

Abstract
NGB (human neuroglobin), a recently discovered haem protein of the globin family containing a six-co-ordinated haem, is expressed in nervous tissue, but the physiological function of NGB is currently unknown. As well as playing a role in neuronal O2 homoeostasis, NGB is thought to act as a scavenger of reactive species. In the present study, we report on the reactivity of metNGB (ferric-NGB), which accumulates in vivo as a result of the reaction of oxyNGB (oxygenated NGB) with NO, towards NO2− and H2O2. NO2− co-ordination of the haem group accounts for the activity of metNGB in the nitration of phenolic substrates. The two different metNGB forms, with and without the internal disulfide bond between Cys46 (seventh residue on the inter-helix region between helices C and D) and Cys55 (fifth residue on helix D), exhibit different reactivity, the former being more efficient in activating NO2−. The kinetics of the reactions, the NO2−-binding studies and the analysis of the nitrated products from different substrates all support the hypothesis that metNGB is able to generate an active species with the chemical properties of peroxynitrite, at pathophysiological concentrations of NO2− and H2O2. Without external substrates, the targets of the reactive species generated by the metNGB/NO2−/H2O2 system are endogenous tyrosine (resulting in the production of 3-nitrotyrosine) and cysteine (oxidized to sulfinic acid and sulfonic acid) residues. These endogenous modifications were characterized by HPLC-MS/MS (tandem MS) analysis of metNGB after reaction with NO2− and H2O2 under various conditions. The internal S–S bond affects the functional properties of the protein. Therefore metNGB acts not only as scavenger of toxic species, but also as a target of the self-generated reactive species. Self-modification of the protein may be related to or inhibit its postulated neuroprotective activity.
Keywords: cysteine oxidation, haem, human neuroglobin (NGB), oxidative stress, peroxynitrite, tyrosine nitration
Abbreviations: ABTS, 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid); CD, inter-helix region between helices C and D; CD7 etc., seventh residue on CD etc.; CID, collision-induced dissociation; DTT, dithiothreitol; EIC, extracted ion current; ESI, electrospray ionization; GdnHCl, guanidine hydrochloride; F8 etc., eighth residue on helix F etc.; NGB, human neuroglobin; h-NGB, modified NGB at high NO2− and H2O2 concentrations; h′-NGBSH, modified NGBSH at high H2O2 concentrations; HPA, 3-(4-hydroxyphenyl)propionic acid; LC, liquid chromatography; LPO, lactoperoxidase; lpo-NGB, modified NGB in the presence of LPO, NO2− and H2O2; Mb, myoglobin; hMb, human Mb; metNGB, ferric-NGB; NGBSH, NGB without the internal disulfide bond; NGBS-S, NGB with the internal disulfide bond; oxyNGB, oxygenated NGB; 4-PDS, 4,4′-dithiodipyridine; p-NGB, modified NGB at low NO2− and H2O2 concentrations; p′-NGBSH, modified NGBSH at low H2O2 concentrations; RNS, reactive nitrogen species; ROS, reactive oxygen species; TFA, trifluoroacetic acid
INTRODUCTION
NGB (human neuroglobin) was first identified in 2000 as a third globin-type protein, the other types of globins are Mb (myoglobin) and Hb [1]. NGB is expressed at low levels (in the micromolar range) in various regions of the brain, but at much higher levels (∼100 μM) in retinal cells [1,2]. NGB is composed of 151 amino acids and has a high sequence similarity to mouse neuroglobin (94% identity), but little homology with vertebrate Mbs and Hbs (<21% and <25% identity respectively) [1]. NGB differs from the other vertebrate globins in the binding of the six-co-ordinated haem group, with His96[F8 (eighth residue on helix F)] and His64(E7) acting as the fifth/proximal and sixth/distal ligands respectively, in both the ferrous and ferric forms of NGB [3,4] (Scheme 1). The presence of a six-co-ordinated haem reduces the affinity of the protein for ligands, with respect to the expected values, in the absence of competitive co-ordination by the distal histidine residue [3].
Scheme 1. Cysteine residue oxidation states and co-ordination equilibria in metNGB.

Schematic representation of the low-spin/high-spin equilibria for metNGBS-S (left-hand side) and metNGBSH (right-hand side). The disposition of the side chains of the proximal and distal histidine residues [His96(F8) and His64(E7) respectively] is shown. The length of the vertical arrows indicates that, even if the co-ordination equilibrium is always shifted towards the six-co-ordinated species, in the case of metNGBS-S, the amount of high-spin form is larger compared with metNGBSH.
