diffusion coefficients, the values of Dxy (plotted in Fig. 2 B, appearing later in this article), were calculated by plotting the mean-squared displacement (MSD) as a function of the time lag, Δt, between vesicle positions, as in Tran et al. (18). Only short lags (typically up to 1/10th of the maximum lag) were used in analyses, since statistical errors become large at long lag times as the number-averaged positions decline (31). For most vesicles, the MSD curve was linear at short lags, and a two-dimensional diffusion coefficient was well defined as Dxy = m/4, where m is the slope of the linear portion of the MSD curve, obtained by least-squares fitting. For vesicles which displayed constrained or directed motions, the MSD plot had curvature. For these, a short-time diffusion coefficient was obtained from the initial slope of the MSD curve. Vesicles having a diffusion coefficient less than five-times the average value obtained by tracking immobilized fluorescent beads (16), i.e., Dxy < 5 × 10−4 μm2/s, were considered to be immobile.
FIGURE 2.

Cell membrane attachment leads to a dramatic loss of mobility of a vesicle. (A) Left and right panels show the trajectory of a vesicle in the xy and the xz planes, respectively. A red square marks the beginning of the trajectory in both cases and the time interval between successive measurements is 40 ms. Solid circles correspond to vertical positions (z axis) that are away from the cell membrane. When the vesicle arrived at the cell membrane (open circles), its mobility dropped drastically, both along the z axis and the xy plane—probably because of binding to immobile attachment factors. Lateral diffusion coefficients calculated for the cell-membrane unattached (solid circles) and attached (open circles) portions of similar trajectories from 16 vesicles, from seven cells, differed 9 ± 7 times on average. (B) Distributions of lateral (xy) diffusion coefficients of vesicles which did, and did not, undergo fusion in cells stimulated by digitonin permeabilization in the presence of 30 μM calcium. Half the fused vesicles were immobile (Dxy < 0.5 × 10−4 μm2/s), compared to only 5% of the not-fused ones. The average diffusion coefficient for the fused population is 9.3 times lower than that of the not-fused one, a value remarkably close to the loss of mobility of a single vesicle before and after its contact with the cell membrane (A). Thirty-six fused and 45 not-fused vesicles were analyzed, from the same seven cells. Bins are 0.5 × 10−4 μm2/s wide.
Trajectories of vesicles along the z axis (perpendicular to the glass-solution interface) were calculated as follows. We first evaluated the fluorescence intensity of a spot in a given frame by plotting radially averaged pixel values as a function of distance from the spot's center. The intensity of a spot was defined as the area under a Gaussian fit to this profile, with the tail of the Gaussian set to zero amplitude. This amounts to subtracting the local background; upon complete disappearance of a spot, the calculated intensity drops to zero (i.e., to the local background level). Evanescent field profiles at various incidence angles were calibrated experimentally, as described in Huet et al. (16). The measured profiles were well fitted by exponential decays, I = Io exp(−z/δ), where z is a distance along the z axis. This relation was inverted to calculate the trajectory of a fluorescent spot in the z direction: z(t) = −δ ln[I(t)/Io]. Fluorescence intensity traces were rescaled such that I = Io just before a fusion event. We averaged 5–10 points just preceding an exocytosis event to define Io.
Approximately one-half of the newcomers made a nonrandom, directed approach toward the cell membrane, as in the example given later in Fig. 3 A. Apart from high frequency position fluctuations, these vesicles never reversed their course toward the PM. Many had a constant arrival speed. Some slowed down, or accelerated slightly as they approached their final destination. For these, an average velocity was used for the histogram later in Fig. 3 B.
FIGURE 3.

Newcomer vesicles. (A) Trajectory of a newcomer along the z axis. The vesicle approached the cell membrane with a constant speed of 14 nm/s, from the beginning of acquisition until −9 s. It stayed at the cell membrane for  before fusing with it. (B) Distribution of arrival speeds of newcomers which had a clear directional motion (45 vesicles from 22 cells, 45 out of 97 of newcomers, or 46%). Many of them had a linear arrival, like the vesicle in panel A, whereas some others slightly accelerated or decelerated as they approached the PM. For the latter, the average speed between the appearance and PM-arrival is used in calculating the histogram. The average speed for vesicles which had v < 300 nm/s is 67 nm/s. A few vesicles had speeds >400 nm/s, possibly arising from a mechanism different than that for slower ones. Bin size: 70 nm/s. (C) Distribution of membrane residency times,
before fusing with it. (B) Distribution of arrival speeds of newcomers which had a clear directional motion (45 vesicles from 22 cells, 45 out of 97 of newcomers, or 46%). Many of them had a linear arrival, like the vesicle in panel A, whereas some others slightly accelerated or decelerated as they approached the PM. For the latter, the average speed between the appearance and PM-arrival is used in calculating the histogram. The average speed for vesicles which had v < 300 nm/s is 67 nm/s. A few vesicles had speeds >400 nm/s, possibly arising from a mechanism different than that for slower ones. Bin size: 70 nm/s. (C) Distribution of membrane residency times,  of newcomers (circles). The continuous curve is an exponential fit, N(t) = No e−t/τ, with No = 99.6 and τ = 11.2 s as best-fit parameters.
of newcomers (circles). The continuous curve is an exponential fit, N(t) = No e−t/τ, with No = 99.6 and τ = 11.2 s as best-fit parameters.
To characterize a final, stepwise approach toward the cell membrane, we looked for steplike features preceding a fusion event in all z-trajectories. If more than one steplike feature was present, only the final one was considered. A step was usually completed within 1–2 frames (at 10 frames/s acquisition), facilitating identification and characterization. We either used a manual characterization, or fitted a steplike function as in Fig. 5, A–C, shown later in text, to characterize steps. For manual characterization, we typically averaged 5–10 points preceding and following a step, and took the difference to determine the step size. The average timing of the points just preceding and succeeding a step was taken as the time of stepping, and used in the calculations of the timescales t1 and t2 (see Fig. 5, A and B). For characterization using a fit, we used a function of the form zfit = a + Δz/[1 + exp((t − ts)/τ)], where a measures the z-position after stepping down, ts is the time of stepping with respect to the time of fusion (i.e., ts = −t2), τ measures the timescale over which stepping occurs, and Δz is the step size. A minimum of approximately five experimental points before and after a step were used in the fit. Parameters obtained either manually or by fitting agreed to within a few percent: errors due to the choice of the characterization method are much smaller than the intrinsic widths of the distributions of the obtained parameters, namely Δz (see Fig. 5 E), t1 and t2 (see Fig. 6). An exception was τ, for which fitting usually yielded smaller values than manual calculations. This is expected, since in manual characterization, τ cannot be shorter than half the period between frames (i.e., 50 ms for 10 frames/s acquisition). All fits were made using the Curve Fitting Toolbox of MatLab 6.5 (The MathWorks, Natick, MA).
FIGURE 5.

Stepping vesicles. (A–C) Three examples of steppers, i.e., vesicles making a final, stepwise approach to the cell membrane shortly before their exocytoses. Red curves are fits to the experimental vesicle positions, of the form zfit = a + Δz/[1 + exp((t − ts)−τ)], where a measures the z-position after stepping down, ts is the time of stepping with respect to the time of fusion (t = 0 s), τ measures the timescale over which stepping occurs, and Δz is the step size. For panels A–C, the fits produced a = 0.84, 3.6, −2.3 nm, ts = −2.4, −8.5, −0.9 s, τ = 3, 2, 35 ms, and Δz = 25, 42, 21 nm. Time points at which UV pulses were administered are indicated as vertical dash-dotted lines. Times between the last UV pulse preceding a step and the step (t1), and a step and exocytosis (t2) are indicated in panels A and B. The last UV pulse was applied at t = −7.7 s for the vesicle in panel C. (D) A nonstepper vesicle. Incidentally, this was also a resident vesicle, since its z-position remained stable within 50 nm of the cell membrane, for longer than 10 s. The trajectory illustrates a small artifact sometimes caused by UV pulses, namely a short, transient jump of the z-position away from the PM. The effect is mainly due to background fluorescence excited by the UV pulse, which renders the threshold used in the two-dimensional tracking of the vesicle inappropriate, and consequently introduces an error in the xy centroid position of the vesicle. The error propagates to the intensity calculation (by Gaussian fitting to the radial intensity profile), which results in a slightly smaller intensity value, corresponding to a slightly higher z-position. As the tracking program relocates the correct centroid within 0.2–0.3 s, the effect vanishes. (E) Distribution of step sizes, Δz. The curve is a Gaussian fit to the step-size histogram, which yielded 〈Δz〉 = 21 nm, and the standard deviation σ = 12 nm. (F) All stepper trajectories, aligned to the time point just after the step, plotted together. The thick red curve is the average trajectory. Because the time range over which the trajectories can be averaged is determined by the shortest trajectory, we excluded 14 trajectories lasting <0.8 s after a step in calculating the average (including these short trajectories makes no noticeable change in the average step size).
