

Abstract
As charged macromolecules adsorb and diffuse on cell membranes in a large variety of cell signaling processes, they can attract or repel oppositely charged lipids. This results in lateral membrane rearrangement and affects the dynamics of protein function. To address such processes quantitatively we introduce a dynamic mean-field scheme that allows self-consistent calculations of the equilibrium state of membrane-protein complexes after such lateral reorganization of the membrane components, and serves to probe kinetic details of the process. Applicable to membranes with heterogeneous compositions containing several types of lipids, this comprehensive method accounts for mobile salt ions and charged macromolecules in three dimensions, as well as for lateral demixing of charged and net-neutral lipids in the membrane plane. In our model, the mobility of membrane components is governed by the diffusion-like Cahn-Hilliard equation, while the local electrochemical potential is based on nonlinear Poisson-Boltzmann theory. We illustrate the method by applying it to the adsorption of the anionic polypeptide poly-Lysine on negatively charged lipid membranes composed of binary mixtures of neutral and monovalent lipids, or onto ternary mixtures of neutral, monovalent, and multivalent lipids. Consistent with previous calculations and experiments, our results show that at steady-state multivalent lipids (such as PIP2), but not monovalent lipid (such as phosphatidylserine), will segregate near the adsorbing macromolecules. To address the corresponding diffusion of the adsorbing protein in the membrane plane, we couple lipid mobility with the propagation of the adsorbing protein through a dynamic Monte Carlo scheme. We find that due to their higher mobility dictated by the electrochemical potential, multivalent lipids such as PIP2 more quickly segregate near oppositely charged proteins than do monovalent lipids, even though their diffusion constants may be similar. The segregation, in turn, slows protein diffusion, as lipids introduce an effective drag on the motion of the adsorbate. In contrast, monovalent lipids such as phosphatidylserine only weakly segregate, and the diffusions of protein and lipid remain largely uncorrelated.
INTRODUCTION
Many proteins that peripherally adsorb on lipid membranes contain structured domains that target the protein to the bilayer; examples include the C2 (1), PH (2), FERM (3), and BAR (4) domains. Many of these domains act by specifically binding to a particular lipid species, like the PH domain that binds phosphatidylinositol 4,5-bisphosphate (or PIP2) lipids. However, an apparently different type of targeting is achieved by numerous other proteins that contain natively unstructured clusters of basic residues, such as the well-studied examples of the GAP43, GTPase K-Ras, and MARCKS (5–9). The use of positively charged residues for targeting may come as no surprise, as cellular plasma membranes typically contain ≈20% anionic lipids. This affords a simple mechanism for protein-lipid binding that is essentially nonspecific, yet able to confine proteins to membrane interfaces.
This simple molecular picture has been challenged by recent theoretical and experimental evidence suggesting that the major anionic lipid component in many cells, phosphatidylserine (or phosphatidylglycerol), might not be the major participant in peripheral protein binding (10–13). Instead, the typically multivalent phosphoinositides, such as PIP2 or even phosphatidylinositol 3,4,5-bisphosphate, are more likely implicated in segregation close to peripherally adsorbed proteins. This is interesting, because phosphoinositides are known to play an important regulatory role at the plasma membrane. Despite the fact that they constitute typically only ∼1% of membrane composition, these minority lipids can act at sites of regulation at least partly by electrostatic association with peripheral and embedded proteins (14). Concentrating PIP2 at the site of protein adsorption is therefore a likely mechanism for local and specific recruitment. It has been suggested that segregated lipids can subsequently be released upon cellular changes, e.g., in Ca+2 concentrations. This provides a way to control the amount of free PIP2 in the membrane, and hence a mechanism for regulating PIP2 known to participate in cellular signaling processes such as enzyme activation, endocytosis, and ion-channel activation (15).
To begin to understand why electrostatic targeting could primarily be achieved by polyvalent rather than the more abundant monovalent lipids, we must focus on the forces that underlie this protein-lipid interaction. Theory and experiments show that the attraction of positively charged protein domains to the oppositely charged membrane is due not only to Coulombic interaction, but is also entropically driven. This entropic gain is due to the release of counterions that were previously confined locally to the vicinity of the isolated protein or membrane by the requirement to preserve charge neutrality (16–19). Upon protein-membrane binding, these counterions are no longer required and are released to produce a translational entropy gain in the bulk solution, while the protein and membrane neutralize one another. At the same time, to allow maximal counterion release, charged lipids can migrate in the membrane plane toward the protein adsorption site to fully compensate charges on the protein, causing demixing (20). But this local lipid demixing comes at an entropic cost. The lower the membrane charge density, the higher the cost for the necessary lipid segregation and demixing upon protein binding. At equilibrium, the system has reached some compromise between maximal counterion release and minimal demixing.
Experiments have suggested that that PIP2 preferentially segregates at sites of charged protein adsorption (10). This is reasonable because multivalent lipids should incur a smaller demixing penalty and larger counterion release entropy per segregated lipid, simply because each of them carries a larger charge. Recent theoretical studies predict that multivalent lipids should indeed segregate more than monovalent ones, and that the binding free energy to rigid macromolecules as well as to polyelectrolytes is significantly stronger for such lipids (11–13). But recognizing the dynamic nature of the adsorption problem raises the possibility that the kinetic energy of each adsorbing protein allows it to move so quickly on lipid membranes that some lipids rarely manage to segregate at all. Conversely, lipids may rearrange so quickly around an adsorbing protein that the protein appears stationary to them, creating a transient binding site and thus impeding the protein's motion in the membrane plane.
If we consider free lipid diffusion in the membrane plane under the influence of the field exerted by the oppositely charged protein, we might conclude that multivalent lipids are more mobile (move faster) than monovalent ones, even if their diffusion in the absence of the protein field is similar. This conclusion simply follows if we assume lipid diffusion to be directed by spatial (second-order) derivatives of the electrochemical potential that in turn are linearly proportional to the product of charge and local potential. Higher mobility near the charged macromolecule may allow multivalent lipid segregation that can happen on timescales fast enough to follow the protein random motion as it moves on the membrane surface. This not only reflects a stronger adsorption free energy for protein on PIP2-containing membranes, but also highlights the important role of the minority multivalent lipids in lipid segregation. Note, however, that living cells often exhibit diffusion behavior of membrane components (lipids, proteins) that is much different than in vitro (21).
To quantify the combined kinetic effect of many lipid species interacting with peripheral proteins, it is essential to be able to calculate the steady state of adsorbing macromolecules in a way that will include all important degrees of freedom in a self-consistent way. Previous theoretical studies have shown that equilibrium distributions can be predicted from a self-consistent mean-field model based on the modified Poisson-Boltzmann (PB) equation (20,22). But to address the concerted action of protein adsorption and lipid segregation, we extend this strategy here by using a dynamic propagation method to efficiently derive steady-state configurations for membranes interacting with macromolecules. Our model is an application of a time-dependent self-consistent mean-field approach that has been used to address similar problems (for examples, see (23–26)). The numerical scheme we developed provides not only the adsorption free energy and charge density distribution on the membrane at steady state, but also can be used to gain additional dynamic information on the segregation process.
Our method uses an atomic level representation in three dimensions, and takes into account lateral reorganization and demixing of lipids during adsorption. Lipids are allowed to move in the membrane plane according to a diffusion-like Cahn-Hilliard (CH) equation (27), where segregation rates are in proportion to the Laplacian of their chemical potential. The local chemical potentials are derived from the free-energy functional that depends on local lipid component densities and are calculated using results from nonlinear PB theory.
The membranes we consider here are binary or ternary mixtures of neutral (zwitterionic) lipids, as well as negatively charged ones bearing one or more charges. At steady state, solutions of our two-dimensional CH equations in the membrane plane match the equilibrium solutions of the corresponding modified PB equation. We find, in agreement with previous studies (11–13), that static positively charged polypeptides and proteins positioned near net-acidic membranes of biologically relevant composition preferentially recruit to the adsorption zone multivalent lipids such as PIP2, rather than monovalent ones.
We then extend our model by using a dynamic Monte Carlo scheme to consider lipid dynamics combined with adsorbate diffusion on the membranes. This dynamic model allows us to conclude that it is the composition of the membrane on which proteins are diffusing that determines whether lipids will be sequestered. In particular, we find that PIP2 lipids can be expected to segregate near oppositely charged proteins and thereby slow down the diffusion of the protein. An important prediction of our model is that PIP2 lipids will be able to diffuse in concert with the retarded adsorbed proteins, while lipids such as phosphatidylserine (PS) will only weakly segregate and, in this case, both protein and lipid diffusion will be largely uncorrelated.
The implied consequences of the detailed dynamic picture are clear: by virtue of their charge alone, PIP2 lipids can be sequestered and retained for extended periods of time in the vicinity of oppositely charged peripheral proteins. This may serve as a possible mechanism for the formation of mobile lipid microdomains that diffuse slowly in the membrane plane, allowing both the lipid and the protein the time needed to act in concert.
MODEL
Free-energy functional
Consider charged proteins and lipid bilayers immersed in an aqueous solution. The solution also contains a symmetric 1:1 electrolyte of concentration n0, corresponding to a Debye length
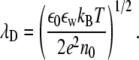 |
(1) |
Here, kB is the Boltzmann constant, T is the temperature, e is the elementary charge, ε0 is the permeability of free space, and εw = 80 is the dielectric constant of the aqueous solution. At the physiological conditions modeled in our calculations, at T = 300 K and n0 = 0.1 M electrolyte (monovalent salt) concentration, λD ≈ 10 Å.
We consider the limit of low surface density of adsorbing proteins, so that interactions between proteins are negligible. We represent the adsorbing protein in full-atomistic three-dimensional details, whereas the membrane is considered as a two-dimensional fluid, allowing us to treat lipid headgroup charges in the continuum representation as usual in regular solution theory (28). Hybridizing these atomistic and continuum representations provides a realistic mesoscale description of the electrostatic problem, while still allowing the entire system to be described at the same mean-field level.