The function of NGB in the human brain remains unclear. In analogy with other globins, several possible physiological roles have been considered [5]. Indirect evidence suggests a role of NGB in neuronal O2 homoeostasis, owing to the correlation between NGB expression and O2 consumption [6]. The O2 affinity of NGB depends on several factors, including the relative binding rates of O2 with the haem sixth position and the competing distal histidine residue [3,7], and the redox state of the cell which controls the formation or cleavage of an internal disulfide bond between Cys46 [CD7 (seventh residue on the inter-helix region between helices C and D)] and Cys55(D5) [8,9]. The presence of the S–S bond could perturb the three-dimensional structure of the CD–D (inter-helix region between helices C and D, extending to the end of helix D) region and affect the location of the neighbouring E-helix, thus modulating the binding of the endogenous His64(E7) ligand to the haem group [9,10]. As a consequence, in ferrous NGB, the distal histidine residue dissociation rate increases by a factor of 10 with respect to the protein form without the disulfide bond [9,11], leading to an effective increase in O2 affinity by the same factor [12] (Scheme 1).
O2 storage or diffusion to or from the haem group of NGB may be assisted by the presence of a large protein matrix cavity (approx. 120 Å3, where 1 Å=0.1 nm), located between the haem distal site and the EF interhelical hinge and connected to the protein surface [10,13]. The cavity may be reshaped after ligand binding, which may allow trapping of harmful ROS (reactive oxygen species) or RNS (reactive nitrogen species) [14–17]. In particular, the increase in the concentration of NO up to the low micromolar range, as observed under ischaemic conditions [18], may be contrasted with its reaction with oxyNGB (oxygenated NGB), yielding metNGB (ferric-NGB) and NO3− through a haem-bound ONOO− (peroxynitrite) intermediate [15]:
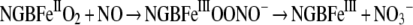 |
(1) |
As a metNGB reductase system has not yet been identified in the brain [15], the in vivo formation of metNGB through this mechanism may be physiologically relevant. Also, metNGB reacts with NO [19] to generate the stable NGBFeIINO species by reductive nitrosylation. This form of NGB is a good scavenger of ONOO−, which oxidizes the protein back to metNGB as follows [19]:
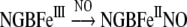 |
(2) |
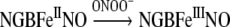 |
(3) |
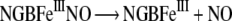 |
(4) |
In contrast with Mb and Hb, the protein does not generate the ferryl form on reaction with H2O2 or ONOO− [20]. Also, no reaction was observed on the addition of NO2− [19].
Therefore even if the FeIII centre of metNGB appeared to be protected by the co-ordinated distal histidine residue from attack by oxidizing species, the competitive co-ordination of exogenous ligands could account for the activation of alternative reactions. In the present study, we report on the reactivity of human metNGB in the presence of NO2− and H2O2, both of which are increased in vivo under conditions of oxidative stress [21]. The activation of NO2−/H2O2 by metNGB was assayed by the nitration of phenolic substrates [22–24] and an unexpectedly high reactivity was observed. Moreover, the two forms of the protein, metNGBS-S (NGB with the internal disulfide bond) and metNGBSH (NGB without the internal disulfide bond), are not equally as efficient in activating NO2− and H2O2. The issue of endogenous modification of metNGB, resulting from the reaction with NO2− and H2O2, at pathophysiological concentrations in the absence of external substrates was also addressed, since the identification of protein residues modified by ROS and RNS is essential for the understanding of the mechanisms involved in the development of various pathologies [25–29] and the postulated neuroprotective function of NGB. The endogenous targets of the reactive species generated by the protein are tyrosine and cysteine residues, and a more extensive pattern of modifications was observed for these residues than observed previously with hMb (human Mb) [24].
EXPERIMENTAL
Reagents
All buffer solutions were prepared using deionized Milli-Q water. ABTS [2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)], DTT (dithiothreitol), GdnHCl (guanidine hydrochloride), 30% (v/v) H2O2 solution, HPA [3-(4-hydroxyphenyl)propionic acid], 4-hydroxybenzonitrile, 4-PDS (4,4′-dithiodipyridine), phenylacetic acid, sodium nitrite and trypsin were obtained from Sigma–Aldrich. All reagents were obtained at the best grade available. The concentration of H2O2 was controlled by monitoring the formation of the ABTS radical cation using a standard enzymatic method as published previously [30]. Recombinant human NGB was expressed and purified as reported previously [12]. In order to obtain a homogeneous sulfur-bridged form of NGB (i.e. NGBS-S), the protein solution was dialysed overnight at 4 °C against 50 mM phosphate buffer, pH 7.5 (17 mM NaH2PO4 and 33 mM Na2HPO4). To reduce the intramolecular disulfide bond, forming NGBSH, the NGB solution was dialysed at 37 °C against 1 mg/ml DTT dissolved in degassed 50 mM Tris/HCl, pH 7.5, and 0.5 mM EDTA under anaerobic conditions. After 30 min of dialysis, three exchanges of degassed 50 mM Tris/HCl, pH 7.5, and 0.5 mM EDTA were performed to eliminate DTT. Protein dialysis was carried out at 4 °C. In all of the experiments described below, when not explicitally stated, the proteins (NGBS-S and NGBSH) were used in their ferric form. All spectrophotometric measurements were performed on a Hewlett Packard HP 8452A diode array spectrophotometer.
