

Abstract
Pre-elafin is a tight-binding inhibitor of neutrophil elastase and myeloblastin; two enzymes thought to contribute to tissue damage in lung emphysema. Previous studies have established that pre-elafin is also an effective anti-inflammatory molecule. However, it is not clear whether both functions are linked to the antipeptidase activity of pre-elafin. As a first step toward elucidating the structure/function relationship of this protein, we describe here the construction and characterization of pre-elafin variants with attenuated antipeptidase potential. In these mutants, the P1′ methionine residue of the inhibitory loop is replaced by either a lysine (pre-elafinM25K) or a glycine (pre-elafinM25G) residue. Both mutated variants are stable and display biochemical properties undistinguishable from WT (wild-type) pre-elafin. However, compared with WT pre-elafin, their inhibitory constants are increased by one to four orders of magnitude toward neutrophil elastase, myeloblastin and pancreatic elastase, depending on the variants and enzymes tested. As suggested by molecular modelling, this attenuated inhibitory potential correlates with decreased van der Waals interactions between the variants and the enzymes S1′ subsite. In elastase-induced experimental emphysema in mice, only WT pre-elafin protected against tissue destruction, as assessed by the relative airspace enlargement measured using lung histopathological sections. Pre-elafin and both mutants prevented transient neutrophil alveolitis. However, even the modestly affected pre-elafinM25K mutant, as assayed in vitro with small synthetic substrates, was a poor inhibitor of the neutrophil elastase and myeloblastin elastolytic activity measured with insoluble elastin. We therefore conclude that full antipeptidase activity of pre-elafin is essential to protect against lung tissue lesions in this experimental model.
Keywords: antipeptidase function, experimental emphysema, human pre-elafin, inhibitory loop mutant, myeloblastin, neutrophil elastase
Abbreviations: aa, amino acids; BAL, bronchoalveolar lavage; Boc-Ala-Ala-Nva-SBzl, Boc-Ala-Ala-norvaline-thiobenzyl ester; COPD, chronic obstructive pulmonary disease; HNE, human neutrophil elastase; Lm, mean linear intercept; LPS, lipopolysaccharide; MMP, matrix metalloprotease; NE, neutrophil elastase; PE, pancreatic elastase; PPE, porcine pancreatic elastase; SLPI, secretory leukocyte protease inhibitor; WAP, whey acidic protein; WT, wild-type
INTRODUCTION
It is now well accepted that in COPD (chronic obstructive pulmonary disease), such as emphysema, an imbalance between destructive peptidases secreted primarily by activated neutrophils and macrophages and their natural inhibitors is a determinant factor in the progressive destruction of pulmonary tissue (reviewed in [1]). The characteristic lesion in emphysema is the irreversible destruction of the alveolar walls, which alters the gasexchanging structures and leads to major impairment in lung function [2]. Among the peptidases with elastinolytic activities, NE (neutrophil elastase; EC 3.4.21.37) and myeloblastin (formerly named Proteinase-3; EC 3.4.21.76) secreted by activated neutrophils, and MMP-12 (matrix metalloprotease-12; EC 3.4.24.65) secreted by infiltrating macrophages are thought to play key roles in the destruction process [3,4]. In addition to elastin degradation, these peptidases contribute to perpetuate inflammation by stimulating the production of different pro-inflammatory mediators [5,6]. Neutrophil-derived peptidases are also involved in zymogen activation of pro-MMPs and inactivation of MMP inhibitors [7]. Similarly, MMPs degrade the most abundant endogenous inhibitor of neutrophil proteinases, α1-peptidase inhibitor (formerly named α1-antitrypsin) [5]. Because of this amplification cascade, and despite the involvement of numerous enzyme activities implicated in the pathogenesis of COPD, targeting key enzymes such as NE or macrophage MMP-12 for inhibition was shown to prevent against the increased influx of inflammatory cells at sites of lung injury and long-term tissue destruction (reviewed in [8]).
Pre-elafin is characterized by an N-terminal moiety [aa (amino acids) 1–38] termed cementoin, and a C-terminal domain (aa 39–95) with homology to WAP (whey acidic protein) [9]. The antipeptidase activity is entirely contained within the WAP domain that is characterized by four disulfide bridges, which confer high thermal and broad pH range stability [9]. Pre-elafin is used to designate the full-length protein, whereas elafin refers to the mature C-terminal domain (aa 39–95). Remarkably, however, pre-elafin and elafin are both effective in vitro inhibitors of NE, myeloblastin and PE (pancreatic elastase; EC 3.4.21.36) [10–12]. This narrow specificity, in addition to the small size of the elafin-derived peptides (6–9 kDa), and their tight-binding inhibition (Ki values ranging from 10−9–10−10 M; [9,12–14]) make them potential candidates for therapeutic use.
We previously tested pre-elafin efficiency in two animal models of pulmonary diseases: NE-induced acute lung injury in hamsters and LPS (lipopolysaccharide)-induced acute lung inflammation in mice. We found that pre-elafin is potent in preventing acute lung injury caused by a single instillation of NE in hamsters [11]. It was also observed that pre-elafin significantly reduces the levels of pro-inflammatory chemokines, as well as the number of leukocytes recovered by BAL (bronchoalveolar lavage), in an LPS-induced mouse model of lung inflammation [15]. This suggests that pre-elafin is a potent anti-inflammatory mediator. Since no elastolytic activity could be recovered in BAL fluid supernatants from the LPS-treated animals, we hypothesized that this property might either result from the inhibition of other uncharacterized serine peptidases or, alternatively, through a mechanism not involving the classical inhibitor/peptidase interaction.
As a first step toward understanding the structure/function of pre-elafin, in the present study we report the construction and characterization of human pre-elafin mutated variants. We show that replacement of Met25 within the inhibitory loop of pre-elafin by Lys25 or Gly25 does neither alter its thermal stability and resistance to proteolytic degradation, nor its proper disulfide-bridge connections and inhibitory profile. Both mutations, however, moderately or drastically alter the inhibitory properties of preelafin toward HNE (human NE), myeloblastin and PPE (porcine PE) in vitro. Molecular modelling analyses of the pre-elafin variants in complex with HNE stress the importance of hydrophobic interactions at the primary inhibitor/enzyme contact site. These variants were also used to evaluate the importance of the pre-elafin inhibitory function in an elastase-induced mice emphysema model. We demonstrate that full pre-elafin antipeptidase activity is essential to protect lung tissue integrity in this model.
EXPERIMENTAL
Materials
HNE, PPE, myeloblastin and Boc-Ala-Ala-Nva-SBzl (Boc-Ala-Ala-norvaline-thiobenzyl ester) were purchased from Elastin Products Co. Mature elafin was from the Peptide Institute. Other enzyme substrates were from Sigma Chemical Co. and all other chemicals were of the purest grade available.