FIGURE 6.

Distributions of stepping times after a UV pulse and post-step survival times. (A) Number of steppers not yet stepped a time t1 after the nearest UV pulse. (B) Number of steppers not yet fused a time t2 after having made a step.
Cell stimulation
Digitonin permeabilization in the presence of calcium
To clamp the calcium concentration to a known and well-controlled value, we placed cells in a potassium glutamate medium containing 30 μM free calcium (150 mM potassium glutamate, 20 mM PIPES, 2 mM HEDTA, 2 mM EGTA, 2 mM ATP, 0.3 mM GTP, 4.1 mM MgCl2, 2.5 mM CaCl2 and 0.3% BSA, pH adjusted to 7.0 using KOH. The free [Ca2+] and [Mg2+] are calculated to be 30 μM and 1 mM, respectively, using the programs MaxChelator (http://www.stanford.edu/∼cpatton/maxc.html, (32)), and Calcv22 (33)) just before TIRFM observations. Single cells were permeabilized by a 10 s superfusion with the same buffer supplemented by 20 μM digitonin (18), using a glass or quartz micropipette (opening diameter 10–100 μm), connected to a perfusion system (model BPS-4; ALA Scientific Instruments, Westbury, NY). A second, larger micropipette, placed at the other side of the cell, was used to aspirate the stimulation solution. This helped avoid stimulating other cells in the dish, so that up to ∼10 cells could be stimulated per dish, one after another. Image acquisition typically started just before the superfusion of the cell under observation. No fusion occurred before digitonin superfusion. Cells could also be suspended in a digitonin-containing medium and stimulated by superfusion of Ca2+.
To assess successful cell permeabilization and calcium entry, the acetoxymethyl ester derivative of the calcium indicator Fluo-4 (Fluo-4-AM, Invitrogen, Carlsbad, CA) was used. Cells were loaded by incubation in Locke buffer supplemented with 2 μM Fluo-4-AM at 37°C for 30 min. Digitonin was applied and data collected as above.
Ultraviolet-uncaging of calcium
Cells were incubated at 37°C for 60 min in Locke buffer with 30 μM o-nitrophenyl-EGTA-acetoxymethyl ester (NP-EGTA-AM, Invitrogen, Carlsbad, CA) followed by 60-min incubation in the culture medium. Under the TIRF microscope, typically eight brief pulses of ultraviolet (UV) light (300–400 nm) were generated every 10 s using a JML-C2 Xenon arc flash lamp (Rapp Optoelectronic, Hamburg, Germany) coupled to the epifluorescence port of the microscope using a liquid light guide and a modified LGA-Z-1M adaptor. UV light was reflected by a 400 nm dichroic toward the sample, and fluorescence was collected through a 515–550 nm band-pass filter. Although a near maximal power output from the JML-C2 Xenon arc flash lamp was used (three capacitors, 370 V discharge), only a small fraction of the flash light reached the sample due to the poor transmission of the LUMPlanFL/IR 60×/0.9 NA objective (Olympus). In these experiments, [Ca2+]i rises almost instantaneously upon photolysis of the NP-EGTA with a UV pulse, then decreases roughly exponentially in 2–15 s (see Supplementary Material). For the calculation of the t2 distributions shown later in Fig. 7 B, an event was considered to belong to UV pulse n if fusion occurred between pulses n and n+1, even if a step occurred in the period n−1 to n.
FIGURE 7.

Distributions of stepping times after a UV pulse and post-step survival times, for vesicles that stepped after early or late pulses. (A) Fraction of steppers not yet undergone a step a time t1 after the nearest UV pulse. Vesicles that stepped after early (pulses 2–4) and late (pulses 6–8) pulses are indicated as squares or circles, respectively. (B) Fraction of steppers not yet fused a time t2 after having made a step, grouped for early and late pulses as in panel A.
Newcomers, residents and steppers were observed with both types of stimulation, with apparently similar characteristics.
Measurements of intracellular calcium levels are described in the online Supplementary Material.
RESULTS
Secretion from BON cells
The original cell line (wild-type, WT) was reported to be heterogeneous (34). We confirmed this, and generated a number of clones. Secretory properties of these, and of a clone previously prepared by B. Gasnier were tested both in cell-population and single-cell assays (see Supplementary Material). A clone named N13 was selected for its improved secretory response compared to the original cell line, and used in all experiments reported here, unless specified otherwise. A brief morphological analysis of N13 cells by electron and light microscopy is also presented in the Supplementary Material.
Under TIRFM observation, secretion was elicited either by permeabilizing single cells in a medium containing 30 μM Ca2+, using superfusion of a 20 μM digitonin solution (26), or by ultraviolet (UV)-induced uncaging of calcium ions from a photolabile Ca2+ chelator, o-nitrophenyl-EGTA (NP-EGTA) (11,35,36). In the former approach, the intracellular Ca2+ concentration, [Ca2+]i, should be clamped at 30 μM, but permeabilization kinetics may obscure the initial kinetic phase. The advantage of the latter approach is that cells can be stimulated repetitively and that [Ca2+]i is raised essentially instantaneously, allowing better kinetic resolution. However, control of the level of [Ca2+]i is not as good as in permeabilization studies. Although widely used in electrophysiological measurements (36,37) and in two-photon microscopy (38–40), application of pulsed Ca2+-uncaging to TIRFM studies of exocytosis is novel, to the best of our knowledge.
Under stimulation, several types of single exocytotic event were observed using a typical acquisition rate of 10 Hz. The most frequent one (normal exocytosis, >70% of all cases) was characterized by a sudden increase of the intensity of a fluorescent spot, followed by its rapid disappearance accompanied by a cloud of dye radially diffusing away (Fig. 1 A), all within ∼100 ms; a phenomenon previously described as single-vesicle exocytosis (3). Remaining types of event, interpreted due to sequential exocytosis of granules queued-up at the cell membrane, are discussed elsewhere (11).
FIGURE 1.

Secretion kinetics by counting single-vesicle exocytosis events. (A) A single-vesicle exocytosis, visualized by TIRFM. A nearly diffraction limited fluorescent spot disappears by giving off a puff of light as the released soluble vesicular content marker, NPY-GFP, diffuses away. (B) Cumulative number of exocytosis events as a function of time, in digitonin-permeabilized cells in the presence of 30 μM calcium. Since the delay for successful permeabilization was different from cell to cell, the timing of the initial event for every cell (marking successful permeabilization and calcium rise) was set to t = 0 s. With data from 35 cells pooled together, this procedure causes the 35 initial events to artificially cluster together at t = 0 s. This artifact is removed by excluding the 35 initial events from the plot. Dots are experimental data, while the continuous red curve is a fit to the data, up to t = 45 s (see text). Inset shows data and the fit for short times. (C) Cumulative number of exocytosis events as a function of time from 59 cells from caged-calcium experiments in which eight UV pulses were applied at 10 s intervals to increase the [Ca2+]i abruptly. Peak [Ca2+]i generated per pulse is estimated to be ∼1 μM (see Supplementary Material). The value t = 0 s marks the moment the first pulse was applied. Every pulse caused a burst of activity, but with different efficiency. Pulse 1 only caused four events in one cell. Inset shows the number of exocytosis events as a function of time after a UV pulse was applied, for data from all pulses combined (1138 events). Bins are 0.8 s wide. The maximum rate of exocytosis is reached ∼2 s after a UV pulse. (D) Average number of exocytosis events per pulse. On a per-cell basis, early pulses (pulses 2–4) were approximately two-times more efficient as late ones (pulses 6–8), on average.