Kinetic studies of phenol nitration catalysed by NGBS-S and NGBSH
The kinetic experiments were carried out in 200 mM phosphate buffer, pH 7.5 (68 mM NaH2PO4 and 132 mM Na2HPO4), using a quartz cuvette with a 1 cm-path-length, thermostated at 25.0±0.1 °C and equipped with a magnetic stirrer. The initial solution, containing protein and variable substrate and NO2− concentrations in a final volume of 1.6 ml, was obtained by mixing solutions of the reagents (at an appropriate concentration) into the buffer. The reaction was started by the addition of H2O2 and was followed by monitoring A450 during the initial 10–15 s of the reaction. The conversion of the rate data from change in absorbance/s into [nitrophenol]/s was performed using the known molar absorption coefficient of HPA at 450 nm, ϵ=3350 M−1·cm−1 [22]. The kinetic parameters were obtained from fitting of the plots of experimental rates at different substrate/NO2− concentrations to the appropriate equations.
For each substrate, the change in the reaction rate in the presence of various reactant concentrations was studied through a series of steps. First, a suitable concentration of H2O2 was found which would maximize the reaction rate, but avoided the use of excess oxidant. Secondly, this H2O2 concentration was used to study the dependence of the reaction rate on the concentration of HPA, and, finally, the dependence of the reaction rate on the concentration of NO2− was examined. These steps were performed by following the iterative procedure described previously [23]. The concentration of NGBS-S or NGBSH used in these reactions was 1 μM, whereas the concentration of the other reactants for each of the steps is detailed as follows. Optimization of H2O2 concentration: using NGBS-S, [HPA]=1 mM, [NO2−]=0.3 M and [H2O2]=0.09–0.8 M; using NGBSH, [HPA]=0.4 mM, [NO2−]=1.0 M and [H2O2]=0.09–0.8 M. Dependence of the rate on varying HPA concentration: using NGBS-S, [H2O2]=0.6 M, [NO2−]=0.3 M and [HPA]=0.031–1.2 mM; using NGBSH, [H2O2]=0.6 M, [NO2−]=1.0 M and [HPA]=0.0096–0.96 mM. Dependence of the rate on varying the concentration of NO2−: using NGBS-S, [H2O2]=0.6 M, [HPA]=0.4 mM and [NO2−]=0.0062–0.44 M; using NGBSH, [H2O2]=0.6 M, [HPA]=0.3 mM and [NO2−]=0.037–2.5 M.
The reaction rates observed in the absence of the protein (non-catalytic reaction) or without H2O2 were completely negligible.
Binding of NO2−
In order to test the binding of NO2− to NGBs, a 1 cm-path-length quartz cuvette containing either 4.8 μM NGBS-S or 5.2 μM NGBSH in 200 mM phosphate buffer, pH 7.5, was incubated in increasing concentrations of NO2− in 200 mM phosphate buffer, pH 7.5 (final concentration from 0 to 0.42 M for NGBS-S and from 0 to 0.64 M for NGBS). The reaction was thermostatically controlled at 25.0±0.1 °C, and UV–visible spectra were recorded after each increase in NO2− concentration. Blank spectra were recorded in the same way, but in the absence of protein. After subtracting the corresponding blank from each spectrum, the resulting spectra were corrected for dilution and then transformed into difference spectra by subtracting the native protein spectrum. A plot was constructed demonstrating the difference between A414 and A434 (these wavelengths had the maximum variation in difference spectra) against the ligand concentration.
For the binding of NO2− to NGBS-S, the constants K1 and K2 were obtained by interpolation of the absorbance data with the binding isotherm for low-affinity binding of two consecutive ligands:
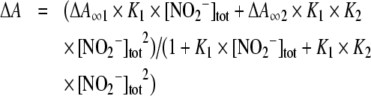 |
where ΔA∞1 and ΔA∞2 represent the absorbance changes owing to the binding of one and two ligand molecules respectively, and [NO2−]tot is the total NO2− concentration, i.e. free plus bound NO2− concentrations. For the binding of NO2− to NGBSH, the constant KB was obtained by interpolation of the absorbance data with the binding isotherm for low-affinity binding of a single ligand:
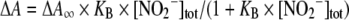 |
where ΔA∞ represents the absorbance change as a result of binding NO2−, and [NO2−]tot is the total NO2− concentration.