Fig. 1 shows a unit simulation cell containing a single charged macromolecule (poly-Lysine 13 residues long) adsorbed on a membrane. Both the macromolecule and the lipid membrane are treated as a low dielectric material, with dielectric constant εm = 2 within adsorbate and membrane. Each atom on the adsorbing protein is assigned a radius and a partial charge. The oppositely charged membrane consists of mixtures of net-neutral, mono-, and multivalent lipids. Each membrane bilayer is composed of two adjoined lipid monolayers, forming a slab of thickness d. We assume that the membrane is stiff with respect to any weak-deforming forces exerted by the protein, and we therefore keep the membrane flat in all our calculations.
FIGURE 1.

Schematic view of a simulated unit cell containing a protein (or peptide) adsorbed on a membrane. For illustration, we use the basic poly-peptide Lysine, 13 residues long (Lys13). The membrane is represented by a rectangular-shaped slab of thickness d. Charges on the lipid headgroups are represented by a continuous surface charge density. The distance of nearest approach between protein and membrane surfaces is h. In all our calculations the dielectric constant for the membrane interior as well as protein is εm = 2, the dielectric constant of the aqueous environment is εm = 80, and the Debye length in the electrolyte solution is λD = 10 Å.
Using the continuum representation, we consider a single lipid layer as an incompressible, infinite, flat, two-dimensional surface, composed of a mixture of m different lipid species. We define φi as the local mole fraction of the ith lipid species in the membrane plane, and set i = 1 for neutral lipid. Assuming membrane incompressibility, the constraint on material conservation requires that
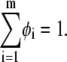 |
(2) |
For simplicity, we assume here that all lipids have the same lateral area per headgroup, a (though the model can easily be extended to include different headgroup areas as in Andelman et al. (29)). Denoting the valency of the ith lipid species by zi, we define the local surface charge density as
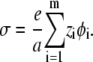 |
(3) |
The adsorbing macromolecule is first considered as fixed in space at a distance h from the membrane surface, where h is defined as the shortest distance between the van der Waals surfaces of atoms on the adsorbate and lipid headgroups (Fig. 1).
The free-energy functional for the system can be written as a sum of the electrostatic energy, salt ion translational entropy, and lipid mixing entropy in the membrane plane (20):
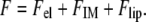 |
(4) |
The system's electrostatic energy is given, as usual, by
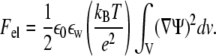 |
(5) |
Here Ψ = eΦ/kBT is the dimensionless (reduced) electrostatic potential, Φ being the electrostatic potential. The integration of this term must be carried out over the entire space. The contribution from the translational entropy of mobile (salt) ions in solution is
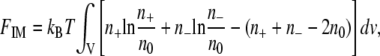 |
(6) |
where n+ and n– represent local concentrations of positive and negative mobile electrolyte ions, respectively, and n0 is the electrolyte concentration in the bulk solution.
The two-dimensional mixing entropy of mobile lipid molecules contribution is given by
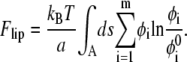 |
(7) |
The sum extends over m lipid species, 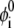 represents the average composition of the ith lipid species, and the integral is taken over the membrane surface.
represents the average composition of the ith lipid species, and the integral is taken over the membrane surface.
Functional minimization of F with respect to the mobile ion concentrations leads to the nonlinear PB equation (22,30–35):
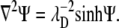 |
(8) |
Solving this equation yields the electrostatic potential Ψ. The additional minimization of F with respect to the lipid compositional degrees of freedom leads to a second differential equation on the bilayer boundary that should be solved simultaneously with Eq. 8 (20,22). Here, however, we do not solve this boundary equation. Instead, we use a dynamic propagation scheme that reaches the equilibrium lipid distribution in the long-time limit, as shown in the next section.
Lipid propagation in time
We now derive the dynamic equations that govern the time-dependent lipid rearrangement in our model. The steady-state solutions to these equations correspond to the lipid distribution in the membrane plane that minimizes the free-energy functional F with respect to all φi-values. The starting point is the continuity equations for all lipid species. The equation for the ith lipid species has the form (36)
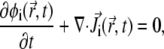 |
(9) |
where  is the local (lipid) current at position
is the local (lipid) current at position  corresponding to a mole fraction
corresponding to a mole fraction 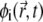 of the ith lipid species at time t, and
of the ith lipid species at time t, and 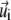 represents the flow velocity of the ith lipid species at time t and located at
represents the flow velocity of the ith lipid species at time t and located at 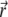 on the membrane surface.
on the membrane surface.
We next relate the local current of the ith lipid species and the local gradients of its electrochemical potential. The idea is to assume that gradients in the chemical potential determine the velocities of lipids migrating in the membrane plane, in the spirit of Diamant and Andelman (26), and references therein. We define the electrochemical potential within our model:
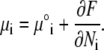 |
(10) |
Here Ni is the number of lipids of species i, and μ°i represents the standard chemical potential for the ith lipid species that is independent of φi. Using Eqs. 2 and 4 in Eq. 10, we find (12)
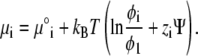 |
(11) |
We assume that the above expression is valid not only in the equilibrium state of the system, but can be used as well to obtain local instantaneous chemical potentials for lipids as the system evolves in time toward the steady state. Then, the generalized force exerted locally on the ith lipid species is given by −∇μi (37). This force is balanced by the friction experienced by all lipid species. Hence, we can write the following set of balance equations (37),
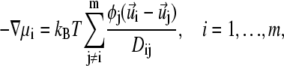 |
(12) |
where Dij values are the so-called diffusivities, or the elements of the diffusion matrix. Based on the experimental findings by Golebiewska et al. (10), showing that the effective diffusion coefficients for uncharged, mono-, and multivalent acidic lipids are similar, we assume here that all diffusivities are equal and simplify Eq. 12 by setting Dij ≡ Dlip for all lipid types. With this approximation, the only distinguishing characteristic between the net-neutral, mono-, and multivalent lipids in our model is their net headgroup charge.
Combining Eq. 12 with the definition of currents, the lipid incompressibility constraint, and with the flux neutrality condition leads to the relationship between currents and gradients in electrochemical potentials:
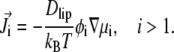 |
(13) |
With the above expression, Eq. 9 simplifies to
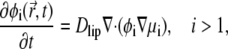 |
(14) |
which is the desired CH equation for the time-evolution of lipid compositions.
Equation 14 is solved for all lipid species i ≠ 1 self-consistently with the nonlinear PB Eq. 8, so that at each iteration the local surface charge density and the electrostatic potential gradient on a membrane surface are linked through the boundary condition,
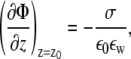 |
(15) |
with z0 describing the position of the charged interface (Fig. 1). The diffusion of i = 1 follows from the material conservation constraint Eq. 2. We note that within the CH formalism, lipid fractions 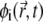 (i = 1, …, m) are globally conserved fields and, for a given time instance t, they obey
(i = 1, …, m) are globally conserved fields and, for a given time instance t, they obey
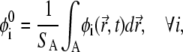 |
(16) |
where SA is the area of a membrane surface A. We stress that, in our model, the lateral motion of lipids is entirely due to the presence of the adsorbed protein. We do not consider any fluctuations, i.e., thermal motions of lipids, which in principle can be introduced into the CH dynamics using a stochastic noise term (27). Describing lateral reorganization of lipid molecules by the CH equations implies that the motion of lipid molecules in the membrane plane obeys the laws of normal (regular) diffusion. In contrast to artificial membranes, lipids in cellular membranes may also undergo anomalous diffusion (see for example (38,21) and reference therein). Therefore, our model does not attempt to describe a priori any anomalous or hop-diffusion dynamics, neither for lipids nor for the adsorbed macromolecule. However, within our model such diffusion could still be the result of lipid-protein interactions.
Mobile protein: hybrid Cahn-Hilliard and dynamic Monte Carlo
To this point, we have assumed that the adsorbing macromolecule remains stationary at a distance h from the membrane surface and only let lipids rearrange laterally in the two-dimensional membrane plane. We can further extend our model to account for the diffusional motion of the protein in the membrane plane within a dynamic Monte Carlo (DMC) scheme (38–44). Within DMC, we start with some particular system configuration as the initial state, and then generate a sequence of other possible trial configurations. Acceptance of these moves represents a stochastic dynamic trajectory of the system.
Our goal is to couple the DMC scheme for the motion of the adsorbing macromolecule on the membrane surface with the CH formalism for lateral rearrangement of lipids (which we term the CHDMC method). Protein and lipids simultaneously diffuse, each with their own typical diffusion rate. The two corresponding timescales in our model are d2/Dlip, for the lateral diffusion motion of lipids, and d2/Dprot, for the diffusion of adsorbing macromolecule, where d is the lattice constant, Dlip is the lipid diffusion coefficient, and Dprot is the diffusion coefficient of the macromolecule on a homogeneous membrane. The ratio of the two diffusion coefficients D′ = Dprot/Dlip determines how close the two relevant timescales are, and therefore also reflects the coupling strength of these different modes of motion. Simply stated—if protein and lipid diffuse at similar rates, their motion is expected to be highly correlated. However, if the protein diffuses much faster than lipid, lipid rearrangement will not achieve complete relaxation, and in the limit of Dprot ≫ Dlip, the motions of lipid and protein can be expected to be largely uncorrelated.
