

Abstract
CBS (cystathionine β-synthase) domains are found in proteins from all kingdoms of life, and point mutations in these domains are responsible for a variety of hereditary diseases in humans; however, the functions of CBS domains are not well understood. In the present study, we cloned, expressed in Escherichia coli, and characterized a family II PPase (inorganic pyrophosphatase) from Moorella thermoacetica (mtCBS-PPase) that has a pair of tandem 60-amino-acid CBS domains within its N-terminal domain. Because mtCBS-PPase is a dimer and requires transition metal ions (Co2+ or Mn2+) for activity, it resembles common family II PPases, which lack CBS domains. The mtCBS-PPase, however, has lower activity than common family II PPases, is potently inhibited by ADP and AMP, and is activated up to 1.6-fold by ATP. Inhibition by AMP is competitive, whereas inhibition by ADP and activation by ATP are both of mixed types. The nucleotides are effective at nanomolar (ADP) or micromolar concentrations (AMP and ATP) and appear to compete for the same site on the enzyme. The nucleotide-binding affinities are thus 100–10000-fold higher than for other CBS-domain-containing proteins. Interestingly, genes encoding CBS-PPase occur most frequently in bacteria that have a membrane-bound H+-translocating PPase with a comparable PPi-hydrolysing activity. Our results suggest that soluble nucleotide-regulated PPases act as amplifiers of metabolism in bacteria by enhancing or suppressing ATP production and biosynthetic reactions at high and low [ATP]/([AMP]+[ADP]) ratios respectively.
Keywords: adenine nucleotide, bidirectional regulation, cystathionine β-synthetase domain (CBS domain), inorganic pyrophosphatase (PPase), Moorella thermoacetica
Abbreviations: CBS, cystathionine β-synthase; DTPA, diethylenetriaminepenta-acetic acid; PPase, inorganic pyrophosphatase; mtCBS-PPase, CBS domain-containing PPase from Moorella thermoacetica
INTRODUCTION
PPi (inorganic pyrophosphate) is produced in vast amounts by biosynthetic reactions, such as protein, RNA and DNA synthesis, and its concentration affects the equilibria of these reactions [1]. In addition, PPi regulates many other cellular processes, including calcification, cell proliferation and iron transport [2]. Consequently, disruption of PPi metabolism can lead to a variety of pathological conditions [2].
PPi is mainly hydrolysed to Pi (orthophosphate) by PPase (inorganic pyrophosphatase) (EC 3.6.1.1), an enzyme that is essential for life [3–6]. There are two known types of PPase: soluble and integral membrane-bound. Soluble PPases are subdivided further into families I and II, which are not homologous [7,8]. Family I PPases are found in all kingdoms of life and are among the best-characterized phosphoryl transfer enzymes [9,10]. Family II PPases, which were discovered more recently [7,8], are found in Bacilli and Clostridia and in some other bacterial lineages, including several human pathogens, and belong to the DHH (Asp-His-His) family of phosphohydrolases [11]. Family II PPases are homodimers of subunits formed by two well-defined domains, whereas family I PPases have two or six compact one-domain subunits. The N- and C-terminal domains of family II PPases are connected by a flexible linker, and the active site is located at the domain interface [12,13]. Also, unlike family I PPases, family II PPases contain a tightly bound transition metal ion, usually Mn2+ or Co2+, although they also require Mg2+ for maximal activity. In addition, bivalent cations, especially Mn2+, promote dimerization of family II PPases [14].
Interestingly, the N-terminal domain of approx. 25% of the known family II PPase sequences contains a large (∼250-amino-acid) insert, comprising two CBS (cystathionine β-synthase) domains. None of these CBS-PPases has been isolated and characterized. CBS domains, originally found in cystathionine β-synthase [15], are widely distributed among proteins in all three kingdoms of life, but their roles are not well understood [16,17]. In some cases, CBS domains are potential targets for regulation by adenosine derivatives [16–21]. Importantly, point mutations in CBS domains cause several hereditary diseases in humans [17].
To elucidate the role of CBS domains in family II PPases, we have cloned, expressed and purified a CBS-PPase from Moorella thermoacetica (formerly known as Clostridium thermaceticum) (mtCBS-PPase), a low-G+C Gram-positive thermophilic acetogen with an optimum growth temperature of 55–60 °C [22]. We found that, unlike all other known PPases, this enzyme is subject to a strong bidirectional regulation by adenine nucleotides.
EXPERIMENTAL
Cloning and mutagenesis
Genomic DNA extracted from M. thermoacetica strain ATCC 35608 was obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH). The mtCBS-PPase open reading frame (GenBank® accession number NC_007644) was amplified by PCR using primers 5′-TTATCATATGGGTAAAGAGATTCTGGTTATCG-3′ (forward) and 5′-TTATCTCGAGTTATCCCTGCAGCAACCGCCG-3′ (reverse). The amplified gene was sequenced in both directions, and one difference compared with the sequence shown in GenBank® was observed (TTG encoding Leu190 was replaced by CTG), but this did not affect the protein sequence. The PCR fragment was inserted via the pET36b vector (Novagen) into pBluescript SK (Stratagene), and site-directed mutagenesis was performed using an overlapping PCR technique (QuikChange®, Stratagene). The mutation was verified by DNA sequencing.
Protein expression and purification
To produce wild-type and variant mtCBS-PPases, the PCR products were cloned into the pET36b vector using NdeI and XhoI. The plasmid construct was transformed into Escherichia coli BL21(DE3) RIL cells (Stratagene), and the transformants were grown in Terrific broth [23] containing 30 μg/ml kanamycin and 30 μg/ml chloramphenicol. mtCBS-PPase expression was induced for 3 h using 0.4 mM IPTG (isopropyl β-D-thiogalactoside).