A difficulty in the digitonin/Ca2+ studies was that the delay between initiation of superfusion and observation of the first exocytosis event was highly variable. In separate experiments, successful cell permeabilization and calcium entry into cells were assessed using Fluo-4, a calcium-sensitive dye, previously loaded into cells. Rise of Fluo-4 fluorescence was again highly variable relative to the start of superfusion with digitonin, taking anywhere from ∼1 s up to several seconds. This suggested that the variable delay between the application of digitonin and the beginning of an exocytotic response was mainly due to cell-to-cell variability of the delay required for permeabilization. To circumvent this variability, we took the first exocytosis event for each cell as the origin of time, considering that it marked successful permeabilization and calcium entry. This also allowed us to pool data from different cells and plot together the number of exocytosis events as a function of time, as shown in Fig. 1 B for WT cells. Remarkably, the data can be described accurately by simple first-order kinetics: a fit of the form N(t) = N∞[1 − exp(−t/τ)] up to t = 45 s yielded N∞ = 431 and τ = 13.4 ± 0.1 s (R2 = 0.999). Beyond 45–50 s after permeabilization, loss of cytosolic factors may become significant (S. Tran, unpublished observations, and (41)). Interestingly, events occurring beyond ∼45 s have faster kinetics than first order. A similar analysis using N13 cells also yielded first-order secretion kinetics (not shown), but with a faster characteristic time (4.9 ± 0.2 s, R2 = 0.99), and a more robust response (∼55 and ∼12 events/cell for N13 and WT cells, respectively). The goodness of the first-order kinetic fits down to the shortest times (Fig. 1 B, inset) suggests that the rise of [Ca2+]i is sufficiently rapid after the first event in every cell to produce a homogeneous response thereafter. That is, BON cells seem to lack ultra-fast modes of response characteristic of adrenal chromaffin cells (37).
For better kinetic resolution at short times, ultraviolet (UV)-induced uncaging of calcium from NP-EGTA was used. Typically 6–8 UV pulses, each lasting ∼1 ms, were administered at 6–10 s intervals to cells under TIRFM observation, and numerous exocytosis events were observed after each pulse. An exception was pulse number 1, which only caused a few events, perhaps due to rapid rebinding of freshly released Ca2+ to excess free NP-EGTA (27). The cumulative number of exocytosis events as a function of time is plotted in Fig. 1 C, using data from 59 cells to which eight UV pulses were applied at 10 s intervals. Fig. 1 D plots the average number of exocytosis events per cell, produced by every pulse. Ignoring the effect of the first UV pulse (applied at t = 0), application of each subsequent pulse caused a burst of exocytotic activity. Looking at short-time kinetics after a UV pulse, it seems that the rate of exocytosis accelerates after a certain lag. This is best seen in the inset of Fig. 1 C where events from all pulses are pooled together, binned into 0.8 s intervals after a UV pulse, and plotted as a histogram. The maximum rate of exocytosis is reached only after ∼2 s. The different efficiency of every pulse to cause exocytosis was correlated with its efficiency to generate [Ca2+]i (Materials and Methods and Supplementary Material).
Alternating detection of vesicle fusions using TIR excitation and of [Ca2+]i using wide-field excitation of a ratiometric Ca2+-sensitive dye, Fura-2, indicated that as little as 0.5 μM [Ca2+]i was sufficient to trigger a robust exocytotic response in BON cells in UV-induced calcium uncaging experiments (Supplementary Material).
Contact of a vesicle with the cell-membrane leads to loss of its lateral mobility
After detection of a normal exocytosis event, the preceding motions of the corresponding vesicle were analyzed by single-particle tracking, backward in time, a method we call retrotracking. Using the exponential variation of the evanescent field along the z axis perpendicular to the imaging plane, motions of a vesicle can be related to changes of its fluorescence intensity using z(t) = −δ ln[I(t)/Io], where δ is the evanescence depth, and I(t) and Io are the vesicle fluorescence intensities corresponding to positions z(t) and z = 0, respectively (3,16). We took Io as the average intensity of 5–10 data points just preceding exocytosis. Since fusion necessarily occurs at the PM, this amounts to setting z = 0 as the position corresponding to a vesicle touching the cell membrane, and calculating all z(t) positions relative to this reference.
Our first objective was to detect vesicle arrival at, and attachment to the cell membrane. We considered that contact between vesicular and cellular membranes occurred either when 1), a vesicle's approach ceased suddenly near z = 0 when the approach had a well-defined direction and speed (as in Fig. 3 A, shown later); or 2), when z decreased to <50 nm and stayed as such until fusion, for more irregular approaches. Most often, contact in the z-direction was accompanied by a concomitant lateral immobilization (in the xy plane), as shown in Fig. 2 A. Two-dimensional diffusion coefficients, Dxy, calculated for the portion of a trajectory before contact (solid circles in Fig. 2 A) were 9 ± 7 times higher than after contact (open circles in Fig. 2 A, 16 vesicles from seven cells). This observation is consistent with previous studies which also reported loss of mobility upon arrival of a vesicle to the cell membrane (4,5,16).
Typically, stimulation caused exocytosis of only a fraction of vesicles near the basal cell membrane, visible by TIRFM, in agreement with many previous reports (4,5,10,42). To see whether the lateral immobilization indicated some commitment of a vesicle toward fusion, the distribution of two-dimensional diffusion coefficients,  of vesicles that underwent exocytosis was compared to that of vesicles which did not fuse,
of vesicles that underwent exocytosis was compared to that of vesicles which did not fuse, 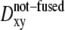 (Fig. 2 B). These diffusion coefficients were obtained by analyzing whole trajectories, lasting 22 s on average, without any attempt to separate contact and noncontact periods. Fig. 2 B indicates marked differences between the two populations: 50% of the fused vesicles were immobile (Dxy < 0.5 × 10−4 μm2/s), compared to only 5% of the not-fused ones. The average diffusion coefficient for the fused population is 9.3 times lower than that of the not-fused vesicles, a value remarkably close to the loss of mobility of a single vesicle before and after its contact with the cell membrane. Evidently, loss of lateral mobility represents some sort of vesicle-attachment to the cell membrane, and this is required for exocytosis. In BON cells, the overall lifetime of the membrane-attached state must be comparable to, or longer than, ∼20 s—the mean duration of a trajectory used to calculate lateral diffusion coefficients.
(Fig. 2 B). These diffusion coefficients were obtained by analyzing whole trajectories, lasting 22 s on average, without any attempt to separate contact and noncontact periods. Fig. 2 B indicates marked differences between the two populations: 50% of the fused vesicles were immobile (Dxy < 0.5 × 10−4 μm2/s), compared to only 5% of the not-fused ones. The average diffusion coefficient for the fused population is 9.3 times lower than that of the not-fused vesicles, a value remarkably close to the loss of mobility of a single vesicle before and after its contact with the cell membrane. Evidently, loss of lateral mobility represents some sort of vesicle-attachment to the cell membrane, and this is required for exocytosis. In BON cells, the overall lifetime of the membrane-attached state must be comparable to, or longer than, ∼20 s—the mean duration of a trajectory used to calculate lateral diffusion coefficients.
Implicit in the foregoing discussion, two subpopulations can be defined among cell-membrane-attached vesicles: those which have become attached during the observation period (which we call newcomers) and those which had been attached all along (named residents), including before stimulation. To avoid any ambiguity, the following strict criteria were used to define the two classes:
A newcomer is a vesicle which was at least 100 nm away from the PM (i.e., for which z > 100 nm) anytime during the observation period. There is little possibility that a vesicle would be physically bound to the cell membrane when such a distance away.