We start the CHDMC simulation with a homogenous distribution of lipids on the membrane, and with the adsorbing macromolecule at distance h from the membrane surface. At the initial step, we allow the adsorbate center of mass to make a random displacement in the two-dimensional membrane plane. In accordance with the fluctuation-dissipation theorem, we treat the random move as a combination of two independent displacements, each of size 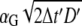 (27), in two membrane plane directions. Here αG is a Gaussian random number with zero mean and unit variance, and the dimensionless time step Δt′ is related by the CH Eq. 14 to the real-time step Δt through
(27), in two membrane plane directions. Here αG is a Gaussian random number with zero mean and unit variance, and the dimensionless time step Δt′ is related by the CH Eq. 14 to the real-time step Δt through
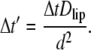 |
(17) |
The move is accepted with a Metropolis-like criterion with the usual transition probability of W = 1 if Fnew ≤ Fold, and  if Fnew > Fold. Here Fold and Fnew are the adsorption free energies of the “old” and trial states of the protein-membrane system, respectively. If a trial move is accepted, the macromolecule is advanced to the “new” position, and the CH equations for lipids are solved taking into consideration the new position of the adsorbate. If, on the other hand, the trial move is rejected, the protein remains at its previous position and lipids rearrange with respect to the old location of the adsorbate. Because time is set by the CH equations, the dimensionless time is updated by Δt′, regardless of the outcome of the DMC move, and the next stochastic step for the protein is attempted. Note that the adsorbate DMC move sizes are not arbitrary, but rather are determined by Δt′. Therefore, in the context of the dynamic scheme, each rejected DMC move can be viewed as a time-interval within which, on average, the protein's position does not change appreciably due to favorable local interactions with underlying charged lipids. Clearly, such an assumption requires the discretization of the CH equations with sufficiently small Δt′ as compared to all relevant timescales in the system. With our choice of Δt′, we found a rejection rate of only ≈1.5–4.6% (depending on the value of D′).
if Fnew > Fold. Here Fold and Fnew are the adsorption free energies of the “old” and trial states of the protein-membrane system, respectively. If a trial move is accepted, the macromolecule is advanced to the “new” position, and the CH equations for lipids are solved taking into consideration the new position of the adsorbate. If, on the other hand, the trial move is rejected, the protein remains at its previous position and lipids rearrange with respect to the old location of the adsorbate. Because time is set by the CH equations, the dimensionless time is updated by Δt′, regardless of the outcome of the DMC move, and the next stochastic step for the protein is attempted. Note that the adsorbate DMC move sizes are not arbitrary, but rather are determined by Δt′. Therefore, in the context of the dynamic scheme, each rejected DMC move can be viewed as a time-interval within which, on average, the protein's position does not change appreciably due to favorable local interactions with underlying charged lipids. Clearly, such an assumption requires the discretization of the CH equations with sufficiently small Δt′ as compared to all relevant timescales in the system. With our choice of Δt′, we found a rejection rate of only ≈1.5–4.6% (depending on the value of D′).
A simple limiting case for the CHDMC algorithm is when lipid distribution on the membrane remains homogenous during the entire simulation. For this case, proteins should perform free diffusion with their effective diffusion coefficient Dprot unaffected by the presence of the membrane. Any protein slowing seen with our model is because of energetic barriers to lateral motion in the membrane plane that arise when charged lipids segregate around the adsorbate. In principle, the adsorbate might be able to overcome these barriers by diffusing away from the membrane and subsequently readsorbing. However, we also show in Results and Discussion that diffusion perpendicular to the membrane plane has an energetic cost. For example, when a globular protein carrying a surface charge of 1e− per 93 Å2 drifts 1 Å away from a membrane with neutral to monovalent lipid ratio of 80:20, we find a loss of ≈kBT in adsorption free energy (see also Fig. 2 a). In contrast, when moving the same distance as a result of a single lateral DMC step in the same system, the protein will face a significantly lower free-energy barrier of, at most, 0.1 kBT. Motion in the perpendicular direction (increasing h) is, therefore, far less likely than lateral motion, where barriers are smaller. This finding allows us to ignore variations in h during the DMC run, as these are associated with prohibitively large energies. Extension to three-dimensional diffusion can be easily incorporated if required in future applications.
FIGURE 2.

Adsorption of a spherical macroion (Rp = 10 Å) onto binary (PC/PS) lipid membranes with 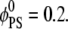 (a) Steady-state (equilibrium) adsorption free energy (in kBT units) as a function of macroion-membrane separation calculated using our method (solid line) and by the method used in May et al. (20) (dashed line). (b) Time sequence of radial profiles of the local fraction of PS lipids, for macroion-membrane separation of h = 3 Å. The steady-state distribution of PS lipids from the calculations by May et al. is shown for comparison (dotted-dashed line).
(a) Steady-state (equilibrium) adsorption free energy (in kBT units) as a function of macroion-membrane separation calculated using our method (solid line) and by the method used in May et al. (20) (dashed line). (b) Time sequence of radial profiles of the local fraction of PS lipids, for macroion-membrane separation of h = 3 Å. The steady-state distribution of PS lipids from the calculations by May et al. is shown for comparison (dotted-dashed line).
Several studies clearly demonstrated that, for the relation between simulated time and real time to be well defined in DMC, the transition probabilities must be calculated from activation energies, rather than energy differences between initial and trial states (38,41). However, if the activation barrier between the initial and trial states is negligible, the so-called Kang-Weinberg probabilities that use barrier heights reduce to the Metropolis probabilities (38,41). Calculation of the activation barrier requires a priori knowledge of all possible pathways the system can follow from some current state. The continuous trajectory of the protein in our model makes this calculation impractical. However, as we show in the Results and Discussion, in our model even the largest possible difference between Fnew and Fold is comparable to kBT, therefore allowing us to assume, following the work of Saxton (38), that we are at the limit of low activation barriers and use the Metropolis criterion for transition probabilities.
Simulation details
We focus on two types of mixed lipid bilayers. The first is composed of binary mixtures (m = 2) of neutral (phosphatidylcholine, i.e., PC) and acidic monovalent (phosphatidylserine, i.e., PS) lipids. The second is a ternary mixture (m = 3) of uncharged (PC), monovalent (PS), and acidic multivalent (PIP2, valency of −4 at neutral pH) components. The discretized version of the CH equation (Eq. 14) for the case of ternary mixtures is presented in the Appendix. For all calculations the lipid membrane was modeled as a low dielectric slab (εm = 2) of dimensions 256 Å × 256 Å × 10 Å. Assuming that a = 65 Å2 is the area per lipid headgroup, the slab dimensions correspond to roughly 1008 lipid molecules in one membrane layer. We note that, in principle, the electrostatic properties on one side of the bilayer may have an impact on those on the other side. Under physiological conditions, when 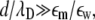 this coupling has been shown to be weak (30,45). We have verified the above inequality by performing calculations on membranes of different thickness, d ≥ 10 Å. We found that the value of the electrostatic potential on the slab surface did not change when we varied the slab thickness. Therefore, to simplify calculations we completely decouple electrostatically the two membrane interfaces, and treat the lipid slab as a leaflet of thickness d = 10 Å (Fig. 1).
this coupling has been shown to be weak (30,45). We have verified the above inequality by performing calculations on membranes of different thickness, d ≥ 10 Å. We found that the value of the electrostatic potential on the slab surface did not change when we varied the slab thickness. Therefore, to simplify calculations we completely decouple electrostatically the two membrane interfaces, and treat the lipid slab as a leaflet of thickness d = 10 Å (Fig. 1).
We note that attempting to describe dielectric properties of the interior of a lipid membrane by a uniform dielectric constant may not always be appropriate. Since solvent molecules generally penetrate deep into the lipid headgroup region, the dielectric constant in this area can reach much higher values than assumed in our work. To test the effect of the εm value on the predictions from our model, we compared the results for a membrane slab of thickness d = 10 Å with uniform εm = 2 to those obtained for a membrane described as two fused slabs, one of d = 5 Å and εm = 20 (interfacial headgroup region), and the other d = 10 and εm = 2 (hydrocarbon tail region). The compared adsorption free energies were within 1.5% of each other. We concluded that treating the entire lipid membrane as a low dielectric media was indeed an adequate representation of the systems investigated here.
At each iteration of the CH Eq. 14, the nonlinear PB Eq. 8 was solved using a modified version of the publicly available open-source software APBS, Ver. 0.4.0 (46). The system was placed on a 256 Å × 256 Å × 256 Å cubic grid with grid spacing of 1 Å, and the nonlinear PB was discretized with the finite-difference method. The APBS software was modified to include periodic boundary conditions in the (xy) bilayer in-plane directions. The charge, ion accessibility, and dielectric maps were configured and supplied to APBS. After each dynamic step, these maps were updated and fed back to the PB solver.
The Cahn-Hilliard equations were discretized on a 256 Å × 256 Å square lattice with 1 Å grid spacing. Convergence of the CH equations to equilibrium was checked by confirming that within numerical uncertainty the lipid electro-chemical potentials for all lipid species at steady state are uniform across the membrane surface. In all simulations, we chose a real time step of 200 ps.
RESULTS AND DISCUSSION
As a first test of the method, we considered the adsorption of a uniformly charged spherical macroion onto an oppositely charged lipid membrane, consisting of binary mixtures of monovalent (PS) and neutral lipids (PC). The equilibrium adsorption free energy of charged macroions on binary membranes, and the steady-state distribution of lipid molecules around the macroion, have previously been derived using the theoretical model of May et al. (20). To validate our numerical methodology, we repeated the calculation performed by May et al. (20), with the expectation that our dynamic minimization scheme of the free energy described in Model should yield a steady-state solution identical, within numerical uncertainty (see Simulation Details), to theirs. The free energies were obtained in May et al. by using the alternative method of solving Eq. 8 with a boundary condition that accounts for lipid mobility. We then considered ternary lipid mixtures of monovalent, multivalent, and neutral lipids, and again applied our model to a uniformly charged spherical macroion (i.e., protein) adsorbing onto such membranes.
Next, we present the model results for the basic poly-peptide (Lysine-13, i.e., Lys13) adsorbing onto similar net acidic membranes consisting of binary and ternary mixtures of lipids.
Finally, we present results from a set of simulations in which the adsorbing macromolecule is allowed to diffuse. We compare diffusion rates of the membrane-bound and free macromolecules, and show that lipid composition plays a crucial role in regulating diffusion properties of peripheral, bound proteins.
Macroion adsorption on mixed neutral and monovalent lipid membranes
Fig. 2 a shows binding (adsorption) free energies (in kBT units) as a function of membrane-macroion separation from our model and as derived from the free-energy functional minimization scheme implemented by May et al. (20). We consider a lipid membrane composition of 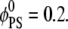 Assuming an area per lipid headgroup of 65 Å2 for both PS and PC lipids, this composition corresponds to one negative charge per 325 Å2 of membrane area. The adsorbing spherical macroion of Rp = 10 Å radius mimics a globular protein carrying a surface charge of 1e− per 93 Å2—that is, 3.5 higher than the average membrane charge density. The macroion was placed at successively higher distances from the membrane surface ranging from 3 Å to 15 Å, and for each separation, lipids were allowed to evolve toward the steady state with the dynamic method described in the previous section.
