

Abstract
Interactions governing protein folding, stability, recognition, and activity are mediated by hydration. Here, we use small-angle neutron scattering coupled with osmotic stress to investigate the hydration of two proteins, lysozyme and guanylate kinase (GK), in the presence of solutes. By taking advantage of the neutron contrast variation that occurs upon addition of these solutes, the number of protein-associated (solute-excluded) water molecules can be estimated from changes in both the zero-angle scattering intensity and the radius of gyration. Poly(ethylene glycol) exclusion varies with molecular weight. This sensitivity can be exploited to probe structural features such as the large internal GK cavity. For GK, small-angle neutron scattering is complemented by isothermal titration calorimetry with osmotic stress to also measure hydration changes accompanying ligand binding. These results provide a framework for studying other biomolecular systems and assemblies using neutron scattering together with osmotic stress.
INTRODUCTION
It is widely acknowledged that the structure and dynamics of proteins are strongly influenced by their interactions with water (1–3). Measurements of the number of water molecules associated with proteins, and particularly of changes in hydration coupled to conformational transitions and binding reactions, are fundamental for furthering our understanding of the role of water. The definition of hydration water typically depends on the nature of the technique probing water structure (4,5). Here, we focus on protein-associated water that excludes small solutes either due to a preferential hydration of exposed protein surface or to a steric exclusion from internal cavities (6–16). The exclusion of solutes from water hydrating biomolecular surfaces or from water sequestered in cavities substantially affects stability, conformational changes, and binding reactions. Systematic measurement of the dependence of exclusion on solute chemistry and size can provide a better understanding of the interactions underlying exclusion and the role of water. The osmotic stress method is a particularly useful approach for this, since it encompasses the many possible effects of an added solute while also allowing the measurement of forces between biopolymers and hydration changes accompanying biomolecular reactions (17,18). Solute exclusion is due to the size and chemical nature of the solute together with the chemical nature of the biomolecular surface under investigation, and the solute thereby acts osmotically on these regions of “excess” water. Steric exclusion can be exploited to probe protein pores and cavities (12–16) as well as to measure forces between macromolecules in liquid crystalline arrays (19).
Protein hydration as defined by solute exclusion can be investigated using a number of techniques, including light, x-ray, and neutron scattering, as well as sedimentation equilibrium (20,21), utilizing the difference in properties of water and solute/water mixtures. Small-angle neutron scattering (SANS) is particularly well suited for examining water that is associated with protein and that excludes small solutes. Three regions of scattering contrast are created with added solute: the protein, the protein-associated water, and the bulk water/solute solution. The layer of excess water surrounding the protein that excludes solutes has a different scattering length density from both the protein and the bulk solution that contains solute. The scattering intensity of the protein therefore contains contributions from both the protein itself and its preferential hydration layer. In experimental terms, SANS measures the dry protein volume, or that inaccessible to the solvent, when in H2O, D2O, and mixtures of the two (22). The protein-associated water volume determined in solute/D2O mixtures is then the volume inaccessible to the solute but accessible by water. Stuhrmann et al. (23) determined the average solvation of apoferritin created by a variety of added small organic molecules from the zero-angle neutron scattering intensity, I(0). The excess contrast variation from these solutes compared to H2O/D2O contrast variation was attributed to restricted solute penetration relative to water, i.e., solutes are excluded from the protein surface relative to water. A similar preferential hydration of RNase in the presence of ethanol and glycerol was also measured with neutron scattering (24).
We investigate here the exclusion of polyethylene glycols (PEGs) varying in molecular mass between 150 and 1000 g/mol (Da) from two proteins, lysozyme and guanylate kinase (GK). These two proteins are similar in size. Whereas lysozyme is compact, however, GK has a large internal water-filled cavity that closes in steps as the ligands GMP and ATP/ADP sequentially bind. Both changes in the zero angle scattering intensity, I(0), and the apparent radius of gyration, Rg, with increasing solute concentration are used to gain information about the number of excess, solute-excluding water molecules associated with these proteins. I(0) is sensitive to the number of excess waters, whereas Rg depends on both the number and location of these waters. These SANS experiments illustrate how such structural differences between the two proteins can be discerned based on their measured solute exclusion.
To ensure that the structure of GK does not change significantly due to the osmotic pressure of excluded solute acting on the water-filled ligand binding cavities, we also probe the dependence of GK ligand binding on osmotic stress using isothermal titration calorimetry (ITC). The ITC measurements allow a more clear interpretation of the SANS results by ruling out any large-scale conformational changes due to solute osmotic stress. Furthermore, the difference in the number of water molecules between the apoenzyme and the ligand-bound forms of GK as measured by the SANS and ITC techniques are in reasonable agreement. This demonstrates the benefit of complementing SANS with other techniques. Overall, the combination of neutron scattering and osmotic stress can lead to new insights into the relationship among protein structure, flexibility, energetics, and function.
In addition, the ITC measurements indicate that the traditional crowding theory, based on hard-sphere interactions, to account for solute exclusion (25,26) is not consistent with the observed effect of PEGs on the thermodynamics of ligand binding. Interactions other than simple entropic hard-sphere steric forces govern solute preferential exclusion and, equivalently, biomolecular preferential hydration. This has also been demonstrated by the exclusion of alcohols from DNA and salts and polar solutes from hydroxypropyl cellulose, where water-structuring forces, not simple sterics, are consistent with the observed behavior (27–30). Likewise, ITC measurements of adamantane binding to cyclodextrin in the presence of several salts and polar solutes show that the entropic contribution to binding is small or, at best, comparable to the enthalpy for all solutes studied (31). Since steric exclusion or “crowding” is strictly entropic, these results indicate that other interactions are significant. The ITC results presented here for protein ligand binding also display enthalpy-entropy compensation for the exclusion of PEG.
EXPERIMENTAL PROCEDURES
Brand names are cited for clarity and do not imply endorsement by the National Institute of Standards and Technology or the National Institutes of Health, nor do they imply that the items are the best choices for their intended uses.
Materials
The nucleotides GMP, ADP, and ATP, coupling enzymes pyruvate kinase and lactic dehydrogenase from rabbit muscle, phosph(enol)pyruvate, and NADH were obtained from Sigma (St. Louis, MO). PEG of molecular mass 400 Da (purum), 1000 Da (BioChemika Ultra), triethylene glycol (puriss.), and betaine monohydrate (BioChemika) were from Fluka (Buchs, Switzerland), and glycerol was from Invitrogen (Carlsbad, CA). The D2O (99.9% D) used for neutron experiments was from Cambridge Isotope Laboratories (Andover, MA). Lysozyme from chicken egg white (3× crystallized, dialyzed, and lyophilized) was purchased from Sigma. All chemicals were used without further purification.
Protein expression, purification, and activity
Protein expression and purification of yeast guanylate kinase (Mr = 20,548 Da) were performed similarly to previously described procedures (32) using the pET17b-YGK plasmid kindly provided to us by Dr. Honggao Yan (Michigan State University, E. Lansing, MI). After expression, cells were lysed using CelLytic B-II bacterial cell lysis/extraction reagent (Sigma) containing, in addition, 1 mM dithiothreitol, 1 mM phenylmethanesulfonyl fluoride, and 500 units DNase, and then homogenized using a tissue grinder. The cell solution was centrifuged (15,000 × g) and the soluble protein fraction recovered. After dialysis (10,000 molecular weight cutoff (MWCO)) into 30 mM Tris-Cl (pH 7.5), 1 mM Na2EDTA (30-1 TE), 1 mM phenylmethanesulfonyl fluoride, 0.1 mM NaN3, the solution was filtered (YM100 Centricon, Amicon, Houston, TX) to remove higher-molecular-weight components. The filtrate was loaded onto Affi-Gel Blue affinity columns (Bio-Rad, Hercules, CA) and eluted with 5 mM GMP, 0.1 M KCl in 30-1 TE. GK fractions were collected, concentrated (YM10 Centriprep, Amicon) and dialyzed (10 kDa MWCO) exhaustively into 10 mM Tris-Cl (pH 7.5) or 10 mM HEPES (pH 7.5) before lyophilization. The purity of GK was determined by SDS-PAGE (see Fig. 1), and the specific activity measured by a coupled enzyme assay (32,33) was ∼370 units/mg, with average kcat = 126 s−1 and Ki,GMP = 0.039 mM.
FIGURE 1.

SDS-PAGE of E. coli expressed and purified yeast guanylate kinase (lane 1) and a protein standards ladder (lane 2). Gels were stained with SYPRO Orange.