A resident is a vesicle which stayed within 50 nm of the PM (z < 50 nm) during its entire observation. To maximize differences between the two types, we further required that a resident be PM-bound for a time longer than the typical dwell-time of newcomers at the PM,
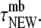 In practice, this amounts to requiring that the observation time of a resident,
In practice, this amounts to requiring that the observation time of a resident, 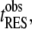 be longer than
be longer than 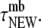
Any vesicle not conforming to these definitions was not classified and not used in the analyses of newcomers and residents. In caged-calcium experiments, out of 1138 vesicles that underwent fusion, 431 could be tracked satisfactorily in the z-direction for classification purposes. One-hundred-and-fifteen of these (∼27% of trackable vesicles) were not classified. We judged that sacrificing these borderline vesicles was justified to keep definitions (and any observed differences) clear between the two classes. All types of vesicle behavior reported below were observed in digitonin/Ca2+ as well as in caged-Ca2+ experiments, with apparently similar characteristics, but the largest set of data was obtained in caged-Ca2+ experiments. Hence, unless specified otherwise, all data presented hereafter are from the latter.
Newcomers
Since the definition of residents depends on an intrinsic property of newcomers, namely the time they spend at the cell membrane, we first classified and analyzed newcomers. A sample z-trajectory is shown in Fig. 3 A. The approach to the PM is followed by attachment (t ≈ −9 s) and fusion (t = 0). The fraction of newcomers among classified vesicles was 31% (97 out of 316). In Fig. 3 A, the motion along the z axis is characterized by a nonrandom, linear approach, with a velocity of 14 nm/s (red line). Approximately fifty-percent of newcomers made a similar, directed approach, with a typical arrival velocity of <100 nm/s (Fig. 3 B).
In Fig. 3 C, we plotted the number of newcomers not yet undergone fusion at time t since their arrival to the cell membrane at t = 0. The distribution is reasonably well fit by an exponential with a characteristic time of  s (R2 = 0.98), suggesting newcomers constitute a homogeneous population. The distribution of dwell times at the cell membrane is broad; it includes vesicles that spent any time from only a fraction of a second up to 50 s. Thus, the minimum time required to become fusion-competent after PM-attachment may be very short (<∼100 ms), but this is accomplished by only a few vesicles which do not constitute a separate pool, in contrast to what had been suggested in chromaffin cells by Allersma et al. (10,43). A typical newcomer needs to spent ∼10 s at the cell membrane before fusing with it. We found no correlation between the arrival speed of a newcomer and its membrane-dwelling time. We characterized fusion kinetics of newcomers by plotting the number of surviving vesicles as a function of the time separating the last UV stimulation-pulse from fusion (Fig. 4 A).
s (R2 = 0.98), suggesting newcomers constitute a homogeneous population. The distribution of dwell times at the cell membrane is broad; it includes vesicles that spent any time from only a fraction of a second up to 50 s. Thus, the minimum time required to become fusion-competent after PM-attachment may be very short (<∼100 ms), but this is accomplished by only a few vesicles which do not constitute a separate pool, in contrast to what had been suggested in chromaffin cells by Allersma et al. (10,43). A typical newcomer needs to spent ∼10 s at the cell membrane before fusing with it. We found no correlation between the arrival speed of a newcomer and its membrane-dwelling time. We characterized fusion kinetics of newcomers by plotting the number of surviving vesicles as a function of the time separating the last UV stimulation-pulse from fusion (Fig. 4 A).
FIGURE 4.

Survival kinetics of newcomers and residents. (A) Number of surviving newcomers as a function of time since the last UV pulse. (B) Number of surviving residents as a function of time since the last UV pulse. (C) Surviving fraction of newcomers (squares) and residents (circles) as a function of time since the last UV pulse. These distributions are equivalent to unnormalized probability density functions for fusion after a UV pulse.
Residents
Sixty-nine percent of classified vesicles were residents (219 out of 316). By definition, these had a much longer dwelling time at the cell membrane than newcomers. Thus, if membrane attachment was rate-limiting for exocytosis, residents should have a kinetic advantage over newcomers. The number of surviving residents as a function of time since the last UV stimulation is shown in Fig. 4 B. When the surviving fraction of newcomers and residents are plotted together, no significant differences can be discerned in their kinetics (Fig. 4 C). Overall, these results suggest that during the last 10 s of their lives, newcomers and residents undergo similar stages of preparation for exocytosis.
Vesicles suddenly approaching the cell membrane shortly before exocytosing: steppers
The results above indicated that one or more stages occurring during the PM-attached period, but not attachment itself, should be rate-limiting for exocytosis. We reasoned that, if one of these stages involves relatively rapid molecular rearrangements such as switching between attachment factors, this may be visualized using TIRFM. Thus, we scrutinized z-trajectories of vesicles more closely, especially shortly before their fusions. In 150 cases out of 356 high quality z-trajectories (42%) we observed a sudden, stepwise approach toward the cell membrane, typically 1–5 s before exocytosis, as shown in Fig. 5, A–C. For comparison, a nonstepper vesicle's z-trajectory is shown in Fig. 5 D, also illustrating a small perturbation sometimes caused by UV pulses. Sixty-eight trajectories were not classified because vesicles did not stay at two stable positions long enough or else the approach was too slow, obscuring a clear, stepwise motion toward the PM. Only 13 vesicles from eight cells (13 out of 356 = 3.6% of all vesicles) displayed a stepwise motion of a few tens of nanometers away from the PM, 3–9 s before fusion. The remainder did not show any clear stepwise motion away from the PM. Without exception, every backstepper vesicle recovered its z = 0 nm position before undergoing exocytosis. An example of a backstepper is given in Fig. 3 A. This vesicle approached the PM with constant velocity. Upon reaching the PM, it seemed to bounce back (t = −6 s) but was held at ∼20 nm from the PM. The vesicle took a 20-nm forward step ∼1 s before exocytosis. The very rare backward steps were not analyzed any further.
The distribution of forward step sizes, Δz, is plotted in Fig. 5 E. A Gaussian fit to the distribution yielded 〈Δz〉 = 21 nm, with a standard deviation of 12 nm. BON cells stimulated by digitonin/Ca2+ yielded similar observations, albeit on a smaller set of data (not shown). We also aligned all stepper trajectories with respect to the first data point after the step and plotted them together in Fig. 5 F. The average stepper trajectory is shown as the thick, red line.
A sudden fluorescence increase of a vesicle, interpreted here as an approach to the PM, may also be caused by transient openings of a fusion pore leading to the neutralization of the intragranular pH, since the EGFP we used is brighter at neutral pH than at intragranular pH (5.5). However, at least three arguments suggest a pH change is not at the origin of our observations. First, a full pH neutralization should lead to an approximately threefold increase in EGFP fluorescence (44,45), corresponding to Δz = 110 nm and 165 nm at the evanescence depths of 100 nm and 150 nm used here, respectively. These values are much higher than typical step sizes (Fig. 5 E), although partial pH neutralization may account for some exceptionally large steps. Second, transient pore openings, observed as small foot currents preceding large spikes in amperometric detection of catecholamines released during single fusion events, typically precede exocytosis by a delay much shorter than the delay between the brightening of a stepper and its fusion (3.3 s, see below). For example, foot currents last ∼100 ms or less in chromaffin cells (46). Similar or shorter durations for foot currents were found in BON cells stimulated by application of 5 μM ionomycin in Locke solution (C. Amatore, S. Arbault, and F. Lemaître, personal communication, 2007). As in the case of digitonin/Ca2+ or caged Ca2+ stimulation, ionomycin causes a direct increase of intracellular calcium, bypassing receptor-mediated mechanisms. Finally, a pH change should occur independently of the evanescence depth δ, yielding the same brightening ratio (If/Ii), but a 1.5 times larger Δz, at δ = 150 nm than δ = 100 nm, since Δz = |−δ ln(If/Ii)|. Contrary to this, essentially the same step size distribution was obtained at δ = 100 and 150 nm (Δz = 25 ± 14 nm, 135 granules/31 cells, and Δz = 23 ± 11 nm, 8 granules/3 cells, respectively).
