

Abstract
In skeletal muscle, the L-type voltage-gated Ca2+ channel (1,4-dihydropyridine receptor) serves as the voltage sensor for excitation-contraction (EC) coupling. In this study, we examined the effects of Rem, a member of the RGK (Rem, Rem2, Rad, Gem/Kir) family of Ras-related monomeric GTP-binding proteins, on the function of the skeletal muscle L-type Ca2+ channel. EC coupling was found to be weakened in myotubes expressing Rem tagged with enhanced yellow fluorescent protein (YFP-Rem), as assayed by electrically evoked contractions and myoplasmic Ca2+ transients. This impaired EC coupling was not a consequence of altered function of the type 1 ryanodine receptor, or of reduced Ca2+ stores, since the application of 4-chloro-m-cresol, a direct type 1 ryanodine receptor activator, elicited myoplasmic Ca2+ release in YFP-Rem-expressing myotubes that was not distinguishable from that in control myotubes. However, YFP-Rem reduced the magnitude of L-type Ca2+ current by ∼75% and produced a concomitant reduction in membrane-bound charge movements. Thus, our results indicate that Rem negatively regulates skeletal muscle EC coupling by reducing the number of functional L-type Ca2+ channels in the plasma membrane.
INTRODUCTION
The skeletal muscle L-type Ca2+ channel (1,4-dihydropyridine receptor (DHPR)) serves as the voltage sensor for excitation-contraction (EC) coupling (1). Conformational changes in the DHPR in response to plasma membrane depolarization are conveyed to the ryanodine-sensitive Ca2+ release channel (RyR1), resulting in efflux of Ca2+ from the stores of the sarcoplamsic reticulum (SR) and in activation of the contractile machinery (2). Because communication between the DHPR and RyR1 is rapid and does not require Ca2+ entry via the L-type channel itself (3–5), it is believed that there is a physical interaction between the two proteins. This view is supported by ultrastructural evidence demonstrating that the tetradic arrangement of L-type channels within skeletal muscle triad junctions is altered by exposure to ryanodine (6). Because the DHPR is a key protein in EC coupling, modulation of its activity may have significant consequences for the frequency, strength, and duration of muscle contraction.
Members of the RGK (Rem, Rem2, Rad, Gem/Kir) family of Ras-related monomeric GTP-binding proteins inhibit high-voltage-activated (HVA) Ca2+ channels via an interaction with Ca2+ channel β subunit isoforms (7–16). Recent evidence indicates that Ca2+ channel α1 subunits and RGK proteins have structurally distinct binding sites within the conserved guanylate-kinase-like domain of β subunits and that such interactions may support formation of a tripartite complex (14,15). Several studies have established that overexpression of an RGK family member ablates, or nearly ablates, HVA Ca2+ currents in a variety of preparations (7–14,16–21). Moreover, intramembrane charge movements of cardiac myocytes were reduced by viral overexpression of Gem (17), indicating that this RGK protein either prevents voltage-driven conformational changes necessary for channel gating or reduces the number of channel proteins in the plasma membrane. Establishing which of these mechanisms accounts for the inhibition of Ca2+ channels by RGK proteins is a matter of ongoing debate. Some biochemical and morphological evidence suggests that the RGK-β subunit interaction causes a reduction in the total number of channel proteins in the plasma membrane (7,9,10,12,13,15,16). However, binding of radiolabeled ω-conotoxin GVIA to heterologously-expressed N-type channels in tsA-201 fibroblasts and surface biotinylation of native L-type channels in HIT-T15 β-islet cells were both found to be unaltered by exogenous expression of RGK proteins (11,14,19). Moreover, Rem2 affects gating kinetics of cloned α1C channels heterologously-expressed in Xenopus laevis oocytes (21). These latter findings point to a mechanism of action by which RGKs may modulate HVA channels at the plasma membrane by altering channel gating. Regardless of the mechanism, overexpression of RGK family proteins negatively regulates physiological processes dependent on the activity of HVA Ca2+ channels including myocardial function (16,17,20) and secretion in pancreatic and neuroendocrine cells (7,11,12).
Rem is endogenously expressed in normal skeletal muscle and Finlin et al. have reported that viral overexpression of Rem inhibits myoplasmic Ca2+ release in C2C12 myoblasts in response to membrane depolarization (8). However, the mechanism for this inhibition of EC coupling was not established. One possibility is that overexpression of Rem impairs the formation of the junctional associations between the plasma membrane and the SR that are required for EC coupling. Although Finlin and colleagues (8) did not find Rem-induced morphological changes, other groups have reported that overexpression of Rem or other RGK family members causes pronounced morphological changes in a number of cell types (9,10,13,18,22–25), including C2C12 myoblasts (9). A second possibility is that Rem directly inhibits the function of the DHPR as voltage sensor for EC coupling. A third possibility is that Rem negatively modulates the EC coupling machinery downstream of the voltage sensor by inhibiting RyR1 or by reducing the SR Ca2+ store. Here, we describe experiments to distinguish these possibilities. We find that expression of YFP-Rem in normal myotubes reduces the frequency of electrically evoked contractions, dampens voltage-dependent Ca2+ release from the SR, and decreases the magnitude of L-type Ca2+ currents and immobilization-resistant charge movements, but does not affect Ca2+ release in response to the RyR activator 4-chloro-m-cresol. Thus, YFP-Rem inhibits EC coupling in skeletal muscle by reducing the number of functional DHPRs in the plasma membrane.