Expression and purification of recombinant human pre-elafin and mutated variants
The Kunkel method [16,17] was used for site-directed mutagenesis of pre-elafin expressed from the yeast pVT-Ela2 phagemid [10]. Oligonucleotides were annealed using a stepwise cooling procedure, and second strand synthesis was performed using the T7 DNA polymerase. Mutagenic oligonucleotide sequences were 5′-TAGGGGGATTCAATTTTGCGCACCGGAT-3′ and 5′-TAGGGGGATTCAACCCTGCGCACCGGAT-3′ for pre-elafinM25K and pre-elafinM25G respectively. All mutated DNA sequences were confirmed by automated DNA sequencing.
WT (wild-type) and mutated pre-elafin expression and purification were performed as described previously [10], except that the ammonium sulfate precipitation step was omitted. Electrophoresis and immunodetection with rabbit polyclonal anti-elafin antiserum recognizing the proper disulfide-bridge arrangement found in functional elafin was performed as described previously [10].
Thermal stability, resistance to proteolytic degradation and inhibitory assays
The thermal stability of WT and pre-elafin mutants was assessed by comparing the inhibitory potential toward PPE of triplicate inhibitor samples previously pre-incubated for 30 min at either 4 °C or 85 °C. Enzyme kinetics in the presence of mock- or heat-treated pre-elafin peptides, and in the absence of inhibitor, was then performed as described below for PPE assays. The enzyme/inhibitor ratios used were 1:0.4, 1:20 and 1:350 for pre-elafin, pre-elafinM25K and pre-elafinM25G respectively. The resistance to proteolytic degradation was assayed by incubating the various pre-elafin peptides with bovine Tos-Phe-CH2Cl (TPCK; tosylphenylalanylchloromethane)-treated trypsin at a ratio of 2000:1 for 2 h at 37 °C. Reactions were stopped by the addition of SDS/PAGE sample buffer (50 mM Tris/HCl, pH 6.8, 100 mM dithiotheitol, 2% SDS, 10% glycerol and 0.1% Bromophenol Blue) and boiling for 10 min. The functional inhibitory activity of pre-elafin toward trypsin, α-chymotrypsin, and cathepsin G was assayed as described previously [10]. The chromogenic substrates were N-benzoyl-Phe-Val-Arg p-nitroanilide for trypsin, and N-succinyl-Ala-Ala-Pro-Phe p-nitroanilide for chymotrypsin and cathepsin G. Inhibition assays of the HNE and myeloblastin elastolytic activity with elastin Congo Red (4 mg/ml) were performed in duplicate, essentially as described by Nobar et al. [18], except the buffer for HNE was buffer A (see below) and the appearance of digestion products was monitored at 495 nm.
Enzyme kinetics
All enzyme kinetics were performed at 25 °C and all reaction mixtures were supplemented with 10% (v/v) DMSO and 0.01% Triton X-100. Kinetics using HNE and PPE were performed in buffer A (0.1 M Tris/HCl, pH 8.0, and 150 mM NaCl) using N-MeO-succinyl-Ala-Ala-Pro-Val-p-nitroanilide and succinyl-(Ala)3-p-nitroanilide as HNE and PPE substrates respectively. Kinetics with myeloblastin were performed in buffer B [0.1 M Mops, pH 7.5, and 0.1 mM DTNB (Ellman's reagent)] using Boc-Ala-Ala-Nva-SBzl ester as substrate. The Michaelis constant (Km) for each enzyme/substrate pair was determined by measuring the enzyme velocity for different substrate concentrations. Data were analysed by non-linear least square fits to the Michaelis–Menten equation (KaleidaGraph, Synergy Software).
Enzyme titration experiments for tight-binding inhibitors were conducted at steady state under second-order conditions ([E]≈[I]). Briefly, enzyme was incubated in the presence of inhibitor for 30 min prior to substrate addition. The reactions were conducted in 200 μl and product formation was monitored at 405 nm for 20 min using a 96-well plate reader (VersaMax model, Molecular Devices). Inhibitor concentrations were determined by ELISA as described previously [19], but using synthetic human elafin as the standard. For WT recombinant pre-elafin, a titration experiment with active site-titrated HNE indicated that >95% of the purified inhibitor was active. Enzyme concentrations were 10 nM for HNE, 5 nM for myeloblastin and 20 nM for PPE. Substrate concentrations were 1.5 mM, 0.1 mM and 4.5 mM for HNE, myeloblastin and PPE assays respectively. The apparent Ki [Ki(app)] was obtained by fitting eqn (1) (taken from [20]) to the data, for at least two independent experiments performed in duplicate, by non-linear regression analysis.
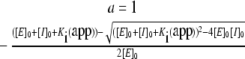 |
(1) |
Experimental Ki values were derived from eqn (2):
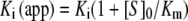 |
(2) |
where a is the fractional enzyme activity, and [I]0 and [S]0 are the initial inhibitor and substrate concentrations respectively. To evaluate the inhibitory constants for classical competitive inhibitors, we measured the enzyme velocity (v) in the presence of various inhibitor and substrate concentrations to determine the observed Km [Km(obs)] and the Ki using eqns (3) and (4):
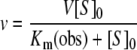 |
(3) |
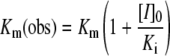 |
(4) |
Stopped-flow kinetics were conducted under pseudo first-order conditions, i.e.[E]≤10[I]. Enzyme concentrations were 5 nM. Substrate concentrations were as described for the titration experiments. At time zero, the inhibitor/substrate solution was mixed to the enzyme solution at a 1:1 ratio using a stopped-flow apparatus (SX.18MV Kinetic Spectrometer Workstation, Applied Photophysics). Product formation was monitored at 405 nm for 300 s, and the data from at least four independent progress curves (<10% variation) were used for data analysis. For slow-binding inhibitors conforming to Scheme 1, product formation [P] against time (t) under pseudo-first order conditions is defined by equation (5):
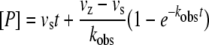 |
(5) |
where kobs is the pseudo-first order rate constant, and vz and vs the initial and final enzyme velocity respectively. The estimated values for kobs were then used to determine the association (k3), and dissociation (k−3) constants, and the calculated Ki values from the following relationships:
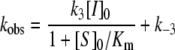 |
(6) |
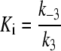 |
(7) |
Scheme 1.

Molecular modelling and energy minimization
Structure modelling was performed using Insight II software (Accelrys). Starting with the known PPE/elafin co-ordinates (PDB accession number 1FLE), modelling of the elafin mutated variants with PPE was performed using the Insight II Biopolymer module. For inhibitor–enzyme complexes involving HNE, the PPE/elafin co-ordinates were modified by the HNE co-ordinates (PDB accession number 1B0F). Energy minimization was achieved to convergence with the Discover module using cvff.frc applied ForceField and Conjugate Gradient method. Molecule representations were generated using MacPyMol [DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, U.S.A.].
