

Abstract
Fortilin, a 172-amino-acid polypeptide present both in the cytosol and nucleus, possesses potent anti-apoptotic activity. Although fortilin is known to bind Ca2+, the biochemistry and biological significance of such an interaction remains unknown. In the present study we report that fortilin must bind Ca2+ in order to protect cells against Ca2+-dependent apoptosis. Using a standard Ca2+-overlay assay, we first validated that full-length fortilin binds Ca2+ and showed that the N-terminus (amino acids 1–72) is required for its Ca2+-binding. We then used flow dialysis and CD spectropolarimetry assays to demonstrate that fortilin binds Ca2+ with a dissociation constant (Kd) of approx. 10 μM and that the binding of fortilin to Ca2+ induces a significant change in the secondary structure of fortilin. In order to evaluate the impact of the binding of fortilin to Ca2+ in vivo, we measured intracellular Ca2+ levels upon thapsigargin challenge and found that the lack of fortilin in the cell results in the exaggerated elevation of intracellular Ca2+ in the cell. We then tested various point mutants of fortilin for their Ca2+ binding and identified fortilin(E58A/E60A) to be a double-point mutant of fortilin lacking the ability of Ca2+-binding. We then found that wild-type fortilin, but not fortilin(E58A/E60A), protected cells against thapsigargin-induced apoptosis, suggesting that the binding of fortilin to Ca2+ is required for fortilin to protect cells against Ca2+-dependent apoptosis. Together, these results suggest that fortilin is an intracellular Ca2+ scavenger, protecting cells against Ca2+-dependent apoptosis by binding and sequestering Ca2+ from the downstream Ca2+-dependent apoptotic pathways.
Keywords: apoptosis, Ca2+, cell death, fortilin, thapsigargin
Abbreviations: AM, acetoxymethyl; BAPTA-AM, acetoxy-methyl-1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; CBB, Ca2+-binding buffer; DMEM, Dulbecco's modified Eagle's medium; EDB, equilibrium dialysis buffer; ER, endoplasmic reticulum; EthD-1, ethidium homodimer-1; FCS, fetal calf serum; GST, glutathione transferase; MEF, mouse embryonic fibroblast; R110, benzyloxycarbonyl-DEVD-rhodamine 110; r-[Ca2+]i-base, baseline relative intracellular Ca2+ concentration; RFI, relative fluorescence intensity; RFU, relative fluorescence unit; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase; siRNA, small-interfering RNA
INTRODUCTION
Fortilin is a 172-amino-acid polypeptide originally identified in our laboratory as a molecule that specifically interacts with MCL1 (myeloid cell leukaemia sequence 1) [1], a member of the Bcl-2 family of anti-apoptotic molecules [2]. Fortilin is also known as TCTP (translationally controlled tumour protein).
The amino acid sequence of fortilin is highly conserved among species ranging from human to rice [3]. Its message is distributed ubiquitously in normal tissue, most abundantly in the liver and kidney [3]. Fortilin is present in both the nucleus and cytosol [3], inducible by serum stimulation [4] and heavy metals [5], and expressed at much higher levels in cancerous cell lines than in non-cancerous cell lines [3]. Fortilin is overexpressed in a variety of human malignancies, including liver, thyroid, laryngeal, skin, uterine, breast, ovarian, prostate and rectal cancers [6]. Fortilin has a potent anti-apoptotic function: its overexpression protects HeLa [3] and U2OS [7] cells against apoptosis. In addition, the depletion of fortilin from cells induces spontaneous cell death in MCF-7 [3] and U937 cells [6]. Although fortilin is likely to make cells cancerous and cancerous cells more resistant to chemotherapy by preventing these cells from undergoing apoptosis, the exact mechanism by which fortilin blocks apoptosis remains unknown.
That fortilin binds calcium was originally shown by Haghighat and Ruben in 1992 [8]. In that work, a large quantity of homogenate of Trypanosoma brucei, the parasitic cause of West African sleeping sickness, was fractionated and probed by 45Ca2+ in a Ca2+-overlay assay to identify Ca2+-binding proteins. One of the proteins shown by the Ca2+-overlay assay to bind Ca2+ was a 22-kDa protein, whose N-terminal sequence was found to be identical with that of T. brucei fortilin [8]. The fact that fortilin binds Ca2+ has since been validated by Ca2+-overlay assays in several laboratories including those of Sanchez et al. [9], Kim et al. [10], Rao et al. [11] and Arcuri et al. [12]. In 1999, Xu et al. [13] reported that Ca2+ up-regulated fortilin transcription, suggesting a role for fortilin in intracellular calcium homoeostasis. However, the biochemistry of the fortilin–Ca2+ interaction has not been extensively studied. In addition, the exact biological consequence of fortilin binding to Ca2+ remains unknown. Furthermore, it is unknown whether fortilin is required to bind Ca2+ in order to block the Ca2+-dependent apoptosis.
The cytosolic concentration of free Ca2+ is maintained at very low levels (∼100 nM) by the continuous pumping of Ca2+ out of the cytosol into the ER (endoplasmic reticulum) by SERCAs (sarcoplasmic/endoplasmic reticulum Ca2+-ATPases) [14]. This explains why the ER, among all organelles, contains the highest concentration of Ca2+ and why the inhibition of SERCAs, for example by thapsigargin, immediately and drastically elevates Ca2+ levels in the cytosol. Even slight elevation of the Ca2+ concentration beyond the tightly regulated range could lead to the most drastic biological phenotype, death of the cell by apoptosis. Cytosolic Ca2+ beyond the normal range could attack and injure mitochondrial membranes, leading to the release of pro-apoptotic molecules such as cytochrome c and AIF (apoptosis-inducing factor) [14,15], and to programmed cell death. Ca2+ can also induce apoptosis by activating pro-apoptotic Ca2+–CaM-dependent enzymes [16–18], Ca2+-dependent endonucleases [19], Ca2+-binding cysteine proteases [20–22], calcineurin [23] and Ca2+-sensitive NO synthases [14,24]. Once Ca2+ levels go beyond a critical threshold to activate one of these death-bound pathways, apoptosis is inevitable.
It is possible, however, that there is a cellular mechanism by which the cell sequesters cytosolic Ca2+ before it activates the downstream cell-death pathways described above. Based on the fact that fortilin blocks apoptosis [3] and the fact that fortilin binds Ca2+[8], we hypothesized that fortilin binds Ca2+ in the cytosol, functions as a Ca2+ scavenger and sequesterer, prevents Ca2+ levels from going beyond the apoptosis threshold and protects cells against Ca2+-dependent cell death. We hypothesized that the anti-apoptotic activity of fortilin was due to its binding to, and consequent sequestration from downstream apoptotic pathways of, Ca2+ that was released in response to apoptotic stimuli. In the present study we report that fortilin indeed binds Ca2+ and scavenges Ca2+ released in response to thapsigargin, preventing cytosolic Ca2+ levels from increasing and activating Ca2+-dependent apoptosis pathways.
MATERIALS AND METHODS
Materials
Calmodulin was a gift from Dr John A. Patkey (The University of Texas Health Science Center at Houston, Houston, TX, U.S.A.). Fortilin peptides were chemically synthesized by GenScript. The sequence of each peptide is shown in Figure 8(A). For all procedures described below, plastic and glass instruments were rinsed several times with 0.1 M HCl and then washed extensively with ultra-pure water to remove contaminating Ca2+.
Figure 8. Short peptides of fortilin, fortilin(43–62), fortilin(57–76) and fortilin(127–146), bind Ca2+ in equilibrium dialysis assays.

(A) Fortilin peptides covering various regions of fortilin were synthesized. Peptides in the highlighted rows are fortilin peptides that exhibited significant Ca2+-binding activity in equilibrium dialysis assays. (B) Equilibrium dialysis of fortilin peptides showed significant binding of fortilin(43–62), fortilin(57–76) and fortilin(127–146) to Ca2+. ****P<0.001, ***P<0.005, **P<0.01, *P<0.05, +P=0.05. CPM, counts per min.