MATERIALS AND METHODS
Construction of YFP-Rem
Rem (accession number U91601) tagged with hemagglutinin, was a kind gift from Dr. D. A. Andres (University of Kentucky, Lexington, KY). To generate Rem tagged with enhanced yellow fluorescent protein (YFP-Rem), PCR-amplified YFP (Clontech, Palo Alto, CA) was cloned into pcDNA4.1 (Invitrogen, Carlsbad, CA) using KpnI and BamHI sites. Subsequently, full-length Rem was amplified by PCR and cloned in-frame downstream of YFP using BamHI and EcoRI sites. All PCR constructs were verified by sequencing.
Myotube culture and expression of cDNA
Primary cultures of normal (+/+ or +/mdg on an outbred Black Swiss background) and dyspedic (RyR1 null on C57BL/6 background) myotubes were prepared from newborn mice, as described previously (26). Altogether, myotubes from nine separate normal cultures and one dyspedic culture were utilized in experiments. Two days after the change from plating to differentiation medium, single nuclei were microinjected with YFP-Rem or pEYFP-C1 (Clontech). The injection solution contained 5–100 ng/μl of YFP-Rem cDNA or 10 ng/μl of YFP cDNA. In the initial stages of the study, concentrations of YFP-Rem cDNA ranging from 5–100 ng/μl were assayed by their ability to reduce L-type current. The effects on L-type current were similar for each concentration tested (analysis of variance (ANOVA), p > 0.05). In the majority of experiments (including the measurement of both Ca2+ transients and charge movements), 10 ng/μl YFP-Rem cDNA was the amount chosen to be used, because at this concentration, YFP-Rem could be detected easily by fluorescence without the potentially deleterious effects of higher YFP-Rem expression levels. N-benzyl-P-toluene sulfonamide (BTS, 20 μM; S949760, Sigma, St. Louis, MO) was added to the culture medium during microinjection to prevent contractions. Fluorescent myotubes were used in experiments 2 days after microinjection.
Contractions
Electrically evoked contractions were elicited by 10-ms, 100-V stimuli applied via an extracellular pipette that contained 150 mM NaCl (see Papadopoulos et al. (27)). The myotubes were bathed in rodent Ringer's solution (in mM, 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 glucose, pH 7.4, with NaOH). Contractions were assayed by the movement of an identifiable portion of a myotube across the visual field.
Measurement of intracellular Ca2+ transients
Changes in intracellular Ca2+ were recorded with Fluo-3 (No. F-3715, Molecular Probes, Eugene, OR). The salt form of the dye was added to the standard internal solution (see below) for a final concentration of 200 nM. After entry into the whole-cell configuration, a waiting period of >5 min was used to allow the dye to diffuse into the cell interior. A 100 W mercury illuminator and a set of fluorescein filters were used to excite the dye present in a small rectangular region of the voltage-clamped myotube. A computer-controlled shutter was used to block illumination in the intervals between 50-ms test pulses. Fluorescence emission was measured by means of a fluorometer apparatus (Biomedical Instrumentation Group, University of Pennsylvania, Philadelphia, PA). The average background fluorescence was quantified before bath immersion of the patch pipette. Fluorescence data are expressed as ΔF/F, where ΔF represents the change in peak fluorescence from baseline during the test pulse and F is the fluorescence immediately before the test pulse minus the average background (non-Fluo-3) fluorescence. Where applicable, the peak value of the fluorescence change (ΔF/F) for each test potential (V) was fitted according to
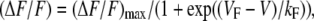 |
(1) |
where (ΔF/F)max is the maximal fluorescence change, VF is the potential causing half the maximal change in fluorescence, and kF is a slope parameter.
Measurements of responses to 4-chloro-m-cresol
Myotubes were washed with Ca2+/Mg2+-free Ringer's (in mM, 146 NaCl, 5 KCl, 10 HEPES, 11 glucose, pH 7.4, with NaOH) twice and subsequently loaded with 5 μM Fluo-3 AM (F-1242, Molecular Probes) dissolved in rodent Ringer's solution for 1 h. Myotubes were then washed three times in rodent Ringer's solution with gentle agitation. Fluo-3-AM-loaded myotubes were then placed on the stage of a Nikon Diaphot inverted microscope and viewed under 40× magnification. A 100 W mercury illuminator and a set of fluorescein filters were used to excite the Fluo-3 AM dye present in an isolated myotube. To facilitate identification of myotubes expressing YFP-Rem in the presence of Fluo-3 AM, yellow fluorescent myotubes were cytoplasmically injected with 1 μM sulforhodamine (S-1307, Molecular Probes) before loading; myotubes expressing YFP-Rem were then identified by red fluorescence. Transients were elicited by application of a bolus of 4-chloro-m-cresol (4-CmC) (C10570, Pfaltz and Bauer, Waterbury, CT) via an extracellular pipette placed near the myotube of interest. 4-CmC was dissolved in EtOH to make a stock solution of 500 mM and was diluted in 150 NaCl to the working concentration (0.5 mM) just before experiments. Fluorescence emission was measured essentially as described above.