Experimental emphysema
In the present study, a well-established experimental model of emphysema was used [21], where PPE is used to trigger the inflammatory process leading to emphysematous changes. Once instilled, the half-life of PPE in rodent lungs is 50 min [22] and 80% of lung injuries are attributable to inflammation and, namely, the release of neutrophil secreted peptidases (HNE and myeloblastin) [23]. Female C57BL/6 mice weighting 18–22 g (Charles River, Canada) were anaesthetized using 3% isoflurane and received a single intranasal instillation of 35 μl of PPE (20 units/100 g of body mass or approx. 1.15 nmol/mouse). Pre-elafin and variants (1 μg/100 g of body mass or approx. 17.5 pmol/mouse) were instilled using the same route three times a week for 2 weeks. The first dose was administered 1 h following PPE instillation. Animals were killed 2 weeks post-PPE instillation using a sodium pentobarbital overdose. For each condition tested, groups were composed of four animals. Emphysematous changes were quantified as mean linear intercept (Lm) on haematoxylin and eosin-stained lung sections as described [24]. Animal experiments were performed according to the Canadian Council on Animal Care guidelines and approved by the ethical committee of Université Laval.
RESULTS
Design, production and characterization of pre-elafin variants
The previously determined three-dimensional structure of elafin complexed with PPE showed that the inhibitory loop of elafin lies within the Leu20–Leu26 segment (elafin numbering), and residues of the scissile peptide bond (Ala24-Met25) make extensive van der Waals contact with residues forming the enzyme subsites S1 and S1′ respectively [25]. We sought to create pre-elafin variants with decreased inhibitory potential, without modifying the narrow specificity of the inhibitor. Because modifying the P1 position might change the pre-elafin enzyme specificity, as previously documented for SLPI (secretory leukocyte protease inhibitor) [26], an analogous elastase inhibitor containing WAP domains, we targeted the P1′ position Met25 (elafin numbering) for site-directed mutagenesis. Given the extensive hydrophobic interaction of this residue with PPE, we expected that the replacement of Met25 by either a positively charged residue of similar size (lysine) or an amino acid with no side chains (glycine) would significantly lower its affinity for PPE, as well as for HNE and myeloblastin; two peptidases that have similar substrate-binding pockets. These pre-elafin variants were therefore produced in yeast and purified from the culture supernatants.
Before determining the kinetic parameters of these variants, however, we first compared their biochemical properties with WT pre-elafin. The four-disulfide bridges that characterize the elafin WAP core domain confer high resistance to thermal and proteolytic inactivation and their integrity is also essential for the antipeptidase function [9]. As shown in Figure 1, WT pre-elafin and both of the mutants retained their antipeptidase activity following a 30 min incubation at 85 °C (Figure 1A). They also resisted complete degradation by trypsin upon a 2 h incubation with this enzyme and instead accumulated a peptide fragment corresponding in size to the WAP core domain (Figure 1B). In contrast, at the same trypsin/peptide ratio, but in the presence of a reducing agent (50 mM dithiothreitol) WT pre-elafin and both mutants were completely digested within minutes (results not shown). Therefore the proper four-disulfide-bridge arrangement characteristic of the WAP domain was maintained in the pre-elafin mutants, which was further supported by the finding that an antiserum recognizing the native disulfide-bridged elafin [10], but neither the reduced form nor multimers indicative of improper disulfide-bridge connections, quantitatively detected WT pre-elafin, pre-elafinM25K and pre-elafinM25G (Figure 1C). Pre-elafin, in contrast to SLPI, possesses a narrow specificity and does not inhibit cathepsin G, or the prototypical serine-peptidases α-chymotrypsin and trypsin. We next addressed whether replacing the P1′ position in pre-elafin modified this narrow specificity. As shown in Figure 2, even at a high inhibitor to enzyme ratio (i.e. 500:1), neither pre-elafin mutant demonstrated inhibiting activity toward these enzymes. Taken together, our data therefore indicate that the point mutations preserved proper disulfide-bonding and stability of the peptide inhibitor and did not broaden its specificity.
Figure 1. Biochemical properties of pre-elafin mutants.

(A) The indicated recombinant pre-elafin peptides were incubated at 85 °C for 30 min. Resistance to heat inactivation was tested by PPE inhibition assay as described in the Experimental section. The enzyme/inhibitor ratios used were 1:0.4, 1:20 and 1:350 for pre-elafin, pre-elafinM25K and pre-elafinM25G respectively. (B) Recombinant pre-elafin peptides were incubated at 37 °C for 2 h in the presence of trypsin at a trypsin/protein ratio of 1:2000. Digestion products at 0 and 2 h were resolved by SDS/PAGE (15% gels) under reducing conditions and revealed by silver staining. Note that the time to process the samples (t=0 h) was sufficient to digest pre-elafin, as evidenced by the decreased intensity of intact pre-elafin and the concomitant appearance of smaller species. (C) Purified WT pre-elafin and mutated variants were resolved by SDS/PAGE (15% gels) under non-reducing conditions, transferred on to PVDF membranes, and immunodetected with a polyclonal rabbit anti-elafin antibody specifically recognizing the native and functional disulfide-bonded elafin domain. The 16.5 kDa mass marker is indicated to the left-hand side of the Figure.
Figure 2. Inhibitory profile of WT and pre-elafin mutants.

The inhibitory profiles of WT pre-elafin, pre-elafinM25K and pre-elafinM25G were compared by measuring the relative activity of trypsin (Tryp) cathepsin G (Cat G) and α-chymotrypsin (Chym) in the presence of an excess (500:1 ratio) of the indicated inhibitor. α1-Peptidase inhibitor (α1-PI) was used as a positive control for peptidase inhibition with a peptidase/α1-peptidase inhibitor ratio of 1:1.
Pre-elafinM25G no longer behaves as slow- and tight-binding inhibitor toward its natural peptidase targets
Previous studies established that elafin and pre-elafin both act as slow- and tight-binding inhibitors of PPE, HNE and myeloblastin [12–14]. The titration method (see Experimental section) was used to determine experimental inhibition constants (Ki) for pre-elafin and the mutated variants toward HNE, myeloblastin and PPE. The fractional activity (a) of HNE expressed as a function of the inhibitor concentration gave typical curves of tight-binding inhibition for pre-elafin and pre-elafinM25K, with significant inhibition when [E]0≈[I]0 (Figure 3). Similar traces were also observed for the inhibition of myeloblastin with the same peptides (results not shown). However, with PPE only WT pre-elafin gave a typical tight-binding titration curve (results not shown). Hence, for the mutated variants pre-elafinM25G with all three enzymes, as well for pre-elafinM25K with PPE, all traces revealed that no significant inhibition could be achieved when [E]0≈[I]0, indicating no tight inhibition (Figure 3 and results not shown).