HPLC analysis of the nitration products of 4-hydroxybenzonitrile and phenylacetic acid
The product mixtures derived from the nitration of 4-hydroxybenzonitrile and phenylacetic acid, promoted by the NGB/NO2−/H2O2 system, were analysed by HPLC using a Jasco MD-1510 instrument with diode array detection equipped with a Supelcosil™ LC18 reverse-phase semipreparative column (5 μm; 250×10 mm). Elution was carried out using 0.1% TFA (trifluoroacetic acid) in distilled water (solvent A) and 0.1% TFA in acetonitrile (solvent B), with a flow rate of 5 ml/min. Elution started with the use of 100% solvent A for 5 min, followed by a linear gradient from 100% solvent A to 100% solvent B over 25 min. Spectrophotometric detection of the eluate was performed in the range of 200–600 nm.
The catalytic nitrations of 4-hydroxybenzonitrile and phenylacetic acid were studied under the following experimental conditions: [NGBS-S]=1 μM, [substrate]=1 mM, [H2O2]=0.6 M and [NO2−]=0.01, 0.05 or 0.15 M in 200 mM phosphate buffer, pH 7.5. The reaction mixtures were incubated for 10 min at room temperature (25 °C) and then were analysed by HPLC. The retention time of 4-hydroxybenzonitrile was 15.7 min, whereas the corresponding nitration product 4-hydroxy-3-nitrobenzonitrile was absent under all the conditions tested. Its retention time (17.4 min) was established using LPO (lactoperoxidase)-catalysed nitration, carried using the following conditions: [LPO]=80 nM, [phenol]=1 mM, [H2O2]=1 mM and [NO2−]=0.01 M in 200 mM phosphate buffer, pH 7.5. The HPLC chromatograms of the mixtures obtained from the nitration of phenylacetic acid showed there was unreacted substrate present at 16.8 min, two main peaks at 16.0 and 16.2 min, and two minor peaks at 14.5 and 14.7 min (obtained at both low and high NO2− concentrations).
HPLC analysis showed that no modification of 4-hydroxybenzonitrile and phenylacetic acid occurred in the absence of the protein. The presence of H2O2 was also demonstrated to be essential for the modification.
Modification of NGB in the presence of NO2− and H2O2
Samples of modified NGBS-S and NGBSH proteins were prepared by the addition of sodium nitrite and H2O2 to 60 μM NGBS-S in 50 mM phosphate buffer, pH 7.5, or 60 μM NGBSH in 50 mM Tris/HCl, pH 7.5, and 0.5 mM EDTA. Four different concentrations of sodium nitrite and H2O2 were added to create a variety of reaction conditions as follows: (i) pathophysiological conditions [21] (p-NGBS-S and p-NGBSH) of 0.1 mM NO2− and 0.15 mM H2O2 (divided into five aliquots of 30 μM each); (ii) harsh conditions [h-NGBS-S (modified NGB at high NO2− and H2O2 concentrations) and h-NGBSH] of 0.1 M NO2− and 1.0 mM H2O2 (divided into five aliquots); (iii) modifications without NO2− [p′-NGBSH and h′-NGBSH (modified NGBSH at high H2O2 concentrations)] of 0.15 mM H2O2 (divided into five ali-quots of 30 μM each) and 1.0 mM H2O2 (divided into five aliquots) respectively, and (iv) LPO-catalysed modifications [lpo-NGBS-S (modified NGB in the presence of LPO, NO2− and H2O2) and lpo-NGBSH] of 0.25 M NO2−, 0.15 mM H2O2 (divided into five aliquots) and 80 nM LPO.
The proteins were allowed to react at 20 °C for 10 min. Excess NO2− and oxidant were removed by overnight dialysis against 20 mM phosphate buffer, pH 7.5 (6.8 mM NaH2PO4 and 13.2 mM Na2HPO4) for NGBS-S derivatives, and against degassed 20 mM Tris/HCl, pH 7.5, and 0.2 mM EDTA for NGBSH derivatives.
Analysis of protein fragments and haem modification
For analysis of protein fragments, approx. 0.5 mg of unmodified NGBS-S and NGBSH and the derivatives p-NGBS-S, h-NGBS-S, lpo-NGBS-S, p-NGBSH, h-NGBSH, p′-NGBSH, h′-NGBSH and lpo-NGBSH was transformed into the apo protein using the standard HCl/2-butanone method [31] and was subsequently hydrolysed by trypsin. Digestion was performed using 1:50 (w/w) trypsin at 37 °C for 3 h in 20 mM ammonium bicarbonate solution, pH 8.0. Prior to HPLC-MS/MS analysis, the samples were incubated with 5 mM DTT for 15 min at 37 °C to prevent coupling of the cysteine-containing peptides during digestion. The reduction of the disulfide bonds was necessary for the quantification of unreacted cysteine residues in NGB derivatives.
Haem modification was studied by direct HPLC-MS/MS analysis of NGB derivatives in 20 mM ammonium bicarbonate solution, pH 8.0, after acidification with HCl to pH∼1.