Small-angle neutron scattering
Small-angle neutron scattering experiments were performed on the NG3 and NG7 30 m SANS instruments at the National Institute of Standards and Technology (NIST) Center for Neutron Research (NCNR) (34). Neutrons of wavelength λ = 5.5 Å with a wavelength spread of Δλ/λ = 0.11 were used. Measurements were made with a 1.5-m sample-to-detector distance and a 20-cm detector offset, with some sample measurements also made at 5.0 m to obtain data at lower wavevector transfer, 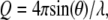 where 2θ is the scattering angle. All measurements in H2O were made with the 5.0 m configuration only. After brief centrifugation, samples were loaded into banjo-shaped quartz cuvettes (Hellma USA, Plainville, NY) of 1- or 2-mm pathlength (for H2O or D2O buffer, respectively). Neutron exposure time was 1 h. Scattered neutrons were detected with a 64 cm × 64 cm two-dimensional position-sensitive detector with 128 × 128 pixels. Data reduction was accomplished using Igor Pro software (WaveMetrics, Lake Oswego, OR) with SANS macros developed at the NCNR (35). The total two-dimensional scattering was corrected for the scattering from the quartz cell and from stray neutrons in the room and for nonuniform detector response. Then, the scattering was normalized by the incident beam flux and radially averaged to obtain absolute scale intensity, I(Q) versus Q. Background scattering from the buffers and solutes, Ibg(Q), and the Q-independent excess incoherent scattering, Iinc(0), were subtracted from the total scattering, Itot(Q), to obtain the protein sample scattering,
where 2θ is the scattering angle. All measurements in H2O were made with the 5.0 m configuration only. After brief centrifugation, samples were loaded into banjo-shaped quartz cuvettes (Hellma USA, Plainville, NY) of 1- or 2-mm pathlength (for H2O or D2O buffer, respectively). Neutron exposure time was 1 h. Scattered neutrons were detected with a 64 cm × 64 cm two-dimensional position-sensitive detector with 128 × 128 pixels. Data reduction was accomplished using Igor Pro software (WaveMetrics, Lake Oswego, OR) with SANS macros developed at the NCNR (35). The total two-dimensional scattering was corrected for the scattering from the quartz cell and from stray neutrons in the room and for nonuniform detector response. Then, the scattering was normalized by the incident beam flux and radially averaged to obtain absolute scale intensity, I(Q) versus Q. Background scattering from the buffers and solutes, Ibg(Q), and the Q-independent excess incoherent scattering, Iinc(0), were subtracted from the total scattering, Itot(Q), to obtain the protein sample scattering, 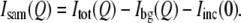 A linear interpolation of the collected Ibg(Q) profiles for each solute was performed to obtain exactly the same solute concentration as in each protein sample. Iinc(0) was determined from Q > 0.35 Å−1, assuming that the residual difference in intensity is completely incoherent scattering. The protein radius of gyration, Rg, was calculated from the neutron scattering at low Q using Guinier plots (36). Computed SANS profiles from protein crystal structures were performed using the program Xtal2sas (37) and references therein.
A linear interpolation of the collected Ibg(Q) profiles for each solute was performed to obtain exactly the same solute concentration as in each protein sample. Iinc(0) was determined from Q > 0.35 Å−1, assuming that the residual difference in intensity is completely incoherent scattering. The protein radius of gyration, Rg, was calculated from the neutron scattering at low Q using Guinier plots (36). Computed SANS profiles from protein crystal structures were performed using the program Xtal2sas (37) and references therein.
GK (4–6 mg/mL) in D2O buffer of either 20 mM Tris-Cl (pH measured, pHm, 7.7), 0.1 M KCl, or 20 mM HEPES (pHm 7.6), 0.1 M K+ ion (using KCl) were used. GK (10 mg/mL) in H2O buffer of 20 mM Tris-Cl (pH 7.5), 0.1 M KCl was used. Binary complexes were formed with 5 mM GMP and ternary complexes had, in addition, 5 mM ADP with 6 mM MgCl2. Measurements of lysozyme (5.5 mg/mL) were in 20 mM potassium acetate (pHm 5.0), 50 mM KCl, and D2O buffer. Protein concentration was determined spectrophotometrically by 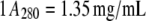 for GK (32) and
for GK (32) and 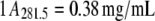 for lysozyme, for a 1-cm pathlength in both cases. All neutron scattering experiments were conducted at 22°C.
for lysozyme, for a 1-cm pathlength in both cases. All neutron scattering experiments were conducted at 22°C.
Fitting procedures for the SANS data are described in the Results section. The scattering-length density of H2O/D2O and solute/D2O mixtures, ρ0, were calculated from the volume fraction and individual scattering-length densities of these components using ρH2O = −0.562 × 10−6 Å−2, ρD2O = 6.404 × 10−6 Å−2, and the pure solute scattering-length densities, ρs, for glycine betaine (8.06 × 10−7 Å−2), glycerol (4.87 × 10−7 Å−2), triethylene glycol (TEG) (4.84 × 10−7 Å−2), PEG 400 (5.82 × 10−7 Å−2), and PEG 1000 (6.55 × 10−7 Å−2). The NIST scattering length density calculator (38) was used to calculate ρs. Solute concentrations are given either as volume fraction, fv, or vol %. The scattering length density of the bulk solution containing solute in D2O is given by 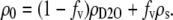
Microcalorimetry
ITC measurements were performed using a VP-ITC microcalorimeter (MicroCal, Northampton, MA). For the GMP binding experiments, 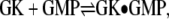 GMP (6 mM) was titrated into GK (25–50 μM) at 30°C using a 12-μcal/s reference power and 310-rpm stir rate. The injection sequence (number, volume) 1, 2 μl; 14, 3 μl; 13, 5 μl; 12, 8 μl was used, for a total of 40 injections, with a 0.5-μl/s injection rate, 2-s filter, and 240-s spacing between injections. GK and GMP were prepared in 20 mM HEPES (pH 7.5), 0.5 mM Na2EDTA, and 0.1 M K+ (using KCl). GK was dialyzed (10 kDa MWCO) into the ITC buffer. Solutes were added to both protein and ligand solutions at the same concentration (v/v), with protein and ligand concentrations remaining fixed per total volume. GMP titrations into buffer at each solute concentration were used for background subtraction.
GMP (6 mM) was titrated into GK (25–50 μM) at 30°C using a 12-μcal/s reference power and 310-rpm stir rate. The injection sequence (number, volume) 1, 2 μl; 14, 3 μl; 13, 5 μl; 12, 8 μl was used, for a total of 40 injections, with a 0.5-μl/s injection rate, 2-s filter, and 240-s spacing between injections. GK and GMP were prepared in 20 mM HEPES (pH 7.5), 0.5 mM Na2EDTA, and 0.1 M K+ (using KCl). GK was dialyzed (10 kDa MWCO) into the ITC buffer. Solutes were added to both protein and ligand solutions at the same concentration (v/v), with protein and ligand concentrations remaining fixed per total volume. GMP titrations into buffer at each solute concentration were used for background subtraction.
The ADP binding experiments, 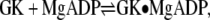 were performed similarly, but with 3–6 mM ADP as the titrant, 7.5 mM MgCl2 in both the protein and ligand solutions, and the reference power set to 15 μcal/s. ADP binding experiments also were performed with saturated GMP (1.325 mM) in both the protein and ligand solutions,
were performed similarly, but with 3–6 mM ADP as the titrant, 7.5 mM MgCl2 in both the protein and ligand solutions, and the reference power set to 15 μcal/s. ADP binding experiments also were performed with saturated GMP (1.325 mM) in both the protein and ligand solutions, 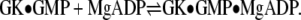 Appropriate titrations without protein were used for background subtraction.
Appropriate titrations without protein were used for background subtraction.
Binding isotherms were fit to a single-site binding model that corrected for the background heat of ligand dilution and the relative enzyme activity between different protein expression batches. Two linear fits across each background run (see Fig. 10) were used to approximate the background within the fitting procedure. Fits were made using the Igor Pro software program with a modified version of the fit equation used by the MicroCal Origin software package (OriginLab, Northampton, MA) to include the background and relative enzyme activity. The binding enthalpy, ΔH, and equilibrium constant, Keq, were directly obtained from the fits for each solute concentration measured. Solute osmolalities were measured using a VAPRO model 5520 vapor pressure osmometer (Wescor, Logan, UT).
FIGURE 10.

Calorimetric heat of GMP binding versus [GMP]/[GK] ratio, both without solute and with 20% PEG 400. Corresponding background titrations of GMP into buffer also are shown (open symbols).