Figure 3. Inhibition of HNE by WT and pre-elafin mutants.

The residual activity (Fractional Activity) of HNE (10 nM) was assayed at 25 °C following a 30 min incubation in the presence of the indicated concentrations of inhibitor. The inhibitors used were: ●, pre-elafin; ■, pre-elafinM25K; dashed line, pre-elafinM25G. The lines are the best fit of eqn (1) to the experimental data. Note that no experimental points are shown for pre-elafinM25G as the smallest concentration showing a significant inhibition was 350 nM.
Compared with pre-elafin (Ki=1.0×10−10 M), Ki values for HNE inhibition by pre-elafinM25K (Ki=6.9×10−10 M) and pre-elafinM25G (Ki=3.9×10−7 M) were approx. 7-fold and 4000-fold higher respectively (Table 1). Pre-elafin also inhibited myeloblastin and PPE with Ki values within the same order of magnitude to that observed for HNE (3.6×10−10 M and 7.8×10−10 M respectively). Similarly, pre-elafinM25G, which poorly inhibited HNE, was also a poor inhibitor of both myeloblastin (Ki=4.2×10−6 M) and PPE (Ki=3.5×10−6 M) with Ki values 12000-fold and 4500-fold greater than those of pre-elafin toward these enzymes. In contrast, pre-elafinM25K, a relatively good inhibitor of HNE, was less efficient toward both myeloblastin (Ki=3.7×10−8 M) and PPE (Ki=3.4×10−7 M). Compared with the Ki values obtained with pre-elafin, those determined for pre-elafinM25K against HNE, myeloblastin and PPE were approx. 7-, 100- and 450-fold higher respectively.
Table 1. Kinetic parameters for the inhibition of serine peptidases by pre-elafin and its variants.
NS, not a slow-binding inhibitor, k−3 and k3 cannot be determined; N.D., not determined.
| Inhibitor | k3 (M−1·s−1) | k−3 (s−1) | Calculated Ki (M)* | Experimental Ki (M)† |
|---|---|---|---|---|
| HNE | ||||
| Pre-elafin | 2.8×106 | 4.9×10−4 | 1.7×10−10 | 1.0×10−10 |
| Pre-elafinM25K | 2.9×106 | 5.8×10−3 | 2.0×10−9 | 6.9×10−10 |
| Pre-elafinM25G | NS | 3.9×10−7 | ||
| PPE | ||||
| Pre-elafin | 1.4×106 | 1.8×10−3 | 1.3×10−9 | 7.8×10−10 |
| Pre-elafinM25K | NS | 3.4×10−7 | ||
| Pre-elafinM25G | NS | 3.5×10−6 | ||
| Myeloblastin | ||||
| Pre-elafin | ND | 3.6×10−10 | ||
| Pre-elafinM25K | ND | 3.7×10−8 | ||
| Pre-elafinM25G | ND | 4.2×10−6 |
To evaluate whether the mutants had retained their slow-binding mode of inhibition, we next used the progress curve method to monitor inhibition of HNE and PPE under pseudo-first order conditions. Typical progress curves for slow-binding inhibitors [i.e. a change from an initial enzyme velocity (vz) to a final enzyme velocity (vs)] were observed when product formation by HNE was recorded continuously in the presence of various concentrations of WT pre-elafin (Figure 4A). Non-linear regression analysis showed that fitting eqn (5) to the kinetic data gave excellent correlation coefficients. Furthermore, kobs was shown to increase linearly with [I]0 (results not shown), indicating that enzyme inhibition was satisfactorily described by Scheme 1 for WT pre-elafin. Similar progress curves were also observed for HNE inhibition by preelafinM25K (Figure 4B), as well as for PPE inhibition by WT pre-elafin (results not shown). However, traces obtained for HNE inhibition by pre-elafinM25G were all linear (Figure 4C), indicating that a fast equilibrium between inhibitor and substrate for HNE binding was reached, and that pre-elafinM25G no longer behaves as a slow-binding inhibitor. Similarly, when product formation under these conditions was monitored for PPE, both pre-elafin mutants were shown to behave as classical Michaelis–Menten competitive inhibitors rather than slow-binding inhibitors (results not shown). Hence, only pre-elafinM25K, but not pre-elafinM25G, retained its slow- and tight-binding characteristics toward HNE, but not PPE.
Figure 4. Progress curves for the inhibition of HNE by WT and pre-elafin mutants.

Assays were conducted at 25 °C with 5 nM HNE and increasing inhibitor concentrations (from top to bottom). (A) Inhibition by WT pre-elafin (0–250 nM). (B) Inhibition by pre-elafinM25K (0–250 nM). (C) Inhibition by pre-elafinM25G (0–1 μM).
The calculated Ki values derived from the progress curve method agreed within a range expected from experimental error to that experimentally determined by the titration curves (Table 1). Most importantly, although the association constants of both pre-elafin and pre-elafinM25K to HNE were nearly identical (k3 of 2.8×106 M−1·s−1 and 2.9×106 M−1·s−1 respectively), pre-elafinM25K was found to dissociate more rapidly from HNE with an approx. 12-fold greater k−3 compared with that of pre-elafin (5.8×10−3 s−1 compared with 4.9×10−4 s−1), therefore explaining its slightly higher Ki value toward HNE. Likewise, the main difference in inhibition between PPE and HNE by pre-elafin appears to rely on a less stable enzyme–inhibitor complex with PPE (k−3 of 1.8×10−3 s−1 for PPE compared with 4.9×10−4 s−1 for HNE).