LC (liquid chromatography)-MS and LC-MS/MS data were obtained using an LCQ ADV MAX ion-trap mass spectrometer equipped with an ESI (electrospray ionization) source and controlled by Xcalibur software 1.3 (Thermo Finnigan). ESI experiments were carried out in positive-ion mode under the following constant instrumental conditions: source voltage of 5.0 kV, capillary voltage of 46 V, capillary temperature of 210 °C and tube lens voltage of 55 V. The system was run in automated LC-MS/MS mode using a Surveyor HPLC system (Thermo Finnigan) equipped with a BioBasic™ C18 column (5 μm; 150×2.1 mm). The elution was performed using 0.1% methanoic acid in distilled water (solvent A) and 0.1% methanoic acid in acetonitrile (solvent B), at a flow rate of 0.2 ml/min. Elution started with 98% solvent A for 5 min, followed by a linear gradient from 98 to 55% solvent A in 65 min for the analysis of tryptic digests. A two-step linear gradient was used to analyse the non-digested protein solutions (98 to 50% solvent A in 5 min, followed a gradient from 50 to 20% solvent A in 40 min). MS/MS spectra obtained by CID (collision-induced dissociation) were performed with an isolation width of 2 m/z, the activation amplitude was approx. 35% of the ejection RF (radio frequency) amplitude of the instrument.
For the analysis of protein fragments derived from NGB derivatives, the mass spectrometer was set such that one full MS scan was followed by a zoomscan and a MS/MS scan on the most intense ion observed from the MS spectrum. To identify the modified residues, the acquired MS/MS spectra were automatically searched against a protein database for NGB, using the SEQUEST® algorithm incorporated into Bioworks 3.1 (Thermo Finnigan).
GdnHCl denaturation assay
The denaturation stability of NGBSH, p-NGBSH and h-NGBSH was determined by monitoring the absorbance variation of the Soret band of the protein (approx. 7 μM) after addition of increasing amounts of 8 M GdnHCl solution (up to 4.5 M final concentration) in degassed 50 mM Tris/HCl, pH 6.0, and 0.5 mM EDTA. Data were corrected for dilution caused by the addition of GdnHCl. The GdnHCl concentration at 50% unfolding of the protein was evaluated from the curves of absorbance against denaturant concentration according to a standard method [32].
RESULTS
The two forms of NGB, with and without the disulfide bond
The purification of human NGB provides the protein in its ferric form, since oxyNGB was unstable in vitro and rapidly autoxidizes to metNGB (t1/2=11 min) [7]. This was confirmed by the UV–visible spectrum characteristics of metNGB (Figure 1), as it was clearly distinguishable in the visible region from the spectrum of the FeII–O2 form [for metNGB, λmax=532 nm, with a shoulder at 554 nm (Figure 1); for oxyNGB, λmax=543, 576 nm (results not shown)] [7,12]. The molar absorption coefficient of the Soret band of metNGB in the disulfide-bridged form (metNGBS-S), obtained using the standard pyridine haemochrome assay [33] and used to determine protein concentration, was 1.29×105 M−1·cm−1 at 414 nm (in 200 mM phosphate buffer, pH 7.5). The phosphate buffer used in our experiments is believed to promote the formation of disulfide bonds (Scheme 1) [12]. Actually, the number of accessible thiol groups per haem, as measured by the 4-PDS assay [12,34], was slightly lower than 1.0. This result is consistent with the presence of the single Cys120(G19) in the reduced form and with the almost quantitative oxidation of the other two cysteines to a S–S bond [9]. These disulfide bonds are thought to play a significant role in the function of the protein [9,35].
Figure 1. UV–visible spectra of metNGBS-S and NGBSH.

UV–visible spectra of 6.7 μM metNGBS-S (solid line) and 6.2 μM metNGBSH (dashed line) in 200 mM phosphate buffer, pH 7.5. The wavelength maxima are indicated (arrows).
Treatment of NGBS-S with DTT under anaerobic conditions leads to the partial reduction of the disulfide bridge, as NGBSH was stable to oxidation when kept in degassed 50 mM Tris/HCl, pH 7.5, and 0.5 mM EDTA at 4 °C. Different experimental conditions (temperature and reaction time) were examined to maximize the reduction of the S–S bond, with 2.4 cysteines per haem titrated in the protein observed after reaction with DTT for 30 min at 37 °C. It should be noted that the 4-PDS assay underestimates the number of thiol groups, since the initial fast reduction of 4-PDS to 4-thiopyridone by the SH groups of cysteine residues overlaps with the subsequent slow formation of the latter product by reaction with some reducing species that can be present in solution; therefore, it can be assumed that, in NGBSH, almost all cysteine residues are in the reduced form.
The UV–visible spectra of the ferric forms of NGBS-S and NGBSH are identical (λmax=414 and 532 nm respectively, with a shoulder at 554 nm) and are characteristic of a six-co-ordinated ferric haem (Figure 1). Actually, even if the ratio between the high-spin and low-spin forms increased on formation of the disulfide bridge, the amount of the high-spin form would remain low (<5% at pH 7.5 [36] and <10% at pH 5 [37]). However, the only species detectable in the UV–visible spectra for both NGBS-S and NGBSH was the predominant six-co-ordinated species.
