

Abstract
Endosialidase (endo-N-acetylneuraminidase) is a tailspike enzyme of bacteriophages specific for human pathogenic Escherichia coli K1, which specifically recognizes and degrades polySia (polysialic acid). polySia is also a polysaccharide of the capsules of other meningitis- and sepsis-causing bacteria, and a post-translational modification of the NCAM (neural cell-adhesion molecule). We have cloned and sequenced three spontaneously mutated endosialidases of the PK1A bacteriophage and one of the PK1E bacteriophage which display lost or residual enzyme activity but retain the binding activity to polySia. Single to triple amino acid substitutions were identified, and back-mutation constructs indicated that single substitutions accounted for only partial reduction of enzymic activity. A homology-based structural model of endosialidase revealed that all substituted amino acid residues localize to the active site of the enzyme. The results reveal the importance of non-catalytic amino acid residues for the enzymatic activity. The results reveal the molecular background for the dissociation of the polySia binding and cleaving activities of endosialidase and for the evolvement of ‘host range’ mutants of E. coli K1 bacteriophages.
Keywords: bacteriophage, catalytic activity, endosialidase (endo-N-acetylneuraminidase), host range mutant, polysialic acid
Abbreviations: GFP, green fluorescent protein; NCAM, neural cell-adhesion molecule; polySia, polysialic acid
INTRODUCTION
PolySia (polysialic acid) is a homopolymer of α-(2,8)-linked NeuNAc and constitutes the capsule of the pathogenic bacteria Escherichia coli K1, Neisseria meningitidis group B, Mannheimia (Pasteurella) haemolytica A2 and Moraxella nonliquefaciens [1–3]. PolySia, the K1 antigen, is a promising target of bacteriophage therapy [4,5]. PolySia is also a post-translational modification of the NCAM (neural cell-adhesion molecule) and promotes complex neurological processes, such as plasticity [6,7]. PolySia synthesis and its role to direct tumour cell growth has been studied extensively [8–10].
E. coli K1-specific bacteriophages encoding K1 depolymerase activity have been isolated [11–13]. The coding genes of the polySia degrading enzyme endosialidase (endo-N-acetylneuraminidase) have been cloned from some of the bacteriophages [13–15]. We have previously isolated host range mutants of the PK1A and PK1E bacteriophages using E. coli mutants with a sparse polySia capsule [16,17]. The bacteriophage mutants have lost the catalytic activity of their endosialidase partly or totally, but still retain their polySia-binding activity [18]. The structural basic for the dissociation of the catalytic and binding activities has remained unresolved.
In the present study, we have cloned and sequenced the PK1A endosialidase gene of the wild-type bacteriophage and its three mutants, as well as a mutant of the PK1E bacteriophage, and identified the amino acid substitutions in their sequence. A homology-based structural model of the endosialidase predicts the spatial arrangement of the mutant amino acids close to each other at the active site of the enzyme, which thus indicates the molecular basis of dissociation of the binding and catalytic activity, and the altered host range of the mutant bacteriophages.
MATERIALS AND METHODS
Bacteriophages and bacterial strains
Bacteriophage PK1A was kindly supplied by Dr B. Rowe (Central Public Health Laboratory, Colindale, London, U.K.) [11]. The mutant bacteriophages PK1A2, PK1A5, PK1A8 and PK1E3 have been described by Pelkonen et al. [16]. The following E. coli strains were used: IH3088 [19] and K1-defective PK1A-resistant mutant strains, EH954 and EH1008, derived from rough E. coli IH3088 [17,19]. IH3088 is the host of the native bacteriophage PK1A and PK1E. EH954 is infected by PK1A2. PK1A5, PK1A8 and PK1E3 have EH1008 host specificity. LB (Luria–Bertani) broth, LB agar plates and soft agar [20] were used for the cultivation of the organisms. The bacteriophages were propagated by standard methods [21].
Purification of the genomic bacteriophage DNA
For the DNA purification, 100–200 μg of bacteriophage was used (measured by the Lowry method [42]). Proteinase K (Promega) and SDS were added to a final concentration of 200 μg/ml and 0.5% respectively in a total volume of 450 μl. After incubating for 2.5 h at 37°C the DNA was extracted with phenol/chloroform and precipitated with 0.1 vol. of 3 M sodium acetate (pH 5.2) and dissolved in water.
Cloning, sequencing and gene analysis
Part of the endosialidase gene, the BglII–BclI fragment, was cloned into pBluescript vector. DNA was sequenced on both strands by using the universal primers T3 and T7. The sequence was completed by primer walking by using synthetic oligonucleotide primers (Table 1) and the total bacteriophage genome as the template. The oligonucleotides were synthesized by Cyber Gene (Novum, Huddinge, Sweden). The same oligonucleotides were used to determine the mutant sequences. The sequences were determined on an ABI PRISM 377 sequencer at the Department of Medical Genetics, University of Turku. The assembly and sequence analysis were performed with DNASTAR (Lasergene package), BLAST [22] and EMBOSS software [23]. The EMBOSS Needle program was used to compare translated amino acid sequences.