Measurement of L-type Ca2+ currents
Pipettes were fabricated from borosilicate glass and had resistances of ∼2.0 MΩ when filled with internal solution, which consisted of (mM) 140 Cs-aspartate, 10 Cs2-EGTA, 5 MgCl2, and 10 HEPES, pH 7.4, with CsOH. The standard external solution contained (mM) 145 TEA (tetraethylammonium)-Cl, 10 CaCl2, 0.003 tetrodotoxin, and 10 HEPES, pH 7.4 with TEA-OH. Linear capacitative and leakage currents were determined by averaging the currents elicited by eleven 30-mV hyperpolarizing pulses from a holding potential of −80 mV. Test currents were corrected for linear components of leak and capacitive current by digital scaling and subtraction of this average control current. Electronic compensation was used to reduce the effective series resistance (usually to <1 MΩ) and the time constant for charging the linear cell capacitance (usually to <0.5 ms). Ionic currents were filtered at 2 kHz and digitized at 10 kHz. To measure macroscopic L-type current in isolation, a 1-s prepulse to −20 mV followed by a 50-ms repolarization to −50 mV was administered before the test pulse (prepulse protocol (28)) to inactivate T-type Ca2+ channels. Cell capacitance was determined by integration of a transient from −80 mV to −70 mV using Clampex 8.0 and was used to normalize current amplitudes (pA/pF). Current/voltage (I/V) curves were fitted using the equation
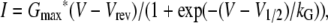 |
(2) |
where I is the current for the test potential, V; Vrev is the reversal potential; Gmax is the maximum Ca2+ channel conductance, V1/2 is the half-maximal activation potential; and kG is the slope factor. The activation phase of macroscopic ionic currents was fitted using the exponential function
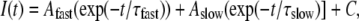 |
(3) |
where I(t) is the current at time t after the depolarization; Afast and Aslow are the steady-state current amplitudes of each component, with their respective time constants of activation (τfast and τslow); and C represents the steady-state peak current (see Avila and Dirksen (29)).
Measurement of charge movements
For measurement of intramembrane charge movements, ionic currents were blocked by the addition of 0.5 mM CdCl2 + 0.1 mM LaCl3 to the standard extracellular recording solution. All charge movements were corrected for linear cell capacitance and leakage currents using a −P/8 subtraction protocol. Filtering was at 2 kHz and digitization was at 20 kHz. Voltage-clamp command pulses were exponentially rounded with a time constant of 50–500 μs and the prepulse protocol (see above (28)) was used to reduce the contribution of gating currents from voltage-gated Na+ channels and T-type Ca2+ channels. The integral of the ON transient (Qon) for each test potential (V) was fitted according to
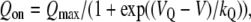 |
(4) |
where Qmax is the maximal Qon, VQ is the potential causing movement of half the maximal charge, and kQ is a slope parameter.
Analysis
To calculate Gmax/Q′ ratios, Gmax was calculated using Eq. 2 and Q′ was derived by subtracting the average maximal charge movement of dysgenic myotubes (Qdys) from the average total Qmax, where Qdys = 1.0 nC/μF ((30) and Bannister and Beam, unpublished observations). Figures were made using SigmaPlot (version 7.0, Systat Software, San Jose, CA). All data are presented as mean ± SE. Statistical comparisons were by ANOVA or by unpaired, two-tailed t-test (as appropriate), with p < 0.05 considered significant.
RESULTS
YFP-Rem inhibits skeletal muscle EC coupling
To assay the effects of YFP-Rem expression on EC coupling, myotube contractions were evaluated in response to electrical stimulation. Expression of YFP-Rem caused an ∼50% reduction in the fraction of contracting myotubes (contractions in 7 of 16 myotubes; Fig. 1 A) in comparison to nontransfected normal and normal myotubes expressing unconjugated YFP (contractions in 60 of 68 and 8 of 10 myotubes, respectively; Fig. 1 A). To determine whether this reduction was a result of an action of YFP-Rem on RyR1 or the SR Ca2+ store, myotubes were loaded with the Ca2+ indicator Fluo-3 AM and challenged with the RyR agonist 4-CmC. Robust responses were observed in nontransfected, normal myotubes and in YFP-Rem expressing myotubes, whereas no obvious response was seen in dyspedic (RyR1 null) myotubes (Fig. 1 B). On average, the responses to 4-CmC (0.5 mM) of control and YFP-REM-expressing myotubes (2.0 ± 0.9 ΔF/F, n = 4 vs. 2.4 ± 1.0 ΔF/F, n = 5, respectively) were not significantly different (p = 0.796; Fig. 1 C). In control experiments, dyspedic myotubes responded negligibly to 0.5 mM 4-CmC (0.2 ± 0.1 ΔF/F, n = 5; Fig. 1 C). These data indicate that YFP-Rem does not cause depletion of SR Ca2+ stores or inhibit the ability of RyR1 to release Ca2+ from the SR.