Cell paste (10 g wet weight) obtained by centrifugation at 6300 g for 10 min was resuspended in 30 ml of ice-cold 25 mM Tris/HCl buffer (pH 7.3), containing 24 mM MgCl2, 1 mM CoCl2, 20 μM DTPA (diethylenetriaminepenta-acetic acid), and was homogenized twice using a French press (SLM Instruments) at 900 p.s.i. (1 p.s.i.=6.9 kPa). The crude extract obtained was loaded on to a 150 ml column containing Fast Flow DEAE-Sepharose (GE Healthcare). The column was washed with the same buffer, and protein was eluted with a 450 ml linear gradient of 0.1–0.3 M NaCl. Fractions containing mtCBS-PPase (150–190 mM NaCl) were pooled, concentrated to 12 ml with Centriprep 10 (Amicon), and purified further by gel filtration on a Superdex 200 26/60 column (GE Healthcare) equilibrated with 50 mM Tris/HCl buffer (pH 7.5), containing 50 mM KCl, 2 mM MgCl2, 0.1 mM CoCl2 and 20 μM DTPA. The fractions containing mtCBS-PPase were pooled, concentrated to 20–40 mg/ml and were stored frozen at −70 °C.
Because the enzyme was most stable in the presence of 0.1 mM Co2+ and 2 mM Mg2+, these cations were routinely added to the solutions used to purify, store and dilute the enzyme. When needed, these metal ions were removed from the enzyme stocks by incubation with 5 mM DTPA for 1 day at 4 °C. The bulk of the chelator was removed by dialysis against three 1 litre changes of 100 mM Mops/KOH buffer (pH 7.5) containing 50 μM DTPA.
The purity of enzyme samples was assessed by electrophoresis on 8–25% gradient polyacrylamide gels in the presence of 0.55% SDS using the Phast System (GE Healthcare). Concentrations of mtCBS-PPase solutions were determined on the basis of a subunit molecular mass of 48.1 kDa and a specific absorption coefficient A1%280 of 4.5, as estimated from the amino acid composition using the ProtParam program [24].
Activity measurements
Except where noted, the activity was measured at 25 °C in 25 ml of 100 mM Mops/KOH buffer (pH 7.2) containing 0.1 mM CoCl2, 5 mM MgCl2 and the nucleotide tested. An aliquot (5–150 μl) of diluted enzyme solution was added to the mixture (0.2–2 μg/ml final enzyme concentration), followed 1 min later by 0.16 mM PPi. The formation of Pi was then monitored for 3–10 min using an automatic Pi analyser [25], and initial reaction rates were estimated from the recorder tracing.
AMP, ADP and ATP were from Fluka. All other nucleotides were from Sigma. AMP was the free acid, ADP was the potassium salt, and all other nucleotides were sodium salts. Immediately before the use of ATP, any contaminating ADP was converted into ATP by treating 5 mM stock solutions of ATP for 1 h at room temperature (23±2 °C) with 10 units/ml rabbit muscle creatine kinase (Roche) in 100 mM Mops/KOH buffer (pH 7.2) containing 10 mM phosphocreatine (Fluka) and 5 mM MgCl2. The AMP preparation was judged to be reasonably pure because a similar treatment with creatine kinase and phosphocreatine had no effect on the PPase activity measured in the presence of AMP. In addition, treating stock ADP solutions with 10 units/ml hexokinase and 10 mM glucose to convert any contaminating ATP into ADP also had no effect on the PPase activity. For competition measurements, in which ADP and ATP were added simultaneously, creatine kinase in stock solutions of ATP was removed by ultrafiltration using 4 ml Vivaspin concentrators (Sartorius AG) with a 10 kDa molecular-mass cut-off PES (polyethersulfone) membrane.
Sedimentation
Analytical ultracentrifugation was carried out at 20 °C in a Spinco E instrument (Beckman Instruments) with scanning at 280 nm. The samples contained 10 μM enzyme and appropriate ligands. Before each run, the samples were incubated for 2–3 h at 20 °C. The sedimentation velocity was measured at 48000 rev./min, and the sedimentation coefficient (s020,w) was calculated as described by Chervenka [26].
Cross-linking and electrophoresis
Enzyme was diluted to 8.3 μM with 25 mM Hepes/KOH (pH 7.5) and then incubated with 26 mM glutaraldehyde for 15 or 30 min at room temperature [27]. The reaction was stopped by addition of 1/10 vol. of 1.0 M Tris/HCl (pH 7.3). The samples were separated by electrophoresis on Phast System SDS/8–25% PAGE gradient gels (GE Healthcare), and the gels were stained with PhastGel Blue R (GE Healthcare). A Perfect Protein Marker kit (Novagen) was used for molecular-mass standards.