Lack of phenol oxidation by NGB
In the presence of H2O2, haem proteins such as Mb in their ferric form exhibit peroxidase-like activity towards phenolic substrates, oxidizing them to phenoxy radicals, which can give rise to dimer formation in solution [38,39]. The formation of these species can be followed spectrophotometrically through a characteristic absorption at a wavelength of approx. 300 nm. Moreover, a protein–ferryl intermediate (named compound II) can be detected in the reactions of both Mb and peroxidases after addition of the oxidant. This was characterized by a red-shift of the protein Soret band from 408 to 424 nm for Mb [40], 403 to 420 nm for horseradish peroxidase [41] and 412 to 422 nm for LPO [42]. In contrast, no spectral change was observed after mixing of NGB (1 μM) with H2O2 (increasing concentrations up to 100 mM) in the absence or presence of a phenol, such as HPA. Indeed, Herold et al. [19] also reported that, in contrast with Mb and Hb, NGB does not generate the ferryl form of the protein, since its reactivity was strongly limited by the co-ordinated distal histidine residue. In fact, when NGB was reacted with H2O2 and HPA, the UV–visual spectrum features that are characteristic of phenolic dimers do not develop, indicating that the reactivity of NGB with H2O2 was negligible.
Phenol nitration is catalysed by NGBS-S and NGBSH
Surprisingly, NGB activates NO2− in the presence of H2O2, and this results in the nitration of the phenolic substrate HPA into the position o-relative to the hydroxy group. An analogous catalytic activity was reported for peroxidases [21,22,43,44] and the haem proteins Mb [23,24,45,46] and Hb [46]. The investigation of the kinetics of phenol nitration catalysed by NGB (both the NGBS-S and NGBSH forms) was not simple, since the reaction rate depends on the concentration of all the reagents. In order to gain an insight into the mechanism of nitration by the metNGB/NO2−/H2O2 system, it was necessary to investigate the reaction by varying the concentration of the reagents in a large range, far beyond the physiological concentrations likely to be encountered in vivo. Using the approximations and procedures followed previously for enzymatic nitration reactions [22], it was possible to obtain the kinetic parameters kcat (the maximum turnover of the proteins), KMPh-OH (the dissociation constant of HPA from the complex formed with the proteins) and kcat/KMPh-OH (used to calculate the dependence of the rate of reaction at low reagent concentrations on HPA) from the rate dependence on the HPA concentration, through the following equation:
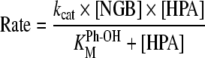 |
(5) |
and kcat, KMnitrite (the dissociation constant of NO2− from the complex formed with the proteins) and kcat/KMnitrite (used to calculate the dependence of the rate of reaction at low reagent concentrations on NO2−) from the rate dependence on NO2− concentration, through the following equation:
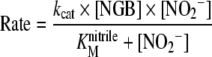 |
(6) |
The data collected are displayed in Table 1, and the corresponding data for the nitration of HPA as catalysed by hMb [24] are also shown.
Table 1. Kinetic data for HPA nitration catalysed by NGB and hMb.
Steady-state kinetic parameters for NGB- and hMb-dependent HPA nitration by NO2−/H2O2 in 200 mM phosphate buffer, pH 7.5, at 25 °C.
| Protein | KMPh-OH (mM) | kcat (s−1) | kcat/KMPh-OH (M−1·s−1) | KMnitrite (M) | kcat (s−1) | kcat/KMnitrite (M−1·s−1) |
|---|---|---|---|---|---|---|
| NGBS-S | 0.078±0.008 | 0.177±0.004 | 2300±200 | 0.043±0.006 | 0.140±0.007 | 3.3±0.3 |
| NGBSH | 0.058±0.008 | 0.132±0.005 | 2300±200 | 0.22±0.03 | 0.139±0.005 | 0.63±0.06 |
| hMb* | 0.12±0.02 | 0.67±0.03 | 5600±900 | 0.24±0.04 | 1.03±0.08 | 4.3±0.5 |
*Data from [24].
It is worth noting that the overall catalytic efficiency of NGB (expressed by kcat, kcat/KMPh-OH and kcat/KMnitrite) was only slightly lower than that of Mb. This was surprising because the reactivity of the protein in the nitration reaction was expected to be negligible, owing to the presence of the six-co-ordinated haem group in NGB, as was observed for the phenol oxidation. Since the fraction of five-co-ordinated high-spin metNGB was less than 5% of total metNGB, its intrinsic reactivity must be very high, much greater than that of Mb. The interpretation of the kinetic parameters is complicated by the presence of the haem–distal histidine residue dissociation equilibrium, since the reported value of k−H (rate constant for histidine residue release) of approx. 1 s−1 [36]) was of the same order of magnitude as the NGB kcat. It follows that the high-spin/low-spin equilibrium is neither slow nor fast, so that, during the kinetic studies, a steady-state equilibrium was not reached in the initial phase and the predominant form of the protein that was present in solution tended to change with time.