Molecular modelling highlights the importance of hydrophobic interaction at the pre-elafin/HNE primary contact site for stabilizing the complex
Replacement of the P1′ position of pre-elafin by a glycine residue was found to modify its slow- and tight-binding inhibition mode against all enzymes tested. However, with a lysine residue occupying the P1′ position, the mutated pre-elafin variant was shown to retain its characteristic slow- and tight-binding inhibition mode with HNE, but not with PPE. To further explore the molecular basis for these findings, we performed molecular modelling analyses on the various inhibitor–enzyme complexes based on the known crystal structure of the elafin–PPE complex [25]. Compared with elafin complexed with PPE, the elafinM25K variant was found to induce major local changes within the active site cleft at the enzyme S1′ position (Figure 5A). The charged guanidine group of Lys25 from the elafin inhibitor (Lys25i) is involved in hydrogen bonding with Asp60 from the enzyme (Asp60e). This induces a shift of 3.7 Å (1 Å=0.1 nm) for the Asp60e γ carbon. This hydrogen bonding also favours the insertion of a water molecule at the protein complex interface. More importantly, charge repulsion occurs between Lys25i and Arg61e. This repulsion forces the terminal carbon of Arg61e to move 1.08 Å away from Lys25i. In contrast to this situation, when elafinM25K is complexed with HNE very little local changes are observed at the enzyme S1′ subsite (Figure 5B). Compared with PPE, the S1′ subsite of HNE is completely devoid of charged residues (i.e. Arg61e and Asp60e in PPE are replaced by Phe61e and Asn60e in HNE), thus avoiding any charge repulsion. In addition, Val62e of HNE further adds to the overall hydrophobic environment at the S1′ subsite. The P1′ position of elafin in this context is primarily involved in van der Waals interactions with His57e and Phe41e. None of these interactions appears to be perturbed by the replacement of Met25 by Lys25. Lys25 from elafinM25K sits well in the active site cleft of HNE and is, in fact, perfectly superimposed to the WT Met25 residue (Figure 5B). These observations provide a satisfactory explanation for the relatively good efficiency of preelafinM25K in HNE, but not PPE, inhibition. The absence of lateral side chains at the P1′ subsite resulting from the replacement of Met25 by Gly25 is similarly detrimental to both HNE and PPE inhibition as it precludes hydrophobic interactions and/or hydrogen-bonding with residues occupying the S1′ subsite in both enzymes. In the PPE–elafin complex, this leaves a cavity of 55 Å3 for the inhibitor–enzyme complex (Figure 5C).
Figure 5. Stereo views of structural models for WT and elafin mutants complexed to HNE and PPE.

(A) and (B) Molecular representations of WT elafin–enzyme complex (white) superimposed on to an elafinM25K–enzyme complex (grey). The WT Met25 residue is yellow and Lys25 blue. Residues making direct contact with residue 25 from the inhibitor are in stick representation. Protein backbones are in ribbon representation. Magenta and black dashed lines represent hydrogen bonds for the complex containing pre-elafin and pre-elafinM25K respectively. (A) Inhibitors complexed to PPE. The yellow dashed line represents the distance between Lys25i side chain nitrogen atom and Arg61e ζ carbon. (B) Inhibitors complexed to HNE. (C) ElafinM25G complexed to PPE. The yellow mesh represents the cavity left by the replacement of Met25 by Gly25.
Full antipeptidase activity of pre-elafin is essential to preserve lung tissue integrity in an animal model of PPE-induced emphysema
Production of stable mutated pre-elafin variants with attenuated inhibitory activity, but with the same inhibitory profile, enabled a direct evaluation of the antipeptidase function of pre-elafin in preventing lung tissue damage in a well-established experimental model of emphysema. Animals were first instilled intranasally with PPE (1.15 nmol/mouse), or mock-treated, then received three times a week for 2 weeks, through the same route, either saline or one of the pre-elafin variants (17.5 pmol/mouse). Histopathological lung sections of PPE-treated animals at the end of the treatment were characterized by larger airspaces (Figure 6A) with a mean distance separating alveolar walls (Lm) significantly greater (57.4±3.9 μm; Figure 6B) than that of sham-exposed mice (33.8±3.8 μm; Figure 6B). In contrast, pre-elafin significantly reduced the PPE-induced emphysematous-like lesions (Figure 6A) and Lm values (41.3±3.5 μm compared with 57.4±3.9; Figure 6B). However, pre-elafin mutated variants did not achieve a similar protection as evidenced on lung sections (Figure 6A) and Lm values (55.6±7.5 μm for pre-elafinM25K and 49.7±4.2 μm for pre-elafinM25G; Figure 6B), which were not statistically different from animals treated with PPE alone. Hence, full antipeptidase function of pre-elafin is essential to preserve lung tissue integrity in this animal model of emphysema.
Figure 6. Effect of WT and pre-elafin mutants on lung histopathology and neutrophil influx in an experimental model of emphysema.

Animals treated with PPE were given the indicated inhibitor, or saline only (PPE), three times a week for 2 weeks, at which time they were killed. Control animals (Ctrl) received vehicle only. (A) Representative lung slices. (B) Lm calculated from lung slices (15 microscopic fields per animal). *, Only control animals and those receiving PPE and WT pre-elafin show statistical differences compared with animals instilled with PPE only (P≤0.003; n=4–12). (C) Neutrophil influx in BAL. Animals received the indicated inhibitor 1 h post-PPE instillation and BAL were collected 24 h post-PPE administration for neutrophil cell counts. *Statistically different from the animals who received only PPE (P≤0.012; n=4). All results are expressed as means±S.E.M. Comparisons were made using ANOVA followed by a Fisher's protected least-significant difference test with Statview for Windows version 5.0 (SAS Institute, Cary, NC, U.S.A.). Pre-elafin, WT pre-elafin; M25K, pre-elafinM25K; M25G, pre-elafinM25G.
Using the same animal model, we recently showed that pre-elafin given 1 h post-PPE instillation leads to a sharp reduction in inflammation, as assessed by the neutrophil counts present in BAL fluids 24 h after animals were given pre-elafin [27]. This was accompanied by much-reduced counts of macrophages in BAL fluids of animals 2 weeks post-PPE administration. We then asked whether the mutants had retained this anti-inflammatory property. As observed previously, the single dose of PPE led to a significant influx of neutrophils after 24 h (approx. 170000/ml; Figure 6C). In contrast, BAL fluids of animals treated with either pre-elafin, pre-elafinM25K or pre-elafinM25G were shown to contain lower neutrophil counts (approx. 20000/ml, 58000/ml and 18000/ml respectively; Figure 6C) and in all cases, this was statistically significant compared with that observed in animals treated with PPE alone. This therefore suggests that the elafin antipeptidase function is not responsible for preventing and/or attenuating transient neutrophil alveolitis. However, at the doses used in the present study, the neutrophil influx at 24 h in the presence of any pre-elafin peptides is noticeably higher than in mock-treated animals. It thus suggests that failure to protect against emphysematous-like lesions by both pre-elafin mutants is linked to their inability to efficiently inhibit in vivo the residual secreted neutrophilic peptidases. Given that pre-elafinM25K did not afford any significant protection despite its tight-binding inhibition of HNE when assayed with a small synthetic substrate, it also suggests that its modest increase in Ki may translate to a very weak inhibition when competing with insoluble elastin, the natural substrate of HNE and myeloblastin. To test this hypothesis, we next compared the in vitro elastolytic activity of HNE and myeloblastin with elastin Congo Red in the presence of WT pre-elafin and pre-elafinM25K. As shown in Table 2, at an inhibitor/enzyme ratio of 1, even WT pre-elafin poorly competed with elastin for inhibition of HNE and myeloblastin with only 54.0% and 35.0% inhibition respectively. Nearly complete quantitative inhibition (>95%) was achieved with both peptidases when WT pre-elafin was present at an inhibitor/enzyme ratio of 5. In contrast, and in support to our hypothesis, no significant inhibition (from 0 to 8.9%) of either peptidase was observed with pre-elafinM25K even at an inhibitor/enzyme ratio of 5.