RESULTS
Lysozyme hydration in the presence of solutes
SANS profiles of lysozyme with no solute and with PEG 400 (fv = 0.08) are shown in Fig. 2. The scattering contribution from the bulk solution has been subtracted as described in the Experimental Procedures. The scattering intensity is also normalized by I(0) for comparison. In the presence of 8% PEG 400, the protein appears smaller; the Rg determined from the slope at small Q values decreases from 13.3 ± 0.1 Å to 12.7 ± 0.1 Å, and there is more scattering intensity at higher Q. This can be attributed either to a conformational change in the protein or to a solute-excluded hydration layer around the protein that has a contrast relative to the bulk solution, ρw − ρ0, that is opposite in sign from the protein, ρp − ρ0. All of the protein-solute solutions show similar trends that become more pronounced with increasing solute concentration. This is illustrated in Fig. 3, where the Rg and zero-angle neutron scattering intensity ratio (solute/no solute), 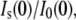 of lysozyme with glycine betaine (A), TEG (equivalent to PEG 150) (B), PEG 400 (C), and PEG 1000 (D) are given. The error in measured Rg increases with increasing solute concentration, since the addition of solute reduces the contrast between protein and solution and also increases the incoherent background scattering that arises mainly due to the solute protons.
of lysozyme with glycine betaine (A), TEG (equivalent to PEG 150) (B), PEG 400 (C), and PEG 1000 (D) are given. The error in measured Rg increases with increasing solute concentration, since the addition of solute reduces the contrast between protein and solution and also increases the incoherent background scattering that arises mainly due to the solute protons.
FIGURE 2.

SANS profiles of lysozyme at 5.5 mg/mL in D2O with no solute and in the presence of 8% PEG 400. For easier comparison, the SANS intensity is normalized by the scattering at zero angle, I(Q)/I(0). The protein appears smaller with added solute; Guinier fits to these curves yield radii of gyration Rg = 13.3 ± 0.1 Å and 12.7 ± 0.1 Å for no solute and 8% PEG 400, respectively.
FIGURE 3.

Lysozyme Rg determined by SANS and the zero-angle neutron scattering intensity ratio with and without solute, 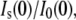 both as a function of solute volume fraction, fv, for (A) glycine betaine, (B) TEG, (C) PEG of molecular weight 400, and (D) PEG 1000. Solid lines are fits made to Rg with a composite model (Eqs. 3 and 4) to calculate the amount of protein-associated water, Nw, and dashed lines show the expected intensity ratio dependence on fv based on the Rg fits.
both as a function of solute volume fraction, fv, for (A) glycine betaine, (B) TEG, (C) PEG of molecular weight 400, and (D) PEG 1000. Solid lines are fits made to Rg with a composite model (Eqs. 3 and 4) to calculate the amount of protein-associated water, Nw, and dashed lines show the expected intensity ratio dependence on fv based on the Rg fits.
The ratio of zero angle intensities with and without added solute, 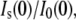 decreases with added solute mostly due to the reduced contrast between the protein and bulk solution. However, protein-associated water that excludes solute creates an additional contribution so that the protein and this water can be treated as a composite particle. The composite Is(0) can be expressed
decreases with added solute mostly due to the reduced contrast between the protein and bulk solution. However, protein-associated water that excludes solute creates an additional contribution so that the protein and this water can be treated as a composite particle. The composite Is(0) can be expressed
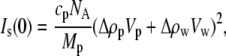 |
(1) |
where cp is the protein concentration, NA is Avogadro's number, Mp is the protein molecular weight, Δρp (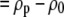 ) is the contrast between protein and bulk solution, Δρw (
) is the contrast between protein and bulk solution, Δρw (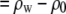 ) is the contrast between protein-associated water (D2O) and bulk solution, and Vp and Vw are the molecular volumes of the protein and protein-associated water, respectively. For fitting, we consider the intensity ratio as a function of fv
) is the contrast between protein-associated water (D2O) and bulk solution, and Vp and Vw are the molecular volumes of the protein and protein-associated water, respectively. For fitting, we consider the intensity ratio as a function of fv
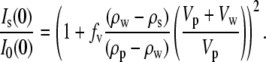 |
(2) |
Likewise, a composite Rg can be defined
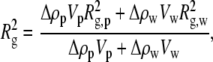 |
(3) |
where Rg,p and Rg,w are the individual radii of gyration of the protein and associated water, respectively, calculated relative to their common center of scattering. Both Eqs. 2 and 3 can be used to fit a series of data with varying solute fv assuming that Vw is a constant across the entire solute concentration range. This is generally found to be true (9,28,31), although excessively high solute concentrations could result in solute penetration and a reduced Vw.
Initially, the lysozyme 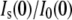 data was fit using Eq. 2, but Vw calculated for all solutes was small relative to Vp and therefore difficult to discern within the error of the fits. To overcome this, we chose to fit Rg to a composite model (Fig. 3, solid lines) since Rg is not dependent on cp and only has a square-root dependence on contrast, compared to the square dependence by I(0). This offers greater sensitivity than I(0), since the errors associated with these parameters are minimized. However, interpretation of a composite Rg requires a geometrical model, so we approximate lysozyme as a sphere with radius rp having a spherical shell of associated water with thickness t. The radius of the composite sphere is then
data was fit using Eq. 2, but Vw calculated for all solutes was small relative to Vp and therefore difficult to discern within the error of the fits. To overcome this, we chose to fit Rg to a composite model (Fig. 3, solid lines) since Rg is not dependent on cp and only has a square-root dependence on contrast, compared to the square dependence by I(0). This offers greater sensitivity than I(0), since the errors associated with these parameters are minimized. However, interpretation of a composite Rg requires a geometrical model, so we approximate lysozyme as a sphere with radius rp having a spherical shell of associated water with thickness t. The radius of the composite sphere is then 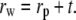 This allows the following substitutions into Eq. 3:
This allows the following substitutions into Eq. 3:
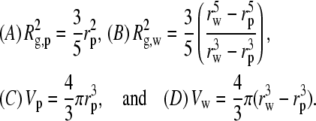 |
(4) |
The spherical core-shell model is reasonable, since hen egg lysozyme is a monomer and lacks large cavities. Also, we find that the SANS from lysozyme without solute can be fit to a spherical model shape up to about Q = 0.2 Å−1 (not shown) while also yielding the correct Rg calculated using Eq. 4 A. Using the average Rg calculated from three SANS runs of lysozyme without solute, the fixed parameter rp = 16.974 Å was determined. Lysozyme ρp = 2.854 × 10−6 Å−2 (4) was calculated from the crystal structure coordinates (6LYZ). Also, since the protein-solute solutions were prepared volumetrically, the water volume around the protein that excludes solute necessarily increases the actual solute concentration in the bulk solution. To account for this, several iterations between Vw and fv, and therefore ρ0, were performed within the fitting routine to obtain the corrected solute volume fraction in the bulk solution,
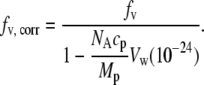 |
(5) |
In all cases, convergence was reached with three iterations or less.
The effective water shell thickness, t, and number of associated water molecules, Nw, per lysozyme molecule determined by the Rg fits are given in Table 1. Nw is calculated from Vw by assuming a standard water molecular volume, νw = 30 Å3. Previously determined Nw values by densitometry (39) are also given in the table for comparison. Upon fitting Rg versus solute fv, the intensity ratio can be calculated:
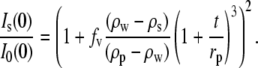 |
(6) |
As seen in Fig. 3 (dashed lines), these calculated ratios for each solute are in good agreement with the experimentally measured values at low fv, with some deviations seen at higher solute fv. This may result from the increased error in determining Rg and 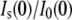 at higher fv or the inadequacy of the shell model to describe lysozyme accurately. It is also possible that the solute begins to penetrate into the protein preferential hydration layer at higher fv, such that Nw decreases with fv.
at higher fv or the inadequacy of the shell model to describe lysozyme accurately. It is also possible that the solute begins to penetrate into the protein preferential hydration layer at higher fv, such that Nw decreases with fv.
TABLE 1.
Lysozyme hydration calculated from Rg fits
| Solute | t (Å) | Nw | Nw densitometry* |
|---|---|---|---|
| Glycine betaine | 0.67 ± 0.04 | 84 ± 5 | — |
| TEG | 0.90 ± 0.18 | 114 ± 24 | 110 ± 100† |
| PEG 400 | 1.21 ± 0.06 | 156 ± 8 | 204 ± 153 |
| PEG 1000 | 2.51 ± 0.07 | 347 ± 11 | 657 ± 95 |
Densitometry data from Bhat and Timasheff (39).