The most interesting information emerged from the catalytic activity studied as a function of NO2− concentration, since the disulfide-bridged NGBS-S exhibited a higher affinity for NO2− with respect to the thiol form NGBSH (KMnitrite=43 and 220 mM respectively). For ferrous NGBSH, the distal histidine residue dissociation rate was nearly one order of magnitude smaller than that of NGBS-S, thus leading to an effective decrease in O2 affinity [9]. A similar effect of the S–S bond can be considered for the metNGBs on the iron affinity towards exogenous ligands such as NO2−. Therefore the larger KMnitrite value observed for NGBSH is in agreement with the requirement of NO2− co-ordination to the iron of NGB in order to promote nitrating activity.
The catalytic efficiency at low concentrations of NO2− (i.e. kcat/KMnitrite) was similar for NGBS-S and hMb, since the lower turnover rate of the former is compensated by its higher affinity for NO2−.
Binding of NO2− to NGBS-S and NGBSH
Small but significant changes can be observed in the spectrum of NGBS-S in solution after the addition of excess NO2−. Difference spectra from optical titration clearly show both a smoothing and a slight shift towards higher energy of the Soret band during the first part of the binding process (NO2− concentration <50 mM), and a slight decrease in the intensity of the band in the second part of the titration (NO2− concentration >50 mM). The fitting of the changes in the Soret band as a function of NO2− concentration was consistent with low-affinity binding of two ligands, yielding the constants K1=51±15 M−1 and K2=5±2 M−1 (Figure 2). The former constant probably refers to the electrostatic interaction of an NO2− ion with the charged amino acid residues of the protein, and the latter constant to the co-ordination of an NO2− ion to the iron centre. As NO2− has to replace the distal histidine residue from the sixth co-ordination site, the affinity of NGBS-S for NO2− results approx. one order of magnitude lower with respect to that previously obtained for hMb under the same conditions (KB=76 M−1) [24].
Figure 2. Binding of NO2− to NGBS-S and NGBSH.

The difference between A414 and A434 in the UV–visible spectra of NGBS-S (NgbS-S) and NGBSH (NgbSH) after addition of NO2− compared with the ligand concentration is shown. The absorbance data were fitted with the binding isotherm for low-affinity binding of two ligands for NGBS-S, and a single ligand for NGBSH.
The NO2−-binding experiment performed with NGBSH showed a different behaviour with respect to NGBS-S, since only the initial spectral changes were similar for the two proteins. Indeed, the data could be fitted with a single binding isotherm, giving the association constant KB=48±5 M−1, which was very similar to the K1 value obtained with NGBS-S (Figure 2). By analogy, the process can be associated with the electrostatic interaction of NO2− to NGBSH. It was impossible to obtain a reliable binding constant for the direct co-ordination of NO2− to the haem iron for NGBSH, thus confirming the lower affinity of the thiol form of the protein for exogenous ligands with respect to the disulfide-bridged NGB, as was also observed from the kinetic studies of phenol nitration.
The interaction of NGB with NO2− was also investigated by Herold et al. [19], but, at the low NO2− concentration employed (<1 mM), the binding of NO2− was not observed. Since metNGB was active in promoting the nitration reaction at low concentrations of NO2− (pathophysiological values), a small and hardly detectable fraction of the haem iron must be accessible to the ligand.
Nitration of 4-hydroxybenzonitrile and phenylacetic acid
The substrates phenylacetic acid and 4-hydroxybenzonitrile were used as mechanistic probes for the nitration reaction catalysed by NGB/NO2−/H2O2. The reaction could proceed through the formation of the same nitrating species involved in the reactions promoted by peroxidases and Mb, i.e. nitrogen dioxide (NO2) or ONOO−, depending on the NO2− concentration [22–24]. Phenylacetic acid reacts with ONOO−, generating nitrated and/or hydroxylated compounds, but it is unreactive to NO2 and thus it is a good probe for ONOO−. The reactivity of 4-hydroxybenzonitrile is different, since it reacts with NO2 to generate 4-hydroxy-3-nitrobenzonitrile, whereas ONOO− is a poor nitrating agent for this substrate [22] (Scheme 2).
Scheme 2. Reactivity of 4-hydroxybenzonitrile and phenylacetic acid with RNS.

Schematic representation of the reactivity of 4-hydroxybenzonitrile and phenylacetic acid with the nitrating agents NO2 and ONOO−, and the catalytic system metNGB/NO2−/H2O2.