Table 2. In vitro inhibition of HNE and myeloblastin elastolytic activity.
| % inhibition of elastolysis | |||
|---|---|---|---|
| Inhibitor | [I]/[E] | HNE | Myeloblastin |
| Pre-elafin | 1 | 54.0 | 35.0 |
| 5 | 95.2 | 98.7 | |
| Pre-elafinM25K | 1 | 0.0 | 0.0 |
| 5 | 4.8 | 8.9 | |
DISCUSSION
As a first step toward understanding the structure/function of preelafin, the aim of the present study was to: (i) engineer recombinant human pre-elafin variants with attenuated antipeptidase activity; and (ii) compare their efficiency in preventing tissue lesions in an experimental model of emphysema. By targeting the P1′ position of the pre-elafin inhibitory loop for replacement by either a lysine or glycine residue, we showed that the resulting variants are stable and adopt the native disulfide bridge connections essential for the antipeptidase function of elafin [28,29]. As judged by inhibition assays performed with the prototypical serine peptidases α-chymotrypsin and trypsin, as well as with the neutrophilic cathepsin G, it was also shown that replacing the P1′ position of pre-elafin does not modify its inhibitory profile. This is in sharp contrast with that observed previously with SLPI mutants where the P1 leucine residue (Leu72) was targeted for mutagenesis. For instance, compared with WT SLPI, SLPILys72 is an excellent trypsin inhibitor, but a very poor HNE inhibitor [30]. In addition to providing support that the P1 position is the main specificity determinant for peptide inhibitors [31], our results suggest that targeting the P1′ residue for mutagenesis may constitute a better approach to modulate the kinetic parameters of peptide inhibitors without modifying their specificity.
The Ki values determined in the present study for pre-elafin are in very good agreement with those previously observed by others with either mature elafin or pre-elafin against HNE [12,13,32], myeloblastin [12,14] and PPE [12,32]. However, both pre-elafin mutants showed reduced inhibitory potential against all three peptidases. Intriguingly, we found that the Met→Gly replacement is more detrimental to the inhibitory activity of pre-elafin than the Met→Lys replacement, despite the introduction of a positive charge. As suggested by molecular modelling of the various elafin variants with PPE and HNE, the cavity left in the enzyme active site by the absence of a side chain in the pre-elafinM25G variant is more destabilizing, due to the loss of hydrophobic interaction, than the Met→Lys replacement. Methionine and lysine residues are similar in size (163 Å3 compared with 167 Å3) and if we exclude the polar group, as proposed by Karplus [33], both residues have similar estimated hydrophobic effect for side-chain burial (2.3 kcal/mol for methionine compared with 1.9 kcal/mol for lysine). In the context of HNE, a favourable molecular environment, i.e. the absence of polar residues within the active-site centre, thus accounts for the modest effect (<10-fold increase in k−3) due to the positively charged Lys25 residue on the stability of the pre-elafinM25K–HNE complex. In contrast, the positive charge of Lys25 is much less well accommodated in PPE, due mainly to charge repulsion with Arg61e and electrostatic interaction with Asp60e of PPE. Along with the previous observation that the active-site cleft of myeloblastin is more polar than that of HNE [34], its closest related enzyme; this highlights the exquisite contribution of hydrophobic interactions at the primary contact site between pre-elafin and HNE, and provides an explanation for the relative increase on Ki of pre-elafinM25K in the following order: HNE≪myeloblastin<PPE.
In most instances, slow-binding inhibition reflects a mechanism proceeding in two steps where an initial inhibitor–enzyme complex is slowly converted into a more stable EI* complex [35]. With the inhibitor concentration range used here, and in agreement with previous studies performed with elafin and/or pre-elafin [12–14], we found that the one-step mechanism of Scheme 1 satisfactorily accommodates the kinetic data measured with HNE and PPE. However, we cannot formally exclude that inhibition by pre-elafin involves a two-step mechanism and our finding that pre-elafinM25G proceeds through a classical fast-equilibrium competitive inhibition argues in favour of this mechanism for pre-elafin. According to Scheme 1, the switch from slow- to fast-binding inhibition implies that formation of the HNE–pre-elafinM25G complex proceeds faster than that involving pre-elafin. As there is no reason to suppose that pre-elafinM25G associates more rapidly to the enzyme than the more efficient native inhibitor from which it is derived, it suggests that the slow-binding characteristics of pre-elafin may rather reflect the slow establishment of a more stable EI* complex by isomerization of an initial inhibitor–enzyme complex.
Most importantly, construction of pre-elafin mutants with attenuated inhibitory activity enabled a direct assessment of the pre-elafin antipeptidase function in elastase-induced emphysema in mice. We found that full inhibitory activity of pre-elafin is essential to preserve lung tissue integrity, as assessed by airspace enlargement at 2 weeks post-PPE instillation. Pre-elafin and both mutants similarly reduced, but did not completely eliminate, the initial influx of neutrophils 24 h post-PPE instillation, which suggests that this pre-elafin anti-inflammatory property is not linked to the antipeptidase function. However, given the numerous direct and indirect actions of HNE and myeloblastin on tissue degradation and inflammation, incomplete inhibition of these neutrophil secreted peptidases by both pre-elafin mutants (see below) probably explains the observed tissue damage. For instance, elastin fragments produced by these peptidases are known to recruit macrophage at sites of lung injury and the release of macrophage elastolytic activity (e.g. MMP-12) further contributes to tissue destruction and to perpetuate inflammation [36]. Moreover, antagonizing the chemotactic activity of the elastin fragments with a mAb (monoclonal antibody) was shown to abrogate both macrophage recruitment and tissue injury in a PPE-induced mice model of emphysema.