Assuming an area per lipid headgroup of 65 Å2 for both PS and PC lipids, this composition corresponds to one negative charge per 325 Å2 of membrane area. The adsorbing spherical macroion of Rp = 10 Å radius mimics a globular protein carrying a surface charge of 1e− per 93 Å2—that is, 3.5 higher than the average membrane charge density. The macroion was placed at successively higher distances from the membrane surface ranging from 3 Å to 15 Å, and for each separation, lipids were allowed to evolve toward the steady state with the dynamic method described in the previous section.
Fig. 2 a demonstrates full agreement in binding free energies resulting from the two calculations for all membrane-macroion separations. Fig. 2 b shows the fraction of acidic lipids as a function of the radial distance r from the projected center of the ion onto the membrane surface. We show radial profiles for membrane-ion separation of h = 3 Å, at different times starting from a protein that is adsorbed on a homogenously charged membrane. For comparison, we also plot the theoretically predicted equilibrium charge distribution calculated in May et al. (20).
By following the dynamic evolution of lipid rearrangement around the macroion, we can identify two timescales corresponding to two processes. The first corresponds to a major recruitment of charged lipids toward the interaction zone. This process, driven by the strong electrostatic interactions between oppositely charged macroion and charged lipids, and subsequent counterion release, occurs on short timescales: during the first 100 ns, the fraction of PS lipids close to the ion increases 2.5-fold, whereas during the remaining part of the simulation only minor changes occur close to the macroion. As a consequence of this initial charged lipid sequestration, a deficiency of PS lipids is created starting from ∼r = 20 Å away from the ion resulting in the formation of a depletion-well in the lipid distribution, where φPS(r) < 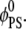 At longer times, the second process of filling up the well begins, as PS lipids from the bulk start flowing into the area of lower electrochemical potential and replace neutral lipids in the process. Fig. 2 b shows the relatively long timescales for the second process, as the depleted zone still persists after 1 μs of simulation.
At longer times, the second process of filling up the well begins, as PS lipids from the bulk start flowing into the area of lower electrochemical potential and replace neutral lipids in the process. Fig. 2 b shows the relatively long timescales for the second process, as the depleted zone still persists after 1 μs of simulation.
Comparison of the lipid distribution after 1 μs of simulation with that of the equilibrium state calculated in May et al. (20) also shows good agreement. The small discrepancy in the two plots at distances r = 20–40 Å is due to the slow filling-up process, and further evolution of the system yields even better agreement with the equilibrium plot. The corresponding free energy at this point is already within numerical accuracy. Close to the ion, our calculation predicts a 2.6% higher fraction of PS lipids, over the prediction by May et al. This minor difference can be attributed to the specific details of the two models and the membrane representation in each. In May et al. (20), the lipid membrane is a continuous planar surface of charge density σmemb. This surface divides space in two with dielectric constant εm = 0 in the nonaqueous volume. The macroion dielectric constant was also taken there to be εm = 0. In contrast, in our model, the lipid bilayer is represented by a rectangular slab of finite thickness d and the desired local charge density is achieved by placing (partial) point charges 1 Å apart on the membrane surface. The dielectric constant inside the slab, as well as inside the adsorbing macroion, is taken as εm = 2 in our model—a value probably more representative of the dielectric properties of macromolecules such as proteins and lipids. This higher εm in the membrane and macroion may allow slightly more charged lipids to segregate (less repulsion from image charges).
To conclude, the set of simulations presented above demonstrates that the numerical solution of Eq. 14 converges at long times to the equilibrium state as it should. Thus, our numerical scheme for calculating free energies is fully validated and allows us to follow lipid diffusion in the membrane plane.
Multivalent versus monovalent lipid segregation
Fig. 3 details lipid segregation in the adsorption process of a spherical macroion on ternary mixtures of neutral (PC), monovalent (PS), and polyvalent (PIP2) lipid membranes containing 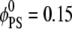 and
and 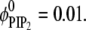 The spherical macroion again has a radius of Rp = 10 Å and a surface charge of 1e−/93 Å2.
The spherical macroion again has a radius of Rp = 10 Å and a surface charge of 1e−/93 Å2.
FIGURE 3.

Adsorption of spherical macroion (Rp = 10 Å) onto ternary (PC/PS/PIP2) lipid membrane with 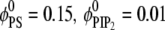 composition. (a) Normalized fraction φ* of PIP2 lipids (upper panel) and PS lipids (lower panel) as a function of the radial distance from the macroion, r, at different times from the initial macroion binding. (b) Time sequence for the electrochemical potentials (μ – μ°), reported in kBT units, of PIP2 (upper panel) and PS (lower panel) lipids upon macroion binding.
composition. (a) Normalized fraction φ* of PIP2 lipids (upper panel) and PS lipids (lower panel) as a function of the radial distance from the macroion, r, at different times from the initial macroion binding. (b) Time sequence for the electrochemical potentials (μ – μ°), reported in kBT units, of PIP2 (upper panel) and PS (lower panel) lipids upon macroion binding.
In Fig. 3 a we plot local lipid fractions reported as ratios of local and average values 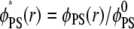 and
and 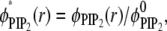 as a function of r at different time steps. In Fig. 3 b, the corresponding time sequence of the electrochemical potentials of PS and PIP2 lipids is shown as radial profiles. Fig. 3 a illustrates a remarkable difference in the organization of the two lipids around the macroion: after 400 ns, the fraction of PIP2 lipids close to the ion increased ∼10-fold, whereas the fraction of PS lipids increased less than twofold. To better quantify the segregation level of PIP2 and PS lipids around the macroion, we define and evaluate the lipid excess for PS and PIP2 at steady state with analogy to the so-called preferential interaction coefficient (or the Gibbs density excess; see, for example, (47,48) and references therein):
as a function of r at different time steps. In Fig. 3 b, the corresponding time sequence of the electrochemical potentials of PS and PIP2 lipids is shown as radial profiles. Fig. 3 a illustrates a remarkable difference in the organization of the two lipids around the macroion: after 400 ns, the fraction of PIP2 lipids close to the ion increased ∼10-fold, whereas the fraction of PS lipids increased less than twofold. To better quantify the segregation level of PIP2 and PS lipids around the macroion, we define and evaluate the lipid excess for PS and PIP2 at steady state with analogy to the so-called preferential interaction coefficient (or the Gibbs density excess; see, for example, (47,48) and references therein):
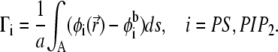 |
(18) |
Here a is the area per lipid headgroup, the integral is taken over the membrane area around the macroion, and 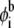 is the lipid composition at the cell boundary (Fig. 3 a). With the above definition, Γi measures the excess number of the ith lipid species around the adsorbate, relative to the respective number in the homogenous membrane. From Eq. 18 we find that after 400 ns the concentration of PIP2 in the first coordination shell is increased by 150%, whereas PS lipid content increased by only 17%. Furthermore, our calculations reveal that during the adsorption process, the macroion attracts −2.1e additional charges (compared to a homogenous membrane) from the acidic lipids, of which PIP2 contributes −1.2e, and PS −0.9e charges. In light of the major role observed for PIP2, despite its very low content in the membrane, we conclude that the binding free energy of the complex is minimized most efficiently by the macroion interacting primarily with PIP2 lipid molecules.
is the lipid composition at the cell boundary (Fig. 3 a). With the above definition, Γi measures the excess number of the ith lipid species around the adsorbate, relative to the respective number in the homogenous membrane. From Eq. 18 we find that after 400 ns the concentration of PIP2 in the first coordination shell is increased by 150%, whereas PS lipid content increased by only 17%. Furthermore, our calculations reveal that during the adsorption process, the macroion attracts −2.1e additional charges (compared to a homogenous membrane) from the acidic lipids, of which PIP2 contributes −1.2e, and PS −0.9e charges. In light of the major role observed for PIP2, despite its very low content in the membrane, we conclude that the binding free energy of the complex is minimized most efficiently by the macroion interacting primarily with PIP2 lipid molecules.
The preferential increase in multivalent phospholipid concentration at the adsorption region over monovalent ones has been reported in several experiments (8,10,49) as well as in theoretical studies (11–13). In our model, the large difference in sequestration of PIP2 versus PS lipids is mainly due to the lower entropic cost associated with recruiting a multivalent lipid. The multivalent lipid carries several charges (better electrostatic interaction) but still loses only the entropy of one free lipid when sequestered compared with four PS lipids that would have to be sequestered instead. In our model, this preference also has an additional dynamic aspect. Highly charged lipids tend to be more mobile and move faster under the influence of an external (protein) electric field. This is because the chemical potential is directly related to the product of the electrostatic potential, and lipids valency (Eq. 11), as are the derivatives of the electrochemical potential that determine lipid mobility. Therefore, PIP2 lipids can be expected to move faster toward the interaction zone, and once sequestered, are expected to remain bound or localized to the protein with preference over PS lipids.
Due to the lower PIP2 content, these lipids might not always segregate quickly enough to the protein's vicinity despite their high mobility, thus being unable to replace the majority of PS lipids that, initially, are already quite abundant. This is expressed in the later time spans of the simulation as a displacement of the initially segregated PS lipids by PIP2 lipids that are thermodynamically favored.
Fig. 3 b follows the changes in local chemical potentials with time. Clearly, the tendency at long times is toward achieving uniform chemical potential throughout the membrane plane for each of the three lipid components (μPC follows from μPS, 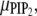 and Eq. 2). The figure also shows clearly that the gradients in initial chemical potentials are much larger for PIP2 than PS, although its membrane content is much smaller. For both lipids, uniform chemical potential can only be achieved by major lipid recruitment to the interaction zone.
and Eq. 2). The figure also shows clearly that the gradients in initial chemical potentials are much larger for PIP2 than PS, although its membrane content is much smaller. For both lipids, uniform chemical potential can only be achieved by major lipid recruitment to the interaction zone.