Kinetic data analysis
Non-linear least squares fitting of the data was performed using SCIENTIST, version 2.01 (Micromath). The dependence of activity on the concentration of the ligand (L) was fitted to eqn (1), where A+L and A−L are activities with and without bound ligand respectively, and Kd is the apparent dissociation constant of the enzyme–ligand complex, all measured at a fixed substrate concentration:
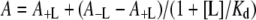 |
(1) |
In competition experiments, the dependence of the apparent Kd (Kd,app) for ligand L1 on the concentration of the second ligand, L2, was fitted to eqn (2), where Kd1 and Kd2 are the dissociation constants for ligands L1 and L2 respectively:
 |
(2) |
RESULTS
Expression, purification and catalytic activity of mtCBS-PPase
SDS/PAGE analysis of crude extracts obtained from recombinant E. coli cells revealed an intense ∼50 kDa band that was absent in cells transformed with empty pET36b vector (results not shown). The SDS/PAGE also indicated that mtCBS-PPase represented ∼10% of the total protein. The recombinant protein was easily purified to homogeneity by ion-exchange chromatography and gel filtration. This procedure yielded 30–40 mg of pure mtCBS-PPase per litre of cell culture (∼6 g of cell paste). E. coli PPase, which belongs to family I of soluble PPases, was absent from the purified mtCBS-PPase, because the former was eluted at a 20 mM lower NaCl concentration during DEAE-Sepharose chromatography.
The purified mtCBS-PPase exhibited a maximal kcat value (1.7 s−1) when assayed in the presence of both 1 mM Mg2+ and 0.1 mM Co2+. Replacement of Co2+ by Mn2+ decreased kcat to 0.65 s−1. The kcat values observed in the presence of Co2+, Mn2+ or Mg2+ alone were 1.15, 0.36 and 0.01 s−1 respectively. According to its cofactor specificity, mtCBS-PPase resembles the common family II PPase of Methanococcus jannaschii [28]. However, the maximal observed kcat value for mtCBS-PPase at 25 °C was three orders of magnitude lower than that for common family II PPases [14] and was only 2-fold higher at 55 °C, the growth temperature of M. thermoacetica.
Effects of nucleotides and nucleosides
Table 1 shows that mtCBS-PPase is strongly inhibited by 100 μM AMP and ADP, whereas ATP activates it 1.6-fold. Some nucleotides, including cAMP, CDP and UDP were less effective at inhibiting mtCBS-PPase, and most other nucleotides and nucleosides did not significantly affect the activity. Thus adenine nucleotides are effective modulators of mtCBS-PPase activity. This is strikingly different from the common family II PPase from Bacillus subtilis and family I PPases from E. coli and other sources, which are not modulated by ATP, ADP or AMP and lack CBS domains.
Table 1. Effects of nucleotides and nucleosides (100 μM) on Co2+/Mg2+-activated mtCBS-PPase.
The activity of mtCBS-PPase measured with no effector added (1.64 s−1) was taken as 100%.
| Effector | Activity (%) |
|---|---|
| None | 100 |
| Adenosine | 94±2 |
| AMP | 4.0±0.1 |
| ADP | <1 |
| ATP | 158±2 |
| cAMP | 42±4 |
| CDP | 50±8 |
| GMP | 82±3 |
| GDP | 81±3 |
| UDP | 65±8 |
| UTP | 96±2 |
| Cytidine, guanosine, uridine, CMP, CTP, GTP, cGMP, UMP | 100±3 |
Kinetic analysis of adenine nucleotide effects
Figure 1 shows the dose-dependencies of AMP, ADP and ATP effects measured at a fixed substrate concentration (160 μM total PPi). Each curve obeyed eqn (1) for activation or non-linear inhibition with a finite activity of the enzyme–inhibitor complex. The relative values of these activities (A+L/A−L) for AMP and ADP were low, but significantly different from zero (Table 2). The apparent binding constants (Kd) derived for each nucleotide from these profiles are also summarized in Table 2. As expected from the appearance of the profiles in Figure 1, the Kd values were similar for AMP and ATP, and an order of magnitude lower for ADP.
Figure 1. Effects of adenine nucleotides on Co2+/Mg2+-activated mtCBS-PPase.

The lines show the best fit to eqn (1).
Table 2. Parameters describing the effects of nucleotides on Co2+- and Mn2+-bound mtCBS-PPase.
| Co2+/Mg2+-activated enzyme | Mn2+/Mg2+-activated enzyme | |||||
|---|---|---|---|---|---|---|
| Enzyme | Nucleotide | A+L/A−L (%) | Kd (μM) | A+L/A−L (%) | Kd (μM) | Source |
| Wild-type | AMP | 3.7±0.4 | 0.22±0.02 | 20±4 | 1.4±0.2 | Inhibition of activity (Figure 1) |
| AMP | 0.17±0.05 | Effect on ATP activation | ||||
| ADP | 0.8±0.3 | 0.012±0.001 | 21±2 | 0.033±0.004 | Inhibition of activity (Figure 1) | |
| ADP | 0.017±0.005 | Effect on ATP activation (Figure 3) | ||||
| ATP | 180±10 | 0.20±0.05 | 400±40 | 7.2±0.8 | Inhibition of activity (Figure 1) | |
| ATP | 0.16±0.01 | Effect on AMP inhibition | ||||
| ATP | 0.6±0.2 | Effect on ADP inhibition (Figure 3) | ||||
| CDP | <10 | 150±20 | Inhibition of activity | |||
| UDP | <10 | 340±90 | Inhibition of activity | |||
| Y169A | AMP | 52±2 | 5.3±0.9 | Inhibition of activity | ||
| ADP | 6.7±0.4 | 0.21±0.02 | Inhibition of activity | |||
| ATP | 143±4 | 2.4±0.9 | Inhibition of activity | |||
The nucleotide effects on activity were analysed further using a Lineweaver–Burk plot. Surprisingly, the inhibition by AMP and ADP was of different types. AMP increased only the Michaelis constant (Figure 2, upper panel) and was thus a competitive inhibitor, whereas ADP additionally decreased the maximal velocity (Figure 2, lower panel) and was thus a mixed-type inhibitor. The effect of ATP on the kinetic parameters was the opposite of that of ADP (Figure 2, upper panel). At 10 μM, ATP decreased the Michaelis constant (from 10.6±0.5 to 7.0±0.6 μM) and increased the maximal velocity (from 1.69±0.05 to 2.3±3 s−1). True inhibition constants governing AMP binding to substrate-free enzyme (Ki=19±2 nM) and ADP binding to substrate-free enzyme (Ki=1.3±0.1 nM), and to the enzyme–substrate complex (Ki′=0.35±0.03 μM), were estimated from the secondary dependencies shown in the insets of Figure 2. The substrate thus afforded partial protection against inhibition by ADP and completely prevented inhibition by AMP. The residual activity of the nucleotide-bound enzyme was ignored in this analysis, as this activity would have only a minor effect on estimated parameter values. For ATP, true binding constants governing binding to substrate-free enzyme (Ka) and to enzyme–substrate complex (Ka′) were not determined separately, but their ratio, Ka′/Ka, should be close to the ratio of the Michaelis constants measured at ATP concentrations of zero and near-saturating (10 μM), i.e. 1.5±0.2. That Ka′/Ka exceeds unity is also evident by the fact that the lines measured in the absence and in the presence of ATP intersected above the x-axis in Figure 2 (upper panel).