Molecular cloning
The cDNA of fortilin and its deletion and point mutants were synthesized by PCR-based strategies as we have described previously [25]. In all cases, the authenticity of cloned constructs was confirmed by automated dideoxynucleotide sequencing (Lark Technologies).
Expression and purification of recombinant fortilin and its mutants
Expression and purification of recombinant fortilin protein and its mutants were performed as previously described [26], using Escherichia coli BL21 cells transfected with pGEX-fortilin (or its mutants; Amersham–Pharmacia) or pQE41-fortilin (Qiagen).
Ca2+-overlay assay
Ca2+-overlay assays were performed as described previously [9,10,12,13,27]. In brief, proteins were size-fractionated by SDS/PAGE and transferred on to a PVDF membrane (Immobilon-P). The membrane was airdried, incubated for 2 h at room temperature (25 °C) with 45CaCl2 (MP Biomedicals) in CBB [Ca2+-binding buffer; 60 mM KCl, 5 mM MgCl2 and 10 mM imidazole (pH 6.8)] at a final concentration of 40 μCi/ml, rinsed in CBB, washed in 50% ethanol for 10 min, air-dried, and exposed to a phosphor imager screen overnight. Signals were detected by a Molecular Scanner-FX and Quantity One software system (Bio-Rad), according to the manufacturer's instructions. Proteins transferred on to the membrane were semi-quantitatively assessed by Ponceau S staining.
CD spectropolarimetry
Far-UV CD spectra of GST (glutathione transferase)–fortilin samples (0.36 μg/μl) were collected on a Jasco J715 spectropolarimeter using a 1-mm-path-length quartz cuvette at room temperature. Spectra were recorded from 260 to 195 nm in 0.5 nm steps at 100 nm/min. Data represent the average of 10 independent spectra. All protein samples were passed through a PD-10 column (Sephadex-25; Pharmacia) equilibrated in 0.1 M phosphate buffer (pH 7.4) for desalting. The samples were then passed twice through a Chelex 100 chelating ion-exchange resin column (Bio-Rad). Data were expressed as mean residue ellipticity (degrees cm2/dmol).
Flow dialysis assay
A flow dialysis assay was performed as previously described [28], at 25 °C, using 45CaCl2 and Spectrapore 6 cellulose dialysis membrane (Mr cut-off 1000 Da, Spectrum Medical Industry). The flow rate was 3 ml/min and the response time of the apparatus was about 0.9 min. Initially, 2.25 μCi of 45Ca2+ was added into 1 ml of the GST–fortilin protein solution in the upper chamber. After the successive titration with unlabelled Ca2+, an 800 μl portion of the dialysate fraction (1 ml/tube) which flowed out through the lower chamber (0.66 ml) was taken, and the radioactivity was determined using a Beckmann LS-600C liquid scintillation counter. The resulting Ca2+-binding data were analysed by fitting to the Adair equation for the 2-site model:
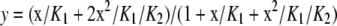 |
where y is the number of bound Ca2+ (mol/mol-protein), x is the concentration of free Ca2+ and K1 and K2 are the macroscopic dissociation constants.
Cell, cell lines and culture conditions
U2OS cells and MEFs (mouse embryonic fibroblasts) were maintained in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% FCS (fetal calf serum) and, when appropriate, antibiotics. MEF cells from passages 4–9 were used in all experiments.
Isolation of MEFs
Western blot analysis
Cells were harvested by the direct addition of Laemmli SDS gel loading buffer [1,3]. When appropriate, cells were harvested into RIPA buffer [50 mM Tris/HCl (pH 7.4), 150 mM NaCl, 1% NP40 (Nonidet P40), 0.1% SDS, 0.5% sodium deoxycholate, and protease inhibitors (Complete Protease Inhibitor Cocktail Tablets; Roche Biochemicals)] for the determination of protein concentrations (using the Bradford method; Bio-Rad). Western blot analysis was performed as described previously [3,25], using anti-fortilin and anti-actin (Roche Molecular Biochemicals) antibodies.
In vivo Ca2+ release assay
U2OS (1×104) or MEF (2×104) cells were seeded in each well of a 96-well plate. The next day, the cells were transfected with either siRNA (small interfering RNA) against luciferase, a non-mammalian protein from Photinus pyralis (American firefly) (siRNAluciferase, control), or siRNA against human fortilin (siRNAfortilin) at final concentrations of 100 nM. Both siRNAs [32,33] were synthesized by Dharmacon Research. Transfection was carried out using the DharmaFECT™ two transfection reagent (Dharmacon) as described previously [1]. The siRNAfortilin consisted of a mixture of four siRNA duplexes targeting four different regions of fortilin mRNA, namely 5′-AGATGTTCTCCGACATCTA-3′, 5′-CGAAGGTACCGAAAGCACA-3′, 5′-GGGAGATCGCGGACGGGTT-3′ and 5′-GGTACCGAAAGCACAGTAA-3′. At 48 h after transfection, cells were exposed to 5 μM Fura 2/AM (fura 2 acetoxymethyl ester) for 1 h at 37 °C under 5% CO2, allowed to recover in DMEM with 10% FCS for 20 min, and transferred to assay buffer consisting of HBSS (Hanks balanced salt solution; Cambrex Bioscience) supplemented with 10 mM Hepes, 200 μM Ca2+, 0.1% BSA, 2.5 mM probenecid and pluronic-F127 (Molecular Probes). A baseline relative intracellular Ca2+ concentration (termed r-[Ca2+]i-base), was calculated by first obtaining a fluorescent signal by excitation at 355 nm and emission at 505 nm [termed RFU355/505 (where RFU is the relative fluorescence unit)] and a fluorescent signal by excitation at 363 nm and emission at 512 nm (termed RFU363/512) on a SpectraMax M2 plate reader (Molecular Probes) and then dividing RFU355/505 by RFU363/512, as described previously [34]. The Fura 2 fluorescence emission ratio [i.e. the ratio of RFI (relative fluorescence intensity) at an excitation at 355 nm and an emission at 505 nm (RFI355/505) to an excitation at 363 nm and emission at 512 nm (RFI363/512)] correlates with intracellular Ca2+ concentration ([Ca2+]i) [34]. At zero time, thapsigargin (Sigma) was added to a final concentration of 400 nM. Then, fluorescent signals (RFU355/505 and RFU363/512) were obtained every 10 s for the next 10 min. The r-[Ca2+]i at a given time point was calculated by dividing RFU355/505 by RFU363/512 minus r-[Ca2+]i-base. The free intracellular Ca2+ index represents the area under the curve between zero time and 600 s and was expressed as an arbitrary unit. Thapsigargin, a sesquiterpene γlactone derived from the plant Thapsia garganica, is a specific and irreversible inhibitor of the SERCAs, including SERCA1, SERCA2a, SERCA2b and SERCA3 [35]. Conversely, thapsigargin has no effect on plasma membrane Ca2+-ATPase, Na,K-ATPase, or other enzymes [35]. In addition, the administration of thapsigargin results in the immediate and drastic elevation of [Ca2+]i [36]. Experiments were performed in quadruplicate and repeated at least three times, with consistent results. After the determination of r-[Ca2+]i, cells were subjected to Western blot analysis as described above.