Table 1. The oligonucleotides used to determine the endosialidase sequence of native bacteriophage PK1A and its three host range mutants and the oligonucleotides for sequencing of the PK1E3 mutant.
| Number of oligonucleotides | 5′–3′ Sequence | |
|---|---|---|
| PK1A | 1 (=T3) | AATTAACCCTCACTAAAGGG |
| 2 (=T7) | TAATACGACTCACTATAGGG | |
| 3 | CTTCAGAGGCTTCACCTTTG | |
| 4 | TGAGATAGAAAGTACCTGCA | |
| 5 | GCTGGAAAGAATTGGCACAT | |
| 6 | ATGTTTGACCATCGTCACCA | |
| 7 | TACCATCGTGCACATCCCTC | |
| 8 | TTACAAAGTATCATCTTTAC | |
| 9 | AAAATGAATGGGAAGCAGGT | |
| 10 | TAACCTCCTTTTCTGAAAGG | |
| 11 | AAGTACAAGTGCTAATATTC | |
| 12 | ACGTTCTGAACCAAACATAA | |
| 13 | TGTGCACCACACTCACTTTG | |
| 14 | CACAAAGTGAGGCGAATCAT | |
| 15 | TTTAGTACACTCTCATCAAT | |
| 16 | AAGGTAGGTATAGCCTGTCC | |
| 17 | TTCCAGCGTTATTCAAATCT | |
| PK1E3 | ||
| Left | 1 | CGTGGGCATCACTTGTATCA |
| 2 | CGGTACTGGTGCTACTTACA | |
| 3 | GTGGTATCACTAAGGCTGCA | |
| 4 | ACCTTATCACTCGTGGCACT | |
| 5 | GCAGATTGCAAATGACAGTC | |
| 6 | GATGCTGAACACAATTACGG | |
| Right | 1 | AGCTGACACACGCCTATAC |
| 2 | TTAGTGGCATACATCTCCTC | |
| 3 | AGCAGGCATGATTGGTGCAC | |
| 4 | GTTGTCACCATAAGTCATTG | |
| 5 | ACTCCAGATGGTATCTGACG | |
| 6 | ATGCACAATTGGTTAGTGCG |
Construction of fusion proteins
The catalytically active and inactive fusion proteins have been described earlier [24]. Constructions of point mutated fusion proteins (417- and 489-mutation) were made from catalytically inactive fusion protein by PCR, with the primers listed (Table 2). N- and C-terminal deletions of inactive EndoA2–GFP (endosialidase of mutant bacteriophage PK1A2–green fluorescent protein)-fusion protein were made by PCR by using the primers listed (Table 2). Constructs were monitored by sequencing. The products were expressed as described earlier [24].
Table 2. Oligonucleotides used in the back-mutations from PK1A2 endosialidase.
| Name | 5′–3′ Sequence | |
|---|---|---|
| Back-mutations | Y417H | CCACATAATGTGCATCATACTAC |
| Y417H | AAATCTTAGTGACTCCCAAGTC | |
| D489N | GACATTGTGAACTCTAGTGTAGG | |
| D489N | ACCCTGATAGATTTGGTCTGTG | |
| N-terminal deletions | PK1A-A | TCAGGTAAGAGAATTGACGG |
| PK1A-B | GCTTGGCCTCAAGACAAAGC | |
| PK1A-C | CTGCATCCAGATTACCCTAC | |
| PK1A-D | GGTGGTATCACTAAGGCTGC | |
| STALKEND | CGTGTACAGCTTATCTAACC | |
| C-terminal deletions | PK1A-1 | TTCGGATGCAAAAGCAGCTAC |
| PK1A-2 | CTTATGAGTGGCATCAGACG | |
| PK1A-3 | AGAAGTACTAGAACCTGCTCC | |
| PK1A-4 | TGAGCGAATATTAGCACTTGTAC | |
| STOP-pri | AGCTGAGCTTGGACTCCTG |
Enzyme activity assays
Thiobarbituric acid assay was used to measure endosialidase activity. The reagent volumes used in this study were half of the volumes of the modified Horgan procedure [25], except that the chromophore was extracted with 0.75 ml of acidified n-butanol. The total reaction volume was 50 μl adjusted with 100 mM sodium phosphate buffer (pH 6.6). Cultured bacteriophage stock (15 μl) or 4 μg of protein was used as enzyme sample. Colominic acid (poly-2,8-N-acetylneuraminic acid sodium salt; Sigma) substrate (1 μg/μl) was added in 30 μl of 100 mM sodium phosphate buffer (pH 6.6) for bacteriophage stocks or 50 mM sodium phosphate buffer (pH 7.5) and 300 mM NaCl for protein samples. Absorbance was measured at 549 nm against phosphate buffer and corrected with a substrate blank containing no enzyme. NeuNAc (Fluka) was used as reference. Results represent mean values from three independent assays. The protein concentration measurements of the bacteriophage stocks were taken into account in calculating the activity results.