Figure 2. Lineweaver–Burk plots for mtCBS-PPase inhibition and activation by adenine nucleotides.

Upper panel: inhibition by AMP and activation by ATP. Numbers on the lines indicate AMP concentrations (in μM). The line marked ATP shows the effect of 10 μM ATP. Lower panel: inhibition by ADP. Numbers on the lines indicate ADP concentrations (in μM). The insets show the dependencies of Km,app (upper panel) or 1/kcat,app and kcat,app/Km,app (lower panel) on nucleotide concentration.
To investigate interactions of inhibiting and activating nucleotides on mtCBS-PPase activity, the profiles of ADP modulation were measured at several fixed ATP concentrations and vice versa (Figure 3). ATP protected mtCBS-PPase from inhibition by ADP by decreasing the sensitivity to ADP (i.e. by shifting the dose–response inhibition curve to the right) and by increasing the basal activity in the absence of ADP. The Kd,app for ADP changed in proportion to the ATP concentration (Figure 3, upper panel inset) up to 100 μM ATP, which is 500 times the Kd for ATP. Similarly, the Kd,app for ATP was a linear function of the ADP concentration up to 0.2 μM ADP (Figure 3, lower panel inset), which is 80 times the Kd for ADP. These results indicate that the binding of ADP and ATP was mutually exclusive in the concentration ranges tested. If a mixed enzyme–ADP/ATP and/or enzyme–substrate–ADP/ATP complex was formed, Kd,app for ADP would have attained a constant level, intermediate between the Kd values for ADP binding to enzyme–ATP and enzyme–substrate–ATP. These considerations also apply to the lower panel of Figure 3. Qualitatively similar inhibition/activation patterns were observed for the AMP/ATP pair (results not shown). The effective concentrations of AMP were, however, higher than those of ADP by an order of magnitude. The binding constants derived for each nucleotide, from results shown in the insets to Figure 3, are summarized in Table 2.
Figure 3. Combined effects of ADP and ATP on the activity of mtCBS-PPase.

Upper panel: effect of various concentrations of ADP at several fixed concentrations of ATP. Lower panel: effect of various concentrations of ATP at several fixed concentrations of ADP. The numbers on the curves denote the fixed concentration of the second nucleotide in μM. The lines show the best fit to eqn (1). The insets show the dependences of Kd,app on the concentration of the corresponding fixed ligand. The lines in the insets show the best fit to eqn (2).
The effects of the adenine nucleotides on Mn2+/Mg2+-activated mtCBS-PPase were qualitatively similar to those observed for the Co2+/Mg2+-activated form (Figure 1). The Kd values for AMP and ADP were also similar, but the Kd value for ATP was 10-fold higher (Table 2). Furthermore, the relative residual activities at infinite AMP or ADP concentrations (A+L/A−L) were markedly higher in the presence of Mn2+. Finally, AMP, ADP and ATP had similar effects on Co2+/Mg2+-activated mtCBS-PPase at 55 °C, which is the physiological temperature for M. thermoacetica (results not shown).
Effect of Y169A substitution on adenine nucleotide binding
Tyr-169 is located in the CBS domain of mtCBS-PPase, in the region corresponding to the nucleotide-binding pocket of the chloride transporter ClC-5, another CBS domain protein whose three-dimensional structure has been determined recently [29]. Consistent with this localization, the replacement of Tyr-169 with alanine markedly decreased the affinity of mtCBS-PPase for the adenine nucleotides (Table 2), while having only a small effect (1.8-fold activation) on enzyme hydrolytic activity measured in their absence. Qualitatively, the Y169A variant behaved similarly to wild-type mtCBS-PPase and exhibited activation by ATP and inhibition by AMP and ADP (Table 2).
Quaternary structure of mtCBS-PPase
Common family II PPases are dimeric proteins in the presence of metal cofactors, but dissociate into nearly inactive monomers in the absence of such cofactors [14,30]. As CBS domains are reported to control oligomerization of CBS [31,32], we determined the oligomeric structure of mtCBS-PPase in the absence and presence of adenine nucleotides and compared it with that of common family II PPases. The sedimentation coefficient (s020,w) at pH 7.2 was 6.4±0.1 S for the metal-depleted mtCBS-PPase and increased to 7.7±0.1, 7.9±0.2 and 7.6±0.1 S in the presence of 100 μM CoCl2, 100 μM MnCl2, or 1 mM MgCl2 respectively. Adenine nucleotides (AMP, ADP and ATP) at 100 μM had no effect on the s020,w value for mtCBS-PPase measured in the presence of 100 μM CoCl2.