Cell death assay in the presence and absence of BAPTA-AM [acetoxy-methyl-1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid]
In brief, 1.5×104 U2OS cells were seeded in each well of a 96-well plate and transfected with siRNAfortilin or siRNAluciferase as described above. A cell death assay using EthD-1 (ethidium homodimer-1; Molecular Probes) was performed as described previously [37,38], with the following modifications. At 12 h after transfection, cells were treated with 15 μM BAPTA-AM-Ca2+-chelator (Molecular Probe) or vehicle (DMSO) for 45 min at 37 °C, washed twice with PBS, challenged with 1 μM thapsigargin or vehicle, and incubated for 18 h at 37 °C. After incubation, cells were washed with PBS and stained with 8 μM EthD-1 for 30 min at room temperature. EthD-1 is excluded from living cells but can cross the compromised plasma and nuclear membranes of dying cells and interact with nucleic acids to give red fluorescence. EthD-1 positivity has been observed in the late-phase of apoptosis or in necrosis [37,38]. Fluorescent signals were obtained by 528 nm excitation and 617 nm emission (RFU528/617). The EthD-1 index, reflecting cell death rate, was calculated using the following equation:
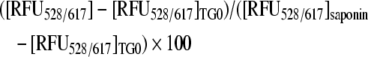 |
where [RFU528/617]TG0 is the RFU528/617 from non-TG-treated cells (background) and [RFU528/617]saponin is RFU528/617 from cells treated with 0.1% saponin (Sigma). Typically, EthD-1 indices were calculated for two groups of cells, one treated and one not treated with BAPTA-AM. The EthD-1 indices of BAPTA-AM-treated cells represented Ca2+-independent cell death rates, whereas those of non-BAPTA-AM-treated cells represented total cell death rates both Ca2+-dependent and -independent. Data from caspase 3 activity assays (Figures 6C and 7C) show that most of thapsigargin-induced Ca2+-dependent cell death represents necrosis, without significant activation of caspase 3. For MEF, 1.5×104 MEFfortilin+/− or MEFfortilin+/+ cells were used. Experiments were normally performed in quadruplicate and repeated at least three times, with consistent results.
Figure 6. Fortilin inhibits thapsigargin-induced Ca2+-dependent cell death in U2OS cells.

(A) Morphology of BAPTA-AM-treated and untreated U2OS cells transfected with either siRNAfortilin or siRNAluciferase. BAPTA-AM-treatment reduces the percentage of dead cells (rounded and detached), especially in siRNAfortilin-treated cells. Cell death preventable by BAPTA-AM (a potent Ca2+-chelator), represented Ca2+-dependent cell death. (B) Fortilin protected cells against thapsigargin-induced cell death (column 1 compared with column 3), most of which was Ca2+-dependent cell death (CDC). **P<0.01; #, cell death blocked by fortilin. (C) The siRNAfortilin-treated cells, but not siRNAluciferase-treated cells, exhibit caspase 3 activation upon thapsigargin stimulation, suggesting that most of the cell death of siRNAluciferase-treated cells in (B) (columns 3 and 4) represented necrosis (cell death lacking caspase 3 activation). **P<0.01.
Figure 7. Fortilin inhibits thapsigargin-induced Ca2+-dependent cell death in MEF cells.

(A) Morphology of BAPTA-AM-treated and -untreated MEFfortilin+/− and MEFfortilin+/+ cells. BAPTA-AM-treatment reduces the percentage of dead cells (rounded and detached), especially in MEFfortilin+/− cells. Cell death preventable by BAPTA-AM (a potent Ca2+-chelator), represented Ca2+-dependent cell death. (B) Fortilin protects cells against thapsigargin-induced, Ca2+-mediated (BAPTA-AM-preventable), cell death (CDC). ***P<0.005; #, cell death blocked by fortilin. (C) MEFfortilin+/− cells, but not MEFfortilin+/+ cells, exhibit caspase 3 activation upon thapsigargin stimulation, suggesting that most of cell death of MEFfortilin+/− cells in (B) (columns 3 and 4) represented necrosis (cell death lacking caspase 3 activation). *P<0.05.
Caspase 3 activity assay
Caspase 3 assays for MEFs and siRNA-treated U2OS cells were performed as previously described [3]. In brief, cells were treated with 1 μM thapsigargin or vehicle as described above. Cytosolic proteins were extracted by three cycles of freezing and thawing in hypotonic cell lysis buffer [25 mM Hepes (pH 7.5), 5 mM MgCl2, 5 mM EDTA, 5 mM dithiothreitol and 0.05% PMSF; all from Sigma]. Cytosolic extracts (20 μg) were added to caspase assay buffer [312.5 mM Hepes (pH 7.5), 31.25% sucrose and 0.3125% CHAPS] with R110 (benzyloxycarbonyl-DEVD-rhodamine 110) as substrates (Molecular Probes). Release of R110 by caspase-3-like activity was quantified, after 2 h of incubation at 37 °C, using a SpectraMax M2 plate reader (Molecular Probes) set to an excitation value of 498 nm and emission value of 521 nm. The results were expressed as relative fluorescence units/μg of protein.
Equilibrium dialysis assay of fortilin peptides and GST–fortilin and its mutants
An equilibrium dialysis assay to assess qualitatively the binding of candidate proteins and peptides to Ca2+ was performed as described previously [39–41], with modifications. Briefly, 100 μl each of fortilin peptides, GST, GST–fortilin, its mutants or His6-tagged fortilin [20 μM in EDB (equilibrium dialysis buffer; 10 mM Mops, pH 7.4 and 100 mM KCl)] was placed in a Tube-O-Dialyzer tube (Mr cut off 1000 Da; Genotech), which was then placed in a beaker containing 200 ml of EDB with 1 μM Ca2+ and 45Ca2+ at 1000 c.p.m./μl. The radioactivity of each 15 μl aliquot was determined in triplicate.
Generation of cells stably overexpressing fortilin and its mutant
U2OS cells were transfected, using FuGENE™ 6 reagent (Roche Molecular Biochemicals), with empty pcDNA4-His-Max (pcDNA4; Invitrogen) or pcDNA4 containing the cDNA encoding either wild-type fortilin (pcDNA4Fortilin) or its mutant (pcDNA4FortilinΔ2). FortilinΔ2 contained two point mutations: the glutamate residues at amino acid positions 58 and 60 were both mutated to alanine residues and sometimes referred to as fortilin(E58A/E60A). Transfected cells were selected in medium containing 400 μg/ml Zeocin (Invitrogen) and characterized by Western blot analyses. The resulting cell lines were named U2OSempty, U2OSfortilin and U2OSfortilinΔ2 respectively.
Statistical analysis
Experiments were performed in duplicate, triplicate or quadruplicate to obtain S.D. Variations in flow dialysis were expressed as S.E.M. Differences between two experimental groups were analysed using the Student's t test. P<0.05 was considered statistically significant.
RESULTS
Fortilin binds Ca2+ in a Ca2+-overlay assay
To test the hypothesis that fortilin binds to Ca2+, standard Ca2+-overlay assays were performed where proteins of interest were resolved by SDS/PAGE and transferred on to PVDF membranes, which were in turn incubated with 45Ca2+, washed and exposed to a phosphor imager screen. As shown in Figure 1(A), 45Ca2+ was not bound at all by albumin (lane 1), was bound robustly by the established Ca2+-binding protein calmodulin (lane 2), and was bound by fortilin–His6 (lane 3). Using the same system, we tested different portions of fortilin for their importance in Ca2+ binding. As shown in Figure 1(B), full-length fortilin [GST–fortilin(1–172)], but not BSA or GST alone, bound 45Ca2+ (lane 3 compared with lanes 1 and 2). In this system, GST–fortilin(1–70), but not GST–fortilin(71–120) or GST–fortilin(121–172), bound 45Ca2+. These results suggest that fortilin in fact binds Ca2+ and that the 70 amino acid residues at the N-terminus of fortilin are critical for the binding of fortilin to Ca2+ (Figure 1C).
Figure 1. Ca2+-overlay assay shows that fortilin binds Ca2+ through its first 70 amino acids.

(A) Ca2+-overlay assay of fortilin–His6. (B) Ca2+-overlay assay of GST–fortilin and its deletion mutants. Exactly 10 μg of proteins were size-fractionated by SDS/PAGE and transferred on to a PVDF membrane, which was then incubated with 45Ca2+, washed and exposed to a phosphor imager screen. Neither albumin nor GST-only bound Ca2+, whereas calmodulin (a known Ca2+-binding protein) did bind Ca2+. In this system, both fortilin–His6 (A) and GST–fortilin (B) bound Ca2+. In addition, GST–fortilin(1–70), but not GST–fortilin(71–120) or GST–fortilin(121–172), bound Ca2+ (B and C). *Degradation products.