FIGURE 1.

YFP-Rem inhibits skeletal muscle EC coupling without affecting Ca2+ release in response to direct activation of RyR1. (A) YFP-Rem reduces the fraction of myotubes contracting in response to a 100-V, 10-ms electrical stimulus. (B) Representative changes in Fluo-3 AM fluorescence in response to application of 4-CmC (0.5 mM) elicited in a normal myotube (upper), a myotube expressing YFP-Rem (middle), and a dyspedic myotube (lower). Black arrows indicate time of 4-CmC application. (C) Summary of myoplasmic Ca2+ transients in response to 4-CmC (0.5 mM) for normal (black bar), YFP-Rem-injected (gray bar) normal, and dyspedic myotubes (white bar). Throughout, error bars represent mean ± SE. For each group in A and C, the total number of myotubes tested is indicated above each bar.
YFP-Rem inhibits voltage-dependent Ca2+ release from the SR
To obtain more precise information regarding the inhibition of EC coupling by YFP-Rem, we measured intracellular Ca2+ transients in the whole-cell patch-clamp configuration. As shown in Fig. 2 A, nontransfected normal myotubes produced robust Ca2+ transients with an amplitude that had a sigmoidal dependence on test potential (Fig. 2 C) and a maximum (ΔF/F)max of 0.46 ± 0.08 (n = 9). Myotubes expressing YFP-Rem yielded Ca2+ transients that were considerably smaller (Fig. 2 B), with a (ΔF/F)max of 0.14 ± 0.11 (n = 8, p < 0.05). Only one of the eight YFP-Rem-expressing myotubes examined had sufficiently large Ca2+ transients to analyze voltage dependence, which appeared to be little affected (this cell dominated the average data illustrated in Fig. 2 C). In control experiments, expression of unconjugated YFP had no significant effect on either the magnitude or voltage dependence of Ca2+ transients relative to nontransfected normal myotubes (Fig. 2 C and Table 1).
FIGURE 2.

YFP-Rem reduces voltage-dependent Ca2+ release from the SR. Recordings of myoplasmic Ca2+ transients elicited by 50-ms depolarizations from −50 mV to the indicated test potentials are shown for a nontransfected normal myotube (A) and a YFP-Rem-expressing myotube (B). (C) Comparison of ΔF/F-V relationships for normal myotubes (•, n = 9) and myotubes expressing either YFP-Rem (○, n = 8) or YFP (▵, n = 4). Sigmoidal ΔF/F-V curves are plotted according to Eq. 1. The best-fit parameters for each plot are presented in Table 1.
TABLE 1.
YFP-Rem inhibits EC coupling
|
ΔF/F-V
|
Contracting cells/ number tested | |||
|---|---|---|---|---|
| ΔF/Fmax (ΔF/F) | VF (mV) | kF (mV) | ||
| Normal | 0.46 ± 0.08 (9) | 6.9 ± 1.2 | 6.4 ± 0.5 | 60/68 |
| Normal + YFP-Rem | 0.14 (8) | 8.4 | 6.8 | 7/16 |
| Normal + YFP | 0.63 ± 0.25 (4) | 4.3 ± 0.9 | 6.5 ± 0.3 | 8/10 |
ΔF/F-V relationships were fitted using Eq. 1. For untransfected normal myotubes and myotubes expressing YFP, data represent the average of the fits for each individual experiment (numbers in parentheses represent the number of myotubes tested) and are given as mean ± SE. Because seven of eight experiments with myotubes expressing YFP-Rem were difficult to fit with Eq. 1, the ΔF/F-V relationship is given as the fit of the average data.
YFP-Rem reduces skeletal muscle L-type Ca2+ currents
Whole-cell patch-clamping was employed to test directly whether Rem affects skeletal muscle L-type Ca2+ currents mediated by the DHPR. Normal myotubes produced large, slowly activating L-type currents (−10.4 ± 0.9 pA/pF, n = 21; Fig. 3 A). By comparison, L-type currents were reduced by 73% in normal myotubes expressing YFP-Rem (−2.8 ± 0.5 pA/pF, n = 15, p < 10−6; Fig. 3 B). YFP-Rem caused a small rightward shift in the voltage dependence of activation (V1/2 = 19.0 ± 1.0 mV vs. 24.1 ± 1.2 mV, for normal and YFP-Rem currents, respectively, p = 0.003), but did not alter the kinetics of activation (see Table 3). In control experiments, expression of unconjugated YFP had no effect on either the voltage dependence (V1/2 = 20.3 ± 1.4 mV, p > 0.05) or the magnitude of L-type current (−12.5 ± 2.0 pA/pF, n = 6, p > 0.05) relative to nontransfected normal myotubes (Fig. 3 C and Table 2).
FIGURE 3.