Fluorescence microscopy of cells and paraffin sections
Human neuroblastoma SHSY-5Y cells (ECACC:94030304) and BHK (baby-hamster kidney) cells, BHK-21[C-13] (ATCC:CCL-10), were cultured on 13-mm glass slides in 24-well plates. The cells were fixed in 600 μl of 3% (w/v) paraformaldehyde in PBS (10 mM potassium phosphate buffer, pH 7.5, and 0.15 M NaCl) at room temperature (20–25°C) for 15 min, washed three times with PBS, and blocked with normal horse serum (Vector Laboratories) for half an hour at room temperature. Then, 250 μl of 10 μg/ml of each fusion protein in the buffer was added to the cells. Alternatively, a dilution series of the deletion fusion protein derivatives (10, 5, 3 and 0.5 μg/ml) in PBS was added to the cells. The cells were incubated for 1 h at room temperature, washed six times with the PBS, mounted on object glasses with Immu-mount (Shandon) and examined by fluorescence microscopy. Controls included staining in the presence of colominic acid (100 μg/ml, 30 min) or pre-incubation on the presence of active endosialidase enzyme (10 μg/ml) in PBS at room temperature for 30 min.
Paraffin-embedded temporal sections of human fetal (14-week) cortex cerebrum were sectioned (4 μm), deparafinized and rehydrated in descending ethanol series. Staining was done as described for the cells. The tissue samples were obtained with the appropriate approval of the Joint Ethical Committee of the University Hospital of Turku and the University of Turku.
Protein homology modelling
The sequence of the PK1A endosialidase was aligned to the sequence of chain D of the PK1F structure (PDB ID: 1V0E [26]) by using the MALIGN [27] algorithm in the computer program Bodil [28]. The default structural families-based scoring matrix [27] was used in the alignment, although – considering the similarity of the compared sequences – selection of a different selection matrix would not change the result significantly. The PK1A and PK1F endosialidases were assumed to contain a conserved active site structure due to the high sequence similarity. The residues corresponding to the point mutations of PK1A2, PK1A5 and PK1A8 endosialidase mutants were located from the PK1F structure via sequence alignment.
Sequence deposition
The PK1A endosialidase sequence data will appear in the GenBank® Nucleotide Sequence Data Library under the accession number EF507428.
RESULTS
Characteristics of PK1A endosialidase sequence
Bacteriophage PK1A is a helical bacterial virus with double-stranded DNA. Endo-α-sialidase (EC 3.2.1.129), a glycosyl hydrolase, is a component of the bacteriophage's short tail structure. The length of the native PK1A endosialidase sequence is 2436 bp and encodes a polypeptide of 811 amino acid residues. According to the amino acid sequence (Figure 1) the molecular mass of the polypeptide would be 89 kDa, but due to post-translational modification the molecular mass of the mature protein monomer is 76 kDa (Figure 2). PK1E endosialidase is similar in size to the PK1A enzyme. The enzymes are of exactly the same length as the PK1-5 endosialidase [13], while the PK1F polypeptide is longer and consists of 1064 amino acid residues [29].
Figure 1. Primary nucleotide sequence and derived amino acid sequence of the endosialidase PK1A gene.

A K1-5-like putative promoter region, the putative Shine–Dalgarno sites, the two sialidase motif sequences and ATP/GTP-binding site motif A (P-loop) are shown as boxed. An asterisk (*) marks the translation stop codon. Nucleotide and amino acid positions are given on both sites. These sequence data will appear in the GenBank® Nucleotide Sequence Data Library under the accession number EF507428.
Figure 2. Molecular size of the native and mutant endosialidases.
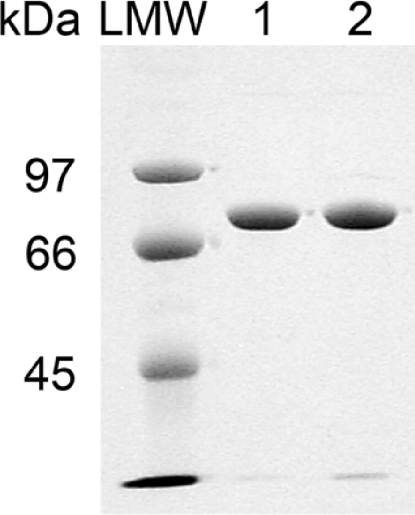
SDS/PAGE of the purified wild-type PK1A (lane 1) and mutant PK1A2 (lane 2) endosialidases. LMW, low-molecular-mass marker proteins.
The bacteriophage K1A endosialidase gene contains two copies of the sialidase (neuraminidase) motif Ser-Xaa-Asp-Xaa-Gly-Xaa-Thr-Trp (Figure 1). This Asp motif, typical of glycosyl hydrolases, probably maintains the correct structural fold and affects the solubility properties [30–33].
Based on sequence alignment an eight-element fingerprint provides a signature for the family 58 hydrolases. The PK1A endosialidase contains the signature at the following amino acid sequence positions: 104–129, 169–198, 288–312, 358–385, 436–463, 492–517, 598–625 and 630–658 (http://www.ebi.ac.uk/interpro/ and http://afmb.cnrs-mrs.fr/CAZY/). At the amino acid positions 38–45 the PK1A endosialidase matches the (Ala or Gly)-Xaa-Xaa-Xaa-Xaa-Gly-Lys-(Ser or Thr) pattern (Figure 1). This is the best-conserved motif of ATP/GTP-binding proteins named ‘A’ consensus sequence (or P-loop) [34,35].