Flow dialysis assays suggest the presence of two high-affinity Ca2+-binding sites in fortilin
In order to characterize further the binding of fortilin to Ca2+, flow dialysis assays were performed. Results of three independent measurements were shown in Figure 2(A). GST–fortilin showed a biphasic Ca2+ binding, binding to approx. two sites with high affinity and to multiple sites with lower affinity. The high-affinity sites were almost saturated at approx. 10 μM Ca2+. Ca2+ binding to the lower-affinity sites disturbed the measurement of the high-affinity sites and the Ca2+-binding data to the latter were scattered. One of the three experimental results was less scattered in this range, and was subjected to curve-fitting analysis (Figure 2B), which revealed two high-affinity sites with macroscopic dissociation constants of K1=1.75×10−5 M (±3.06×10−6 M) and K2=7.58×10−6 M (±1.64×10−6 M). Although the exact nature of the Ca2+ binding to the low-affinity sites is not clear from these experiments, these results again suggest that fortilin binds Ca2+ and that there are two high-affinity binding sites with dissociation constants of 7.58 and 17.5 μM.
Figure 2. Flow dialysis confirms that fortilin binds Ca2+.

(A) Ca2+ binding to GST–fortilin. Ca2+ binding was measured at 25 °C by flow dialysis as described in the Materials and methods section. The results of three independent experiments were shown. Concentrations of GST–fortilin were 24.7 μM (○), 18.4 μM (□) and 13.8 μM (●) in 0.1 M NaCl and 20 mM Mops/NaOH (pH 7.0). (B) Ca2+ binding to the high-affinity sites of fortilin. One of the results in (A) (24.7 μM GST–fortilin) was quantitatively analysed by fitting to an Adair equation (see the Materials and methods section) revealing two Ca2+-binding sites with K1=1.75×10−5 M (±3.06×10−6 M) and K2=7.58×10−6 M (±1.64×10−6 M).
Fortilin changes its secondary structure in the presence of Ca2+
To determine whether fortilin changes its secondary structure in the presence of Ca2+, we subjected fortilin–His6 to CD spectropolarimetry analyses in the absence and presence of Ca2+. As shown in Figure 3(A), fortilin did change its secondary structure in the presence of 10 μM Ca2+, most significantly at a wavelength of ∼200 nm. More important, the secondary structure of fortilin remained the same in the presence of 10 μM, 100 μM and 1 mM Ca2+, suggesting that Ca2+ saturated fortilin at concentrations of approx. 10 μM or greater (Figure 3B). In addition to the fact that the CD spectrum change again suggests the presence of an interaction between fortilin and Ca2+, these results also suggest that the dissociation constant (Kd) of the fortilin–Ca2+ interaction is in the vicinity of 10 μM (Figures 3A and 3B), a finding that is consistent with the flow dialysis data (Figure 2B).
Figure 3. The binding of fortilin to Ca2+ changes its secondary structure as assessed by CD spectra.

Far-UV CD spectra of GST–fortilin (0.36 mg/ml each) in the presence of no Ca2+ and 10 μM, 100 μM, and 1 mM Ca2+, were collected and recorded from 260 to 200 nm in 0.5-nm steps at 100 nm/min. The samples had been passed through a Chelex 100 chelating ion-exchange resin column (Bio-Rad) to remove residual Ca2+ from samples. Data are expressed as mean residue ellipticity (degrees cm2/dmol). The CD spectra of fortilin changed in the presence of 10 μM Ca2+ (A) but changed no further at Ca2+ concentrations greater than 10 μM (B), suggesting that Ca2+-binding sites on fortilin were saturated by approx. 10 μM Ca2+.
siRNA depletion of fortilin augments the thapsigargin-induced increase in [Ca2+]i in U2OS cells
To determine whether the presence of fortilin changes the intracellular Ca2+ concentration in vivo, we transfected U2OS cells with either siRNAfortilin or siRNAluciferase. Treatment with siRNAfortilin reduced the intracellular fortilin concentration to undetectable levels (siRNAfortilin, Figure 4A). Cells were then incubated in the presence of the Ca2+-sensor dye Fura 2 and stimulated with thapsigargin to induce the release of Ca2+ from the ER into the cytosol. The r-[Ca2+]i in both groups of U2OS cells increased rapidly after thapsigargin stimulation (Figure 4B). Strikingly, in this system, the r-[Ca2+]i of siRNAfortilin-treated U2OS cells increased significantly more than that of siRNAluciferase-treated U2OS cells (Figures 4B and 4C). These results suggest that the presence of fortilin blunted the increase in [Ca2+]i in thapsigargin-challenged U2OS cells.
Figure 4. Fortilin blocks the thapsigargin-induced elevation of [Ca2+]i in U2OS cells.

(A) The treatment of U2OS cells with siRNAfortilin (siRNA against fortilin), but not siRNAluciferase (siRNA against luciferase, negative control), decreased cellular fortilin levels. (B) [Ca2+]i of U2OS cells treated with either siRNAfortilin or siRNAluciferase upon thapsigargin challenge (400 nM). U2OS cells lacking fortilin (siRNAfortilin) exhibit the exaggerated elevation of [Ca2+]i in response to thapsigargin. See the Materials and methods section for experimental details. (C) Free intracellular Ca2+ index, the area under the curve, for U2OS cells treated with siRNAluciferase, was significantly smaller than that for U2OS cells treated with siRNAfortilin [160±5.7 and 201.5±4.9 (arbitrary units) for cells treated with siRNAluciferase and siRNAfortilin respectively.]. ****P<0.001.
MEFfortilin+/− with lower levels of fortilin exhibit a greater increase in [Ca2+]i upon thapsigargin challenge than MEFfortilin+/+
To determine whether our observation in thapsigargin-challenged U2OS cells was true in primary culture cells, we compared the r-[Ca2+]i of thapsigargin-challenged MEFfortilin+/− and MEFfortilin+/+. MEFfortilin−/− has not been successfully isolated. The fortilin expression level in MEFfortilin+/− was approx. half that in MEFfortilin+/+, as determined by Western blot analysis (Figure 5A). Upon thapsigargin challenge, MEFfortilin+/− exhibited a significantly higher r-[Ca2+]i than did MEFfortilin+/+ (Figures 5B and 5C). These results suggest that the presence of fortilin interfered with the thapsigargin-induced elevation of [Ca2+]i in MEF. In our previous work [7], we demonstrated that fortilin is present in both the cytosol and the nucleus. In addition, thapsigargin has been shown to increase [Ca2+]i by releasing Ca2+ that is stored in the ER into the cytosol [36,42,43]. Taken together, our present results suggest that fortilin scavenges Ca2+ released into the cytosol from the ER and blunts the elevation of [Ca2+]i in vivo.
Figure 5. Fortilin blocks thapsigargin-induced elevation of free intracellular Ca2+ ([Ca2+]i) in MEF cells.

(A) MEFfortilin+/− cells expressed a significantly smaller amount of intracellular fortilin than MEFfortilin+/+ as measured by Western blot analysis. (B) r-[Ca2+]i concentration of MEFfortilin+/− or MEFfortilin+/+, upon thapsigargin challenge (400 nM). MEFfortilin+/− showed more exaggerated elevation of [Ca2+]i to thapsigargin stimulation than did MEFfortilin+/+. See the Materials and methods section for experimental details. (C) Free intracellular Ca2+ index, the area under the curve for MEFfortilin+/+ cells was significantly smaller than that for MEFfortilin+/− cells (134.09±6.4 arbitrary units and 172.1±3.7 abitrary units for MEFfortilin+/+ and MEFfortilin+/− cells respectively). ****P<0.001.