PEG 200.
GK conformational changes with ligands
Neutron scattering profiles for the apoenzyme and ligand-bound states of purified yeast GK expressed in E. coli were measured in D2O buffer (Fig. 4 A). GK Rg decreases from 17.2 ± 0.1 Å to 16.4 ± 0.1 Å upon binding GMP. The ATP-binding site also can be closed by addition of MgADP, resulting in Rg = 16.2 ± 0.1 Å. Measurements in H2O were in good agreement with samples in D2O (Table 2), indicating that there are no significant isotopic effects on protein structure. Considering the size of GK, these are large conformational changes that occur upon ligand binding. Small-angle scattering measurements of other kinases, including hexokinase (40), phosphoglycerate kinase (41), and creatine kinase (42), have revealed similar ligand-induced, large-scale structural changes.
FIGURE 4.

(A) Experimental SANS from ligand-free GK, the enzyme complex with GMP, and the ternary complex GK/GMP/ADP at 5 mg/mL in D2O (data points) and SANS profiles calculated from the corresponding crystal structures using Xtal2sas (solid lines). Curves are shifted for clarity. (B) The corresponding guanylate kinase crystal structures: 1, GK, 1EX6; 2, GK/GMP, 1EX7; and 3, GK/GMP/MgADP, 1LVG.
TABLE 2.
Guanylate kinase radius of gyration from SANS
| Experimental Rg (Å) | Calculated Rg (Å)
|
Reduced χ2 | ||
|---|---|---|---|---|
| Sample | Xtal2sas | Guinier | ||
| GK, D2O | 17.2 ± 0.1 | 16.7 | 16.8 | 4.7 |
| GK/GMP, D2O | 16.4 ± 0.1 | 16.2 | 16.4 | 2.0 |
| GK/GMP/MgADP, D2O | 16.2 ± 0.1 | 15.8 | 16.0 | 1.5 |
| GK, H2O | 18.1 ± 1.0 | — | — | — |
| GK/GMP, H2O | 16.6 ± 1.1 | — | — | — |
The neutron data for GK are compared to predicted SANS curves calculated from the Brookhaven Protein Data Bank crystal structure coordinates for GK (apo, yeast: 1EX6; GMP, yeast: 1EX7 (43); and GMP/MgADP, mouse: 1LVG (44)) using the Xtal2sas program (see Fig. 4 A). It should be noted that the ternary complex (GK/GMP/MgADP) crystal structure is for mouse GK, but its overall fold is very similar to yeast GK (44) (Fig. 4 B). The crystal structure for yeast GK (molecular mass 20.5 kDa) contains all 186 residues, whereas mouse GK (molecular mass 21.9 kDa) contains 190 of 198 residues. In all three cases, good agreement between the static crystal structure predictions and the experimental data from protein in solution were observed. Applying smearing to the crystal structure calculations for SANS, which accounts for instrument resolution effects, did not affect the model SANS curves. The best fit to the experimental data was obtained for the model SANS curve of the ternary complex GK/GMP/ADP, with a reduced χ2 value of 1.5, whereas reduced χ2 values of the GK/GMP complex and the GK apoenzyme were 2.0 and 4.7, respectively. Also, the calculated Rg values from the crystal structures, performed using the Xtal2sas program, show good agreement with the experimentally determined values but are always smaller (see Table 2). Both the Rg value obtained from Xtal2sas directly from the model structure and the Guinier Rg value obtained from the model SANS curve are given in Table 2. The large change in Rg with ligand binding is due to the closure of the large water-filled binding pocket of the apoenzyme. The χ2 and Rg comparisons may be indicative of the relative flexibility of GK in solution and the reduction in conformational space available to the protein as ligand-binding sites are filled.
GK hydration in the presence of solutes
We also measured SANS of GK in the presence of neutral solutes. Fig. 5 shows SANS profiles of GK without added solute and with PEG 400 (fv = 0.08). The scattering intensity is normalized by I(0) for comparison. Similar to lysozyme, GK appears smaller in 8% PEG 400 with Rg reduced from 17.0 ± 0.1 Å to 16.0 ± 0.2 Å. Fig. 6 shows the dependence of 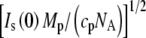 on the bulk solution scattering-length density, ρ0, for H2O/D2O mixtures and for solute/D2O solutions. The difference in slopes indicates a scattering contribution in addition to the contrast difference between protein and bulk solution. We attribute this to protein-associated water acting through the
on the bulk solution scattering-length density, ρ0, for H2O/D2O mixtures and for solute/D2O solutions. The difference in slopes indicates a scattering contribution in addition to the contrast difference between protein and bulk solution. We attribute this to protein-associated water acting through the 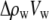 term of Eq. 1. To calculate Nw for GK in the presence of these solutes (Table 3), ρp (3.07 × 10−6 Å−2) and Vp (24,850 Å3) were determined from the crystal structure coordinates and the
term of Eq. 1. To calculate Nw for GK in the presence of these solutes (Table 3), ρp (3.07 × 10−6 Å−2) and Vp (24,850 Å3) were determined from the crystal structure coordinates and the 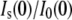 ratios shown in Fig. 7 A were fit (solid lines) to Eq. 2 also using an iterative fv correction, Eq. 5. We also performed SANS measurements of GK/GMP and of GK/GMP/MgADP in the presence of TEG and PEG 400 (data not shown). The Nw values calculated from fitting the I(0) ratios also are given in Table 3, using ρp = 3.103 × 10−6 Å−2, Vp = 25,273 Å3 (including GMP) for GK/GMP, and ρp = 3.151 × 10−6 Å−2, Vp = 25,773 Å3 (including both GMP and ADP) for the ternary complex.
ratios shown in Fig. 7 A were fit (solid lines) to Eq. 2 also using an iterative fv correction, Eq. 5. We also performed SANS measurements of GK/GMP and of GK/GMP/MgADP in the presence of TEG and PEG 400 (data not shown). The Nw values calculated from fitting the I(0) ratios also are given in Table 3, using ρp = 3.103 × 10−6 Å−2, Vp = 25,273 Å3 (including GMP) for GK/GMP, and ρp = 3.151 × 10−6 Å−2, Vp = 25,773 Å3 (including both GMP and ADP) for the ternary complex.
FIGURE 5.

SANS profiles for GK at 5 mg/mL in D2O with no solute and in the presence of 8% PEG 400. For comparison, the SANS intensity is normalized by the scattering at zero angle, I(Q)/I(0). The protein appears smaller with added solute; Guinier fits to these curves yield Rg = 17.0 ± 0.1 Å and 16.0 ± 0.2 Å for no solute and 8% PEG 400, respectively.
FIGURE 6.

Contrast variation for GK by H2O/D2O and solute/D2O. Plotted is the square root of the zero-angle scattering intensity, 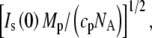 versus bulk solution scattering-length density, ρ0. (Inset) Detailed view of the solute/D2O contrast variation region. The different slopes from the H2O/D2O contrast variation line with added solutes are attributed to protein-associated water.
versus bulk solution scattering-length density, ρ0. (Inset) Detailed view of the solute/D2O contrast variation region. The different slopes from the H2O/D2O contrast variation line with added solutes are attributed to protein-associated water.
TABLE 3.
Guanylate kinase hydration calculated from I(0) fits
|
Nw
|
|||
|---|---|---|---|
| Solute | GK | GK/GMP | GK/GMP/MgADP |
| Glycerol | ∼30 | — | — |
| TEG | 301 ± 60 | 224 ± 126 | 208 ± 123 |
| PEG 400 | 807 ± 47 | 777 ± 113 | 690 ± 108 |
| PEG 1000 | ∼600 | — | — |
FIGURE 7.

(A) The GK zero-angle neutron scattering intensity ratio, 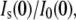 as a function of solute volume fraction, fv. Solid lines are fits using Eq. 2. (B) Measured change in GK Rg versus solute fv. Dashed lines are given for glycerol and PEG 1000 to guide the eye. Glycerol has a negligible effect on GK Rg, whereas the effects of the other solutes become more pronounced according to their size. Glycerol has greater access to the water molecules within the GK cavity, whereas the other solutes are partially excluded.
as a function of solute volume fraction, fv. Solid lines are fits using Eq. 2. (B) Measured change in GK Rg versus solute fv. Dashed lines are given for glycerol and PEG 1000 to guide the eye. Glycerol has a negligible effect on GK Rg, whereas the effects of the other solutes become more pronounced according to their size. Glycerol has greater access to the water molecules within the GK cavity, whereas the other solutes are partially excluded.