The values of s020,w for mtCBS-PPase measured in the presence of the metal ions were somewhat higher than that predicted assuming a spherical protein with a molecular mass of 96 kDa (dimer of 48 kDa monomers). Therefore we performed cross-linking experiments to confirm that this enzyme is a dimer. Specifically, we reacted mtCBS-PPase with glutaraldehyde and separated the products by SDS/PAGE. Almost all of the protein migrated as the cross-linked dimer, although a small amount of unreacted monomer was observed (Figure 4). Similar electrophoresis patterns were obtained for mtCBS-PPase treated with glutaraldehyde in the presence of different metal ions or at various concentrations of mtCBS-PPase. The relative amount of the monomer, however, increased slightly in the absence of the metal ions or at low enzyme concentrations. Collectively, our results indicate that the oligomeric structure of mtCBS-PPase is similar to that of common family II PPases, which lack CBS domains, and is not affected by nucleotide binding.
Figure 4. SDS/PAGE analysis of mtCBS-PPase.

Lane a, molecular-mass markers; lane b, intact enzyme; lane c, enzyme cross-linked with glutaraldehyde for 15 min; lane d, enzyme cross-linked with glutaraldehyde for 30 min. Protein load for lanes b–d was 1 μg/lane.
DISCUSSION
CBS domains are found in both cytosolic enzymes and membrane-associated enzymes and channels from all species. They usually occur as tandem repeats, which are independent units that retain their structure and binding properties even when separated from the bulk protein [17]. The importance of CBS domains is emphasized by the observations that mutations of their conserved residues impair AMP, ATP and S-adenosylmethionine binding to the CBS domains and cause a variety of human hereditary diseases [16,17]. Although they are thought to be regulatory domains, the experimental evidence for this is scarce. Because CBS domains bind adenine nucleotides with varying affinities, they may act as sensors of cellular energy status [16,17]. More recently, CBS domains of the chloride channel ClC-5 have been implicated in the control of intracellular trafficking [33] and the gating of the osmoregulatory transporter OpuA by osmotic strength [34].
CBS domains as regulators of mtCBS-PPase
Several lines of evidence suggest that the modulation of mtCBS-PPase by nucleotides is also mediated by CBS domains. First, isolated CBS domains from several proteins can bind adenine nucleotides [16,17,20], whereas common family II PPases that lack CBS domains do not bind adenine nucleotides [35]. Secondly, a single Y169A replacement in the CBS domain suppressed nucleotide binding to mtCBS-PPase, but increased enzyme catalytic activity. Finally, the mixed-type inhibition of mtCBS-PPase by ADP, and the activation by ATP, indicate formation of a ternary enzyme–substrate–nucleotide complex and are therefore inconsistent with nucleotide binding at the active site.
The observed competition between ATP binding and the binding of both AMP and ADP, as well as the structural similarities between these compounds, imply a common binding site for all nucleotides on mtCBS-PPase. This conclusion is supported by structural data reported for other proteins. To date, a high-resolution three-dimensional structure is available only for a single intact CBS domain-containing protein, namely IMP dehydrogenase, but it does not contain bound nucleotides in the CBS domain [36]. However, three-dimensional structures have been determined for several truncated CBS domain-containing proteins [29,37,38]. The most recent structures of the cytoplasmic part of the chloride transporter ClC-5 [29] contain bound ADP or ATP, and those of the C-terminal domain of AMP-activated protein kinase (Protein Data Bank codes 2OOX and 2OOY) contain bound AMP or ATP, at the same site, formed between two CBS domains. Importantly, the new structures suggest that the nucleotides cause a marked conformational change in the CBS domains, thus explaining nucleotide regulatory action.
Although all polar active-site residues found in common family II PPases [12,13] are conserved in mtCBS-PPase, even in the presence of the best metal cofactor, Co2+, mtCBS-PPase is less active than common family II PPases by three orders of magnitude. Therefore the insert containing two CBS domains appears to severely disrupt the normal catalytic cycle. Importantly, such an ‘internally inhibited’ system should be very sensitive to structural changes caused by nucleotide binding to the CBS domains and could easily be inhibited or activated. In this context, ATP partially compensates for the effect of the CBS insertion, whereas AMP and ADP increase the effect.
Nucleotide specificity of mtCBS-PPase
Nucleotide binding to mtCBS-PPase was highly specific with respect to both the base and the phosphate moieties. At a concentration of 100 μM, adenine nucleotides were the most potent regulators of the enzyme. When guanine and uridine nucleotides were examined, only the diphosphates had significant effects on the activity of mtCBS-PPase; this may be because these nucleotides bound with weaker affinities.