Fortilin blocks Ca2+-dependent apoptosis in U2OS cells
To evaluate the biological consequence of fortilin-blunted [Ca2+]i elevation in thapsigargin-challenged cells, we treated U2OS cells with either siRNAfortilin or siRNAluciferase, stimulated them with thapsigargin in the presence or absence of the potent cell-membrane-permeant intracellular Ca2+ chelator BAPTA-AM, and then evaluated these cells for cell morphology (Figure 6A), cell death rate (Figure 6B) and for caspase 3 activation (Figure 6C). First, fortilin protected cells against thapsigargin-induced cell death because we observed far more rounded dead cells in siRNAfortilin-treated cells that those treated by siRNAluciferase (Figure 6A; top panels) and because the EthD-1 index, representing the late phase of apoptosis or necrosis, was significantly higher in siRNAfortilin-treated cells than in siRNAluciferase-treated cells upon thapsigargin challenge (Figure 6B, column 1 compared with column 3; 5.10±0.94% compared with 1.65±1.28%; **P<0.01). Secondly, most of the cell death caused by the lack of fortilin was preventable by BAPTA-AM and therefore Ca2+-dependent in nature. This was because the frequency of rounded dead cells in siRNAfortilin-treated cells were reduced by BAPTA-AM almost to that in siRNAluciferase-treated cells (Figure 6A, lower left-hand and upper right-hand panels respectively) and because most of cell death preventable by fortilin (indicated with #, Figure 6B) was preventable by BAPTA-AM and thus Ca2+-dependent (Figure 6B). We then measured the caspase 3 activities of these cells. As shown in Figure 6(C), siRNAfortilin-treated cells, but not siRNAluciferase-treated cells, exhibited significant caspase 3 activation upon thapsigargin stimulation (1219±359 compared with 28±213 relative units/μg of protein; **P<0.01). The absence of significant thapsigargin-induced caspase 3 activity in siRNAluciferase-treated cells (Figure 6C, siRNAluciferase) suggests that most of cell death of siRNAluciferase-treated cells in Figure 6(B) (column 3) represented necrosis (cell death lacking caspase 3 activation). The results in turn suggest that most of BAPTA-AM-preventable cell death in siRNAfortilin-treated cells is apoptosis. Taken together, the results presented in Figure 6 suggest that fortilin prevents the thapsigargin-induced elevation of [Ca2+]i from inducing apoptosis in U2OS cells.
Fortilin blocks Ca2+-dependent apoptosis in MEF cells
To determine whether the inhibition by fortilin of Ca2+-dependent apoptosis seen in U2OS also occurs in primary culture cells, we compared the Ca2+-dependent apoptosis rates in MEFfortilin+/− and MEFfortilin+/+. MEF cells were stimulated with thapsigargin in the presence or absence of BAPTA-AM and evaluated for cell morphology (Figure 7A), cell death rate (Figure 7B) and caspase 3 activation (Figure 7C). Fortilin again protected cells against thapsigargin-induced cell death because we observed far more rounded dead cells in MEFfortilin+/− cells than in MEFfortilin+/+ cells (Figure 7A, upper left-hand and upper right-hand panels respectively) and because the EthD-1 index was significantly higher in MEFfortilin+/− cells than in MEFfortilin+/+ cells upon thapsigargin challenge (Figure 7B, column 1 compared with column 2; 15.28±2.0% compared with 3.02±1.75%; ***P<0.005). Secondly, most of the cell death caused by the lack of fortilin was again preventable by BAPTA-AM and therefore Ca2+-dependent in nature. This was because the frequency of rounded dead MEFfortilin+/− cells were reduced by BAPTA-AM almost to that of rounded dead cells in MEFfortilin+/+ (Figure 7A, lower left-hand and upper right-hand panels respectively) and because most of cell death preventable by fortilin (indicated by #, Figure 7B) was preventable by BAPTA-AM and thus Ca2+-dependent (Figure 7B). We then measured the caspase 3 activities of these cells. As shown in Figure 7(C), MEFfortilin+/− cells, but not MEFfortilin+/+ cells, exhibited significant caspase 3 activation upon thapsigargin stimulation (Figure 7C; 1110±15.3 compared with 26±22.5 relative units/μg of protein; *P<0.05). The absence of significant thapsigargin-induced caspase 3 activity in MEFfortilin+/+ cells (Figure 7C, fortilin+/+) suggests that most of the cell death of MEFfortilin+/+ cells in Figure 7(B) (column 3) represented necrosis. The results also suggest that most of BAPTA-AM-preventable cell death in MEFfortilin+/− cells is apoptosis. Taken together, these results suggest that fortilin robustly protects MEFs against thapsigargin-induced apoptosis. Consistent with the data presented in Figure 6, the data presented in Figure 7 suggest that fortilin blocks the apoptosis resulting from thapsigargin-induced elevation of [Ca2+]i.
Glu58 and Glu60 of fortilin are critical for binding to Ca2+
To determine what region(s) of fortilin are critical for Ca2+ binding, we synthesized 12 overlapping fortilin peptides (Figure 8A) and subjected them to equilibrium dialysis. As shown in Figure 8(B), CBB alone in the tube had the exact same radioactivity as CBB in the beaker (Buffer and thick line; Figure 8B). CBBs containing calmodulin, fortilin–His6, and GST–fortilin were all significantly more radioactive than the CBB control (Figure 8B), consistent with the data presented in Figure 1(A). In this system, only three polypeptides, namely fortilin(43–62), fortilin(57–76) and fortilin(127–146), exhibited significantly more radioactivity than did the CBB control, suggesting that the binding of fortilin to Ca2+ occurred through amino acids within these three regions. There are eleven charged amino acids within these regions. We systematically mutated these charged amino acids to generate several mutants of full-length fortilin: fortilinΔ1 (D44A/D45A), fortilinΔ2 (E58A/E60A), fortilinΔ3 (E55A/E58A/E60A/E63A), fortilinΔ4 (D71A), fortilinΔ5 (E138A) and fortilinΔ6 (D143A) (Figure 9A). These six mutant proteins of fortilin were expressed as GST-fusion proteins, purified and characterized (Figure 9B) and subjected to the same equilibrium dialysis. In this experiment, the radioactivities of CBB-only and GST-only solutions were identical with the radioactivity of the CBB in the beaker (Buffer, GST-only; Figure 9C). As expected, calmodulin and GST–fortilin exhibited significantly more radioactivity than did CBB alone and GST-only (Figure 9C). In this system, the solutions containing GST–fortilinΔ5 and Δ6 were significantly more radioactive than the controls (P<0.005 and P<0.05 respectively), suggesting that neither Glu138 nor Asp143 played a critical role in the Ca2+ binding of fortilin (Figure 9C). Strikingly, however, the mutations introduced to Glu58 and Glu60 abolished almost completely the Ca2+-binding capability of fortilin (GST–fortilinΔ2, Figure 9C). The finding that Glu58 and Glu60, but not Glu138 or Asp143, were critical to the Ca2+ binding of fortilin is entirely consistent with our finding in the Ca2+-overlay assay that the 70 amino acid residues at the N-terminus of fortilin were critical for Ca2+ binding (Figure 1B). Based on these observations, we chose fortilinΔ2 to test the biological consequence of the inability of fortilin to bind Ca2+ in vivo. Intriguingly, the crystal structure of Schizosaccharomyces pombe fortilin (MMDB: 16785; PDB: 1H6Q) [44] shows that Glu58 and Glu60 were both localized in the loose loop of the molecule that protrudes from the surface of the more tightly packed portion of the molecule (Figure 9D).
Figure 9. FortilinΔ2(E58A/E60A) fails to bind Ca2+ in equilibrium dialysis assays.