In the caged-Ca2+ experiments, a step was separated from the most recent UV pulse by a delay t1 and from fusion by a delay t2 (Fig. 5, A and B). Distributions of these delays are shown in Fig. 6. The distribution of t1 is relatively complex, showing several regimes (Fig. 6 A), whereas t2 values are distributed nearly exponentially, with a characteristic time of 3.3 s (Fig. 6 B). An important question is the [Ca2+]i-dependence of these timescales. With our setup, this is difficult to investigate with great precision because we cannot simultaneously follow vesicle and calcium dynamics. However, some conclusions may be drawn from the average [Ca2+]i generated by every UV pulse, measured in separate experiments.
We used the calcium-sensitive dye Fluo-4 to measure relative [Ca2+]i variations in cells coloaded with NP-EGTA, under photolysis conditions identical to those of exocytosis studies. Typically, each brief UV pulse caused an instantaneous increase in the Fluo-4 signal, consistent with previous reports (36,38,47). Averaged signals decayed roughly exponentially with a characteristic time of 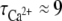 s, with
s, with 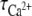 that varied from cell-to-cell and pulse-to-pulse between ∼2 and >15 s. On average, early pulses produced higher peak Ca2+ signals and number of events per cell than late ones (see Supplementary Material). Thus, in Fig. 1 D early pulses (2–4) probably generated approximately two times more [Ca2+]i compared to late pulses (6–8), as they did number of events/cell, providing a means to assess the sensitivity of the delays t1 and t2 to calcium by pooling events generated by early and late UV pulses.
that varied from cell-to-cell and pulse-to-pulse between ∼2 and >15 s. On average, early pulses produced higher peak Ca2+ signals and number of events per cell than late ones (see Supplementary Material). Thus, in Fig. 1 D early pulses (2–4) probably generated approximately two times more [Ca2+]i compared to late pulses (6–8), as they did number of events/cell, providing a means to assess the sensitivity of the delays t1 and t2 to calcium by pooling events generated by early and late UV pulses.
For pooled data, the fraction of steppers which have not yet stepped at time t1 after a UV pulse is shown in Fig. 7 A, while the fraction of steppers which have already made a step but not yet fused a time t2 after stepping is displayed in Fig. 7 B. Fig. 7 A shows clearly that vesicles step-down faster for early (higher [Ca2+]i) than for late (lower [Ca2+]i) pulses (P = 0.007, Kolmogorov-Smirnov test). For early pulses, approximately half the vesicles have stepped down within ∼1 s after a UV pulse, whereas this figure is only ∼35% for late pulses. Thereafter, a plateau is reached for data from late pulses, which lasts until ∼5.5 s, whereas, for early pulses, vesicles continue to step, albeit at a slower rate, until ∼7 s. The acceleration of the stepping rate as t → 10 s both for late and early pulses is artifactual, as the maximum delay between the last UV pulse and stepping cannot exceed 10 s for UV pulses separated by 10 s (except, of course, for the ultimate pulse, but for which no such events were observed). In sharp contrast to the data concerning t1, Fig. 7 B shows that after making a step, survival kinetics are nearly superposable for vesicles from early and late pulses. They can be well described by an exponential decay with a characteristic time of 3.3 s (Fig. 6 B). Evidently, once a vesicle steps-down to the cell membrane, it undergoes exocytosis with an exponential probability distribution, independent of [Ca2+]i, at least within the small, approximately twofold calcium variation probed here. Survival kinetics after stepping do not suffer from the aforementioned artifact concerning the kinetics of stepping after a UV pulse (Fig. 7 A), because t2 measures the delay between a step and exocytosis, independent of when a UV pulse is applied.
If steppers underwent distinct stages of maturation before fusion compared to nonsteppers, this should be reflected in the kinetics of exocytosis of the two types of vesicle. The surviving fraction of all nonstepper and stepper vesicles as a function of time since the last applied UV pulse is plotted in Fig. 8 A: most, if not all, nonsteppers are kinetically equivalent to steppers, since their survival kinetics are very similar.
FIGURE 8.

Comparison of fusion kinetics between steppers and nonsteppers. (A) Fraction of stepper (circles) or nonstepper (squares) vesicles not yet fused a time t since the nearest UV pulse. The two distributions are almost identical. (B) Fraction of newcomer stepper (circles) or newcomer nonstepper (squares) vesicles not yet fused a time t since the nearest UV pulse. Although the two distributions are not superposable, they are not significantly different (P = 0.511, Kolmogorov-Smirnov test). (C) Fraction of resident stepper (circles) or resident nonstepper (squares) vesicles not yet fused a time t since the nearest UV pulse.
Essentially the same fraction of steppers were observed among newcomer and resident vesicles, with 36 out of 97 newcomers (37%) and 78 out of 219 residents (36%) displaying a step. Although not superposable, survival kinetics of stepper and nonstepper newcomers do not differ significantly (Fig. 8 B). Again, no significant differences can be discerned between the survival kinetics of stepper and nonstepper residents (Fig. 8 C).
DISCUSSION
It is well established that attachment of a secretory vesicle to the PM is not a sufficient condition for fusion-competence, i.e., for undergoing rapid fusion in response to elevated Ca2+ concentrations. PM-bound vesicles must go through a number of biochemical maturation stages, collectively known as priming, to become fusion-competent (1,2,12). Despite identification of a number of molecules involved in binding vesicles to the PM and priming them, priming still remains a molecularly ill-defined set of events. A major difficulty has been the lack of assays that can link biochemical priming reactions with corresponding changes in the physical state of a vesicle, such as its mode of PM-attachment, or recruitment of a factor, or a conformational change of a member of the attachment machinery. Electron microscopy, which has high spatial resolution, is of limited use in such studies, since it cannot distinguish which of the morphologically docked vesicles are primed and which are not. As for live imaging studies, TIRFM, which is by far the method of choice, has largely been ineffective in identifying and characterizing substages of vesicle attachment to the PM despite a spatial resolution often surpassing ∼10 nm in the z-direction perpendicular to the imaging plane and its widespread use in the study of single-vesicle exocytosis over the past decade.
Here, using TIRFM to scrutinize trajectories of secretory vesicles shortly before their exocytoses, we have provided a physical description of secretory vesicle-PM interactions at an unprecedented level of detail, including characterization of a ∼21-nm final, stepwise approach of vesicles toward the PM shortly before their fusion with it. Two aspects of this work have been crucial in overcoming past difficulties and allowing these observations: 1), acquisition and analysis of a large body of data; and 2), a simple and robust method we call retrotracking that allows our measuring the axial distance between a vesicle and the PM with high resolution (see below). Approach 2, crucial to our analysis, has rarely been exploited in TIRFM studies despite its power, perhaps because it has been difficult to automate and thus is still very time-consuming. Approach 1 was also tedious, but equally essential, since it allowed us to make statistical analyses, particularly to calculate distributions of step sizes, lifetimes, etc. In the absence of molecular characterization in a novel cell-line model, a characteristic lifetime or a step size that arises from such analyses and the internal consistency of the ensemble of data (see below) argue strongly that what is measured is due to some particular molecular event, rather than an artifact.
Interactions of secretory vesicles with the PM
Retrotracking has been crucial in studying interactions between secretory vesicles and the PM. Although in TIRFM, changes in the fluorescence intensity of a vesicle can be related to variations in its z-position, it is usually difficult to know the distance of a vesicle from the cell membrane. For vesicles which underwent exocytosis upon permeabilizing cells by digitonin in the presence of Ca2+, or by releasing Ca2+ directly into the cytosol from a photolabile chelator, we devised a simple method to measure such distances as a function of time (11,16,18). We reasoned that a vesicle had to be at the cell membrane just before its fusion, and took its average fluorescence intensity 5–10 frames preceding its exocytosis as the reference intensity (setting z = 0 as when it was in contact with the cell membrane). We then tracked positions of the vesicle backward in time.