Poly-peptide adsorption on binary and ternary lipid membranes
A more realistic peptide-membrane interaction is modeled by the adsorption of Lys13 on an oppositely charged (mixed) membrane. For this calculation, Lys13 was represented in all-atom detail, without blocked ends. Atomic radii and partial charges for each peptide atom (including the C-terminus) were derived from the CHARMM force field (50). Following the findings of Ben-Tal et al. (51), we placed Lys13 in a flat conformation next to the membrane such that its plane was parallel to the membrane surface and the minimal distance between the peptide interface and the membrane was h = 3 Å, as shown in Fig. 1. Based purely on the electrostatic calculations, Ben-Tal et al. showed that the free energy of Lys5 binding to 2:1 PC/PS membrane was lowest for this particular configuration (51).
We consider two different lipid compositions with the same surface charge density: a binary mixture with 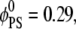 and a ternary mixture with
and a ternary mixture with 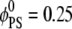 and
and 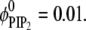 Fig. 4 shows the charged lipid organization for the ternary mixture (Fig. 4, a and b) and binary mixture (Fig. 4 c) upon Lys13 binding. Fig. 4, a and b, show local lipid fractions
Fig. 4 shows the charged lipid organization for the ternary mixture (Fig. 4, a and b) and binary mixture (Fig. 4 c) upon Lys13 binding. Fig. 4, a and b, show local lipid fractions 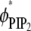 and
and 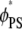 for the ternary system, and Fig. 4 c shows a similar plot for
for the ternary system, and Fig. 4 c shows a similar plot for 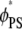 in the binary mixture. Snapshots are taken after 500 ns starting from the homogenous distribution, and the green shades (corresponding to φ* = 1) represent locations on the membrane where the lipid distributions are unaffected by the adsorbing peptide. Darker colors (φ* > 1) show areas with excess lipids, and lighter colors (φ* < 1) identify locations with deficiency in the corresponding lipid species.
in the binary mixture. Snapshots are taken after 500 ns starting from the homogenous distribution, and the green shades (corresponding to φ* = 1) represent locations on the membrane where the lipid distributions are unaffected by the adsorbing peptide. Darker colors (φ* > 1) show areas with excess lipids, and lighter colors (φ* < 1) identify locations with deficiency in the corresponding lipid species.
FIGURE 4.

Adsorption of Lys13 onto ternary (PC/PS/PIP2) lipid membrane with 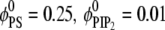 (a and b), and onto binary (PC/PS) lipid membrane with
(a and b), and onto binary (PC/PS) lipid membrane with 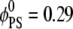 (c). (a) Normalized local fraction of PIP2 lipids in the ternary system. (b) Local PS lipid fractions in the ternary system after 0.5 μs. (c) Local PS lipid fraction in the binary mixture.
(c). (a) Normalized local fraction of PIP2 lipids in the ternary system. (b) Local PS lipid fractions in the ternary system after 0.5 μs. (c) Local PS lipid fraction in the binary mixture.
From Fig. 4 a we learn that the fraction of PIP2 lipid increases up to 4.5-fold (shown in blue) near the Lys13 side chains, where the positive charge is greatest. This area is surrounded by a region with lower PIP2 content (red and purple), showing only 2.5–3-fold increase in multivalent lipid fraction. Because the peptide backbone is rich in both positive and negative charges, there are only minor changes in PIP2 content along the Lys13 backbone with respect to the bulk concentration. Note that the primary donor of PIP2 lipids is the membrane region closest to the C-terminal of the peptide, where the highly acidic carbonyl group repels the negatively charged lipids. Comparing Fig. 4, a and b, reveals that there is almost no sequestration of PS by the peptide. The highest increase in PS lipid is only 1.5-fold, observed, as expected, along the Lys13 side chains.
For comparison, Fig. 4 c shows that even in PIP2-free membranes, the segregation of PS lipids around Lys13 is marginal. There are regions on the membrane where the fraction of PS has increased only approximately twofold. But mostly the plot shows very weak PS sequestration compared to that seen for PIP2.
To better assess the degree of multivalent lipid segregation around the polypeptide, in Fig. 5 we plot the electrostatic potential isosurfaces for Lys13 adsorbed on a ternary (PC/PS/PIP2) lipid membrane. Fig. 5, a and b, show side and top views, respectively, of the system in the initial configuration, with homogenous lipid distribution on the membrane, and Fig. 5, c and d, provide similar views of the final state of the system, after 500 ns. Van der Waals radii for Lys13 are colored in gray. Isocontours are for Φ = −1.5 kBT/e (−37.5 mV) (red surface), and Φ = +1.5 kBT/e (+37.5 mV) (blue mesh). For clarity, the lipid membrane is not shown. Fig. 5 reveals significant change in the electrostatic potential near Lys13 after 500 ns of dynamics. Comparing Fig. 5, a and b (or Fig. 5, c and d), illustrates the growth of the negative electrostatic potential isosurface close to Lys13 as PIP2 lipids segregate around the polypeptide (Fig. 4 a). This increase is because accumulation of PIP2 near Lys13 reduces gradients in the local electrochemical potential, which, in turn, is proportional to the electrostatic potential. Similar plots for Lys13-PC/PS membrane (data not shown) reveal much weaker electrostatic potential near the adsorbate, indicative of lower charge accumulation around Lys13.
FIGURE 5.

Electrostatic potential isosurfaces for Lys13 adsorbed on a ternary (PC/PS/PIP2) lipid membrane. (a and b) Side and top views, respectively, of the system in the initial configuration. (c and d) Similar views for the final state of the system, after 500 ns. Lys13 van der Waals surfaces are colored in gray. The red surface represents Φ = −1.5 kBT/e (−37.5 mV) equipotential contour, and the blue mesh depicts the Φ = +1.5 kBT/e (+37.5 mV) equipotential contour. For clarity, the lipid membrane is not shown.
In conclusion, our model predicts sequestration primarily of PIP2 lipids by the adsorbing basic peptide, and very weak sequestration of PS lipids. This result is consistent with experimental observations by Golebiewska et al. (10), and with the model predictions by Wang et al. (13) and Tzlil et al. (11).
Diffusion of macroion on binary and ternary lipid membranes
Above we showed that stationary basic macromolecules will preferentially sequester PIP2 lipids. Because this picture might change if the adsorbate is allowed to diffuse, we considered the effect of protein mobility. By introducing this degree of freedom we allow the system access to states around the mean field free-energy minimum, and with that we pose several subtly related questions: how are the macromolecule diffusion rates affected by the acidic lipids in the membrane, and how will different lipids influence the apparent protein diffusion rates? To address these questions, we performed a set of calculations in which a (model) spherical macroion (Rp = 10 Å radius and uniform surface charge of 1e−/93 Å2 kept at h = 3 Å from the membrane) was allowed to move concomitantly with lipid diffusion. By performing CHDMC simulations, as detailed in Model, we studied mixed membranes of two different lipid compositions: binary mixtures with 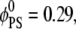 and ternary mixtures with
and ternary mixtures with 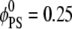 and
and 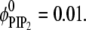
For protein mobility, we focus on two typical cases. In the first, the model protein has a diffusion constant much faster than that of lipids in the unperturbed (bare) membrane, D′ ≡ Dprot/Dlip = 10, while in the second the diffusion constant is comparable to that of the lipids, D′ = 2. As we show, these two scenarios lead to different lipid and protein diffusion characteristics.
Modeling a fast protein diffusing over binary versus ternary membranes
Using color grade, Fig. 6 c shows the local lipid fractions 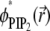 after 0.6 μs of dynamic time evolution. The model protein's trajectory for the entire time interval is shown as a black line that follows the protein projected center-of-mass. The dotted red line indicates the size of the macroion projected onto the membrane, with the arrow indicting the initial protein position. For clarity, we zoom in on the membrane surface region explored by the macroion. The entire trajectory can also be found as an animation file in Supplementary Material, file No. TM-10D.avi.
after 0.6 μs of dynamic time evolution. The model protein's trajectory for the entire time interval is shown as a black line that follows the protein projected center-of-mass. The dotted red line indicates the size of the macroion projected onto the membrane, with the arrow indicting the initial protein position. For clarity, we zoom in on the membrane surface region explored by the macroion. The entire trajectory can also be found as an animation file in Supplementary Material, file No. TM-10D.avi.
FIGURE 6.

Diffusion of charged spherical macroion on mixed membranes. The panels show the local surface charge densities after 0.6 μs of simulations (color scale) and the entire macroion trajectories in that time (connected black lines) for binary (PC/PS) mixture, D′ = 10 (a); for binary (PC/PS) mixture, D′ = 2 (b); for ternary (PC/PS/PIP2) mixture, D′ = 10 (c); and for ternary (PC/PS/PIP2) mixture, D′ = 2 (d). The red-dashed circles on each panel represent the projected size of the macroion with black arrows indicating the starting position of macroion center of mass. For clarity, the figures zoom in on the relevant membrane surface region explored by the macroion.
Following the time evolution of the system reveals local lipid rearrangements on the membrane as the macroion moves, and PIP2 lipids segregate around it. Moreover, there is a prominent retardation in the macroion's movement (see quantitative discussion below). Due to lipid rearrangement, the adsorbate diffusion becomes confined to the area rich in PIP2 for a limited time. However, due to the model protein's high mobility compared to that of lipids, the adsorbate occasionally and temporarily escapes, leaving behind the multivalent lipid cloud that had segregated around it.
The free diffusion of the macroion does not last too long, because PIP2 lipids quickly segregate again around the new position. This segregation is due to the large forces acting on the PIP2 lipids by the electrostatic field emanating from the adsorbate. Concomitantly, the local lipid composition in the region of the membrane just abandoned by the macroion is restored to that of the homogenous mixture. Essentially, the macroion diffuses and drags PIP2 lipids along the way, while the PIP2 units that are segregated retard the free diffusion of the protein. We found that PS segregation for ternary mixtures is very weak, in accordance with our previous findings (Fig. 4 b). Therefore, we show here only the local changes in PIP2 lipids.