(A and B) Generation and characterization of GST–fortilin mutants. Fortilin mutants containing point mutations within full-length fortilin, namely fortilinΔ1 (D44A/D45A), fortilinΔ2 (E58A/E60A), fortilinΔ3 (E55A/E58A/E60A/E63A), fortilinΔ4 (D70A), fortilinΔ5 (E138A) and fortilinΔ6 (D143A) (A), were generated, expressed with a GST-fusion tag, and characterized by SDS/PAGE and Coomassie Blue staining (B). The mutant in the yellow row (fortilinΔ2) exhibited significant Ca2+-binding activity in equilibrium dialysis assays. (C) Equilibrium dialysis of GST–fortilin containing mutations showed the lack of significant binding by fortilinΔ2. *P<0.05, +P=0.073, ***P<0.005. (D) Structure of S. pombe fortilin (PDB: 1H6Q) showing the localization of Glu58 and Glu60. C, C-terminus; N, N-terminus.
FortilinΔ2(E58A/E60A), a fortilin point mutant lacking Ca2+-binding activity, is more susceptible to thapsigargin-induced cell death than its wild-type counterpart
To establish that the protection by fortilin of thapsigargin-challenged cells against cell death is mediated via the binding of fortilin to Ca2+ and sequestration of Ca2+ from Ca2+-dependent downstream apoptosis pathways, we established U2OS cell lines stably expressing wild-type fortilin (U2OSfortilin-wild) and fortilinΔ2 (U2OSfortilinΔ2) as well as U2OS cells harbouring empty plasmids (U2OSempty). Western blot analysis showed that exogenous fortilin and fortilinΔ2 were robustly expressed in U2OSfortilin-wild and U2OSfortilinΔ2 respectively (columns 2 and 3, Figure 10A). We then subjected these cell lines to thapsigargin and determined the EthD-1 indices. As expected, the EthD-1 index was significantly lower in U2OSfortilin-wild than it was in U2OSempty cells (U2OSfortilin-wild compared with U2OSempty; 2.31±0.57% compared with 4.58±0.39% respectively; *P<0.05) (Fortilin compared with Empty, Figure 10B). Strikingly, the EthD-1 index of U2OSfortilinΔ2 cells was no different than that of U2OSempty cells (U2OSfortilinΔ2 compared with U2OSempty; 3.96±1.11 compared with 4.58±0.39% respectively; not significant) (FortilinΔ2 compared with Empty, Figure 10B). These results suggest that fortilin, but not fortilinΔ2, was capable of protecting cells against thapsigargin-induced cell death and that the ability of fortilin to bind Ca2+ was required for fortilin to protect cells against thapsigargin-induced cell death. Since most of the thapsigargin-induced cell death was Ca2+-dependent (Figures 6B and 7B) and represented apoptosis (Figures 6C and 7C), it is suggested that the ability of fortilin to bind Ca2+ is required for fortilin to protect cells against Ca2+-dependent apoptosis. These results further suggest that fortilin is a cytosolic Ca2+ scavenger and that anti-apoptotic activity of fortilin is at least partly due to its ability to sequester ER-derived Ca2+ from downstream Ca2+-dependent apoptotic pathways.
Figure 10. FortilinΔ2 (E58A/E60A), a non-Ca2+-binding mutant of fortilin, fails to prevent thapsigargin-induced cell death.

(A) Western blot analysis of U2OS cells. Actin, loading control; Native fortilin, fortilin natively expressed in U2OS cells, Fortilin/fortilinΔ2, over-expressed fortilin or fortilinΔ2(E58A/E60A) detected by an anti-fortilin antibody. (B) EthD-1 indices of U2OS cells stably harbouring empty plasmid (Empty), stably expressing wild-type fortilin (Fortilin) and stably expressing fortilinΔ2(E58A/E60A)(FortillinΔ2). *P<0.05 in comparison with Empty; NS, no statistically significant difference in comparison with Empty. The overexpression of fortilin, but not fortilinΔ2, a non-Ca2+-binding mutant of fortilin, protects cells against thapsigargin-induced cell death.
DISCUSSION
For over a decade now, fortilin has been known to bind Ca2+ [8–12], but its biological significance has remained purely speculative. Since fortilin is an anti-apoptotic protein and Ca2+ plays a critical role in apoptosis, we hypothesized that fortilin binds and scavenges Ca2+, thus preventing the ion from activating downstream apoptotic execution pathways. In the present study, we set out to test the hypothesis in a series of experiments. There are four key findings in the present study that are novel. First, we have employed four different assays, namely Ca2+-overlay (Figure 1), flow dialysis (Figure 2), CD spectropolarimetry (Figure 3) and equilibrium dialysis (Figure 9C) assays to unequivocally demonstrate the presence of the binding of fortilin to Ca2+. This was significant because previous studies relied entirely on Ca2+-overlay assays to show the binding of fortilin to Ca2+ [8–12]. Importantly, the binding of fortilin to Ca2+ was associated with the change in secondary structure of fortilin as detected by CD spectropolarimetry (Figure 3). Secondly, we have been able to show, using both U2OS cells and MEFs and a standard Fura 2 Ca2+-detection system, that the lack of fortilin led to an exaggerated elevation of free intracellular calcium levels ([Ca2+]i) upon thapsigargin stimulation (Figures 4 and 5). This is significant because it has not been reported previously that fortilin can block the elevation of [Ca2+]i in vivo. Thirdly, we have demonstrated that fortilin protects cells against thapsigargin-induced Ca2+-dependent apoptosis (Figures 6 and 7) and that the binding of fortilin to Ca2+ is required for fortilin's protection against such apoptosis (Figure 10), both of which have not been reported in the literature. Finally, we have identified amino acids of fortilin critical for its binding to Ca2+, namely Glu58 and Glu60 and generated a double-point mutant of fortilin [fortilin(E58A/E60A) or fortilinΔ2] lacking Ca2+-binding (Figure 9). Strikingly, wild-type fortilin, but not fortilin(E58A/E60A) (fortilinΔ2), protected cells against thapsigargin-induced cell death; fortilin is required to bind Ca2+ in order to block Ca2+-dependent apoptosis. Based on these findings, we conclude the hypothesis above to be correct.
Others besides ourselves have also tried to identify the Ca2+-binding site of fortilin. Using fortilin–His6 and fortilin fragments in a 45Ca2+-overlay assay, Kim et al. [10] found that wild-type fortilin and fortilin(1–112), but not fortilin(1–80) or fortilin(1–52), bound 45Ca2+. They therefore concluded that the Ca2+-binding site of fortilin resides in amino acid residues 81–112. However, our experiments with GST–fortilin and its mutants suggest that the Ca2+-binding site of fortilin resides in the polypeptide consisting of amino acids 1–70 (Figures 1B and 1C). In addition, our equilibrium dialysis assays showed no significant binding between Ca2+ and any of several fragments including fortilin(71–90), fortilin(85–104), fortilin(99–118) or fortilin(113–132) peptide. Furthermore, it did show very significant Ca2+ binding to fortilin(43–62) and fortilin(57–76) polypeptides (Figure 8). Consistently, there was no binding between Ca2+ and fortilinΔ2, a double-point mutant in which glutamate residues at amino acid positions 58 and 60 were mutated to alanine residues (Figure 9C). The apparent discrepancy between our findings and those of Kim et al. [10] may possibly be due to their use of fortilin(1–80), which carries a His6-tag at its C-terminus and may not have folded properly. It is also possible that Glu58 and Glu60 consist in one of multiple Ca2+-binding pockets within fortilin. The presence of at least two Ca2+-binding pockets within fortilin is supported by our flow dialysis data presented in Figure 2. The co-crystallization data of fortilin and Ca2+ would allow us to determine the relative spatial contributions of these amino acid residues to Ca2+ and help to resolve this apparent discrepancy.