The K1-5 promoter sequence has similarity to the consensus promoter of SP6. The putative PK1A endosialidase promoter (Figure 1) matches exactly with the K1-5 consensus promoter-like sequence predicted by Scholl et al. [36].
The PK1A nucleotide sequence upstream from the endosialidase coding region and Shine–Dalgarno site has another putative Shine–Dalgarno site (Figure 1). According to the standard genetic code translation there is one putative but rare start codon, GUG, which initiates to code a short peptide (18 amino acids). Downstream from the PK1A endosialidase coding sequence a putative Shine–Dalgarno site and a start codon are found, but the putative peptide (51 amino acids) has no significant similarity to EMBL database sequences. The other published coliphage endosialidase sequences and the K5 lyase (coliphage K5) have the highest similarity to the 3840 bp PK1A sequence reported here. The translated native endosialidase PK1A amino acid sequence is very similar to the PK1E and K1-5 translations, 90 and 89% respectively.
Identification of point mutations in the mutant endosialidase
The three PK1A mutant endosialidases and the PK1E mutant endosialidase are equal in size as compared with the wild-type of the enzyme. Each mutant displays one to three amino acid substitutions at different locations (Table 3). However, the substitution R614G in PK1A5 and PK1A8 is a conserved normal allele present in the wild-type bacteriophages PK1E, PK1-5 and PK1F displaying full endosialidase activity.
Table 3. Amino acid substitutions in mutant endosialidases.
Substitutions are highlighted by bold italic letters.
| Position in sequence | ||||||
|---|---|---|---|---|---|---|
| Bacteriophage | 118 | 332 | 370 | 417 | 489 | 614 |
| PK1A* | W | H | A | H | N | R |
| PK1A2* | W | H | A | Y | D | R |
| PK1A5* | W | N | A | H | D | G |
| PK1A8* | C | H | A | H | D | G |
| PK1E† | W | H | A | H | N | G |
| PK1E3* | W | H | E | H | N | G |
| K1-5‡ | W | H | A | H | N | G |
| PK1F§ | W | H | S | H | N | G |
Effects of point mutations on the catalytic and binding activities
Quantitative determination of the enzymatic activities revealed that PK1A2 and PK1A5 mutant bacteriophage endosialidases are catalytically inactive, whereas the PK1A8 and PK1E3 mutants exhibit greatly reduced activity (Figure 3). The results correlate well with the previously published qualitative estimation of the enzymic activities and binding properties [16].
Figure 3. Endosialidase activity of the wild-type bacteriophages PK1A and PK1E and the mutants PK1A2, PK1A5, PK1A8 and PK1E3.

The results are presented in nanomoles of released NeuNAc per minute per milligram of bacteriophage protein.
In order to study the role of the two point mutations in the PK1A2 endosialidase, the gene was back-mutated at the individual locations and the mutant proteins harbouring single amino acid substitutions were expressed as GFP-fusion proteins. Both back-mutants were able to cleave polySia, but with less relative activity than the native endosialidase (Figure 4). Thus both of the two amino acids were needed for complete inactivation of the enzyme.
Figure 4. Endosialidase activities of PK1A2 single amino acid mutants.

Endosialidase derivatives back-mutated for either of the two amino acid substitutions in PK1A2 endosialidase were constructed (Back a and Back b). The endosialidase activities of the GFP-fusion proteins were determined and expressed in relation to that of the native PK1A enzyme.
The influence of the single amino acid substitutions on the polySia binding activity of the enzyme was tested by binding to polySia-expressing neuroblastoma cells (binding defined as sustained binding). Of the GFP-fusion protein derivatives, only the catalytically inactive PK1A2 endosialidase remained bound to the cells (Figure 5).
Figure 5. Sustained binding of endosialidase derivatives to polySia-expressing cells.

Fluorescence microscopy of SHSY-5Y cells with endosialidase and endosialidase derivatives fused to GFP. Paraformaldehyde-fixed SHSY-5Y cells stained with catalytically active native endosialidase PK1A, catalytically inactive double mutant endosialidase PK1A2 or with the catalytically partially active single back-mutants Back a and Back b. Scale bars, 20 μm.
Effects of N- and C-terminal deletions on the catalytic and binding activities of endosialidase
In order to investigate whether deletions in terminal areas have an effect on the binding or catalysis, we made N- and C-terminal deletions to the wild-type and the mutant PK1A endosialidases (Table 4). The catalytic activity of wild-type PK1A endosialidase was very sensitive to deletions: only the first N-terminal deletion derivate (1–47) was catalytically active, whereas all C-terminally truncated derivatives were inactive. In the case of the PK1A mutant endosialidase only the two first N-terminally deleted derivatives, Δ1–47 and 1–107, bound polySia in a sustained manner, but also in these cases, the expression levels were low. Interestingly, truncation of residues 1–107 of the wild-type PK1A enzyme resulted in a derivate which was catalytically inactive but which retained its binding activity to polySia (Figure 6). Unlike the PK1A2 mutant endosialidase, the wild-type truncated derivative with the same catalytic and binding properties was, however, expressed in reduced quantities.