There are many more waters associated with GK in the presence of these solutes compared to lysozyme, likely due to a high exclusion of solute from the deep binding cavity of GK. The benefit of having such a large quantity of associated water, where Vw becomes comparable to Vp, is that I(0) can be used reliably to measure Nw without a geometrical model as required for calculations of Rg. Indeed, it becomes difficult to fit GK Rg by treating this protein as a composite particle since the large binding cavity makes the simple core-shell model completely inadequate. The fits of I(0) ratios for glycerol and PEG 1000 (Fig. 7 A, dashed lines) include only a single solute point, so Nw values for these solutes should only be taken as estimates. However, their trends in terms of solute size are found to be significant when compared to the ITC results described later.
The dependence of Rg for GK on solute concentration is shown in Fig. 7 B for the four solutes examined. The dashed lines are linear fits to the glycerol and PEG 1000 data to guide the eye. No change in GK Rg is observed with added glycerol, consistent with the negligible Nw determined from the intensity ratio for this solute. There is a monotonic decrease in Rg with added EGs that becomes more pronounced with increasing molecular weight. To ensure that the observed effects were relatively insensitive to protein concentration within the range of study, experiments with PEG 400 were repeated for varying GK concentration (4–6 mg/mL). Negligible concentration dependence is found for I(0) based on the overlapping points seen in Fig. 7 A. A slightly larger spread in Rg data is observed for PEG 400 (Fig. 7 B), but no trend with GK concentration was observed.
Based on the Nw values obtained from 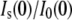 for GK with TEG and PEG 400, we developed simple models of protein and associated water using the program VMD (45). GK with 319 water molecules located uniformly around the protein surface and within 3.3 Å of any protein atom (Fig. 8 A) was used to model GK-TEG. The default water density used by VMD to solvate a structure was used. GK with 808 waters located uniformly within 4.5 Å of any protein atom and completely filling the cavity (Fig. 8 B) was the model for GK-PEG 400. A third model that has 250 waters localized within the GK cavity was also made to determine the effects of cavity waters excluding solute. Using these models, Rg as a function of solute fv could be calculated using Xtal2sas (Fig. 9 A). For these calculations, the coordinates of the water oxygen atoms were used to represent the water molecules and assigned the contrast ρw − ρ0. Considering the simplicity of these models, they manage to capture the approximate magnitude change in Rg with fv for both TEG and PEG 400. As mentioned previously, even in the absence of osmolyte, the Rg of the GK apoenzyme crystal structure (16.8 Å) shows a more compact conformation than that measured by SANS: Rg = 17.2 ± 0.1 Å (see Table 2). The models, however, do underestimate the actual decrease in Rg seen experimentally. The model-predicted SANS curves for GK hydration by PEG 400 are shown in Fig. 9 B (using the model in Fig. 8 B). Intensity is normalized by I(0) for comparison. The change in the Q-dependence of the scattering intensity with added solutes is qualitatively similar to that observed experimentally for lysozyme (Fig. 2) and GK (Fig. 5), although the effect of protein-associated water on the SANS profile is less pronounced experimentally. Possible reasons for this are discussed below.
for GK with TEG and PEG 400, we developed simple models of protein and associated water using the program VMD (45). GK with 319 water molecules located uniformly around the protein surface and within 3.3 Å of any protein atom (Fig. 8 A) was used to model GK-TEG. The default water density used by VMD to solvate a structure was used. GK with 808 waters located uniformly within 4.5 Å of any protein atom and completely filling the cavity (Fig. 8 B) was the model for GK-PEG 400. A third model that has 250 waters localized within the GK cavity was also made to determine the effects of cavity waters excluding solute. Using these models, Rg as a function of solute fv could be calculated using Xtal2sas (Fig. 9 A). For these calculations, the coordinates of the water oxygen atoms were used to represent the water molecules and assigned the contrast ρw − ρ0. Considering the simplicity of these models, they manage to capture the approximate magnitude change in Rg with fv for both TEG and PEG 400. As mentioned previously, even in the absence of osmolyte, the Rg of the GK apoenzyme crystal structure (16.8 Å) shows a more compact conformation than that measured by SANS: Rg = 17.2 ± 0.1 Å (see Table 2). The models, however, do underestimate the actual decrease in Rg seen experimentally. The model-predicted SANS curves for GK hydration by PEG 400 are shown in Fig. 9 B (using the model in Fig. 8 B). Intensity is normalized by I(0) for comparison. The change in the Q-dependence of the scattering intensity with added solutes is qualitatively similar to that observed experimentally for lysozyme (Fig. 2) and GK (Fig. 5), although the effect of protein-associated water on the SANS profile is less pronounced experimentally. Possible reasons for this are discussed below.
FIGURE 8.

Models of GK with (A) 319 associated water molecules located uniformly around the protein surface, (B) 808 waters around the protein surface and filling the binding cavity, and (C) 250 waters localized within the protein cavity. Protein is cyan and water oxygen atoms are red. The models were created using VMD and the coordinates were used for calculating Rg values and SANS Is(Q) profiles with changing bulk-solution scattering-length density.
FIGURE 9.

(A) Calculated Rg values for the GK-water models shown in Fig. 8. Bulk-solution scattering-length density was varied according to solute fv, with TEG as the solute for the Nw = 319 model (Fig. 8 A) and PEG 400 for the models with Nw = 808 (Fig. 8 B) and Nw = 250 (Fig. 8 C). (B) Calculated SANS profiles for the GK-water model incorporating 808 waters (Fig. 8 B) and with contrast variation by added PEG 400.
Hydration changes accompanying GK ligand binding
The calculation of Nw from 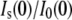 and the consequent modeling of Rg changes with solute concentration implicitly assume that GK structure does not change significantly with solute concentration. To confirm this, we have also measured changes in hydration associated with ligand binding. The osmotic pressure of a solute will act to perturb the equilibrium between two states or conformations if there is a difference in the number of solute-excluding waters between them. GK ligand-binding constants can be measured using calorimetry. Sample ITC curves are given in Fig. 10, showing GK with no solute and 20% PEG 400 along with the corresponding buffer runs. GMP binds relatively weakly in both cases; saturation is not reached until the molar ratio [GMP]/[GK] exceeds 10. However, binding affinity, Keq, is doubled from (1.01 ± 0.04) × 104 M−1 with no solute to (2.27 ± 0.07) × 104 M−1 with 20% PEG 400. For the former, ΔH = −8.1 ± 0.2 kcal/mol and the entropy, ΔS = −8.5 ± 0.6 cal/mol/K; for the latter, ΔH = −7.4 ± 0.1 kcal/mol and ΔS = (−4.6 ± 0.3) cal/mol/K.
and the consequent modeling of Rg changes with solute concentration implicitly assume that GK structure does not change significantly with solute concentration. To confirm this, we have also measured changes in hydration associated with ligand binding. The osmotic pressure of a solute will act to perturb the equilibrium between two states or conformations if there is a difference in the number of solute-excluding waters between them. GK ligand-binding constants can be measured using calorimetry. Sample ITC curves are given in Fig. 10, showing GK with no solute and 20% PEG 400 along with the corresponding buffer runs. GMP binds relatively weakly in both cases; saturation is not reached until the molar ratio [GMP]/[GK] exceeds 10. However, binding affinity, Keq, is doubled from (1.01 ± 0.04) × 104 M−1 with no solute to (2.27 ± 0.07) × 104 M−1 with 20% PEG 400. For the former, ΔH = −8.1 ± 0.2 kcal/mol and the entropy, ΔS = −8.5 ± 0.6 cal/mol/K; for the latter, ΔH = −7.4 ± 0.1 kcal/mol and ΔS = (−4.6 ± 0.3) cal/mol/K.
For a solute osmotic pressure, Π, acting on a difference in sequestered water volumes between the two conformations, ΔVw, then Keq can be expressed in terms of the equilibrium constant in the absence of osmotic pressure, K0, the thermal energy, RT, and the osmotic work
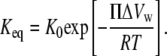 |
(7) |
This expression assumes solute exclusion. The slope of ln(Keq) versus Π,
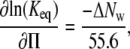 |
(8) |
yields the change in the number of water molecules accompanying ligand binding, ΔNw, where Π is in units of osmolality.