The phosphate moiety of the nucleotide is also important and determines both binding affinity (adenosine does not bind) and whether the nucleotide acts as an activator or inhibitor. A diphosphate seems to result in the highest binding affinity, but a triphosphate is needed for activation. Moreover, the phosphate moiety determines the relative affinities of the nucleotide for free enzyme and the enzyme–substrate complex. AMP, ADP and ATP can all combine with substrate-free enzyme, whereas only ADP and ATP can bind, although with lower affinity, to the enzyme–substrate complex. Accordingly, the inhibition type is competitive with AMP and mixed with ADP, and ATP is a mixed-type activator. Substrate thus interferes with adenine nucleotide binding and vice versa, and the effect decreases with increases in phosphate chain length, especially when comparing AMP effects with ADP effects. A likely structural explanation of these data is that both the α-phosphate and the β-phosphate are involved in strong, thermodynamically favourable, interactions that stabilize inactive conformations of mtCBS-PPase, whereas the interaction with the γ-phosphate is thermodynamically unfavourable, but gives rise to an active enzyme conformation. The conformations elicited by AMP and ADP are different because their complexes bind substrate differently. Although a direct contact of the nucleotide phosphate chain with the active site cannot presently be excluded, it seems unlikely, because ATP, which has the longest phosphate chain, facilitated substrate binding by lowering Km (Figure 2). The assumed conformational flexibility of mtCBS-PPase is consistent with the known structures of common family II PPases, in which active sites lie between two domains connected by a highly flexible linker [12,13]. The dependence of the nucleotide effect on the length of the phosphate moiety may be due, but only in part, to Mg2+ binding; in the presence of 5 mM Mg2+, ATP and ADP predominantly exist as complexes with Mg2+, whereas AMP exists as a mixture of free and Mg2+-bound forms [39].
The effects of the adenine nucleotides vary in different CBS domain proteins. With the exception of the chloride channel ClC-1, which is inhibited by both AMP and ATP, generally AMP and ATP have opposite effects on these proteins [20]. For example, AMP activates and ATP inhibits AMP-activated protein kinase and the chloride channel ClC-2 [16,19], and, as in the case of mtCBS-PPase, ATP activates and AMP inhibits IMP dehydrogenase-2 and the chloride channel ClC-5. These findings have led to the hypothesis that CBS domains act as energy-sensing modules in proteins [16]. Generally, AMP, ADP and ATP bind with similar affinities to these proteins, in contrast with adenine nucleotide interactions with mtCBS-PPase, where ADP binding is strongly preferred. Furthermore, AMP and ATP bind at least 100-fold, and ADP 10000-fold, more tightly (in terms of Kd) to mtCBS-PPase than to ClC-5 [29], or to other CBS domain proteins [16].
Distribution, phylogeny and possible role of CBS-PPases
Common family II PPases predominate in Bacilli, whereas CBS-PPases predominate in Clostridia and sulfate-reducing δ-proteobacteria (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/408/bj4080327add.htm). Both types of family II PPases occur sporadically in other bacterial lineages. In contrast, structurally distinct family I PPases are more widespread and occur in most of the bacterial phyla as well as in archaea and eukaryotes. Interestingly, a majority of the CBS-PPases contain an ∼120-amino-acid DRTGG domain [40] between two CBS domains. The function of DRTGG domains is unknown, and they are found exclusively in bacterial and archaeal proteins. CBS-PPases from M. thermoacetica and Syntrophomonas wolfei are unique in that they possess CBS domains, but lack a DRTGG domain.
Phylogenetic analysis of family II PPases suggests that all CBS-PPases are closely related, but do not form a monophyletic group (Supplementary Figure S1). Some groups of common family II PPases, notably Bacilli PPases, have probably arisen due to the secondary loss of the CBS and DRTGG domain insertion. In addition, M. thermoacetica and S. wolfei are two independent cases where the DRTGG domain appears to have been lost. Finally, the clustering of CBS-PPases near the centre of the tree suggests that the insertion was acquired very early in the evolution of family II PPases.
CBS-PPases are found in 16 of the 329 prokaryotic species with complete genome sequences in the Kyoto Encyclopedia of Genes and Genomes (http://www.genome.jp/kegg/catalog/org_list.html). Thirteen of these 16 species also have a gene encoding a membrane-bound H+-translocating PPase (H+-PPase). Interestingly, none of the 83 prokaryotic species with a gene for H+-PPase has a gene for the common family II PPase. H+-PPase may be unnecessary when there are family II PPases because the latter are three orders of magnitude more active and would therefore deplete the cell of PPi, which H+-PPase uses to establish a transmembrane pH gradient. In contrast, the activities of CBS-PPase and membrane-bound-PPase in M. thermoacetica are similar (A. Malinen, personal communication), permitting control of the PPi concentration by the levels of adenine nucleotides via mtCBS-PPase.
Biosynthetic reactions are the major source of PPi. Accordingly, in M. thermoacetica, the intracellular level of PPi peaks sharply during the mid-exponential phase of growth [41]. To avoid inhibition of biosynthetic enzymes and to provide Pi for ATP synthesis, this PPi must be efficiently degraded by PPase. In fact, during the mid-exponential phase, mtCBS-PPase is expected to be most active because of a high [ATP]/([ADP]+[AMP]) ratio. Interestingly, Co2+, the best cofactor for mtCBS-PPase, is required for the growth of M. thermoacetica [22,42]. Metabolic stresses that decrease ATP production inhibit biosynthetic reactions and cell growth by decreasing the [ATP]/([ADP]+[AMP]) ratio. This, in turn, should inhibit mtCBS-PPase and allow accumulation of PPi, which would further inhibit biosynthetic reactions. Instead of or in conjunction with the action of H+-ATPase, the accumulated PPi is apparently used by H+-PPase to sustain the pH gradient across the cell membrane and therefore maintain cell vitality. This two-PPase system may allow the cell to combat stress more efficiently than cells with only soluble or membrane-bound PPases. Thus CBS-PPase may act as a survival factor under conditions of low energy supply.