In the present study, flow dialysis assays provided a new insight into the binding of fortilin to Ca2+ where fortilin exhibited a biphasic Ca2+ binding (Figure 2), suggesting the presence of more than one, most probably two, high-affinity Ca2+-binding sites, and the possible presence of multiple lower-affinity Ca2+-binding sites. The high-affinity sites were almost saturated at approx. 8–18 μM Ca2+, a finding consistent with CD spectropolarimetry data (Figure 3). Ca2+ binding to the lower-affinity sites seemed to disturb the measurement of the high-affinity sites and the Ca2+-binding data to the latter were scattered. Although the high-affinity binding sites are likely to be responsible for the conformational change of fortilin observed in CD spectra (Figure 3), the exact nature of the Ca2+ binding to the low-affinity sites remains unclear. It is tempting to speculate that fortilin binds Ca2+ in a step-wise fashion in vivo: as the intracellular Ca2+ concentration ([Ca2+]i) starts to rise in response to apoptotic stimuli, fortilin binds and scavenges Ca2+, using its higher-affinity Ca2+-binding sites, to maintain [Ca2+]i at a baseline level. The ligation of higher-affinity Ca2+-binding sites may result in the conformational changes, as detectable by CD spectropolarimetry (Figure 3), to make lower-affinity Ca2+-binding sites available for further Ca2+ binding. Nevertheless, the exact biological role of lower-affinity Ca2+-binding sites calls for further biochemical studies.
Several groups of investigators have also investigated the link between the thapsigargin-induced increase in [Ca2+]i and apoptosis. Thastrup et al. [45] showed that thapsigargin induces rapid and dose-dependent release of stored Ca2+ from ER-rich microsomes, resulting in a robust increase in [Ca2+]i, and that thapsigargin does so by inducing the acute and highly specific arrest of SERCA in the ER and consequent rapid leakage of Ca2+ into the cytosol [45]. Srivastava et al. [46] demonstrated that [Ca2+]i elevation is the primary mechanism that thapsigargin uses to induce apoptosis by showing that the ability of thapsigargin to induce apoptosis was effectively blocked by buffering [Ca2+]i with BAPTA-AM [46]. They also observed that [Ca2+]i elevation was associated not only with the release of cytochrome c from mitochondria but also with caspase 3 activation [46]. In the present study, we consistently observed that BAPTA-AM drastically reduced the rate of cell death in both fortilin-deficient cells (U2OS cells treated with siRNAfortilin and MEFfortilin+/−) and control cells, suggesting the majority of thapsigargin-induced cell death is Ca2+-dependent. Importantly, thapsigarin-induced cell death in siRNAluciferase-treated cells and MEFfortilin+/+ (column 3 of Figures 6 and 7) was not associated with caspase activation (Figures 6C and 7C), suggesting that EthD-1 positivity in these cells represented necrosis, rather than apoptosis. In that light, most of the cell death blocked by the presence of fortilin was both Ca2+-dependent and apoptotic in nature. Strikingly, fortilin(E58A/E60A) (fortilinΔ2), a double-point mutant of fortilin lacking Ca2+-binding activity, was not capable of protecting cells against thapsigargin-induced cell death (Figure 10). It follows that the binding of fortilin to Ca2+ is required if fortilin is to function as an anti-apoptotic agent in thapsigargin-induced, Ca2+-dependent apoptosis. In addition, the difference seen in the indices of EthD-1 for control samples among experiments (Figures 6 and 10) most probably represents the fact that U2OS cells used in Figure 6 were treated with siRNAluciferase, whereas U2OSempty cells used in Figure 10 had been stably transfected with pcDNA4-empty plasmid vector. It is thus safe to conclude that the primary mechanism by which fortilin blocks thapsigargin-induced Ca2+-dependent apoptosis is through its binding to Ca2+. In future investigations of possible roles of fortilin in Ca2+-independent apoptosis, a double-point mutant fortilin(E58A/E60A) will prove to be a highly effective reagent.
Acknowledgments
The present study was supported in part by grants from the NIH (National Institutes of Health; HL04015 and HL68024) (to K. F.), the Roderick Duncan MacDonald General Research Fund at St. Luke's Episcopal Hospital (to K. F.) and the Royal Golden Jubilee Graduate Program of Thailand Research Fund (PhD/0200/2545 to P. G.; PhD/0200/2685 to M. T.) and by an Established Investigator Award from the American Heart Association (to K. F.).
References
- 1.Zhang D., Li F., Weidner D., Mnjoyan Z. H., Fujise K. Physical and functional interaction between MCL1 and fortilin. The potential role of MCL1 as a fortilin chaperone. J. Biol. Chem. 2002;277:37430–37438. doi: 10.1074/jbc.M207413200. [DOI] [PubMed] [Google Scholar]
- 2.Kozopas K. M., Yang T., Buchan H. L., Zhou P., Craig R. W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li F., Zhang D., Fujise K. Characterization of fortilin, a novel anti-apoptotic protein. J. Biol. Chem. 2001;276:47542–47549. doi: 10.1074/jbc.M108954200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang C., Cai Y., Adachi M. T., Oshiro S., Aso T., Kaufman R. J., Kitajima S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001;276:35867–35874. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- 5.Sturzenbaum S. R., Kille P., Morgan A. J. Identification of heavy metal induced changes in the expression patterns of the translationally controlled tumour protein (TCTP) in the earthworm Lumbricus rubellus. Biochim. Biophys. Acta. 1998;1398:294–304. doi: 10.1016/s0167-4781(98)00077-3. [DOI] [PubMed] [Google Scholar]
- 6.Tuynder M., Susini L., Prieur S., Besse S., Fiucci G., Amson R., Telerman A. Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14976–14981. doi: 10.1073/pnas.222470799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graidist P., Phongdara A., Fujise K. Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-FU-induced cytotoxicity. J. Biol. Chem. 2004;279:40868–40875. doi: 10.1074/jbc.M401454200. [DOI] [PubMed] [Google Scholar]
- 8.Haghighat N. G., Ruben L. Purification of novel calcium binding proteins from Trypanosoma brucei: properties of 22-, 24- and 38-kilodalton proteins. Mol. Biochem. Parasitol. 1992;51:99–110. doi: 10.1016/0166-6851(92)90205-x. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez J. C., Schaller D., Ravier F., Golaz O., Jaccoud S., Belet M., Wilkins M. R., James R., Deshusses J., Hochstrasser D. Translationally controlled tumor protein: a protein identified in several nontumoral cells including erythrocytes. Electrophoresis. 1997;18:150–155. doi: 10.1002/elps.1150180127. [DOI] [PubMed] [Google Scholar]
- 10.Kim M., Jung Y., Lee K., Kim C. Identification of the calcium binding sites in translationally controlled tumor protein. Arch. Pharm. Res. 2000;23:633–636. doi: 10.1007/BF02975253. [DOI] [PubMed] [Google Scholar]
- 11.Rao K. V., Chen L., Gnanasekar M., Ramaswamy K. Cloning and characterization of a calcium-binding, histamine-releasing protein from Schistosoma mansoni. J. Biol. Chem. 2002;277:31207–31213. doi: 10.1074/jbc.M204114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arcuri F., Papa S., Carducci A., Romagnoli R., Liberatori S., Riparbelli M. G., Sanchez J. C., Tosi P., del Vecchio M. T. Translationally controlled tumor protein (TCTP) in the human prostate and prostate cancer cells: expression, distribution, and calcium binding activity. Prostate. 2004;60:130–140. doi: 10.1002/pros.20054. [DOI] [PubMed] [Google Scholar]