Upon fitting all titration curves to a single-site binding model, plots of ln(Keq) versus solute osmolality were made for GMP-binding with added solutes (Fig. 11 A). The linearity indicates that ΔNw is independent of solute concentration for each osmolyte. Calculated ΔNw values are given in Table 4, where negative values indicate that water release accompanies ligand binding. The insensitivity of ΔNw to solute concentration supports the conclusion that the conformations of the apoenzyme and nucleotide bound protein are not significantly affected by osmolyte osmotic pressure. The change in hydration with ligand binding, ΔNw, however, does depend on the solute identity. No significant change is observed with glycerol, but progressively larger water volumes are seen for the series TEG, PEG 400, and PEG 1000. This is consistent with the protein hydration measurements with SANS (Table 3). Changes in ln(Keq) with solute osmolality for ADP binding in the presence of Mg2+, and also in the absence and presence of GMP, with added PEG 400 are given in Fig. 11 B. ADP binds to GK with higher affinity once GMP has been bound. Likewise, GMP will bind with a similar magnitude increase in affinity if ADP is bound first (data not shown). Calculated ΔNw for ADP binding is about the same without (–97 ± 5) and with GMP bound (–102 ± 4), giving an average ΔNw = −100 ± 3. With the solute PEG 400, almost twice the amount of water is released upon ADP binding compared to GMP binding (Table 4). The difference in the number of solute-excluding waters between the GK apoenzyme and the ternary complex is ∼160 from calorimetry for PEG 400 and an estimated ΔNw ≈ 120 from SANS (Table 3).
FIGURE 11.

(A) Osmotic stress effects on GMP binding to GK. The slope of the curves is proportional to the change in hydration, ΔNw, accompanying ligand binding. (B) Osmotic stress effects on ADP binding to GK in the presence of Mg2+ and in the presence and absence of GMP.
TABLE 4.
Guanylate kinase hydration changes with ligand binding from ITC
| Solute | ΔNw GMP binding | ΔNw ADP binding |
|---|---|---|
| Glycerol | ≈ 0 | — |
| TEG | –46 ± 1 | — |
| PEG 400 | –58 ± 1 | –102 ± 4 |
| PEG 1000 | –116 ± 6 | — |
The exclusion of PEG results in changes in ΔH and −TΔS for ligand binding that are comparable in magnitude but opposite in sign and much larger than the resultant ΔG. This is observed by the compensation between ΔH and TΔS shown in Fig. 12. Taking all the different ligand-binding experiments and solutes used gives a slope, 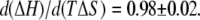 This compensating effect is similar to that observed for certain solutes studied before (31) and references therein.
This compensating effect is similar to that observed for certain solutes studied before (31) and references therein.
FIGURE 12.

Compensation between the enthalpic, ΔH, and entropic, TΔS, contributions to GK ligand binding in the presence of solutes. The different ligand-binding experiments and solutes used are plotted together and give a slope, 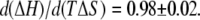
DISCUSSION
Neutron scattering is a particularly useful probe for investigating protein structure and hydration, mostly owing to the inherent contrast between hydrogen and deuterium. The use of solutes or osmolytes that are excluded from water associated with protein enhances the efficacy of SANS by creating three regions of contrast: the protein, the solute-excluding water associated with protein, and the bulk solution. Stuhrmann and co-workers (23) and Lehmann and Zaccai (24) have pioneered the use of changes in Is(0) with added solute to determine the preferential hydration of the surface of small compact proteins. Combining changes in Is(0) with accompanying changes in Rg as we have done here can yield even more information. Changes in Is(0) with solute concentration are dependent on the number of associated waters. The sensitivity of Rg to solute concentration depends both on the number of associated waters and their location. From initial analysis of the SANS data, however, it can be difficult to discern whether the observed changes in protein I(0) and Rg with added solute, occurring for both lysozyme and GK, are due to three regions of neutron contrast, a protein structural change, or possibly something more subtle like the onset of interparticle interference. Correlations between protein molecules in a concentrated solution will result in the formation of a correlation peak in the SANS profile related to the average distance between particles (46). For repulsive interactions, scattering at low angles is suppressed, thereby reducing the measured Is(0) and Rg, since it is not accounted for in the Guinier fits. Without an observed formation of a correlation peak in I(Q) and corresponding depression in I(0), however, interparticle interference mediated by solute exclusion seems at least minimal. It is important to note that the independence of Is(0) and Rg on GK concentration found for PEG 400 (Fig. 7) suggests that the observed changes can be attributed to protein hydration effects. Any cross-correlation between protein and solute is difficult to assess and is also assumed to be negligible.
Just as excluded solutes can act to stabilize the native structure relative to the expanded denatured form, excluded solutes can also act to compact native structures to less hydrated conformations. In fact, our initial effort was to close the water-filled ligand binding cavity of GK using osmotic stress alone to measure the energetics associated with this transition apart from the ligand binding energy. Such measurements were previously performed but utilized indirect methods (13,47). In the case of a protein like GK, which has a volume of associated water comparable to Vp in the presence of solutes, careful analysis of Is(0) is instructive for demonstrating that at least more is occurring than simply a change in conformation. If the Rg decrease is solely due to protein conformational change, then Is(0) should predictably decrease with diminishing contrast, according to Eq. 2 with Vw = 0. However, for a small protein having relatively little Vw, like lysozyme, Is(0) does seem to vary in this way within the error of measured values. Compaction of lysozyme by several Ångstroms in Rg (Fig. 3), however, is not likely.
It is difficult to differentiate between all possible effects of solutes, particularly osmotic-pressure-induced conformational changes by SANS alone. It is therefore necessary to use complementary techniques to aid in excluding some effects and provide more certainty in others. The ITC results allow us to conclude that protein hydration created by the added solutes is the predominate effect on Is(0) and Rg observed in our SANS experiments, similar to the analysis made in previous neutron studies (23,24). It is also worth noting that the ITC experiments are performed at a protein concentration similar to that used for SANS (1 mg/mL for ITC and 5 mg/mL for SANS), thus making comparisons between the two methods more reasonable.
ITC measurements of GK ligand binding discount an osmotically induced closure of the ligand-binding cavity over the range of solutes and solute concentrations used in both the ITC and SANS experiments. This is because the ligand-binding free energy varies linearly with osmolality over each solute concentration range and for both GMP and ADP binding (Fig. 11). This linear dependence indicates that ΔNw, the difference in the numbers of water molecules that exclude solute between the apoenzyme and ligand bound form, is a constant in each case. If a ligand-binding site was osmotically closed, then the corresponding plot in Fig. 11 would show a nonlinear behavior with decreasing slope near the point of osmotic closure. This behavior was observed for hexokinase but required up to 2 Osm PEG 400 (fv ≈ 0.24) and 1 Osm for PEG of molecular mass 1000 Da and greater (13). In comparison to our conditions for a similar kinase, it can be better appreciated that we have not approached the point of osmotically closing GK.
We conclude that the observed dependences of Is(0) and Rg on solute concentration for both lysozyme and GK are due to the additional scattering contribution from protein-associated water. Even though changes in Is(0) for lysozyme are within the error of measurement compared to H2O/D2O contrast variation, Rg changes appreciably with added solute (Fig. 3). With its greater sensitivity to protein hydration compared to Is(0), we use a composite Rg with a spherical core-shell model to interpret the results. We did not include the possibility that the water in contact with the protein may be denser than bulk water (4). As an effective preferential hydration layer surrounding the protein, the calculated thickness, t, is less than a water layer (∼3 Å) for all of these solutes (see Table 1). However, converting thickness to Nw shows that these values are reasonable compared to those obtained previously by densitometry (39). By calculating the solvent-accessible surface area (SASA) of lysozyme with the msms program (48) using a 1.4-Å probe radius and assuming 9 Å2 per water molecule, ∼900 water molecules can cover the lysozyme surface. This is more water than measured with any of the solutes, which again indicates that there is not full exclusion from a complete water layer surrounding the protein. When comparing with other techniques, it also may be important to consider the sensitivity of SANS to preferential hydration, since there are not sharp interfaces between protein, hydrating water, and bulk solution. Considering its size, it is notable that glycine betaine is almost as excluded from lysozyme surface water as TEG. Glycine betaine is highly excluded from many different biomolecular surfaces, though, and is widely used to stabilize native protein structure (6,9,10,49). For glycerol and EGs, Nw increases with solute size, and this trend is consistently observed in all our SANS experiments and the ITC experiments with GK.
In contrast to lysozyme, guanylate kinase has a significant amount of internal water in the ligand-binding cavities. From the calculated Nw, GK has two to three times the amount of associated water for the same solute (Table 3). Also, GK hydration increases by a factor of ∼3 going from TEG to PEG 400 for all GK ligand states. This is a much more significant change with solute size compared to lysozyme. SASA calculation for GK indicates that ∼1100 waters can be accommodated on the surface. The measured GK hydration approaches this for the solutes studied. However, just as with lysozyme, there likely is not a complete 3-Å solute-excluding water layer surrounding the protein surface. In addition, the solute size dependence for exclusion from GK, observed in SANS and ITC measurements, suggests that a large portion of Nw resides in the cavity. The comparison of lysozyme and GK illustrates the potential use of SANS and osmotic stress for resolving structural details based on solute exclusion.