We defined two subpopulations among cell membrane-attached vesicles: newcomers, which became attached during the observation period and stayed PM-bound for an average time of 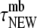 ; and residents, which had been attached for
; and residents, which had been attached for 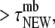 including before stimulation.
including before stimulation.
Newcomers constituted 31% of the classified vesicles. Remarkably, approximately half of these approached the cell membrane in a clearly directed manner, typically with a speed <100 nm/s. In comparison, Steyer et al. (4,6), had found all vesicles arrived at the cell membrane in a directed manner in bovine chromaffin cells under stimulation, but this was based on a very limited number of vesicles. Such active motion may involve molecular motors such as myosins V or II, both implicated in late stages of exocytosis (48,49).
The dwell time of a newcomer at the cell membrane before its exocytosis is, by definition, the time required for priming. We found that dwell times are distributed exponentially ( ), indicating a homogeneous population. Interestingly, Xu and co-workers (36) had found an average priming time of ∼10 s for granules in adrenal chromaffin cells, using electrophysiological methods.
), indicating a homogeneous population. Interestingly, Xu and co-workers (36) had found an average priming time of ∼10 s for granules in adrenal chromaffin cells, using electrophysiological methods.
Our most spectacular finding was that ∼40% of vesicles (150 out of 356) were steppers, i.e., they made a ∼21-nm final, stepwise approach toward the PM shortly before fusing with it. A few vesicles made >50 nm steps, which likely arose from a different mechanism than a rearrangement of the attachment machinery, perhaps due to a partial pH neutralization. Our analysis of the characteristics of a large number of steppers has led us to the following precise model concerning maturation of PM-bound vesicles.
Mechanistic implications
What do our findings imply about maturation stages occurring at the cell membrane? Consider first steppers only. For these, we can rigorously define two modes of attachment to the cell membrane: a tethered (T) state before and a docked (D) state after making a step (Fig. 9 A). Docking invariably leads to fusion (F), since we could only follow distances of fused vesicles from the cell membrane. In contrast, fusion is not permitted directly from the T state. A resident stepper is attached to the cell membrane before stimulation. But before stimulation no fusion occurs, implying that a clamp must be preventing T vesicles from converting to D (i.e., a T-clamp). In addition, we found higher calcium levels promote stepping (T → D). Thus, stimulation (a calcium rise) should cause the clamp to be released, allowing the T → D transition (i.e., stepping down). Once the step is made, fusion occurs with calcium-independent, exponential kinetics, with a characteristic time of 3.3 s, at least within the calcium variation that could be probed. A newcomer stepper, once attached to the cell membrane, spends, on average, ∼10 s before fusion. During that time, fusion kinetics of newcomers were found to be similar to those of residents, suggesting that a newcomer must go through the same stages as a resident, i.e., T → D → F. The release of the brake, in addition to requiring high [Ca2+]i, most likely involves an interaction between vesicular and cell membrane factors; otherwise newcomers could undergo the T → D transition faster or dock directly to the cell membrane, bypassing the T state, and display faster fusion kinetics. These conclusions follow directly from our observations, without any assumption.
FIGURE 9.

Model of maturation stages occurring at the cell membrane. (A) Stepper vesicles are tethered (T) before, and docked (D) after making a stepwise approach toward the cell membrane. By definition, all steppers are initially in the T state. Resident vesicles are already attached to the cell membrane before stimulation, whereas newcomers become so during stimulation. Since stepping inevitably leads to fusion and no fusion occurs in the absence of stimulation, a T-clamp must prevent residents at rest from undergoing the T → D transition (stepping). Stimulation increases the intracellular calcium concentration, which leads to the release of the clamp. This, in turn, allows an ∼20-nm stepwise approach toward the cell membrane, getting the vesicles into the D state. Thereafter, a vesicle undergoes fusion with an exponential probability distribution, on a characteristic timescale of 3.3 s, independent of calcium, at least within the modest range explored here. Because newcomers and residents have indistinguishable fusion kinetics, measured over 10 s, they probably undergo the same preparation stages within the last 10 s of their existence (see text for details). This model, rigorously applicable to steppers, directly follows from our data, without making any assumption. The only speculations in this figure concern the identity of the docking factors (SNARE proteins) and location of the clamps, which are based on other work (see text). The model is most likely applicable to nonsteppers as well, since their kinetics are identical to those of steppers, implying they undergo the same stages as steppers. (B) Several possibilities as to why a T → D transition may not always lead to a detectable step. A tethering factor located off the z axis going through the center of the vesicle (top left), or membrane folds (bottom left) may cause a T → D transition to be accompanied by a small displacement that may not be detectable (docking factors not shown for clarity). In addition, any 20-nm approach that occurs with an oblique angle may lead to a projected z displacement that may be too small to be detected (right). See text for details.
What about nonsteppers? Do they not undergo a T → D transition before fusion? We found that steppers and nonsteppers have indistinguishable kinetics. Thus, the two types should undergo similar stages of maturation. In other words, nonsteppers should also undergo a T → D transition, but this escapes detection for some reason. There are actually a number of reasons why all T → D transitions should not be detectable:
First, unless a vesicle stayed at two stable z-positions sufficiently long before and after stepping, it was not easy to tell with confidence that a step had occurred. We eliminated 68 such doubtful cases out of 357 high quality z-trajectories.
Second, even in many good quality z-trajectories in which the vesicle position was stable for several seconds before fusion, we could not detect a clear step (e.g., as in Fig. 5 D). This can be due to the tethering/docking machineries not being located directly below a vesicle, or the cell membrane not being flat, as schematically drawn in Fig. 9 B, or some other configurations that do not give rise to a detectable transition.
Third, if vesicles approached the cell membrane with an oblique angle, steps along the z axis would only be detected for sufficiently large approach angles (Fig. 9 B, right).
Under our experimental conditions, this last argument alone may be sufficient to explain why only approximately half of the T → D transitions would be detected. To illustrate this, suppose T → D transitions occur with a randomly distributed approach angle, and have exactly Δrc = 20 nm amplitude in three dimensions. With our minimum detectable step size in z, Δzmin ≈ 10 nm, only steps occurring with an angle larger than αmin = arcsin(Δzmin/Δrc) = arcsin(10 nm/20 nm) = π/6 would be detected. The volume fraction of the hemisphere of radius Δrc corresponding to this critical angle can readily be shown to be 1/2, i.e., 1/2 of the T → D transitions would go undetected. It is unlikely that our step-detection efficiency would be much improved were we to calculate steps in three dimensions (i.e., Δr = {Δx2 + Δy2 + Δz2}1/2), since our resolution in either the x or y direction is ∼20 nm (16).
In conclusion, although we cannot exclude the possibility that a small fraction of nonsteppers may undergo other mechanisms, the simplest explanation of our results is that all vesicles undergo the T → D → F stages sequentially. This is supported by the fact that steppers and nonsteppers occurred with essentially the same fraction among newcomer and resident vesicles, because an alternative mechanism would likely affect them differentially. We emphasize that we only analyzed z-motions of vesicles which fused, since only for them could we tell accurately the z-positions with respect to the cell membrane. For these, stepping inevitably led to fusion; that is, the T → D was irreversible. Under resting conditions, or for vesicles which did not fuse, we cannot tell whether docked vesicles can undock and become tethered.
The model implies that when the mean lifetimes in the T and D states are comparable, the overall secretion kinetics should exhibit an initial lag, required for converting nonfusogenic T-vesicles to fusogenic D-vesicles. In contrast, at sufficiently high Ca2+, the highly Ca2+-sensitive T-lifetime (t1) should be rendered much shorter than the D-lifetime (t2), and only a single timescale dominated by the D-lifetime should be detected. Consistent with these considerations, at low [Ca2+]i (∼1 μM) we observed sigmoidal kinetics with a ∼2 s lag to reach the maximum rate of secretion (Fig. 1 C, inset), whereas first-order kinetics were found at high calcium (30 μM), with a characteristic time of 4.9 s.