In summary, unlike other PPases, mtCBS-PPase is subject to strong inhibition by AMP and ADP as well as activation by ATP. Remarkably, this enzyme binds adenine nucleotides much more tightly than other reported CBS proteins. Furthermore, an effect of ADP, which is the most efficient inhibitor of mtCBS-PPase, is uncommon in other CBS proteins. Further studies, including determination of the three-dimensional structure of mtCBS-PPase with a bound adenine nucleotide, are needed to elucidate the structural basis for nucleotide regulation of this interesting enzyme and other CBS proteins.
Online data
Acknowledgments
This work was supported by Academy of Finland Grants 201611 and 114706, a grant for the National Graduate School in Informational and Structural Biology from the Ministry of Education and the Academy of Finland, and Russian Foundation for Basic Research Grant 06-04-48887. We thank University of Oslo Bioportal for providing CPU (central processing unit) time and P. V. Kalmykov (A. N. Belozersky Institute of Physico-Chemical Biology) for his assistance with analytical centrifugation.
References
- 1.Kornberg A. On the metabolic significance of phosphorolytic and pyrophosphorolytic reactions. In: Kasha M., Pullman B., editors. Horizons in Biochemistry. New York: Academic Press; 1962. pp. 251–264. [Google Scholar]
- 2.Heinonen J. Boston: Kluwer Academic Publishers; 2001. Biological Role of Inorganic Pyrophosphate. [Google Scholar]
- 3.Chen J., Brevet A., Formant M., Leveque F., Schmitter J.-M., Blanquet S., Plateau P. Pyrophosphatase is essential for growth of Escherichia coli. J. Bacteriol. 1990;172:5686–5689. doi: 10.1128/jb.172.10.5686-5689.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lundin M., Baltscheffsky H., Ronne H. Yeast PPA2 gene encodes a mitochondrial inorganic pyrophosphatase that is essential for mitochondrial function. J. Biol. Chem. 1991;266:12168–12172. [PubMed] [Google Scholar]
- 5.Sonnewald U. Expression of E. coli inorganic pyrophosphatase in transgenic plants alters photoassimilate partioning. Plant J. 1992;2:571–581. [PubMed] [Google Scholar]
- 6.Ogasawara N. Systematic function analysis of Bacillus subtilis genes. Res. Microbiol. 2000;151:129–134. doi: 10.1016/s0923-2508(00)00118-2. [DOI] [PubMed] [Google Scholar]
- 7.Shintani T., Uchiumi T., Yonezawa T., Salminen A., Baykov A. A., Lahti R., Hachimori A. Cloning and expression of a unique inorganic pyrophosphatase from Bacillus subtilis: evidence for a new family of soluble inorganic pyrophosphatases. FEBS Lett. 1998;439:263–266. doi: 10.1016/s0014-5793(98)01381-7. [DOI] [PubMed] [Google Scholar]
- 8.Young T. W., Kuhn N. J., Wadeson A., Ward S., Burges D., Cooke G. D. Bacillus subtilis ORF yybQ encodes a manganese-dependent inorganic pyrophosphatase with distinctive properties: the first of a new class of soluble pyrophosphatase? Microbiology. 1998;144:2563–2571. doi: 10.1099/00221287-144-9-2563. [DOI] [PubMed] [Google Scholar]
- 9.Cooperman B. S., Baykov A. A., Lahti R. Evolutionary conservation of the active site of soluble inorganic pyrophosphatase. Trends Biochem. Sci. 1992;17:262–266. doi: 10.1016/0968-0004(92)90406-y. [DOI] [PubMed] [Google Scholar]
- 10.Baykov A. A., Cooperman B. S., Goldman A., Lahti R. Cytoplasmic inorganic pyrophosphatases. Prog. Mol. Subcell. Biol. 1999;23:127–150. doi: 10.1007/978-3-642-58444-2_7. [DOI] [PubMed] [Google Scholar]
- 11.Aravind L., Koonin E. V. A novel family of predicted phoshoesterases includes Drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem. Sci. 1998;23:17–19. doi: 10.1016/s0968-0004(97)01162-6. [DOI] [PubMed] [Google Scholar]
- 12.Merckel M. C., Fabrichniy I. P., Salminen A., Kalkkinen N., Baykov A. A., Lahti R., Goldman A. Crystal structure of Streptococcus mutans pyrophosphatase: a new fold for an old mechanism. Structure. 2001;9:289–297. doi: 10.1016/s0969-2126(01)00587-1. [DOI] [PubMed] [Google Scholar]
- 13.Ahn S., Milner A. J., Fütterer K., Konopka M., Ilias M., Young T. W., White S. A. The “open” and “closed” structures of the type-C inorganic pyrophosphatases from Bacillus subtilis and Streptococcus gordonii. J. Mol. Biol. 2001;313:797–811. doi: 10.1006/jmbi.2001.5070. [DOI] [PubMed] [Google Scholar]
- 14.Parfenyev A. N., Salminen A., Halonen P., Hachimori A., Baykov A. A., Lahti R. Quaternary structure and metal-ion requirement of family II pyrophosphatases from Bacillus subtilis, Streptococcus gordonii and Streptococcus mutans. J. Biol. Chem. 2001;276:24511–24518. doi: 10.1074/jbc.M101829200. [DOI] [PubMed] [Google Scholar]
- 15.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 16.Scott J. W., Hawley S. A., Green K. A., Anis M., Stewart G., Scullion G. A., Norman D. G., Hardie D. G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ignoul S., Eggermont J. CBS domains: structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005;289:C1369–C1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 18.Kemp B. E. Bateman domains and adenosine derivatives form a binding contract. J. Clin. Invest. 2004;113:182–184. doi: 10.1172/JCI20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niemeyer M. I., Yusef Y. R., Cornejo I., Flores C. A., Sepúlveda F. V., Cid L. P. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol. Genomics. 2004;19:74–83. doi: 10.1152/physiolgenomics.00070.2004. [DOI] [PubMed] [Google Scholar]