Although the inhibitory potential of pre-elafinM25K toward HNE and myeloblastin is only modestly affected, as estimated with small synthetic substrates (approx. 10-fold and approx. 100-fold increase in Ki values respectively), we showed that it poorly inhibits the activity of both peptidases when assayed with elastin Congo Red. A similar finding was recently reported with the oxidized form of elafin and pre-elafin, both retaining reasonable inhibitory potential (approx. 100-fold increase in Ki values) in vitro with synthetic substrates [18]. As observed in the present study with pre-elafinM25K, the higher Ki was mostly due to an increase in k−3, the dissociation constant and, in the presence of insoluble elastin, this decreased stability of the enzyme–oxidized inhibitor complex led to very poor inhibition of the HNE and myeloblastin elastolytic activity. Hence, despite a significant difference between the affinity of pre-elafinM25K or pre-elafinM25G for the neutrophil peptidases in vitro, using small synthetic substrates, both mutants were inefficient in vivo and the extent of tissue damage was similar for both inhibitors at 2 weeks post-PPE instillation. Alternatively, although WT elafin inhibits the orthologous mouse and human myeloblastin and NE peptidases as efficiently [37], we cannot exclude the possibility that the pre-elafin mutants are significantly less active toward the mouse peptidases both in vitro and in vivo.
As a first step toward understanding the structure/function of the multifunctional pre-elafin peptide, we have constructed and characterized mutants with attenuated antipeptidase activity. We have demonstrated that an intact antipeptidase function is required to confer protection against tissue degradation in an experimental animal model of emphysema. Other known functions of pre-elafin include its anti-inflammatory and antimicrobial properties [15,27,38–41]. We provided evidence that its anti-inflammatory role, as measured by the early neutrophil influx, can be dissociated from the antipeptidase function. The availability of stable pre-elafin mutants with attenuated antipeptidase activity, but with the same inhibitory profile, should help determine whether peptidase inhibition is also dispensable for the antimicrobial action of pre-elafin. Experiments toward that goal are currently ongoing in our laboratory.
Acknowledgments
This work was supported by a Canadian Institutes of Health Research grant #MOP-57743 (G.M.T. and Y.B.) and a Bayer/Talecris/CBS/Hema-Quebec Partnership Fund (Y.B., G.M.T. and S.G.). A.D. thanks the Natural Sciences and Engineering Research Council of Canada and the Fonds Québécois de Recherche sur la Nature et les Technologies for their financial support. G.M.T. and D.B. acknowledge the Fonds de la Recherche en Santé du Québec for its financial support and M.F.J. thanks the Chaire de pneumologie de la Fondation J.-D. Bégin for a studentship. We thank Dr Michel Guertin (Université Laval, Quebec, Canada) who gave us access to his stopped-flow apparatus and Dr Manon Couture (Université Laval, Quebec, Canada) for critical reading of the manuscript.
References
- 1.Spurzem J. R., Rennard S. I. Pathogenesis of COPD. Semin. Respir. Crit. Care Med. 2005;26:142–153. doi: 10.1055/s-2005-869535. [DOI] [PubMed] [Google Scholar]
- 2.Thurlbeck W. M. Pathology of chronic airflow obstruction. In: Cherniack N. S., editor. Chronic Obstructive Pulmonary Disease. Philadelphia: W.B. Saunders Co.; 1991. pp. 3–20. [Google Scholar]
- 3.Reid P. T., Sallenave J. M. Neutrophil-derived elastases and their inhibitors: potential role in the pathogenesis of lung disease. Curr. Opin. Investig. Drugs. 2001;2:59–67. [PubMed] [Google Scholar]
- 4.Belvisi M. G., Bottomley K. M. The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs? Inflamm. Res. 2003;52:95–100. doi: 10.1007/s000110300020. [DOI] [PubMed] [Google Scholar]
- 5.Parks W. C., Shapiro S. D. Matrix metalloproteinases in lung biology. Respir. Res. 2001;2:10–19. doi: 10.1186/rr33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witherden I. R., Vanden Bon E. J., Goldstraw P., Ratcliffe C., Pastorino U., Tetley T. D. Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 2004;30:500–509. doi: 10.1165/rcmb.4890. [DOI] [PubMed] [Google Scholar]
- 7.Ferry G., Lonchampt M., Pennel L., de Nanteuil G., Canet E., Tucker G. C. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS Lett. 1997;402:111–115. doi: 10.1016/s0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- 8.Churg A., Wright J. L. Proteases and emphysema. Curr. Opin. Pulm. Med. 2005;11:153–159. doi: 10.1097/01.mcp.0000149592.51761.e3. [DOI] [PubMed] [Google Scholar]
- 9.Molhuizen H. O., Schalkwijk J. Structural, biochemical, and cell biological aspects of the serine proteinase inhibitor SKALP/elafin/ESI. Biol. Chem. 1995;376:1–7. doi: 10.1515/bchm3.1995.376.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Bourbonnais Y., Larouche C., Tremblay G. M. Production of full-length human pre-elafin, an elastase specific inhibitor, from yeast requires the absence of a functional yapsin 1 (Yps1p) endoprotease. Protein Expr. Purif. 2000;20:485–491. doi: 10.1006/prep.2000.1338. [DOI] [PubMed] [Google Scholar]
- 11.Tremblay G. M., Vachon E., Larouche C., Bourbonnais Y. Inhibition of human neutrophil elastase-induced acute lung injury in hamsters by recombinant human pre-elafin (trappin-2) Chest. 2002;121:582–588. doi: 10.1378/chest.121.2.582. [DOI] [PubMed] [Google Scholar]
- 12.Zani M. L., Nobar S. M., Lacour S. A., Lemoine S., Boudier C., Bieth J. G., Moreau T. Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre-elafin (trappin-2) expressed in Pichia pastoris. Eur. J. Biochem. 2004;271:2370–2378. doi: 10.1111/j.1432-1033.2004.04156.x. [DOI] [PubMed] [Google Scholar]
- 13.Ying Q. L., Simon S. R. Kinetics of the inhibition of human leukocyte elastase by elafin, a 6-kDa elastase-specific inhibitor from human skin. Biochemistry. 1993;32:1866–1874. doi: 10.1021/bi00058a021. [DOI] [PubMed] [Google Scholar]
- 14.Ying Q. L., Simon S. R. Kinetics of the inhibition of proteinase 3 by elafin. Am. J. Respir. Cell Mol. Biol. 2001;24:83–89. doi: 10.1165/ajrcmb.24.1.4300. [DOI] [PubMed] [Google Scholar]