Table 4. Catalytic and binding activities of deletion derivatives of wild-type PK1A and mutant PK1A2 endosialidases.
The derivatives were expressed and purified as their GFP-fusion proteins, and assayed for catalytic and binding activities as described in Figures 3 and 5. Binding is defined as sustained binding, distinct from the transient binding during catalysis. Endosialidases with the deletions 1–158, 1–211, 616–811 and 565–811 were insoluble.
| Deleted sequence | Catalytic activity | Observed binding | |
|---|---|---|---|
| PK1A (wild-type) | Unmodified | + | – |
| Δ1–47 | + | – | |
| Δ1–107 | – | + | |
| Δ719–811 | – | – | |
| Δ668–811 | – | – | |
| PK1A2 (mutant) | Unmodified | – | + |
| Δ1–47 | – | + | |
| Δ1–107 | – | + | |
| Δ719–811 | – | – | |
| Δ668–811 | – | – |
Figure 6. Binding of N-terminally truncated wild-type PK1A endosialidase to polySia.

Paraffin-embedded temporal sections of fetal (14-week) cortex cerebrum were stained by using wild-type PK1A endosialidase with the terminal truncation Δ1–107. (a) Staining with GFP-fusion protein of the endosialidase derivative; (b) as in (a) but pretreated with active endosialidase before staining. Scale bars, 100 μm.
Endosialidase derivatives with longer truncations were all insoluble. Retrospectively, based on comparison with the PK1F structure [26], the deletions that produced insoluble enzyme obviously removed essential parts of the central (catalytic) domain or the C-terminal portion, which is important for trimerization.
Structural model of PK1A endosialidase
The crystal structure of PK1F endosialidase complexed with oligomeric sialic acid has been reported [26]. This endosialidase assembles into a homotrimer, and each monomer has three distinct domains: β-propeller, β-barrel and tailspike. The PK1F sequence contains an N-terminal extension, which is not found in PK1A, PK1E and K1-5 endosialidases. The central regions of the enzymes are highly conserved, including two ‘Asp-box’ motifs and all four enzymes are post-translationally processed from their C-terminus.
The sequence alignment of the PK1A and PK1F endosialidases indicates homology of the proteins: out of 666 residues in PK1F 663 were aligned with PK1A residues and 555 of those pairs are identical. Residues 1–37 of PK1A did not align with any residue of PK1F. The PK1F endosialidase structure lacks its N-terminal residues and thus no correspondence can be established. However, the aligned sections of the PK1A and PK1F sequences have 83% identity. Thus the PK1F endosialidase structure can be considered a viable template for building of a homology-based structural model of residues 38–706 of PK1A endosialidase.
The high identity of the homologues caused us to initially abandon the actual building of the homology model and study the residues of the PK1F endosialidase structure that correspond to the point mutations we observed in PK1A endosialidase. The distribution of conserved residues within the alignment supports this approach: the β-propeller domain, which contains the active site of the enzyme, is well conserved; the sequences are 93% identical. Furthermore, the β-barrel domain is 100% identical in sequence. Thus most differences in the sequences are located in the tailspike domain and in the N-terminal fragment, which forms a ‘cap’ for the β-propeller domain.
Four spontaneous point mutations implicated in the reduced activity of the PK1A mutants are located on or near the putative active site, where cleavage of polySia has been proposed to occur (Figure 7). Also the amino acid substitution in the PK1E3 endosialidase localizes to this site. Interestingly, the amino acid substitutions do not correspond to any of the three residues (Glu371, Arg386, and Arg437 in PK1A) that in PK1F were identified as active site residues. Furthermore, the four residues are all conserved between the PK1A and PK1F endosialidases. A common feature for all the residues involved in PK1A and PK1E activity is that single amino acid substitution did not inactivate the catalytic activity totally. For complete inactivation a combination of two substitutions, H417Y and N489D in PK1A2, H332N and N489D in PK1A5, or a combination of two of the three active site residues in PK1F, are required.
Figure 7. A structural model of PK1A endosialidase active site based on the PK1F endosialidase structure (PDB [41] ID: 1V0E [26]).

The molecular surface is shown with cyan, except that the three residues corresponding to those identified in endosialidase of bacteriophage PK1A2 as active site residues are drawn with grey and the mutated residues with transparent red colour and as space-filling models. Ala370, representing the mutation in PK1E3 (Table 3), is shown with a green surface. The residue numbers refer to the PK1A endosialidase sequence.
Trp118 forms one side of the hydrophobic pocket of the active cleft. A cysteine in the same position would clearly change the hydrophobic interactions that may occur between the substrate and the enzyme. Thus this mutation is likely to change specificity and affinity.
His332 is located next to Glu315, which in turn is next to the Glu371 identified as an active site residue in PK1F endosialidase. A histidine to asparagine mutation (as in PK1A5) is likely to change the co-ordination of Glu371 and thus affect its interaction with polySia.