- 20.Bennetts B., Rychkov G. Y., Ng H.-L., Morton C. J., Stapleton D., Parker M. W., Cromer B. A. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J. Biol. Chem. 2005;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- 21.Wellhauser L., Kuo H.-H., Stratford F. L. L., Ramjeesingh M., Huan L.-J., Luong W., Li C., Deber C. M., Bear C. E. Nucleotides bind to the C-terminus of ClC-5. Biochem. J. 2006;398:289–294. doi: 10.1042/BJ20060142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drake H. L., Daniel S. L. Physiology of the thermophilic acetogen Moorella thermoacetica. Res. Microbiol. 2004;155:869–883. doi: 10.1016/j.resmic.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J., Russel D. W. 3rd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 24.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker J. M., editor. The Proteomics Protocols Handbook. Totowa: Humana Press; 2005. pp. 571–607. [Google Scholar]
- 25.Baykov A. A., Avaeva S. M. A simple and sensitive apparatus for continuous monitoring of orthophosphate in the presence of acid-labile compounds. Anal. Biochem. 1981;116:1–4. doi: 10.1016/0003-2697(81)90313-4. [DOI] [PubMed] [Google Scholar]
- 26.Chervenka C. H. Palo Alto: Spinco Division of Beckman Instruments; 1972. Methods for the Analytical Ultracentrifuge; pp. 23–33. [Google Scholar]
- 27.Baykov A. A., Dudarenkov V. Y., Käpylä J., Salminen T., Hyytiä T., Kasho V. N., Husgafvel S., Cooperman B. S., Goldman A., Lahti R. Dissociation of hexameric Escherichia coli inorganic pyrophosphatase into trimers on His136→Gln or His140→Gln substitution and its effect on enzyme catalytic properties. J. Biol. Chem. 1995;270:30804–30812. doi: 10.1074/jbc.270.51.30804. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn N. J., Wadeson A., Ward S., Young T. W. Methanococcus jannaschii ORF mj0608 codes for a class C inorganic pyrophosphatase protected by Co2+ or Mn2+ ions against fluoride inhibition. Arch. Biochem. Biophys. 2000;379:292–298. doi: 10.1006/abbi.2000.1860. [DOI] [PubMed] [Google Scholar]
- 29.Meyer S., Savaresi S., Forster I. C., Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007;14:60–66. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 30.Zyryanov A. B., Vener A. V., Salminen A., Goldman A., Lahti R., Baykov A. A. Rates of elementary catalytic steps for different metal forms of the family II pyrophosphatase from Streptococcus gordonii. Biochemistry. 2004;43:1065–1074. doi: 10.1021/bi0357513. [DOI] [PubMed] [Google Scholar]
- 31.Kery V., Poneleit L., Kraus J. P. Trypsin cleavage of cystathionine-β-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch. Biochem. Biophys. 1998;355:222–232. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 32.Jhee K. H., McPhie P., Miles E. W. Domain architecture of the heme-independent yeast cystathionine-β-synthase provides insights into mechanisms of catalysis and regulation. Biochemistry. 2000;39:10548–10556. doi: 10.1021/bi001020g. [DOI] [PubMed] [Google Scholar]
- 33.Carr G., Simmons N., Sayer J. A role for CBS domain 2 in trafficking of chloride channel CLC-5. Biochem. Biophys. Res. Commun. 2003;310:600–605. doi: 10.1016/j.bbrc.2003.09.057. [DOI] [PubMed] [Google Scholar]
- 34.Bieman-Oldehinkel E., Mahmood N. B. N., Poolman B. A sensor for intracellular ionic strength. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10624–10629. doi: 10.1073/pnas.0603871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zyryanov A. B., Shestakov A. S., Lahti R., Baykov A. A. Mechanism by which metal cofactors control substrate specificity in pyrophosphatase. Biochem. J. 2002;367:901–906. doi: 10.1042/BJ20020880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang R., Evans G., Rotella F. J., Westbrook E. M., Beno D., Huberman E., Joachimiak A., Collart F. R. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:4691–4700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- 37.Miller M. D., Schwarzenbacher R., von Delft F., Abdubek P., Ambing E., Biorac T., Brinen L. S., Canaves J. M., Cambell J., Chiu H. J., et al. Crystal structure of a tandem cystathionine-β-synthase (CBS) domain protein (TM0935) from Thermotoga maritima at 1.87 Å resolution. Proteins. 2004;57:213–217. doi: 10.1002/prot.20024. [DOI] [PubMed] [Google Scholar]
- 38.Dutzler R., Meyer S. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14:299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Phillips R. Adenosine and the adenine nucleotides: ionization, metal complex formation and conformation in solution. Chem. Rev. 1966;66:501–527. doi: 10.1021/cr60243a002. [DOI] [PubMed] [Google Scholar]
- 40.Bateman A., Coin L., Durbin R., Finn R. D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E. L., et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinonen J., Drake H. L. Comparative assessment of inorganic pyrophosphate and pyrophosphatase levels in Escherichia coli, Clostridium pasterianum and Clostridium thermoaceticum. FEMS Microbiol. Lett. 1988;52:205–208. [Google Scholar]
- 42.Lundie L. L., Drake H. L. Development of a minimally defined medium for the acetogen Clostridium thermoaceticum. J. Bacteriol. 1984;159:700–703. doi: 10.1128/jb.159.2.700-703.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.