We contrast these conditions with the same rapid model protein (D′ = 10) diffusing on a binary membrane containing monovalent (PS) lipids. The corresponding trajectory for the binary mixture after 0.6 μs is shown in Fig. 6 a, and also in Supplementary Material, file No. BM-10D.avi. The color grade here describes the local PS fraction 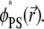 Clearly, acidic (PS) lipids segregate around the macroion to a much lesser extent compared to the ternary mixture, resulting in low energetic barriers to adsorbate motion. Hence, the diffusion of the macroion here is less restricted compared to that seen for the ternary mixture.
Clearly, acidic (PS) lipids segregate around the macroion to a much lesser extent compared to the ternary mixture, resulting in low energetic barriers to adsorbate motion. Hence, the diffusion of the macroion here is less restricted compared to that seen for the ternary mixture.
Slow protein diffusing over binary versus ternary membranes
Diffusion of a slower model protein, with D′ = 2, on the same binary and ternary membranes (Fig. 6, b and d, respectively, and also in Supplementary Material, files No. BM-2D.avi and No. TM-2D.avi) shows qualitatively similar behavior to that observed for D′ = 10. However, due to the lower mobility of the macroion, the acidic lipids have more time to segregate near the adsorbate, and therefore segregate more strongly. The result is that the majority of macroion moves are restricted to the acidic lipid-rich patch that forms close to the protein. This is particularly noticeable for the ternary system, where the macroion practically never escapes to go beyond the circular patch formed by PIP2 lipids, but rather diffuses within it. Whereas for the fast protein on ternary mixtures we observe creation and destruction of macroion/PIP2 “binding sites”, for the slower protein this lipid-protein “complex” stays intact for the entire trajectory. In a sense there are always PIP2 lipids associated with the macroion as it diffuses on the membrane.
Diffusion analysis
We now turn to the quantitative analysis of these simulation results. The final snapshots in Fig. 6 show that, on the simulation timescales, the adsorbate explores a more extended region of space when it diffuses on binary, rather than ternary membrane mixtures. To quantify this finding, we calculated the mean-square-displacement (MSD) 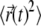 of the macroion as a function of time for the systems shown in Fig. 6. Fig. 7 shows the MSD for a given time lag Δt, obtained by averaging over all pairs of points Δt time-steps apart (52,53). For each system, we only analyzed the last 400 ns of the trajectories. In Fig. 7, we plot
of the macroion as a function of time for the systems shown in Fig. 6. Fig. 7 shows the MSD for a given time lag Δt, obtained by averaging over all pairs of points Δt time-steps apart (52,53). For each system, we only analyzed the last 400 ns of the trajectories. In Fig. 7, we plot 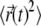 for the first 150 ns of the productive runs, as sampling becomes poor for longer time lags (52,53). If we assume that, in all cases, the relationship between the MSD and elapsed time is linear,
for the first 150 ns of the productive runs, as sampling becomes poor for longer time lags (52,53). If we assume that, in all cases, the relationship between the MSD and elapsed time is linear, 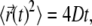 we can derive the effective (observed) protein diffusion constant from a linear regression analysis of the data. For all simulations, the diffusion coefficient of membrane-bound macroion, Dbound, was calculated first, and then we obtained D′bound = Dbound/Dlip, which describes the draglike effect that lipids can have on the protein. The results are summarized in the table inset shown in Fig. 7.
we can derive the effective (observed) protein diffusion constant from a linear regression analysis of the data. For all simulations, the diffusion coefficient of membrane-bound macroion, Dbound, was calculated first, and then we obtained D′bound = Dbound/Dlip, which describes the draglike effect that lipids can have on the protein. The results are summarized in the table inset shown in Fig. 7.
FIGURE 7.

Mean-square-displacement (MSD) as a function of time for D′ = 10 (upper panel) and D′ = 2 (lower panel). The table inset shows the apparent macroion and lipid diffusion coefficient ratios for free and for membrane-bound macroion, the latter calculated from linear regression analysis of the MSD plots.
We find that PIP2-containing membranes have a stronger (relative) impact on the diffusion rate of the macroion, especially when the protein is fast D′ = 10, with the adsorbate being slowed down to D′bound = 3.4. In contrast, for the binary mixtures we found D′bound = 7.5. For the slower proteins, D′ = 2, diffusion rates of the macroion bound to binary versus ternary membranes are similar, but still PIP2 containing membranes have a slightly stronger effect. Differences in MSD plots between binary and ternary mixtures in our model result from different rejection rates of macroion-DMC moves in the respective systems. Consistent with our MSD data, we found a range of 4.6% and 2.5% rejection rates for the fast moving proteins on ternary versus binary mixtures, and 1.8% and 1.5% rejection rates for the corresponding D′ = 2 systems.
Why do multivalent lipids influence the adsorbate diffusion rates more than monovalent lipids? And what role does the inherent (or free) diffusion rate of the macroion (diffusion rate on homogeneous surfaces) play in this process? Assume that the adsorbate inherently travels with high diffusion rate, D′ = 10. Then, only highly mobile lipids would be able to segregate around the protein. Our simulations of ternary mixtures show that PIP2 can be fast enough. These multivalent lipids experience strong electrostatic forces from the adsorbate because of their strongly charged headgroups and therefore are quickly sequestered near the macroion. Once sequestered, PIP2 lipids act to confine the motion of the adsorbate, as any protein motion leaves behind an exposed charged patch that is highly unfavorable. The protein thus finds it difficult to overcome and escape this electrostatic-well created by sequestered PIP2 molecules (8,10,49), and the retarded macroion has a lower diffusion rate.
In contrast, monovalent (PS) lipids segregate only weakly around a quick macroion. Hence, the energetic barriers created by these sequestered acidic lipids are low and the adsorbate, due to its high mobility, finds it easy to consistently escape them. The preferential interaction coefficients show on average −1.2e membrane surface charges for the fast protein on ternary mixtures versus −1.0e in the binary mixture accumulated near the macroion. Furthermore, our calculations reveal that PIP2 and PS lipids contribute with their headgroup charges almost equally in the ternary mixtures, −0.7e charge coming from PIP2 and −0.5e from PS. Based on these findings and in light of substantial difference in mono- and multivalent lipid composition, we conclude that localized electronic charge on multivalent PIP2 lipid plays a major role in regulating the macroion diffusion rate.
As the free macroion diffusion rate decreases, monovalent and multivalent lipids tend to affect adsorbate diffusion rates to a similar extent, as revealed from the MSD plots for D′ = 2, Fig. 7. This is because the underlying acidic lipids are able to rearrange around such slow-diffusing adsorbates, resulting in lipid segregation rather similar to that observed in equilibrium for the immobile protein. Under these conditions, even the less mobile PS lipids can retard the adsorbate's diffusion. Conversely, since the protein is slow enough so that all lipids can relax for all protein steps, the relative drag experienced by the protein and its slowing down are smaller than for fast proteins.
From Figs. 6 and 7 it is evident that there is some level of confinement with possible binding and unbinding events of interacting lipid and proteins, and hence the MSD plots need not be interpreted as strictly linear. In fact, the higher the protein diffusion rate D′, the more anomalous the observed macroion diffusion on the ternary membranes. In the limit of the unrealistically high D′ = 50, our simulations (data not shown) show adsorbate motion on ternary membranes that includes extended local motions with occasional rapid diffusion, resembling a hop-diffusion-type mechanism (52,53). Importantly, even for such extreme D′, multivalent lipids are still capable of affecting protein diffusion rate. On the other hand, for large D′ the diffusion on the binary mixtures is still close to linear, and for D′ = 50 we see practically no slowing-down of the macroion diffusing on a binary PC/PS membrane. Obviously, in the limit of 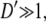 protein and lipid motion will become largely uncorrelated regardless of whether the protein is diffusing on ternary or binary membrane.
protein and lipid motion will become largely uncorrelated regardless of whether the protein is diffusing on ternary or binary membrane.
Because the DMC method allows thermal fluctuations on the order of kBT away from the mean-field free energy, it is interesting to follow these changes for different membrane-protein interactions. In Fig. 8, a and b, we plot the instantaneous adsorption free energies (in thermal energy units) given by Eq. 4 as the macroion diffuses on binary and ternary mixtures, respectively. In both panels, the horizontal lines (D′ = 0) represent the calculated equilibrium mean-field binding free energies for the respective stationary macroion and membrane complexes (no protein motion is allowed), measured with respect to the unbound protein and bare membrane in solution. However, lipids are still mobile and are allowed to segregate around the stationary macroion.
FIGURE 8.

Value of the instantaneous adsorption free-energy functional ΔF (in kBT units) as a function of time in the CHDMC simulations for (a) D′ = 2 systems and for (b) D′ = 10. The horizontal lines show the calculated equilibrium adsorption free energies for the respective systems when the macroion is stationary (see text for details).
Fig. 8 shows that the adsorption energies on ternary mixtures are always stronger (more favorable) than for the corresponding binary mixtures. This is due to the stronger segregation of charged lipids around the macroion in the ternary mixtures (see also Fig. 6). Within fluctuations, the binding free energies for the ternary mixtures are similar for different protein diffusion rates, but are somewhat different between the two binary mixtures. This indicates that the macromolecule diffusing on ternary mixed membranes on average sequesters PIP2 lipids to a similar extent, whether traveling as slow as D′ = 2 or as fast as D′ = 10. For the binary system, in contrast, segregation of lipids is more tightly correlated with the diffusion rate of the macroion.
Note that the largest possible difference in binding free energies between any two states of the mobile protein/membrane complex is ∼1.5 kBT, as expected for thermal fluctuations. In one state the membrane is homogenous, and the lipids are ideally mixed (adsorption free energy at t = 0 in Fig. 8, a and b), and in the other state lipids are completely relaxed around the immobile adsorbed protein (D′ = 0 blue lines in Fig. 8, a and b). Because a stationary macroion sequesters charged lipids most efficiently, the equilibrium-binding free energies for the stationary macroion/membrane complexes in Fig. 8 provide a lower-bound for binding free energies of the macroion/membrane systems. The plots reveal that the adsorption free energies for the ternary mixtures are close to the lowest possible values at multiple timepoints in the simulation, indicating that at those instances the mobile protein sequestered multivalent lipids to an extent similar to a stationary protein. The picture is much different when the macroion diffuses on mixed binary membranes: the adsorption free energies never reach the limiting value, because lipids are too slow to relax locally around the protein.