His417 is buried and therefore not in a position to directly bind to the substrate. Thus the mutation to tyrosine (PK1A2) may have an indirect effect, since the surrounding residues have to compensate for the change of size, shape and hydrogen-bonding tendencies.
Asn489 is on the edge of the putative ‘cup-like’ active site. Asn489 forms a part of a ridge that is between the putative active site and polySia binding site formed by another chain of the trimer [26]. Thus it is possible that when the polySia chain is bound into the active site for cleavage, the chain forms interactions with asparagine at position 489. However, when the asparagine has mutated into aspartic acid, the charge of the side chain has changed so that the carbohydrate chain cannot form all the required interactions.
The two endosialidases from PK1E and PK1E3 differ only by one amino acid residue. The PK1E3 endosialidase has a glutamic acid in position 370, while the PK1E, PK1A and all its spontaneous mutations have alanine in that position. The effect of this single amino acid substitution on the enzyme activity is large (Figure 4). The magnitude of the effect can be explained by the rather radical change from a hydrophobic residue to an acidic one, the difference of the side-chain size, and in particular by the position. Residue 370 precedes Glu371, which in PK1F has been identified to have a major influence on activity [26] (Figure 7). The PK1F structure contains a serine residue at the equivalent position [26] and is in fact the only residue in the active site area that differs between the native endosialidase PK1A and PK1F.
DISCUSSION
Bacteriophages have a strong tendency to produce point mutations in the host identifying tail structure, in order to extend their host range [37]. Head proteins are usually more stable [38]. The spontaneous point mutations in endosialidase reflect Nature's susceptibility to create new wider host specificities. By keeping the binding property while decreasing the catalytic efficiency the mutants acquire the property to infect E. coli with a sparse polySia capsule. Sustained binding apparently provides the time required for the infection to occur in an environment of decreased number of substrate chains.
The spontaneous point mutations detected in the present study significantly reduce the ability of the endosialidase enzyme to cleave polySia. The point mutations reside around the active site residues and thus are likely to affect the shape of the active cleft, co-ordination of the putative catalytic residues, and the interactions with the polySia substrate. In the mutant bacteriophage endosialidases the substrate-binding property of the mutants remains, but the binding of polySia does not promote the transfer to the transition state as in the native enzyme, and thus the mutant enzymes cannot act as effective catalysts.
The dissociation constant, Kd, determined for the PK1A2 mutant fusion endosialidase and polySia earlier, is high [24]. The value 191×10−9 M is comparable with and even higher than typical values, 10−6 to 10−8, for carbohydrate antibodies [39]. Anyway, the binding in the case of mutant endosialidase PK1A2 is sustained and multiple conformational changes characteristic of the enzymatic activity are hindered. It is not possible to determine the dissociation constant for catalytically active endosialidase and polySia.
The effects of the relatively conservative spontaneous mutations resemble the effects of engineered alanine-substitutions in endosialidases [26]. In PK1F the substitutions E581A, R596A or R647A reduce catalytic activity by 95% [26], and in PK1E G138A by 80% [31]. In the present study, the activities of the mutants N489D (Back a) and H417Y (Back b) have decreased by 70 and 50% respectively. In all cases, one substitution alone is not enough to abolish catalytic activity completely. Double mutants are needed for total inactivation.
The ‘host range’ mutant bacteriophages studied were isolated by using E. coli mutants with a highly reduced amount of polySia capsule. The low or absent activity of the mutant endosialidases seems to be an essential requirement for the mutant bacteriophages to bind the sparse capsule of their host bacteria. Identification of individual amino acid substitutions in the active site of endosialidases characterized the molecular background of the changes that contributed to the development of the host range bacteriophage mutants of E. coli K1. Identification of the molecular changes that lead to the evolution of ‘host range’ bacteriophages infecting bacteria resistant to the parent bacteriophage contributes to our understanding of the molecular background of bacteriophage evolution and may also contribute to the development of practical applications, such as bacteriophage therapies to control pathogenic bacterial infections [40].
Acknowledgments
This study was supported by grants from the Finnish Academy and the Finnish Cultural Foundation. We thank Dr Mika Venojärvi (Department of Physiology, University of Kuopio, Kuopio, Finland) for help with confocal microscopy, Dr Olli Pentikäinen (Department of Biological and Environmental Science, University of Jyväskylä, Jyväskylä, Finland) for insights into molecular modelling, and Mrs T. Jompero, Mr J. Karhu and Ms A. Mikkola for technical assistance.