YFP-Rem reduces skeletal muscle L-type Ca2+ current. Recordings of L-type Ca2+ currents elicited by 200-ms depolarizations from −50 mV to the indicated test potentials are shown for a normal, control myotube (A) or a normal myotube expressing YFP-Rem (B). (C) Comparison of I/V relationships for normal, control myotubes (•, n = 21), normal myotubes expressing YFP-Rem (○, n = 15), and normal myotubes expressing unconjugated YFP (Δ, n = 6). Currents were evoked at 0.1 Hz by test potentials ranging from −20 mV through +80 mV in 10-mV increments, following a prepulse protocol (28). The YFP-Rem I/V relationship represents pooled data from myotubes injected with 5, 10, 20, 50, and 100 ng/μl YFP-Rem cDNA; no significant difference in peak current density was found between these groups (p > 0.05, ANOVA). The smooth curves are plotted according to Eq. 2. The best fit parameters for each plot are presented in Table 2.
TABLE 3.
L-type Ca2+ current activation fit parameters
|
Islow
|
Ifast
|
|||||
|---|---|---|---|---|---|---|
| τslow (ms) | Aslow (pA/pF) | Fraction (%) | τfast (ms) | Afast (pA/pF) | Fraction (%) | |
| Normal | 94.1 ± 7.7 (11) | 10.0 ± 1.1 | 87 ± 3 | 8.6 ± 1.1 | 1.6 ± 0.4 | 13 ± 3 |
| Normal + YFP-Rem | 108.8 ± 9.0 (6) | 3.3 ± 0.7* | 84 ± 4 | 6.1 ± 0.3 | 0.5 ± 0.1 | 16 ± 4 |
Activation was fitted with a double exponential function (Eq. 3) yielding time constants τfast and τslow with absolute or fractional amplitudes Aslow and Afast (see Avila and Dirksen (29)). Data are given as mean ± SE; numbers in parentheses indicate the number of myotubes tested. The asterisk indicates a significant difference (p < 0.001). The analysis of activation kinetics excluded cells with very small currents and cells in which tail current decay was obviously not monoexponential.
TABLE 2.
L-type Ca2+ current conductance and charge movement
|
G-V
|
Q-V
|
||||||
|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | V1/2 (mV) | kG (mV) | Qmax (nC/μF) | VQ (mV) | kQ (mV) | Gmax/Q′ (nS/pC) | |
| Uninjected normal | 238 ± 16 (21) | 19.0 ± 1.0 | 4.6 ± 0.3 | 4.8 ± 0.5 (7) | −3.3 ± 0.5 | 9.3 ± 0.8 | 63 |
| Normal + YFP-Rem | 92 ± 10‡ (12) | 24.1 ± 1.2* | 5.3 ± 0.8 | 2.7 ± 0.5† (7) | −3.4 ± 3.4 | 8.6 ± 1.3 | 61 |
| Normal + YFP | 268 ± 39 (6) | 20.3 ± 1.4 | 4.7 ± 0.7 | ND | ND | ||
Data are given as mean ± SE; numbers in parentheses indicate the number of myotubes tested. Three myotubes expressing YFP-Rem had no appreciable L-type current and, for that reason, were not able to be fit by Eq. 2; these cells were included in the calculation of total current density (see Results), but were omitted from the data presented in Table 2. Gmax was calculated according to Eq. 2, whereas the Q′ was derived by subtracting the average maximal charge movement of dysgenic myotubes (Qdys) from the average total Qmax, where Qdys = 1.0 nC/μF (see Bannister and Beam (30)). Symbols indicate significant differences (*p < 0.05; †p < 0.005; ‡p < 10−6) compared to normal myotubes by t-test. I/V and Q/V curves are plotted according to Eqs. 2 and 4, respectively (see Materials and Methods). For all the data given, the calculated average voltage error was <5 mV.
YFP-Rem reduces skeletal muscle charge movement
To determine whether the reduction in L-type current by YFP-Rem was a consequence of a reduced number of functional DHPRs in the plasma membrane, immobilization-resistant charge movements were measured. Charge movements in YFP-Rem-expressing myotubes were reduced by 48% relative to normal, control myotubes (Qmax = 2.5 ± 0.4 nC/μF, n = 7 vs. 4.8 ± 0.5 nC/μF; n = 7, respectively; p = 0.004; Fig. 3, A–C). Under the assumption that these Qmax values include a component unrelated to the DHPR (Qdys = 1.0 nC/μF) ((30) and Bannister and Beam, unpublished observations), the ratio of L-type conductance (Gmax) to charge (Q′ = Qmax − Qdys) was nearly identical in control and YFP-Rem-expressing myotubes (63 vs. 61 nS/pC, respectively; Table 2). Thus, the reduction in L-type current resulting from expression of YFP-Rem can be accounted for by the decrease in functional DHPRs.
DISCUSSION
The aim of this study was to determine the mechanism by which Rem, an RGK family GTP-binding protein, inhibits EC coupling in skeletal muscle. We found that YFP-Rem reduces the fraction of myotubes displaying electrically evoked contractions and attenuates voltage-dependent myoplasmic Ca2+ transients (Figs. 1 A and 2), consistent with the finding of Finlin et al. that virally overexpressed Rem inhibits KCl-induced myoplasmic Ca2+ responses (8). Moreover, we found that this impairment of EC coupling was not a consequence of a reduced SR Ca2+ store or of direct inhibition of RyR1 (Fig. 1, B and C). Instead, it appears that Rem inhibits EC coupling by reducing the number of functional DHPRs, since both L-type Ca2+ currents and membrane charge movements were reduced substantially in myotubes expressing YFP-Rem (Figs. 3 and 4, respectively). This is the first demonstration that skeletal muscle L-type (α1S-mediated) currents can be modulated by an RGK family member. On average, the skeletal muscle L-type channel was not inhibited by Rem to the extent that has been observed for other L-type channels in native and heterologous systems (8,13,14,20,21).