It is thought that a trans-complex formed between four parallel helical domains, belonging to two soluble n-ethyl maleimide attachment protein receptors (SNAREs) of the PM and one of the vesicle membrane, is involved in the attachment of vesicles to the PM and their fusion with it (1). Cleavage of the SNARE proteins by clostridial neurotoxins (50) or genetic inactivation of the corresponding genes (51) block exocytosis, but not the correct targeting of vesicles to the cell membrane. Therefore, a SNARE-independent attachment step, called tethering, should occur upstream of SNARE-dependent docking and fusion (52–54). Known tethering factors are large proteins, having multiple interaction partners to integrate a number of functions such as vesicle targeting, cytoskeleton remodeling, and recruiting docking factors and calcium sensors. As for docking, it is generally accepted that it involves at least partially assembled SNARE complexes, which should bring apposed membranes to close proximity, perhaps to within a few nm (55). Thus, when a vesicle goes from a tethering-factor-attached to a SNARE-docked state, an approach of the order of a few tens of nanometers between the vesicular and cellular membranes may be expected, consistent with our findings.
We found that once the clamp is released, docking occurs, and fusion follows with slow, calcium-independent, first-order kinetics, at least within the limited calcium variation that we could probe. This surprising finding is actually consistent with a large number of in vitro experiments that have shown that when artificial vesicles containing either synaptic t- or v-SNAREs are mixed together, they fuse slowly and independently of calcium (56,57). Thus, unless specialized scaffolding machineries and fast calcium sensors are present as in cells capable of fast secretion, SNARE-mediated fusion from the docked state is not necessarily expected to be fast.
Our ability to directly visualize different modes of PM-attachment and the model we have proposed provide a solid framework for future studies for clarifying the exact role of various molecules implicated in attachment and priming of granules.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank J. M. Townsend (University of Texas, Medical Branch, Galveston, TX) for the original BON cell line; B. Gasnier (CNRS UPR 1929, Paris) for generating the N13 clone; and E. Farge and N. Desprat (UMR 168, I. Curie, Paris) for lending the Olympus UApo340 40×/1.15 NA high UV transmission objective lens. We also thank J.-P. Denizot (Institut Albert Fessard, CNRS Gif-sur-Yvette) and Takeshi Shimahara for help with electron microscopy; B. Gasnier and F. Darchen (CNRS UPR 1929, Paris) for critically reading the manuscript; and Dr. Gert Rapp (Rapp OptoElectronic GmbH, Germany) for technical advice on coupling ratiometric calcium measurements with UV-pulse calcium-uncaging and TIRFM observations.
This work was supported by the Centre Nationale de la Recherche Scientifique and the Université Paris 7 Denis Diderot. S.H. was supported by the Direction Générale de l'Armement and the Association pour la Recherche sur le Cancer, and V.S.T. was the recipient of a fellowship by the “Ministère de l'Education Nationale, de la Recherche et des Nouvelles Technologies”.
Erdem Karatekin and Viet Samuel Tran contributed equally to this work.
Editor: Joshua Zimmerberg.
References
- 1.Jahn, R., T. Lang, and T. C. Sudhof. 2003. Membrane fusion. Cell. 112:519–533. [DOI] [PubMed] [Google Scholar]
- 2.Burgoyne, R. D., and A. Morgan. 2003. Secretory granule exocytosis. Physiol. Rev. 83:581–632. [DOI] [PubMed] [Google Scholar]
- 3.Steyer, J. A., and W. Almers. 2001. A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell Biol. 2:268–275. [DOI] [PubMed] [Google Scholar]
- 4.Steyer, J. A., H. Horstmann, and W. Almers. 1997. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 388:474–478. [DOI] [PubMed] [Google Scholar]
- 5.Oheim, M., and W. Stuhmer. 2000. Tracking chromaffin granules on their way through the actin cortex. Eur. Biophys. J. 29:67–89. [DOI] [PubMed] [Google Scholar]
- 6.Steyer, J. A., and W. Almers. 1999. Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys. J. 76:2262–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johns, L. M., E. S. Levitan, E. A. Shelden, R. W. Holz, and D. Axelrod. 2001. Restriction of secretory granule motion near the plasma membrane of chromaffin cells. J. Cell Biol. 153:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taraska, J. W., D. Perrais, M. Ohara-Imaizumi, S. Nagamatsu, and W. Almers. 2003. Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc. Natl. Acad. Sci. USA. 100:2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuboi, T., H. T. McMahon, and G. A. Rutter. 2004. Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J. Biol. Chem. 279:47115–47124. [DOI] [PubMed] [Google Scholar]
- 10.Allersma, M. W., L. Wang, D. Axelrod, and R. W. Holz. 2004. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol. Biol. Cell. 15:4658–4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran, V. S., S. Huet, I. Fanget, S. Cribier, J. P. Henry, and E. Karatekin. 2007. Characterization of sequential exocytosis in a neuroendocrine cell line using evanescent wave microscopy and “virtual trajectory” analysis. Eur. Biophys. J. 37:55–69. [DOI] [PubMed] [Google Scholar]
- 12.Klenchin, V. A., and T. F. J. Martin. 2000. Priming in exocytosis: attaining fusion-competence after vesicle docking. Biochimie. 82:399–407. [DOI] [PubMed] [Google Scholar]
- 13.Toonen, R. F., O. Kochubey, H. de Wit, A. Gulyas-Kovacs, B. Konijnenburg, J. B. Sorensen, J. Klingauf, and M. Verhage. 2006. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 25:3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nofal, S., U. Becherer, D. Hof, U. Matti, and J. Rettig. 2007. Primed vesicles can be distinguished from docked vesicles by analyzing their mobility. J. Neurosci. 27:1386–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zenisek, D., J. A. Steyer, and W. Almers. 2000. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 406:849–854. [DOI] [PubMed] [Google Scholar]
- 16.Huet, S., E. Karatekin, V. S. Tran, I. Fanget, S. Cribier, and J. P. Henry. 2006. Analysis of transient behavior in complex trajectories: application to secretory vesicle dynamics. Biophys. J. 91:3542–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence, J. P., J. Ishizuka, B. Haber, C. M. Townsend, Jr., and J. C. Thompson. 1990. The effect of somatostatin on 5-hydroxytryptamine release from a carcinoid tumor. Surgery. 108:1131–1134 (discussion 1134–1135). [PubMed] [Google Scholar]
- 18.Tran, V. S., A.-M. Marion-Audibert, E. Karatekin, S. Huet, S. Cribier, K. Guillaumie, C. Chapuis, C. Desnos, F. Darchen, and J. P. Henry. 2004. Serotonin secretion by human carcinoid BON cells. Ann. N. Y. Acad. Sci. 1014:179–188. [DOI] [PubMed] [Google Scholar]
- 19.John, M., B. Wiedenmann, M. Kruhoffer, K. Adermann, I. Ankorina-Stark, E. Schlatter, G. Ahnert-Hilger, W. G. Forssmann, and M. Kuhn. 1998. Guanylin stimulates regulated secretion from human neuroendocrine pancreatic cells. Gastroenterology. 114:791–797. [DOI] [PubMed] [Google Scholar]
- 20.Mergler, S., B. Wiedenmann, and J. Prada. 2003. R-type Ca2+-channel activity is associated with chromogranin A secretion in human neuroendocrine tumor BON cells. J. Membr. Biol. 194:177–186. [DOI] [PubMed] [Google Scholar]
- 21.Parekh, D., J. Ishizuka, C. M. Townsend, Jr., B. Haber, R. D. Beauchamp, G. Karp, S. W. Kim, S. Rajaraman, G. Greeley, Jr., and J. C. Thompson. 1994. Characterization of a human pancreatic carcinoid in vitro: morphology, amine and peptide storage, and secretion. Pancreas. 9:83–90. [DOI] [PubMed] [Google Scholar]