Steric exclusion of solutes is useful for investigating protein pores and cavities (12–16). Glycerol has a negligible preferential hydration effect on GK and as such, protein Rg and GMP binding constant are unaffected. Again, this is likely due to both the chemical nature and size of glycerol compared to the other solutes. PEG 400 is more excluded from GK, and its ligand-bound forms, than TEG, showing a higher Nw and also more water released upon GMP binding (see Table 3). Likewise, PEG 1000 generates the largest ΔNw with GMP binding of all the solutes and has an estimated Nw comparable to that of PEG 400. Changes in GK Rg (Fig. 7 A) also mirror these trends by showing a more drastic decrease in Rg with greater solute exclusion. Although we did not try larger-molecular-weight PEG, studies on hexokinase showed that PEG polymers with molecular weight >1000 yielded the same ΔNw. This was taken to indicate that many of the waters released upon glucose binding were located in the protein cavity (13).
It is interesting to note that the ΔH and TΔS contributions to GK ligand binding from exclusion of the PEGs (Fig. 12) are comparable and compensating. As previously discussed (31), steric exclusion or crowding is strictly entropic. The observed compensating enthalpy indicates that interactions other than a simple steric repulsion are important in determining PEG exclusion. ITC measurements of ΔNw accompanying GK ligand binding can be compared to the change in protein SASA. Using a 1.4-Å probe sphere, ΔNw = −74 waters with GMP binding and −117 waters upon ADP binding to the binary complex GK•GMP. The measured hydration changes are concurrent with these changes in SASA, and both indicate a larger effect from ADP binding compared to GMP binding. Also, the change in hydration that occurs with GK ligand binding can be roughly estimated from the difference in Nw between states obtained by SANS (Table 3). For PEG 400, the ITC results give ΔNw = −58 and −102 for GMP and ADP binding, respectively, whereas SANS gives ΔNw ≈ −30 and −87 for GMP and ADP binding, respectively. Although ITC is a better method for measuring ΔNw directly in these cases, it is encouraging that the SANS estimates indicate water loss upon ligand binding and also are the same order of magnitude as the ITC values.
To better understand how the composite Rg is affected by the amount and location of solute-excluding water associated with GK, we developed models to calculate changes in Rg. The models for GK with TEG (Fig. 8 A) and PEG 400 (Fig. 8 B) provide reasonable approximations for the change in Rg with added solute (Fig. 9 A) compared to experiment (Fig. 7 B). This provides extra support that our Nw values are reasonable. Preferential hydration of the protein surface will have a larger effect on Rg than cavity water. This is seen with the models for PEG 400, where a given amount of cavity water (we just use 250 waters) is a small contribution to the overall change in Rg found for the total hydration (Fig. 9 A). It also can be understood more simply based on a shell of water having a larger Rg than the same volume as a solid sphere. A larger Rg,w makes its contribution to the composite Rg greater.
SANS Is(Q) profiles also were calculated from the GK-PEG 400 model with Nw = 808 (Fig. 9 B). They qualitatively demonstrate the same change in scattering-intensity profile as observed experimentally for lysozyme (Fig. 2) and GK (Fig. 5) with added PEG 400. The calculated change in Is(Q) with added solute can be understood by considering I(Q) as the Fourier transform of the difference between pair-distribution functions for the protein and protein-associated water, since Δρp and Δρw are of opposite sign. This makes the change in scattering-intensity profile appear similar to a shift toward smaller intramolecular correlations (more scattering at higher Q). The effect is more pronounced in the model than in the actual scattering data. This is likely because the shape of the scattering profile has an increased sensitivity to background subtraction at high scattering angle, Q. As a result, it is difficult to resolve such structural features due to errors in incoherent background subtraction. However, it is anticipated that by studying larger proteins and protein complexes with added solutes, the SANS profiles would be more amenable to quantitative Is(Q) analysis since these features will arise at lower Q. This would provide added structural detail that can be used along with Is(0) and Rg in determining both the amount and location of protein hydration using SANS.
CONCLUSIONS
We have demonstrated methods of using SANS combined with osmotic stress to probe the preferential protein hydration seen with a variety of solutes. The roles of solute size and chemistry, as well as protein structure, in determining the extent of solute exclusion, and therefore protein hydration, have been investigated. ITC measurements of GK ligand binding corroborate the SANS results. The main limitations in using SANS to reliably resolve protein-associated water are the size of the protein (larger structures being more amenable due to a greater surface area) and the added incoherent scattering from solute protons. We have essentially explored the lower limit in protein size with lysozyme and GK. In contrast with lysozyme, the large internal cavity of GK, which contains the bulk of the solute-excluding waters, makes these measurements more reliable. From the model calculations incorporating protein-associated waters, it is found that additional information can be obtained from the SANS I(Q) profiles, provided that the protein structure is sufficiently large. Indeed, larger proteins and complexes will have a greater total surface area available for preferentially hydrating water and changes in the SANS profile will occur at lower Q, where background is less important. The shape of the SANS curve then can be used quantitatively in conjunction with Is(0) and Rg to aid in determining both the quantity and location of protein-associated waters. Careful evaluation of effects like polymer partitioning into pores and cavities (50,51) also might become realizable with this approach.
Another opportunity for this research is to use deuterated solutes to unambiguously measure protein structure with solute addition. This can provide assurance that there are no structural changes for an added solute or it can be purposely used at high concentration to induce structural changes and measure the osmotic work. In this way, combining SANS and osmotic stress offers new opportunities for studying protein conformational changes by allowing the contribution of hydration to be measured directly and in the absence of ligand. This study forms the basis for promising future research in that direction, so that structural information about biological systems can be directly connected with their corresponding thermodynamics.
Acknowledgments
We thank Dr. Honggao Yan (Michigan State University, East Lansing, Michigan) for kindly providing us with the pET17b-YGK plasmid. We gratefully acknowledge K. A. Rubinson, B. A. Todd, D. Harries, and H. Nanda for valuable discussions and suggestions. Small-angle neutron scattering experiments were performed on the NG3 and NG7 30 m SANS instruments at the NIST Center for Neutron Research.
This research was supported by the National Research Council, the National Science Foundation under Agreement No. DMR-0454672 (NCNR), and the Intramural Research Program of the National Institute of Child Health and Human Development and National Institutes of Health.
Editor: Marcia Newcomer.
References
- 1.Levy, Y., and J. N. Onuchic. 2006. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 35:389–415. [DOI] [PubMed] [Google Scholar]
- 2.Prabhu, N., and K. Sharp. 2006. Protein-solvent interactions. Chem. Rev. 106:1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raschke, T. M. 2006. Water structure and interactions with protein surfaces. Curr. Opin. Struct. Biol. 16:152–159. [DOI] [PubMed] [Google Scholar]
- 4.Svergun, D. I., S. Richard, M. H. Koch, Z. Sayers, S. Kuprin, and G. Zaccai. 1998. Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA. 95:2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, S. H., L. Liu, E. Fratini, P. Baglioni, A. Faraone, and E. Mamontov. 2006. Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA. 103:9012–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arakawa, T., and S. N. Timasheff. 1985. The stabilization of proteins by osmolytes. Biophys. J. 47:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timasheff, S. N. 1993. The control of protein stability and association by weak interactions with water: how do solvents affect these processes? Annu. Rev. Biophys. Biomol. Struct. 22:67–97. [DOI] [PubMed] [Google Scholar]
- 8.Timasheff, S. N. 1998. Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated. Adv. Protein Chem. 51:355–432. [DOI] [PubMed] [Google Scholar]
- 9.Courtenay, E. S., M. W. Capp, C. F. Anderson, and M. T. Record, Jr. 2000. Vapor pressure osmometry studies of osmolyte-protein interactions: implications for the action of osmoprotectants in vivo and for the interpretation of “osmotic stress” experiments in vitro. Biochemistry. 39:4455–4471. [DOI] [PubMed] [Google Scholar]
- 10.Santoro, M. M., Y. F. Liu, S. M. A. Khan, L. X. Hou, and D. W. Bolen. 1992. Increased thermal stability of proteins in the presence of naturally occurring osmolytes. Biochemistry. 31:5278–5283. [DOI] [PubMed] [Google Scholar]
- 11.Colombo, M. F., D. C. Rau, and V. A. Parsegian. 1992. Protein solvation in allosteric regulation.: a water effect on hemoglobin. Science. 256:655–659. [DOI] [PubMed] [Google Scholar]
- 12.Vodyanoy, I., S. M. Bezrukov, and V. A. Parsegian. 1993. Probing alamethicin channels with water-soluble polymers: size-modulated osmotic action. Biophys. J. 65:2097–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid, C., and R. P. Rand. 1997. Probing protein hydration and conformational states in solution. Biophys. J. 72:1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidorova, N. Y., and D. C. Rau. 1996. Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences. Proc. Natl. Acad. Sci. USA. 93:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sidorova, N. Y., S. Muradymov, and D. C. Rau. 2006. Differences in hydration coupled to specific and nonspecific competitive binding and to specific DNA binding of the restriction endonuclease BamHI. J. Biol. Chem. 281:35656–35666. [DOI] [PubMed] [Google Scholar]
- 16.Rau, D. C. 2006. Sequestered water and binding energy are coupled in complexes of λ Cro repressor with non-consensus binding sequences. J. Mol. Biol. 361:352–361. [DOI] [PubMed] [Google Scholar]
- 17.Parsegian, V. A., R. P. Rand, and D. C. Rau. 1995. Macromolecules and water: probing with osmotic stress. Methods Enzymol. 259:43–94. [DOI] [PubMed] [Google Scholar]
- 18.Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leikin, S., V. A. Parsegian, D. C. Rau, and R. P. Rand. 1993. Hydration forces. Annu. Rev. Phys. Chem. 44:369–395. [DOI] [PubMed] [Google Scholar]
- 20.Eisenberg, H. 1976. Biological Macromolecules and Polyelectrolytes in Solutions. Oxford University Press, New York.