FIGURE 4.

YFP-Rem reduces the number of functional L-type Ca2+ channels (DHPRs) in the plasma membrane. Recordings of immobilization-resistant charge movements elicited by 20-ms depolarizations from −50 mV to −30 mV, −10 mV, +10 mV, and +30 mV are shown for a normal, control myotube (A) or a normal myotube expressing YFP-Rem (B). (C) Comparison of Q-V relationships for normal, control myotubes (•, n = 7) and normal myotubes expressing YFP-Rem (○, n = 7). Charge movements were evoked at 0.1 Hz by test potentials ranging from −40 mV through +60 mV in 10-mV increments, following a prepulse protocol (28). Sigmoidal Q-V curves are plotted according to Eq. 4. The best-fit parameters for each plot are presented in Table 2.
Recent evidence indicates that RGK proteins interact with β subunits via a subdomain of the highly conserved guanylate kinase domain that is distinct from the subdomain that binds the I-II loop of the α1 subunit (14,15). This finding is particularly important when considered in the context of skeletal muscle, because the β1a isoform is required for skeletal-type EC coupling (31–37). The β1-null zebrafish mutant relaxed lacks the characteristic arrays of DHPR tetrads typically observed in freeze-fracture replicas of normal skeletal muscle, indicating that β1a is important for the linkage of DHPRs to RyR1 (37). However, the reduced number of functional DHPRs that are present in YFP-Rem-expressing myotubes appears to be normally linked to RyR1, as indicated both by an unaltered Gmax/Q′ ratio (Table 2) and normal activation kinetics (Table 3), both of which are substantially changed when skeletal DHPRs are not coupled to RyR1 (29,38,39).
Measurement of immobilization-resistant charge movements indicated that YFP-Rem reduced the total number of functional α1S-DHPRs in the plasma membrane (Fig. 3 and Table 2). Our data do not allow us to distinguish whether this reduction is a consequence of decreased channel insertion/increased channel internalization (7,9,10,12,13,15,16) or of the ability of Rem to lock the channel in a nongating state (11,14,19,20). A similar reduction in gating charge movement, associated with α1C-DHPRs, has been observed in ventricular myocytes after viral expression of Gem (17). In contrast to the observed reduction in gating charge after maintained expression of RGK proteins, recent work indicates that acutely applied Rem can inhibit L-type Ca2+ currents via α1C-DHPRs expressed in HEK293 cells, without reducing gating charge movement (40). Possibly, Rem has both acute and chronic effects, such that a rapid inhibition of Ca2+ current is followed by a slower reduction in the number of functional channels in the plasma membrane.
It is clear that an important goal of future studies will be to resolve the mechanisms whereby RGK proteins inhibit voltage-gated Ca2+ channels. Muscle cells would appear to offer several advantages for such studies. For example, because they are concentrated in discrete foci (27,30,41), membrane-associated, fluorescently-tagged L-type channels in myotubes can be identified with less ambiguity than the more diffusely distributed channels typical of cultured neurons or heterologously expressed channels. Thus, in myotubes it should be possible to determine whether YFP-Rem targets to channels already present in the plasma membrane and whether this targeting depends on the isoforms of the α1 and β subunits. Similarly, it will be of interest to determine whether the subcellular targeting of YFP-Rem is altered in myotubes lacking β subunits and whether exogenous expression of Rem alters the tetradic arrangement of DHPRs as seen with freeze-fracture electron microscopy.
In summary, we have shown that the RGK GTP-binding protein Rem inhibits EC coupling in skeletal muscle as a consequence of reducing the number of functional DHPRs. In addition to inhibition of EC coupling, this reduction in functional DHPRs implies that Rem may modulate other physiological processes in muscle cells that are dependent on voltage-driven conformational changes of the DHPR, including excitation-transcription coupling and excitation-coupled Ca2+ entry (42,43).
Acknowledgments
We thank Drs. N. M. Lorenzon, A. M. Payne, and D. C. Sheridan, and Mr. J. D. Ohrtman for insightful discussions.
This work was supported in part by National Institutes of Health grants NS24444 and AR44750 (to K.G.B.). R.A.B. was supported by a developmental grant from the Muscular Dystrophy Association (MDA4155).
Editor: Robert Hsiu-Ping Chow.
References
- 1.Tanabe, T., K. G. Beam, J. A. Powell, and S. Numa. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. [DOI] [PubMed] [Google Scholar]
- 2.Beam, K. G., and P. Horowicz. 2004. Excitation-contraction coupling in skeletal muscle. In Myology. A. G. Engel and C. Franzini-Armstrong, editors. McGraw-Hill, New York. 257–280.