- 22.Aubrey, K., F. Rossi, R. Ruivo, S. Alboni, G. C. Bellenchi, A. Le Goff, B. Gasnier, and S. Supplisson. 2007. The transporters GlyT2 and VIAAT cooperate to determine the vesicular glynergic phenotype. J. Neurosci. 27:6273–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeng, Y. J., C. M. Townsend, Jr., S. Nagasawa, S. Chuo, K. Kern, N. Yanaihara, R. S. Ferrar, F. L. Hill, J. C. Thompson, and G. H. Greeley, Jr. 1991. Regulation of pancreastatin release from a human pancreatic carcinoid cell line in vitro. Endocrinology. 128:220–225. [DOI] [PubMed] [Google Scholar]
- 24.Zhang, T., C. M. Townsend, Jr., V. Udupi, N. Yanaihara, S. Rajaraman, R. D. Beauchamp, J. Ishizuka, B. M. Evers, G. Gomez, and J. C. Thompson. 1995. Phorbol ester-induced alteration in the pattern of secretion and storage of chromogranin A and neurotensin in a human pancreatic carcinoid cell line. Endocrinology. 136:2252–2261. [DOI] [PubMed] [Google Scholar]
- 25.Kim, M., N. H. Javed, J. G. Yu, F. Christofi, and H. J. Cooke. 2001. Mechanical stimulation activates Gα signaling pathways and 5-hydroxytryptamine release from human carcinoid BON cells. J. Clin. Invest. 108:1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham, M. E., R. J. Fisher, and R. D. Burgoyne. 2000. Measurement of exocytosis by amperometry in adrenal chromaffin cells: effects of clostridial neurotoxins and activation of protein kinase C on fusion pore kinetics. Biochimie. 82:469–479. [DOI] [PubMed] [Google Scholar]
- 27.Heinemann, C., R. H. Chow, E. Neher, and R. S. Zucker. 1994. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+. Biophys. J. 67:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang, T., I. Wacker, J. Steyer, C. Kaether, I. Wunderlich, T. Soldati, H. H. Gerdes, and W. Almers. 1997. Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron. 18:857–863. [DOI] [PubMed] [Google Scholar]
- 29.Herzog, E., G. C. Bellenchi, C. Gras, V. Bernard, P. Ravassard, C. Bedet, B. Gasnier, B. Giros, and S. El Mestikawy. 2001. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J. Neurosci. 21:RC181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheezum, M. K., W. F. Walker, and W. H. Guilford. 2001. Quantitative comparison of algorithms for tracking single fluorescent particles. Biophys. J. 81:2378–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian, H., M. P. Sheetz, and E. L. Elson. 1991. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60:910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patton, C., S. Thompson, and D. Epel. 2004. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 35:427–431. [DOI] [PubMed] [Google Scholar]
- 33.Fohr, K. J., W. Warchol, and M. Gratz. 1993. Calculation and control of free divalent cations in solutions used for membrane fusion studies. Methods Enzymol. 221:149–157. [DOI] [PubMed] [Google Scholar]
- 34.Evers, B. M., C. M. Townsend, Jr., J. R. Upp, E. Allen, S. C. Hurlbut, S. W. Kim, S. Rajaraman, P. Singh, J. C. Reubi, and J. C. Thompson. 1991. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 101:303–311. [DOI] [PubMed] [Google Scholar]
- 35.Ellis-Davies, G. C., and J. H. Kaplan. 1994. Nitrophenyl-EGTA, a photolabile chelator that selectively binds Ca2+ with high affinity and releases it rapidly upon photolysis. Proc. Natl. Acad. Sci. USA. 91:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu, T., T. Binz, H. Niemann, and E. Neher. 1998. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat. Neurosci. 1:192–200. [DOI] [PubMed] [Google Scholar]
- 37.Voets, T. 2000. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 28:537–545. [DOI] [PubMed] [Google Scholar]
- 38.Kishimoto, T., R. Kimura, T. T. Liu, T. Nemoto, N. Takahashi, and H. Kasai. 2006. Vacuolar sequential exocytosis of large dense-core vesicles in adrenal medulla. EMBO J. 25:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemoto, T., R. Kimura, K. Ito, A. Tachikawa, Y. Miyashita, M. Iino, and H. Kasai. 2001. Sequential-replenishment mechanism of exocytosis in pancreatic acini. Nat. Cell Biol. 3:253–258. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi, N., H. Hatakeyama, H. Okado, A. Miwa, T. Kishimoto, T. Kojima, T. Abe, and H. Kasai. 2004. Sequential exocytosis of insulin granules is associated with redistribution of SNAP25. J. Cell Biol. 165:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarafian, T., D. Aunis, and M. F. Bader. 1987. Loss of proteins from digitonin-permeabilized adrenal chromaffin cells essential for exocytosis. J. Biol. Chem. 262:16671–16676. [PubMed] [Google Scholar]
- 42.Ohara-Imaizumi, M., Y. Nakamichi, T. Tanaka, H. Ishida, and S. Nagamatsu. 2002. Imaging exocytosis of single insulin secretory granules with evanescent wave microscopy: distinct behavior of granule motion in biphasic insulin release. J. Biol. Chem. 277:3805–3808. [DOI] [PubMed] [Google Scholar]
- 43.Allersma, M. W., M. A. Bittner, D. Axelrod, and R. W. Holz. 2006. Motion matters: secretory granule motion adjacent to the plasma membrane and exocytosis. Mol. Biol. Cell. 17:2424–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patterson, G. H., S. M. Knobel, W. D. Sharif, S. R. Kain, and D. W. Piston. 1997. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73:2782–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou, Z., S. Misler, and R. H. Chow. 1996. Rapid fluctuations in transmitter release from single vesicles in bovine adrenal chromaffin cells. Biophys. J. 70:1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasai, H., T. Kishimoto, T. Nemoto, H. Hatakeyama, T. T. Liu, and N. Takahashi. 2006. Two-photon excitation imaging of exocytosis and endocytosis and determination of their spatial organization. Adv. Drug Deliv. Rev. 58:850–877. [DOI] [PubMed] [Google Scholar]
- 48.Varadi, A., T. Tsuboi, and G. A. Rutter. 2005. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 β-cells. Mol. Biol. Cell. 16:2670–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neco, P., D. Giner, S. Viniegra, R. Borges, A. Villarroel, and L. M. Gutierrez. 2004. New roles of myosin II during vesicle transport and fusion in chromaffin cells. J. Biol. Chem. 279:27450–27457. [DOI] [PubMed] [Google Scholar]
- 50.Hunt, J. M., K. Bommert, M. P. Charlton, A. Kistner, E. Habermann, G. J. Augustine, and H. Betz. 1994. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 12:1269–1279. [DOI] [PubMed] [Google Scholar]
- 51.Broadie, K., A. Prokop, H. J. Bellen, C. J. O'Kane, K. L. Schulze, and S. T. Sweeney. 1995. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 15:663–673. [DOI] [PubMed] [Google Scholar]
- 52.Kavalali, E. T. 2002. SNARE interactions in membrane trafficking: a perspective from mammalian central synapses. Bioessays. 24:926–936. [DOI] [PubMed] [Google Scholar]
- 53.Wang, S., and S. C. Hsu. 2006. The molecular mechanisms of the mammalian exocyst complex in exocytosis. Biochem. Soc. Trans. 34:687–690. [DOI] [PubMed] [Google Scholar]
- 54.Whyte, J. R., and S. Munro. 2002. Vesicle tethering complexes in membrane traffic. J. Cell Sci. 115:2627–2637. [DOI] [PubMed] [Google Scholar]
- 55.Sutton, R. B., D. Fasshauer, R. Jahn, and A. T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- 56.Weber, T., B. V. Zemelman, J. A. McNew, B. Westermann, M. Gmachl, F. Parlati, T. H. Sollner, and J. E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- 57.Schuette, C. G., K. Hatsuzawa, M. Margittai, A. Stein, D. Riedel, P. Kuster, M. Konig, C. Seidel, and R. Jahn. 2004. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. USA. 101:2858–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]