References
- 1.Jennings H. J., Katzenellenbogen E., Lugowski C., Michon F., Roy R., Kasper D. L. Structure, conformation and immunology of sialic acid-containing polysaccharides of human pathogenic bacteria. Pure Appl. Chem. 1984;56:893–905. [Google Scholar]
- 2.Devi S. J. N., Schneerson R., Egan W., Vann W. F., Robbins J. B., Shiloach J. Identity between polysaccharide antigens of Moraxella nonliquefaciens, group B Neisseria meningitidis, and Escherichia coli K1 (non-O acetylated) Infect. Immun. 1991;59:732–736. doi: 10.1128/iai.59.2.732-736.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adlam C., Knights J. M., Mugridge A., Williams J. M., Lindon J. C. Production of colominic acid by Pasteurella haemolytica serotype A2 organism. FEMS Microbiol. Lett. 1987;42:23–26. [Google Scholar]
- 4.Mushtaq N., Redpath M. B., Luzio J. P., Taylor P. W. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob. Agents Chemother. 2004;48:1503–1508. doi: 10.1128/AAC.48.5.1503-1508.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mushtaq N., Redpath M. B., Luzio J. P., Taylor P. W. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J. Antimicrob. Chemother. 2005;56:160–165. doi: 10.1093/jac/dki177. [DOI] [PubMed] [Google Scholar]
- 6.Rutishauser U. Polysialic acid at the cell surface: biophysics in service of cell interactions and tissue plasticity. J. Cell. Biochem. 1998;70:304–312. doi: 10.1002/(sici)1097-4644(19980901)70:3<304::aid-jcb3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Finne J. Occurrence of unique polysialosyl carbohydrate units in glycoproteins of developing brain. J. Biol. Chem. 1982;257:11966–11970. [PubMed] [Google Scholar]
- 8.Mühlenhoff M., Manegold A., Windfuhr M., Gotza B., Gerardy-Schahn R. The impact of N-glycosylation on the functions of polysialyltransferases. J. Biol. Chem. 2001;276:34066–34073. doi: 10.1074/jbc.M101022200. [DOI] [PubMed] [Google Scholar]
- 9.Horstkorte R., Mühlenhoff M., Reutter W., Nohring S., Zimmermann-Kordmann M., Gerardy-Schahn R. Selective inhibition of polysialyltransferase ST8SiaII by unnatural sialic acids. Exp. Cell Res. 2004;298:268–274. doi: 10.1016/j.yexcr.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Seidenfaden R., Krauter A., Schertzinger F., Gerardy-Schahn R., Hildebrandt H. Polysialic acid directs tumor cell growth by controlling heterophilic neural cell adhesion molecule interactions. Mol. Cell. Biol. 2003;23:5908–5918. doi: 10.1128/MCB.23.16.5908-5918.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross R. J., Cheasty T., Rowe B. Isolation of bacteriophages specific for the K1 polysaccharide antigen of Escherichia coli. J. Clin. Microbiol. 1977;6:548–550. doi: 10.1128/jcm.6.6.548-550.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machida Y., Miyake K., Hattori K., Yamamoto S., Kawase M., Iijima S. Structure and function of a novel coliphage-associated sialidase. FEMS Microbiol. Lett. 2000;182:333–337. doi: 10.1111/j.1574-6968.2000.tb08917.x. [DOI] [PubMed] [Google Scholar]
- 13.Scholl D., Rogers S., Adhya S., Merril C. R. Bacteriophage K1-5 encodes two different tail fiber proteins, allowing it to infect and replicate on both K1 and K5 strains of Escherichia coli. J. Virol. 2001;75:2509–2515. doi: 10.1128/JVI.75.6.2509-2515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long G. S., Bryant J. M., Taylor P. W., Luzio J. P. Complete nucleotide sequence of the gene encoding bacteriophage E endosialidase: implication for K1E endosialidase structure and function. Biochem. J. 1995;309:543–550. doi: 10.1042/bj3090543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerardy-Schahn R., Bethe A., Brennecke T., Mühlenhoff M., Eckhardt M., Ziesing S., Lottspeich F., Frosch M. Molecular cloning and functional expression of bacteriophage PK1E-encoded endoneuraminidase Endo NE. Mol. Microbiol. 1995;16:441–450. doi: 10.1111/j.1365-2958.1995.tb02409.x. [DOI] [PubMed] [Google Scholar]
- 16.Pelkonen S., Aalto J., Finne J. Differential activities of bacteriophage depolymerase on bacterial polysaccharide: binding is essential but degradation is inhibitory in phage infection of K1-defective Escherichia coli. J. Bacteriol. 1992;174:7757–7761. doi: 10.1128/jb.174.23.7757-7761.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelkonen S. Capsular sialyl chains of Escherichia coli K1 mutants resistant to K1 phage. Curr. Microbiol. 1990;21:23–28. [Google Scholar]
- 18.Aalto J., Pelkonen S., Kalimo H., Finne J. Mutant bacteriophage with non-catalytic endosialidase binds to both bacterial and eukaryotic polysialic acid and can be used as probe for its detection. Glycoconj. J. 2001;18:751–758. doi: 10.1023/a:1021147316647. [DOI] [PubMed] [Google Scholar]
- 19.Pelkonen S., Finne J. A rapid turbidimetric assay for the study of serum sensitivity of Escherichia coli. FEMS Microbiol. Lett. 1987;42:53–57. [Google Scholar]