- 13.Xu A., Bellamy A. R., Taylor J. A. Expression of translationally controlled tumour protein is regulated by calcium at both the transcriptional and post-transcriptional level. Biochem. J. 1999;342:683–689. [PMC free article] [PubMed] [Google Scholar]
- 14.Hajnoczky G., Davies E., Madesh M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 15.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 16.Fladmark K. E., Brustugun O. T., Mellgren G., Krakstad C., Boe R., Vintermyr O. K., Schulman H., Doskeland S. O. Ca2+/calmodulin-dependent protein kinase II is required for microcystin-induced apoptosis. J. Biol. Chem. 2002;277:2804–2811. doi: 10.1074/jbc.M109049200. [DOI] [PubMed] [Google Scholar]
- 17.Tan K. M., Chan S. L., Tan K. O., Yu V. C. The Caenorhabditis elegans sex-determining protein FEM-2 and its human homologue, hFEM-2, are Ca2+/calmodulin-dependent protein kinase phosphatases that promote apoptosis. J. Biol. Chem. 2001;276:44193–44202. doi: 10.1074/jbc.M105880200. [DOI] [PubMed] [Google Scholar]
- 18.Kruidering M., Schouten T., Evan G. I., Vreugdenhil E. Caspase-mediated cleavage of the Ca2+/calmodulin-dependent protein kinase-like kinase facilitates neuronal apoptosis. J. Biol. Chem. 2001;276:38417–38425. doi: 10.1074/jbc.M103471200. [DOI] [PubMed] [Google Scholar]
- 19.Aagaard-Tillery K. M., Jelinek D. F. Differential activation of a calcium-dependent endonuclease in human B lymphocytes. Role in ionomycin-induced apoptosis. J. Immunol. 1995;155:3297–3307. [PubMed] [Google Scholar]
- 20.Blanchard H., Grochulski P., Li Y., Arthur J. S., Davies P. L., Elce J. S., Cygler M. Structure of a calpain Ca2+-binding domain reveals a novel EF-hand and Ca(2+)-induced conformational changes. Nat. Struct. Biol. 1997;4:532–538. doi: 10.1038/nsb0797-532. [DOI] [PubMed] [Google Scholar]
- 21.Lin G. D., Chattopadhyay D., Maki M., Wang K. K., Carson M., Jin L., Yuen P. W., Takano E., Hatanaka M., DeLucas L. J., Narayana S. V. Crystal structure of calcium bound domain VI of calpain at 1.9 Å resolution and its role in enzyme assembly, regulation, and inhibitor binding. Nat. Struct. Biol. 1997;4:539–547. doi: 10.1038/nsb0797-539. [DOI] [PubMed] [Google Scholar]
- 22.Mandic A., Viktorsson K., Strandberg L., Heiden T., Hansson J., Linder S., Shoshan M. C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol. Cell Biol. 2002;22:3003–3013. doi: 10.1128/MCB.22.9.3003-3013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H. G., Pathan N., Ethell I. M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T. F., Reed J. C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 24.Teubl M., Groschner K., Kohlwein S. D., Mayer B., Schmidt K. Na+/Ca2+ exchange facilitates Ca2+-dependent activation of endothelial nitric-oxide synthase. J. Biol. Chem. 1999;274:29529–29535. doi: 10.1074/jbc.274.41.29529. [DOI] [PubMed] [Google Scholar]
- 25.Fujise K., Zhang D., Liu J., Yeh E. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J. Biol. Chem. 2000;275:39458–39465. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]
- 26.Gong L., Kamitani T., Fujise K., Caskey L. S., Yeh E. T. Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J. Biol. Chem. 1997;272:28198–28201. doi: 10.1074/jbc.272.45.28198. [DOI] [PubMed] [Google Scholar]
- 27.Guo H., Mockler T., Duong H., Lin C. SUB1, an Arabidopsis Ca2+-binding protein involved in cryptochrome and phytochrome coaction. Science. 2001;291:487–490. doi: 10.1126/science.291.5503.487. [DOI] [PubMed] [Google Scholar]
- 28.Yazawa M. Quantitative analysis of Ca2+-binding by flow dialysis. Methods Mol. Biol. 2002;173:3–14. doi: 10.1385/1-59259-184-1:003. [DOI] [PubMed] [Google Scholar]
- 29.Mishra-Gorur K., Singer H. A., Castellot J. J., Jr Heparin inhibits phosphorylation and autonomous activity of Ca2+/calmodulin-dependent protein kinase II in vascular smooth muscle cells. Am. J. Pathol. 2002;161:1893–1901. doi: 10.1016/S0002-9440(10)64465-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Florian J. A., Watts S. W. Integration of mitogen-activated protein kinase kinase activation in vascular 5-hydroxytryptamine2A receptor signal transduction. J. Pharmacol. Exp. Ther. 1998;284:346–355. [PubMed] [Google Scholar]
- 31.Hsieh C. C., Lau Y. Migration of vascular smooth muscle cells is enhanced in cultures derived from spontaneously hypertensive rat. Pflugers Arch. 1998;435:286–292. doi: 10.1007/s004240050514. [DOI] [PubMed] [Google Scholar]
- 32.Elbashir S. M., Lendeckel W., Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 34.Heinrich J. N., O'Rourke E. C., Chen L., Gray H., Dorfman K. S., Bravo R. Biological activity of the growth factor-induced cytokine N51: structure–function analysis using N51/interleukin-8 chimeric molecules. Mol. Cell. Biol. 1994;14:2849–2861. doi: 10.1128/mcb.14.5.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lytton J., Westlin M., Hanley M. R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 36.Foyouzi-Youssefi R., Arnaudeau S., Borner C., Kelley W. L., Tschopp J., Lew D. P., Demaurex N., Krause K. H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer J. P., Rice-Evans C., Williams R. J. Modulation of pro-survival Akt/protein kinase B and ERK1/2 signaling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. J. Biol. Chem. 2003;278:34783–34793. doi: 10.1074/jbc.M305063200. [DOI] [PubMed] [Google Scholar]
- 38.Shi Q., Chen H. L., Xu H., Gibson G. E. Reduction in the E2K subunit of the α-ketoglutarate dehydrogenase complex has effects independent of complex activity. J. Biol. Chem. 2005;280:10888–10896. doi: 10.1074/jbc.M409064200. [DOI] [PubMed] [Google Scholar]
- 39.Furukawa R., Maselli A., Thomson S. A., Lim R. W., Stokes J. V., Fechheimer M. Calcium regulation of actin crosslinking is important for function of the actin cytoskeleton in Dictyostelium. J. Cell Sci. 2003;116:187–196. doi: 10.1242/jcs.00220. [DOI] [PubMed] [Google Scholar]
- 40.Furuyama T., Dzelzkalns V. A. A novel calcium-binding protein is expressed in Brassica pistils and anthers late in flower development. Plant Mol. Biol. 1999;39:729–737. doi: 10.1023/a:1006169808171. [DOI] [PubMed] [Google Scholar]
- 41.Lee B. I., Mustafi D., Cho W., Nakagawa Y. Characterization of calcium binding properties of lithostathine. J. Biol. Inorg. Chem. 2003;8:341–347. doi: 10.1007/s00775-002-0421-8. [DOI] [PubMed] [Google Scholar]
- 42.Jiang S., Chow S. C., Nicotera P., Orrenius S. Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase inhibitor thapsigargin. Exp. Cell Res. 1994;212:84–92. doi: 10.1006/excr.1994.1121. [DOI] [PubMed] [Google Scholar]
- 43.Lam M., Dubyak G., Chen L., Nunez G., Miesfeld R. L., Distelhorst C. W. Evidence that BCl-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thaw P., Baxter N. J., Hounslow A. M., Price C., Waltho J. P., Craven C. J. Structure of TCTP reveals unexpected relationship with guanine nucleotide-free chaperones. Nat. Struct. Biol. 2001;8:701–704. doi: 10.1038/90415. [DOI] [PubMed] [Google Scholar]
- 45.Thastrup O., Cullen P. J., Drobak B. K., Hanley M. R., Dawson A. P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava R. K., Sollott S. J., Khan L., Hansford R., Lakatta E. G., Longo D. L. Bcl-2 and Bcl-X(L) block thapsigargin-induced nitric oxide generation, c-Jun NH2-terminal kinase activity, and apoptosis. Mol. Cell. Biol. 1999;19:5659–5674. doi: 10.1128/mcb.19.8.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]