- 3.Armstrong, C. M., F. M. Bezanilla, and P. Horowicz. 1972. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetraacetic acid. Biochim. Biophys. Acta. 267:605–608. [DOI] [PubMed] [Google Scholar]
- 4.Tanabe, T., K. G. Beam, B. A. Adams, T. Niidome, and S. Numa. 1990. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–569. [DOI] [PubMed] [Google Scholar]
- 5.Dirksen, R. T., and K. G. Beam. 1999. Role of calcium permeation in dihydropyridine receptor function. Insights into channel gating and excitation-contraction coupling. J. Gen. Physiol. 114:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paolini, C., J. D. Fessenden, I. N. Pessah, and C. Franzini-Armstrong. 2004. Evidence for conformational coupling between two calcium channels. Proc. Natl. Acad. Sci. USA. 101:12748–12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Béguin, P., K. Nagashima, T. Gonol, T. Shibasaki, K. Takahashi, N. Ozaki, K. Geering, T. Iwanaga, and S. Seino. 2001. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 411:701–706. [DOI] [PubMed] [Google Scholar]
- 8.Finlin, B. S., S. M. Crump, J. Satin, and D. A. Andres. 2003. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc. Natl. Acad. Sci. USA. 100:14469–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Béguin, P., R. N. Mahalakshmi, K. Nagashima, D. H. K. Cher, N. Kuwamura, Y. Yamada, Y. Seino, and W. Hunziker. 2005. Roles of 14-3-3 and calmodulin binding in subcellular localization and function of the small G-protein Rem2. Biochem. J. 390:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Béguin, P., R. N. Mahalakshmi, K. Nagashima, D. H. K. Cher, A. Takahashi, Y. Yamada, Y. Seino, and W. Hunziker. 2005. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J. Cell Sci. 118:1923–1934. [DOI] [PubMed] [Google Scholar]
- 11.Finlin, B. S., S. M. Crump, R. N. Correll, S. Özcan, J. Satin, and D. A. Andres. 2005. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J. Biol. Chem. 280:41864–41871. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki, T., T. Shibasaki, P. Béguin, K. Nagashima, M. Miyazaki, and S. Seino. 2005. Direct inhibition of the interaction between α-interaction domain and β-interaction domain of voltage-dependent Ca2+ channels by Gem. J. Biol. Chem. 280:9308–9312. [DOI] [PubMed] [Google Scholar]
- 13.Béguin, P., R. N. Mahalakshmi, K. Nagashima, D. H. K. Cher, H. Ikeda, Y. Yamada, Y. Seino, and W. Hunziker. 2006. Nuclear sequestration of β-subuits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J. Mol. Biol. 355:34–46. [DOI] [PubMed] [Google Scholar]
- 14.Finlin, B. S., R. N. Correll, C. Pang, S. M. Crump, J. Satin, and D. A. Andres. 2006. Analysis of the complex between Ca2+ channel β-subunit and the Rem GTPases. J. Biol. Chem. 281:23557–23566. [DOI] [PubMed] [Google Scholar]
- 15.Béguin, P., Y. J. Ng, C. Krause, R. N. Mahalakshmi, M. Y. Ng, and W. Hunziker. 2007. RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+ channel β-subunits via an uncommon effector-binding domain. J. Biol. Chem. 282:11509–11520. [DOI] [PubMed] [Google Scholar]
- 16.Yada, H., M. Murata, K. Shimoda, S. Yuasa, H. Kawaguchi, M. Ieda, T. Adachi, S. Ogawa, and K. Fukuda. 2007. Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L-type Ca2+ channels in the heart. Circ. Res. 101:69–77. [DOI] [PubMed] [Google Scholar]
- 17.Murata, M., E. Cingolani, A. D. McDonald, K. Donahue, and E. Marbán. 2004. Creation of a genetic calcium channel blocker by targeted Gem gene transfer in the heart. Circ. Res. 95:398–405. [DOI] [PubMed] [Google Scholar]
- 18.Ward, Y., B. Spinelli, M. J. Quon, H. Chen, S. R. Ikeda, and K. Kelly. 2004. Phosporylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol. Cell. Biol. 24:651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen, H., H. L. Puhl, S.-L. Niu, D. C. Mitchell, and S. R. Ikeda. 2005. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J. Neurosci. 25:9762–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crump, S. M., R. N. Correll, E. A. Schroder, W. C. Lester, B. S. Finlin, J. Satin, and D. A. Andres. 2006. L-type calcium channel α-subunit and protein kinase inhibitors modulate Rem-mediated regulation of current. Am. J. Physiol. 291:H1959–H1971. [DOI] [PubMed] [Google Scholar]
- 21.Seu, L., and G. S. Pitt. 2006. Dose-dependent and isoform-specific modulation of Ca2+ channels by RGK GTPases. J. Gen. Physiol. 128:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan, J. Y., W. E. Fieles, A. M. White, M. M. Egerton, and D. S. Silberstein. 2000. Ges, a human GTPase of the Rad/Gem/Kir family, promotes endothelial cell sprouting and cytoskeletal reorganization. J. Cell Biol. 149:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leone, A., N. Mitsiades, Y. Ward, B. Spinelli, V. Poulaki, M. Tsokos, and K. Kelly. 2001. The Gem GTP-binding protein promotes morphological differentiation in neuroblastoma. Oncogene. 20:3217–3225. [DOI] [PubMed] [Google Scholar]