- 21.Eisenberg, H. 1994. Protein and nucleic acid hydration and cosolvent interactions: establishment of reliable baseline values at high cosolvent concentrations. Biophys. Chem. 53:57–68. [DOI] [PubMed] [Google Scholar]
- 22.Jacrot, B. 1976. The study of biological structures by neutron scattering from solution. Rep. Prog. Phys. 39:911–953. [Google Scholar]
- 23.Stuhrmann, H. B., J. Haas, K. Ibel, M. H. Koch, and R. R. Crichton. 1976. Low angle neutron scattering of ferritin studied by contrast variation. J. Mol. Biol. 100:399–413. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann, M. S., and G. Zaccai. 1984. Neutron small-angle scattering studies of ribonuclease in mixed aqueous solutions and determination of the preferentially bound water. Biochemistry. 23:1939–1942. [DOI] [PubMed] [Google Scholar]
- 25.Minton, A. P. 1998. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295:127–149. [DOI] [PubMed] [Google Scholar]
- 26.Hall, D., and A. P. Minton. 2003. Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta. 1649:127–139. [DOI] [PubMed] [Google Scholar]
- 27.Hultgren, A., and D. C. Rau. 2004. Exclusion of alcohols from spermidine-DNA assemblies: probing the physical basis of preferential hydration. Biochemistry. 43:8272–8280. [DOI] [PubMed] [Google Scholar]
- 28.Stanley, C., and D. C. Rau. 2006. Preferential hydration of DNA: the magnitude and distance dependence of alcohol and polyol interactions. Biophys. J. 91:912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnet-Gonnet, C., S. Leikin, S. Chi, D. C. Rau, and V. A. Parsegian. 2001. Measurement of forces between hydroxypropylcellulose polymers: temperature favored assembly and salt exclusion. J. Phys. Chem. B. 105:1877–1886. [Google Scholar]
- 30.Chik, J., S. Mizrahi, S. Chi, V. A. Parsegian, and D. C. Rau. 2005. Hydration forces underlie the exclusion of salts and of neutral polar solutes from hydroxypropylcellulose. J. Phys. Chem. B. 109:9111–9118. [DOI] [PubMed] [Google Scholar]
- 31.Harries, D., D. C. Rau, and V. A. Parsegian. 2005. Solutes probe hydration in specific association of cyclodextrin and adamantane. J. Am. Chem. Soc. 127:2184–2190. [DOI] [PubMed] [Google Scholar]
- 32.Li, Y., Y. L. Zhang, and H. G. Yan. 1996. Kinetic and thermodynamic characterizations of yeast guanylate kinase. J. Biol. Chem. 271:28038–28044. [DOI] [PubMed] [Google Scholar]
- 33.Agarwal, K. C., R. P. Miech, and R. E. Parks, Jr. 1978. Guanylate kinases from human erythrocytes, hog brain, and rat liver. Methods Enzymol. 51:483–491. [DOI] [PubMed] [Google Scholar]
- 34.Glinka, C. J., J. G. Barker, B. Hammouda, S. Krueger, J. J. Moyer, and W. J. Orts. 1998. The 30 m small-angle neutron scattering instruments at the National Institute of Standards and Technology. J. Appl. Cryst. 31:430–445. [Google Scholar]
- 35.Kline, S. R. 2006. Reduction and analysis of SANS and USANS data using IGOR Pro. J. Appl. Cryst. 39:895–900. [Google Scholar]
- 36.Guinier, A., and G. Fournet. 1955. Small-Angle Scattering of X-Rays. Wiley, New York.
- 37.Krueger, S., I. Gorshkova, J. Brown, J. Hoskins, K. H. McKenney, and F. P. Schwarz. 1998. Determination of the conformations of cAMP receptor protein and its T127L,S128A mutant with and without cAMP from small angle neutron scattering measurements. J. Biol. Chem. 273:20001–20006. [DOI] [PubMed] [Google Scholar]
- 38.Scattering Length Density Calculator. http://www.ncnr.nist.gov/resources/sldcalc.html.
- 39.Bhat, R., and S. N. Timasheff. 1992. Steric exclusion is the principal source of the preferential hydration of proteins in the presence of polyethylene glycols. Protein Sci. 1:1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonald, R. C., T. A. Steitz, and D. M. Engelman. 1979. Yeast hexokinase in solution exhibits a large conformational change upon binding glucose or glucose 6-phosphate. Biochemistry. 18:338–342. [DOI] [PubMed] [Google Scholar]
- 41.Pickover, C. A., D. B. McKay, D. M. Engelman, and T. A. Steitz. 1979. Substrate binding closes the cleft between the domains of yeast phosphoglycerate kinase. J. Biol. Chem. 254:11323–11329. [PubMed] [Google Scholar]
- 42.Forstner, M., M. Kriechbaum, P. Laggner, and T. Wallimann. 1998. Structural changes of creatine kinase upon substrate binding. Biophys. J. 75:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blaszczyk, J., Y. Li, H. G. Yan, and X. H. Ji. 2001. Crystal structure of unligated guanylate kinase from yeast reveals GMP-induced conformational changes. J. Mol. Biol. 307:247–257. [DOI] [PubMed] [Google Scholar]
- 44.Sekulic, N., L. Shuvalova, O. Spangenberg, M. Konrad, and A. Lavie. 2002. Structural characterization of the closed conformation of mouse guanylate kinase. J. Biol. Chem. 277:30236–30243. [DOI] [PubMed] [Google Scholar]
- 45.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 46.Longeville, S., W. Doster, and G. Kali. 2003. Myoglobin in crowded solutions: structure and diffusion. Chem. Phys. 292:413–424. [Google Scholar]
- 47.Sidorova, N. Y., and D. C. Rau. 2004. Differences between EcoRI nonspecific and “star” sequence complexes revealed by osmotic stress. Biophys. J. 87:2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanner, M. F., A. J. Olson, and J. C. Spehner. 1996. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 38:305–320. [DOI] [PubMed] [Google Scholar]
- 49.Felitsky, D. J., J. G. Cannon, M. W. Capp, J. Hong, A. W. Van Wynsberghe, C. F. Anderson, and M. T. Record, Jr. 2004. The exclusion of glycine betaine from anionic biopolymer surface: why glycine betaine is an effective osmoprotectant but also a compatible solute. Biochemistry. 43:14732–14743. [DOI] [PubMed] [Google Scholar]
- 50.Bezrukov, S. M., I. Vodyanoy, R. A. Brutyan, and J. J. Kasianowicz. 1996. Dynamics of free energy of polymers partitioning into a nanoscale pore. Macromolecules. 29:8517–8522. [Google Scholar]
- 51.Merzlyak, P. G., L. N. Yuldasheva, C. G. Rodrigues, C. M. Carneiro, O. V. Krasilnikov, and S. M. Bezrukov. 1999. Polymeric nonelectrolytes to probe pore geometry: application to the α-toxin transmembrane channel. Biophys. J. 77:3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]