CONCLUDING REMARKS
We have described a self-consistent dynamic mean-field model to study the process of adsorption of charged macromolecules onto oppositely charged lipid membranes. Extending earlier thermodynamic calculations (20), our model adds the dynamic aspects of lipid lateral reorganization and demixing when adsorbed charged macromolecules diffuse upon membranes. In particular, to our knowledge this is a first attempt to study the kinetic mechanisms of acidic monovalent and multivalent lipid segregation around model basic proteins.
The model allows us to solve the full three-dimensional electrostatic problem within the nonlinear Poisson-Boltzmann theory rapidly and efficiently, and at the same time to use the diffusion-like CH equation for information about the time-evolution of lateral reorganization of the membrane components in the adsorption process. On a practical note, we register significant gains in computational time compared to the atomistic MD method. With only modest CPU time requirements, our method explores the behavior of protein-membrane complexes on the microsecond timescale; with rapid progress in PB solvers in three dimensions, the method should become a powerful tool for the study of lipid membrane patches of ∼0.1 μm in lateral dimensions, interacting with proteins of any size. Based entirely on measurable properties derived either from experiments or atomic level simulations (such as lipid diffusion constants, atomic charges, and dielectric constants), results from our model are in good agreement with experimental data and provide valuable predictions about the dynamic aspects of protein adsorption on lipid membranes.
While we have focused here on fully mixed lipid membranes that are far from the critical point, it is important to note that the present formulation of the model makes it possible to also introduce enhanced descriptions, e.g., for nonideal lipid mixing. Such contributions could be incorporated by addition of a phenomenological interaction term to the free-energy functional (Eq. 4) as done in regular solution theory (28) and similar to May et al. (54), which then enables the description of such processes as lipid kinetics in phase-separating mixed membranes upon protein binding. Alternatively, phase separation can be realized here by coupling the lipid fraction field φi to an additional order parameter field that describes lipid-lipid interactions within the membrane (55,56). These extensions would implicitly introduce additional degrees of freedom and would add an additional distinction between different lipids, which may differ not only by their headgroup charge (as in the current model) but also, say, by their hydrocarbon tail content, important for elastic and nonideal mixing properties.
The method presented here is somewhat similar to the Poisson-Nernst-Planck (PNP) formalism (57). PNP combines the Nernst-Planck theory of electrodiffusion with the Poisson equation for electrostatics. The latter is used to compute the electrostatic potential in space, while the time evolution of the local concentrations of mobile ions is described by the diffusion equation, and solved self-consistently with the Poisson equation. With analogy to PNP theory, the steady-state distribution of lipids in the lipid diffusion process is obtained here through the diffusion-like CH equation (Eq. 14) (27). Solving the CH equation requires knowledge of local gradients in electrochemical potentials for different lipids. In our model this is achieved self-consistently through the free-energy functional (yielding the electrochemical potential) and the solution of nonlinear Poisson-Boltzmann equation. As common to mean-field theories, it is an assumption of our model that the functional form of the free energy does not change during the dynamic evolution of the system, and that the instantaneous local electrochemical potentials for different lipids at each step can be obtained from the free-energy functional through Eq. 10. In fact, our model is simply a realization of a time-dependent mean-field approach where forces are derived from a free energy that is a functional of local densities (23–26).
We note that this formalism presents important advantage over an alternative approach in which a free-energy functional could be minimized with respect to the local concentrations of all lipid species. Thus, the solution of the CH equation leads not only to the equilibrium distribution of lipid molecules, but also informs us about the kinetics of the segregation process. In addition, our method uses existing and publicly available high-power PB solvers in three dimensions, and incorporates complex boundary conditions without the need to explicitly couple additional differential equations for the boundary, as was done previously (20).
One simplifying assumption in this work pertains to the static conformational rearrangements in the protein and membrane components. Thus, the free-energy functional describing our system does not include contributions from protein internal degrees of freedom, and we treat adsorbing proteins as rigid constellations of fixed partial charges. Our model can be extended to allow for additional protein flexibility, for instance, by sampling different conformations of the protein, similar to Ben-Tal et al. (51), and for each conformation minimize the free energy further with our dynamic scheme. Because we do not consider here any intramolecular motions in the protein, using a low dielectric value of εm = 2 inside the adsorbing macromolecule is more appropriate compared to the higher values typically used to describe additional polarization due to molecular reorganizations inside the protein (58). We also do not explicitly account for hydration interactions, but this can be introduced, for example, using additional phenomenological forces (22,59).
The results from the application of the model are in good agreement with previous studies, e.g., by showing that the adsorption of positively charged model proteins onto binary (PC/PS) and ternary (PC/PS/PIP2) lipid membranes with composition of biological relevance result in significant segregation of PIP2 but not PS lipids around the stationary basic macromolecules (10–13). Our method revealed as well that the time evolution ultimately leading to the steady-state distribution of lipids consists of at least two distinct processes. The first is an electrostatically driven segregation of charged lipids around the adsorbing macromolecule, and the second involves replenishing of charged lipid in the depletion belt that is due to the initial sequestration. Because we combined our model with dynamic Monte Carlo simulation of the propagation of an adsorbed macromolecule in time, we were able to show that multivalent lipids quickly segregate and remain sequestered near the oppositely charged macromolecules, slowing down adsorbate diffusion in the process. This allows us to conclude that PIP2 lipids can diffuse in concert with adsorbed molecules even when the diffusion of the adsorbate is much faster than lipid diffusion. In contrast, monovalent PS lipids segregate only weakly so that macromolecule and lipid diffusion will remain largely uncorrelated. The difference in behavior of the lipid species arises because PIP2 lipids, in the presence of the protein electric field, are much more mobile than PS, due to their higher charge and hence larger chemical potential.
Predictions from our model bear interesting implications for the role of PIP2 lipids in anchoring natively unstructured domains (and other peripherally bound proteins to lipid membranes). Clearly, to carry out their function, peripheral proteins must often remain localized in certain regions on the membrane for some duration of time. This requires a mechanism that would slow down diffusion across the membrane in the region in which they must act. In agreement with recent experimental observations (9,10), our model finds segregation of PIP2 lipids around the diffusing charged protein which keeps these lipids effectively bound to the protein vicinity, and retards the protein's diffusion.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank Nathan Baker, Michael Holst, and Todd Dolinsky for their advice on modifying the APBS code. We are also grateful to Jim Sethna, Adrian Parsegian, and Brian Todd for valuable comments.
G.K. and H.W. are supported by National Institutes of Health grants No. P01 DA012408 and No. P01 DA012923. D.H. thanks the Israeli Council of Higher Education for its support through an Alon Fellowship, as well as a Career Development Award from the Hebrew University Intramural Research Fund. The Fritz Haber Research Center is supported by the Minerva Foundation, Munich, Germany.
APPENDIX
In this Appendix we rewrite the continuity equations from Model for the case of a protein absorbing onto a lipid membrane consisting of a ternary mixture of neutral (PC, i = 1), monovalent (PS, i = 2), and multivalent (PIP2, i = 3) lipids, and describe the procedure for the discretization of the continuity equations.
We write the two relevant equations corresponding to Eq. 14 for charged lipids in the mixture:
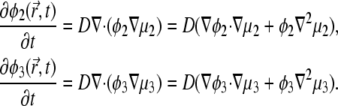 |
(19) |
Electrochemical potentials for PS and PIP2 lipids are given by
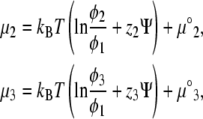 |
(20) |
where z2 = −1 and z3 = −4 are the valences of PS and PIP2 lipids, respectively.
We introduce the lattice constant d and the dimensionless time step,
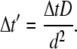 |
(21) |
The expressions in Eq. 19 are expressed using the dimensionless units and are discretized in the following manner:
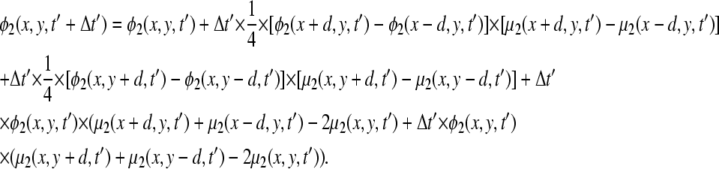 |
(22) |
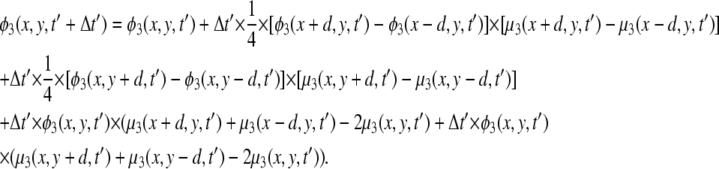 |
(23) |
Editor: Lukas K. Tamm.