- 24.Piddini, E., J. A. Schmid, R. de Martin, and C. G. Dotti. 2001. The Ras-like GTPase Gem is involved in cell shape remodeling and interacts with the novel kinesin-like protein KIF9. EMBO J. 20:4076–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward, Y., S. F. Yap, V. Ravichandran, F. Matsumura, M. Ito, B. Spinelli, and K. Kelly. 2002. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 157:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beam, K. G., and C. Franzini-Armstrong. 1997. Functional and structural approaches to the study of excitation-contraction coupling. Methods Cell Biol. 52:283–306. [DOI] [PubMed] [Google Scholar]
- 27.Papadopoulos, S., V. Leuranguer, R. A. Bannister, and K. G. Beam. 2004. Mapping sites of potential proximity between the DHPR and RyR1 in muscle using a CFP-YFP tandem as a FRET probe. J. Biol. Chem. 279:44046–44056. [DOI] [PubMed] [Google Scholar]
- 28.Adams, B. A., T. Tanabe, A. Mikami, S. Numa, and K. G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. [DOI] [PubMed] [Google Scholar]
- 29.Avila, G., and R. T. Dirksen. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca2+ channel. J. Gen. Physiol. 115:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bannister, R. A., and K. G. Beam. 2005. The α1S N-terminus is not essential for bi-directional coupling with RyR1. Biochem. Biophys. Res. Commun. 336:134–141. [DOI] [PubMed] [Google Scholar]
- 31.Gregg, R. G., A. Messing, C. Strube, M. Beurg, R. Moss, M. Behan, C. Sukhareva, S. Haynes, J. A. Powell, and R. Coronado. 1996. Absence of the β1a subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 93:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strube, C., M. Beurg, C. Sukhareva, C. A. Ahern, J. A. Powell, P. A. Powers, R. G. Gregg, and R. Coronado. 1996. Reduced Ca2+ current, charge movement and absence of Ca2+ transients in skeletal muscle deficient in dihydorpyridine receptor β1 subunit. Biophys. J. 75:2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beurg, M., C. A. Ahern, P. Vallejo, M. W. Conklin, P. A. Powers, R. G. Gregg, and R. Coronado. 1999. Involvement of the carboxy-terminus region of the dihydropyridine receptor β1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 77:2953–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheridan, D. C., L. Carbonneau, C. A. Ahern, P. Nataraj, and R. Coronado. 2003. Ca2+-dependent excitation-contraction coupling triggered by the heterologous cardiac/brain β2a-subunit in skeletal muscle. Biophys. J. 85:3739–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheridan, D. C., W. Cheng, C. A. Ahern, L. Mortensen, D. Alsammarae, P. Vallejo, and R. Coronado. 2003. Truncation of the carboxyl terminus of the dihydropyridine receptor β1a subunit promotes Ca2+ dependent excitation-contraction coupling in skeletal myotubes. Biophys. J. 84:220–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheridan, D. C., W. Cheng, L. Carbonneau, C. A. Ahern, and R. Coronado. 2004. Involvement of a heptad repeat in the carboxyl terminus of the dihydropyridine receptor β1a subunit in the mechanism of excitation-contraction coupling in skeletal muscle. Biophys. J. 87:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schredelseker, J., V. Di Biase, G. J. Obermaier, E. T. Felder, B. E. Flucher, C. Franzini-Armstrong, and M. Grabner. 2005. The β1a subunit is essential for the assembly of dihydropyridine-receptor arrays in skeletal muscle tetrad formation. Proc. Natl. Acad. Sci. USA. 102:17219–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakai, J., R. T. Dirksen, H. T. Nguyen, I. N. Pessah, K. G. Beam, and P. D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- 39.Sheridan, D. C., H. Takekura, C. Franzini-Armstrong, K. G. Beam, P. D. Allen, and C. F. Perez. 2006. Bi-directional signaling between calcium channels of skeletal muscle requires, multiple, direct and indirect interactions. Proc. Natl. Acad. Sci. USA. 103:19760–19765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suhail, Y., and H. M. Colecraft. 2007. GEMIICCs: genetically encoded molecules for inducibly inactivating CaV channels. Biophys. J., 92:354a. (Abstr.) [DOI] [PubMed] [Google Scholar]
- 41.Grabner, M., R. T. Dirksen, and K. G. Beam. 1998. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc. Natl. Acad. Sci. USA. 95:1903–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherednichenko, G., A. M. Hurne, J. D. Fessenden, E. H. Lee, P. D. Allen, K. G. Beam, and I. N. Pessah. 2004. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA. 101:15793–15798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurne, A. M., J. J. O'Brien, D. Wingrove, G. Cherednichenko, P. D. Allen, K. G. Beam, and I. N. Pessah. 2005. Ryanodine receptor type I (RyR1) mutations C4958S and C4961S reveal excitation-coupled calcium entry (ECCE) is independent of sarcoplasmic reticulum store depletion. J. Biol. Chem. 280:36994–37004. [DOI] [PubMed] [Google Scholar]