- 20.Maniatis T., Fritsch E. F., Sambrook J., editors. Plainview, NY: Cold Spring Harbor Laboratory Press; 1982. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 21.Mäkelä P. H. The use of bacteriophagesin the study of bacterial cell surface structures. In: Korhonen T. K., Dawes E. A., Mäkelä P. H., editors. Enterobacterial Surface Antigenes: Methods for Molecular Characterization. Amsterdam: Elsevier Science Publishers; 1985. pp. 155–178. [Google Scholar]
- 22.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Apweiler R., Attwood T. K., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M. D., et al. InterPro – an integrated documentation resource for protein families, domains and functional sites. Bioinformatics. 2000;16:1145–1150. doi: 10.1093/bioinformatics/16.12.1145. [DOI] [PubMed] [Google Scholar]
- 24.Jokilammi A., Ollikka P., Korja M., Jakobsson E., Loimaranta V., Haataja S., Hirvonen H., Finne J. Construction of antibody mimics from a noncatalytic enzyme-detection of polysialic acid. J. Immunol. Methods. 2004;295:149–160. doi: 10.1016/j.jim.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Horgan I. E. A modified spectrophotometric method for determination nanogram quantities of sialic acid. Clin. Chim. Acta. 1981;116:409–415. doi: 10.1016/0009-8981(81)90062-0. [DOI] [PubMed] [Google Scholar]
- 26.Stummeyer K., Dickmanns A., Mühlenhoff M., Gerardy-Schahn R., Ficner R. Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 2005;12:90–96. doi: 10.1038/nsmb874. [DOI] [PubMed] [Google Scholar]
- 27.Johnson M. S., Overington J. P. A structural basis for sequence comparisons. An evaluation of scoring methodologies. J. Mol. Biol. 1993;233:716–738. doi: 10.1006/jmbi.1993.1548. [DOI] [PubMed] [Google Scholar]
- 28.Lehtonen J. V., Still D.-J., Rantanen V.-V., Ekholm J., Björklund D., Iftikhar Z., Huhtala M., Repo S., Jussila A., Jaakkola J., et al. BODIL: a molecular modeling environment for structure–function analysis and drug design. J. Comput. Aided Mol. Des. 2004;18:401–419. doi: 10.1007/s10822-004-3752-4. [DOI] [PubMed] [Google Scholar]
- 29.Mühlenhoff M., Stummeyer K., Grove M., Sauerborn M., Gerardy-Schahn R. Proteolytic processing and oligomerization of bacteriophage-derived endosialidases. J. Biol. Chem. 2003;278:12634–12644. doi: 10.1074/jbc.M212048200. [DOI] [PubMed] [Google Scholar]
- 30.Chien C. H., Shann Y. J., Sheu S. Y. Site-directed mutations of the catalytic and conserved amino acids of the neuraminidase gene, nanH, of Clostridium perfingens ATCC 10543. Enzyme Microb. Technol. 1996;19:267–276. doi: 10.1016/0141-0229(95)00245-6. [DOI] [PubMed] [Google Scholar]
- 31.Leggate D. R., Bryant J. M., Redpath M. B., Head D., Taylor P. W., Luzio J. P. Expression, mutagenesis and kinetic analysis of recombinant K1E endosialidase to define the site of proteolytic processing and requirements for catalysis. Mol. Microbiol. 2002;44:749–760. doi: 10.1046/j.1365-2958.2002.02908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crennell S., Garman E., Laver G., Vimr E., Taylor G. Crystal structure of Vibrio cholerae neuraminidase reveals dual lectin-like domains in addition to the catalytic domain. Structure. 1994;2:535–544. doi: 10.1016/s0969-2126(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 33.Gaskell A., Crennell S. J., Tailor G. L. The tree domains of a bacterial sialidase: a beta-propeller an immunoglobulin module and a galactose-binding jelly-roll. Structure. 1995;3:1197–1205. doi: 10.1016/s0969-2126(01)00255-6. [DOI] [PubMed] [Google Scholar]
- 34.Walker J. E., Saraste M., Runswick M. J., Gay N. J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saraste M., Sibbald P. R., Wittinghofer A. The P-loop – a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 36.Scholl D., Kieleczawa J., Kemp P., Rush J. C. R. C., Merril C., Adhya S., Molineux I. J. Genomic analysis of bacteriophages SP6 and K1-5, an estranged subgroup of the T7 supergroup. J. Mol. Biol. 2004;335:1151–1171. doi: 10.1016/j.jmb.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 37.Haggård-Ljungquist E., Halling C., Calendar R. DNA sequences of the tail fiber genes of bacteriophage P2: evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J. Bacteriol. 1992;174:1462–1477. doi: 10.1128/jb.174.5.1462-1477.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chibani-Chennoufi S., Canchaya C., Bruttin A., Brüssow H. Comparative genomics of the T4-like Escherichia coli phage JS98: implications for the evolution of T4 phages. J. Bacteriol. 2004;186:8276–8286. doi: 10.1128/JB.186.24.8276-8286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellequer J. L., Van Regenmortel M. H. Affinity of monoclonal antibodies to large multivalent antigens: influence of steric hindrance on antibody affinity constants calculated from Scatchard plots. Mol. Immunol. 1993;30:955–958. doi: 10.1016/0161-5890(93)90022-4. [DOI] [PubMed] [Google Scholar]
- 40.Qimron U., Marintcheva B., Tabor S., Richardson C. C. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. U.S.A. 2006;103:19039–19044. doi: 10.1073/pnas.0609428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]