References
- 1.Hurley, J. H., and S. Misra. 2000. Signaling and subcellular targeting by membrane binding domains. Annu. Rev. Biophys. Biomol. Struct. 29:49–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemon, M. A., and K. M. Ferguson. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domain. Biochem. J. 350:1–18. [PMC free article] [PubMed] [Google Scholar]
- 3.Hamada, K., T. Shimizu, T. Matsui, S. Tsukita, and T. Hakoshima. 2000. Structural basis of the membrane-targeting and unmasking mechanisms of radixin FERM domain. EMBO J. 19:4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peter, B. J., H. M. Kent, I. G. Mills, Y. Vallis, P. J. G. Butler, P. R. Evans, and H. T. McMahon. 2004. BAR domains as sensors of membrane curvature: the ampiphysin BAR structure. Science. 303:495–499. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin, S., and D. Murray. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 438:605–611. [DOI] [PubMed] [Google Scholar]
- 6.Heo, W. D., T. Inoue, W. S. Park, M. L. Kim, B. O. Park, T. J. Wandless, and T. Meyer. 2006. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 314:1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeung, T., M. Terebiznik, L. Yu, J. Silvius, W. M. Abidi, M. Phillips, T. Levine, A. Kapus, and S. Grinstein. 2006. Receptor activation alters inner surface potential during phagocytosis. Science. 313:347–351. [DOI] [PubMed] [Google Scholar]
- 8.Wang, J., A. Gambhir, G. Hangyas-Mihalyne, D. Murray, U. Golebiewska, and S. McLaughlin. 2002. Lateral sequestration of phosphatidylinositol 4,5-biphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277:34401–34412. [DOI] [PubMed] [Google Scholar]
- 9.Nomikos, M., A. Mulgrew-Nesbitt, P. Pallavi, G. Mihalyne, I. Zaitseva, K. Swann, F. A. Lai, D. Murray, and S. McLaughlin. 2007. Binding of phosphoinositide-specific phospholipase C-ζ (PLC-ζ) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J. Biol. Chem. 282:16644–16653. [DOI] [PubMed] [Google Scholar]
- 10.Golebiewska, U., A. Gambhir, G. Hangyas-Mihalyne, I. Zaitseva, J. Radler, and S. McLaughlin. 2006. Membrane-bound basic peptides sequester multivalent (PIP2), but not monovalent (PS), acidic lipids. Biophys. J. 91:588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzlil, S., and A. Ben-Shaul. 2004. Flexible charged macromolecules on mixed fluid lipid membranes: theory and Monte Carlo simulations. Biophys. J. 89:2972–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haleva, E., N. Ben-Tal, and H. Diamant. 2004. Increased concentration of polyvalent phospholipids in the adsorption domain of a charged protein. Biophys. J. 86:2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J., A. Gambhir, S. McLaughlin, and D. Murray. 2004. A computational model for the electrostatic sequestration of PI(4,5)P2 by membrane-adsorbed based peptides. Biophys. J. 86:1969–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaughlin, S., J. Wang, A. Gambhir, and D. Murray. 2002. PIP2 and proteins: interactions, organization, and information flaw. Annu. Rev. Biophys. Struct. 31:151–175. [DOI] [PubMed] [Google Scholar]
- 15.Gomperts, B., P. Tatham, and I. Kramer. 2003. Signal Transduction. Academic Press, San Diego, CA.
- 16.Record, M. T., Jr., C. F. Anderson, and T. M. Lohman. 1978. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 11:103–178. [DOI] [PubMed] [Google Scholar]
- 17.Parsegian, V. A., and D. Gingell. 1972. On the electrostatic interaction across a salt solution between two bodies bearing unequal charges. Biophys. J. 81:1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner, K., D. Harries, S. May, V. Kahl, J. O. Raedler, and A. Ben-Shaul. 2000. Direct evidence for counterion release upon cationic lipid-DNA condensation. Langmuir. 16:303–306. [Google Scholar]
- 19.Sharp, K. A., R. A. Friedman, V. Misra, J. Hecht, and B. Honig. 1995. Salt effects on polyelectrolyte-ligand binding: comparison of Poisson-Boltzmann, and limiting law/counterion binding models. Biopolymers. 36:245–262. [DOI] [PubMed] [Google Scholar]
- 20.May, S., D. Harries, and A. Ben-Shaul. 2000. Lipid demixing and protein-protein interactions in the adsorption of charged proteins on mixed membrane. Biophys. J. 79:1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiwara, T., K. Ritchie, H. Murakoshi, K. Jacobson, and A. Kusumi. 2002. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harries, D., S. May, W. M. Gelbart, and A. Ben-Shaul. 1998. Structure, stability, and thermodynamics of lamellar DNA-lipid complexes. Biophys. J. 75:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marconi, U. M. B., and P. Tarazona. 1999. Dynamic density functional theory of fluids. J. Chem. Phys. 110:8032–8044. [Google Scholar]
- 24.Fang, F., and I. Szleifer. 2003. Competitive adsorption in model charged protein mixtures: equilibrium isotherms and kinetic behavior. J. Chem. Phys. 119:1053–1065. [Google Scholar]
- 25.Fang, F., and I. Szleifer. 2006. Controlled release of proteins from polymer-modified surfaces. Proc. Natl. Acad. Sci. USA. 103:5769–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamant, H., and D. Andelman. 1997. Adsorption kinetic of surfactants at fluid-fluid interfaces. Prog. Colloid Polym. Sci. 103:51–59. [Google Scholar]
- 27.Chaikin, P. M., and T. C. Lubensky. 2000. Principles of Condensed Matter Physics. Cambridge University Press, Cambridge, UK.
- 28.Hill, T. L. 1987. An Introduction to Statistical Thermodynamics. Dover Publications, Mineola, NY.
- 29.Andelman, D., M. M. Kozlov, and W. Helfrich. 1994. Phase transitions between vesicles and micelles driven by competing curvature. Europhys. Lett. 25:231–236. [Google Scholar]
- 30.Sharp, K. A., and B. Honig. 1990. Electrostatic interactions in macromolecules: theory and applications. Annu. Rev. Biophys. Chem. 19:301–332. [DOI] [PubMed] [Google Scholar]
- 31.Reiner, E. S., and C. J. Radke. 1990. Variational approach to the electrostatic free energy in charged colloidal suspensions: general theory for open systems. J. Chem. Soc., Faraday Trans. 86:3901–3912. [Google Scholar]
- 32.Honig, B., and A. Nicholls. 1995. Classical electrostatics in biology and chemistry. Science. 268:1144–1149. [DOI] [PubMed] [Google Scholar]
- 33.Borukhov, I., D. Andelman, and H. Orland. 1997. Steric effects in electrolytes: a modified Poisson-Boltzmann equation. Phys. Rev. Lett. 79:435–438. [Google Scholar]
- 34.Fogolari, F., and J. M. Briggs. 1997. On the variational approach to the Poisson-Boltzmann free energies. Chem. Phys. Lett. 281:135–139. [Google Scholar]
- 35.Murray, D., A. Arbuzova, B. Honig, and S. McLaughlin. 2002. The role of electrostatic and nonpolar interactions in the association of peripheral proteins with membranes. Current Topics Membr. 52:277–307. [Google Scholar]
- 36.Chandrasekar, S. 1943. Stochastic problems in physics and astronomy. Rev. Mod. Phys. 15:1–89. [Google Scholar]
- 37.Krishna, R., and J. A. Wesselingh. 1997. The Maxwell-Stefan approach to mass transfer. Chem. Eng. Sci. 52:861–911. [Google Scholar]
- 38.Saxton, M. 1996. Anomalous diffusion due to binding: a Monte Carlo study. Biophys. J. 70:1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Binder, K., and D. W. Heermann. 2002. Monte Carlo Simulation in Statistical Physics, 4th Ed. Springer-Verlag, Berlin.
- 40.Fichthorn, K. A., and W. H. Weinberg. 1991. Theoretical foundations of dynamical Monte Carlo simulations. J. Chem. Phys. 95:1090–1096. [Google Scholar]
- 41.Kang, H. C., and W. H. Weinberg. 1992. Monte Carlo simulations of surface-rate processes. Acc. Chem. Res. 25:253–259. [Google Scholar]
- 42.Baumgartner, A. 1980. Statics and dynamics of the freely jointed polymer chain with Lennard-Jones interaction. J. Chem. Phys. 72:871–879. [Google Scholar]
- 43.Graf, P., A. Nitzan, M. G. Kurnikova, and R. D. Coalson. 2000. A dynamic lattice Monte Carlo model of ion transport in inhomogeneous dielectric environments: method and implementation. J. Phys. Chem. B. 104:12324–12338. [Google Scholar]
- 44.Chern, S.-S., A. E. Cardenas, and R. D. Coalson. 2001. Three-dimensional dynamic Monte Carlo simulations of driven polymer transport through a hole in a wall. J. Chem. Phys. 115:7772–7782. [Google Scholar]
- 45.Andelman, D. 1995. Electrostatic properties of membranes: the Poisson-Boltzmann theory. In Handbook of Biological Physics. Elsevier Science, Amsterdam, The Netherlands.
- 46.Baker, N. A., D. Sept, S. Joseph, M. J. Holst, and J. A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schurr, J. M., D. P. Rangel, and S. R. Aragon. 2005. A contribution to the theory of preferential interaction coefficient. Biophys. J. 89:2258–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gambhir, A., G. Hangyas-Mihalyne, I. Zaitseva, D. S. Cafiso, J. Wang, D. Murray, S. N. Pentyala, S. O. Smith, and S. McLaughlin. 2004. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86:2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 51.Ben-Tal, N., B. Honig, R. M. Peitzsch, G. Denisov, and S. McLaughlin. 1996. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys. J. 71:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saxton, M. J. 1997. Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 72:1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kusumi, A., Y. Sako, and M. Yamamoto. 1993. Confined lateral diffusion of membrane receptors as studies by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys. J. 65:2021–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.May, S., D. Harries, and A. Ben-Shaul. 2002. Macroion-induced compositional instability of binary fluid membranes. Phys. Rev. Lett. 89:268102. [DOI] [PubMed] [Google Scholar]
- 55.Khelashvili, G., S. A. Pandit, and H. L. Scott. 2005. Self-consistent mean-field model based on molecular dynamics: application to lipid-cholesterol bilayers. J. Chem. Phys. 123:034910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandit, S. A., G. Khelashvili, E. Jakobsson, A. Grama, and H. L. Scott. 2007. Lateral organization in lipid-cholesterol mixed bilayers. Biophys. J. 92:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardenas, A. E., R. D. Coalson, and M. G. Kurnikova. 2000. Three-dimensional Poisson-Nernst-Planck theory studies: influence of membrane electrostatic on Gramicidin A channel conductance. Biophys. J. 79:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rocchia, W., E. Alexov, and B. Honig. 2001. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 105:6507–6514. [Google Scholar]
- 59.Sitkoff, D., K. A. Sharp, and B. Honig. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]