- 15.Vachon E., Bourbonnais Y., Bingle C. D., Rowe S. J., Janelle M. F., Tremblay G. M. Anti-inflammatory effect of pre-elafin in lipopolysaccharide-induced acute lung inflammation. Biol. Chem. 2002;383:1249–1256. doi: 10.1515/BC.2002.138. [DOI] [PubMed] [Google Scholar]
- 16.Kunkel T. A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U.S.A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunkel T. A., Roberts J. D., Zakour R. A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 18.Nobar S. M., Zani M. L., Boudier C., Moreau T., Bieth J. G. Oxidized elafin and trappin poorly inhibit the elastolytic activity of neutrophil elastase and proteinase 3. FEBS J. 2005;272:5883–5893. doi: 10.1111/j.1742-4658.2005.04988.x. [DOI] [PubMed] [Google Scholar]
- 19.Tremblay G. M., Sallenave J. M., Israel-Assayag E., Cormier Y., Gauldie J. Elafin/elastase-specific inhibitor in bronchoalveolar lavage of normal subjects and farmer's lung. Am. J. Respir. Crit. Care Med. 1996;154:1092–1098. doi: 10.1164/ajrccm.154.4.8887613. [DOI] [PubMed] [Google Scholar]
- 20.Bieth J. G. Theoretical and practical aspects of proteinase inhibition kinetics. Methods Enzymol. 1995;248:59–84. doi: 10.1016/0076-6879(95)48007-2. [DOI] [PubMed] [Google Scholar]
- 21.Tuder R. M., McGrath S., Neptune E. The pathobiological mechanisms of emphysema models: what do they have in common? Pulm. Pharmacol. Ther. 2003;16:67–78. doi: 10.1016/S1094-5539(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 22.Stone P. J., Lucey E. C., Calore J. D., McMahon M. P., Snider G. L., Franzblau C. Defenses of the hamster lung against human neutrophil and porcine pancreatic elastase. Respiration. 1988;54:1–15. doi: 10.1159/000195496. [DOI] [PubMed] [Google Scholar]
- 23.Lucey E. C., Keane J., Kuang P. P., Snider G. L., Goldstein R. H. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-α and interleukin-1β receptor-deficient mice. Lab. Invest. 2002;82:79–85. doi: 10.1038/labinvest.3780397. [DOI] [PubMed] [Google Scholar]
- 24.Thurlbeck W. M. Internal surface area and other measurements in emphysema. Thorax. 1967;22:483–496. doi: 10.1136/thx.22.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsunemi M., Matsuura Y., Sakakibara S., Katsube Y. Crystal structure of an elastase-specific inhibitor elafin complexed with porcine pancreatic elastase determined at 1.9 Å resolution. Biochemistry. 1996;35:11570–11576. doi: 10.1021/bi960900l. [DOI] [PubMed] [Google Scholar]
- 26.Mulligan M. S., Lentsch A. B., Huber-Lang M., Guo R. F., Sarma V., Wright C. D., Ulich T. R., Ward P. A. Anti-inflammatory effects of mutant forms of secretory leukocyte protease inhibitor. Am. J. Pathol. 2000;156:1033–1039. doi: 10.1016/S0002-9440(10)64971-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janelle M. F., Doucet A., Bouchard D., Bourbonnais Y., Tremblay G. M. Increased local levels of granulocyte colony-stimulating factor are associated with the beneficial effect of pre-elafin (SKALP/trappin-2/WAP3) in experimental emphysema. Biol. Chem. 2006;387:903–909. doi: 10.1515/BC.2006.114. [DOI] [PubMed] [Google Scholar]
- 28.Tsunemi M., Kato H., Nishiuchi Y., Kumagaye S., Sakakibara S. Synthesis and structure-activity relationships of elafin, an elastase-specific inhibitor. Biochem. Biophys. Res. Commun. 1992;185:967–973. doi: 10.1016/0006-291x(92)91721-2. [DOI] [PubMed] [Google Scholar]
- 29.Alkemade H., van de Kerkhof P., Schalkwijk J. Demonstration of skinderived antileukoproteinase (SKALP) in urine of psoriatic patients. J. Invest. Dermatol. 1992;99:3–7. doi: 10.1111/1523-1747.ep12611375. [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg S. P., Hale K. K., Heimdal P., Thompson R. C. Location of the protease-inhibitory region of secretory leukocyte protease inhibitor. J. Biol. Chem. 1990;265:7976–7981. [PubMed] [Google Scholar]
- 31.Bode W., Huber R. Ligand binding: proteinase–protein inhibitor interactions. Curr. Opin. Struct. Biol. 1991;1:45–52. [Google Scholar]
- 32.Wiedow O., Schroder J. M., Gregory H., Young J. A., Christophers E. Elafin: an elastase-specific inhibitor of human skin: purification, characterization, and complete amino acid sequence. J. Biol. Chem. 1990;265:14791–14795. [PubMed] [Google Scholar]
- 33.Karplus P. A. Hydrophobicity regained. Protein Sci. 1997;6:1302–1307. doi: 10.1002/pro.5560060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujinaga M., Chernaia M. M., Halenbeck R., Koths K., James M. N. The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener's granulomatosis antibodies. J. Mol. Biol. 1996;261:267–278. doi: 10.1006/jmbi.1996.0458. [DOI] [PubMed] [Google Scholar]
- 35.Morrison J. F., Walsh C. T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 36.Houghton A. M., Quintero P. A., Perkins D. L., Kobayashi D. K., Kelley D. G., Marconcini L. A., Mecham R. P., Senior R. M., Shapiro S. D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiesner O., Litwiller R. D., Hummel A. M., Viss M. A., McDonald C. J., Jenne D. E., Fass D. N., Specks U. Differences between human proteinase 3 and neutrophil elastase and their murine homologues are relevant for murine model experiments. FEBS Lett. 2005;579:5305–5312. doi: 10.1016/j.febslet.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 38.Tremblay G. M., Janelle M. F., Bourbonnais Y. Anti-inflammatory activity of neutrophil elastase inhibitors. Curr. Opin. Investig. Drugs. 2003;4:556–565. [PubMed] [Google Scholar]
- 39.Simpson A. J., Wallace W. A., Marsden M. E., Govan J. R., Porteous D. J., Haslett C., Sallenave J. M. Adenoviral augmentation of elafin protects the lung against acute injury mediated by activated neutrophils and bacterial infection. J. Immunol. 2001;167:1778–1786. doi: 10.4049/jimmunol.167.3.1778. [DOI] [PubMed] [Google Scholar]
- 40.Meyer-Hoffert U., Wichmann N., Schwichtenberg L., White P. C., Wiedow O. Supernatants of Pseudomonas aeruginosa induce the Pseudomonas-specific antibiotic elafin in human keratinocytes. Exp. Dermatol. 2003;12:418–425. doi: 10.1034/j.1600-0625.2002.120409.x. [DOI] [PubMed] [Google Scholar]
- 41.McMichael J. W., Roghanian A., Jiang L., Ramage R., Sallenave J. M. The antimicrobial antiproteinase elafin binds to lipopolysaccharide and modulates macrophage responses. Am. J. Respir. Cell Mol. Biol. 2005;32:443–452. doi: 10.1165/rcmb.2004-0250OC. [DOI] [PubMed] [Google Scholar]