4-Hydroxybenzonitrile was reacted with NGB and H2O2 at different NO2− concentrations (10–150 mM) and the reaction mixtures were analysed by HPLC. The nitrated product was absent under all of the conditions tested, indicating that NO2 was not formed, or was formed in negligible amounts, by NGB under these conditions. In contrast, NGB/NO2−/H2O2 induced the modification of phenylacetic acid, and by HPLC it was possible to separate four different products, regardless of the NO2− concentration used. These derivatives shared spectral features and retention times with the products obtained in the reaction of phenylacetic acid with ONOO− [22]. These results suggest that the nitration catalysed by NGB in the presence of NO2− and H2O2 proceeds through the formation of a reactive species with the chemical properties of ONOO− (Scheme 2).
Tandem MS/MS analysis of NGBS-S and NGBSH modified by H2O2 and NO2−
To investigate the endogenous modifications undergone by NGB in the presence of NO2− and H2O2, NGB (both in the NGBS-S and NGBSH forms) was reacted with varying quantities of NO2− and H2O2 in the absence of an external substrate. Depending on the concentration of the reagents used, a series of protein derivatives was prepared under very mild conditions (similar to those that can occur under pathophysiological conditions [21]), and NGBS-S and NGBSH were reacted with 0.1 mM NO2− and 0.15 mM H2O2, obtaining p-NGBS-S and p-NGBSH respectively. Using less physiological and more harsh conditions, NGBS-S and NGBSH were reacted with 100 mM NO2− and 1 mM H2O2, obtaining h-NGBS-S and h-NGBSH respectively. The reactivity of the cysteine residues in the presence of H2O2 alone was studied by reacting NGBSH with H2O2 at low (0.15 mM) and high (1 mM) oxidant concentrations, obtaining p′-NGBSH and h′-NGBSH respectively.
Moreover, generation of the NGB derivatives by NO2− and H2O2 was performed in the presence of LPO, as an external source of the nitrating agent. The protein derivatives obtained under these conditions were named as lpo-NGBS-S and lpo-NGBSH.
The UV–visible spectra of the p-NGB derivatives were indistinguishable from that of native metNGB, whereas the spectra for the h-NGB and lpo-NGB derivatives both exhibited a slight broadening of the Soret band and the appearance of a shoulder at approx. 440–450 nm was also observed. These spectral features were associated with haem nitration and reflected the colour change of the protein solutions from brown to greenish-brown, which accompanied the formation of h-NGBS-S, h-NGBSH, lpo-NGBS-S and lpo-NGBSH derivatives [24]. All of the protein derivatives were analysed by HPLC-ESI-MS/MS, with the aim of investigating the endogenous modifications and the results are shown in Tables 2 and 3. The potential targets of the oxidizing and nitrating species are, besides the haem prosthetic group, the protein tyrosine residues Tyr44(CD3), Tyr88 (F3), Tyr115(G14) and Tyr137(H12), and the cysteine residues Cys46(CD7), Cys55(D5) and Cys120(G19) in NGBSH, with Cys120(G19) being the only cysteine residue target in NGBS-S (Figure 3).
Table 2. Haem and tyrosine residue modifications in NGB derivatives.
Nitration of haem and tyrosine residues in NGB derivatives obtained under the following conditions: for p-NGBs, [NGB]=60 μM, [NO2−]=0.1 mM and [H2O2]=0.15 mM; for h-NGBs, [NGB]=60 μM, [NO2−]=0.1 M and [H2O2]=1 mM; for lpo-NGBs, [LPO]=80 nM, [NGB]=60 μM, [NO2]=0.25 M and [H2O2]=0.15 mM. All reactions were performed in 200 mM phosphate buffer, pH 7.5.
| Nitration (%) | |||||
|---|---|---|---|---|---|
| Protein derivatives | Haem-NO2 | Tyr44-NO2 | Tyr88-NO2 | Tyr115-NO2 | Tyr137-NO2 |
| p-NGBS-S | 0 | 0 | 0.6 | 0 | 0 |
| p-NGBSH | 0 | 0 | 1 | 0 | 0 |
| h-NGBS-S | 38 | 50 | 11 | 65 | 38 |
| h-NGBSH | 14 | 11 | 7 | 36 | 14 |
| lpo-NGBS-S | 25 | 5 | 1.3 | 3.5 | 15 |
| lpo-NGBSH | 27 | 2.3 | 0.9 | 7 | 11 |
Table 3. Modification of cysteine residues in NGB derivatives.
Oxidation of cysteine residues in NGB derivatives obtained by protein modification under the following conditions: for p-NGBs, [NGB]=60 μM, [NO2−]=0.1 mM and [H2O2]=0.15 mM; for h-NGBs, [NGB]=60 μM, [NO2−]=0.1 M and [H2O2]=1 mM; for lpo-NGBs, [LPO]=80 nM, [NGB]=60 μM, [NO2−]=0.25 M and [H2O2]=0.15 mM; for p′-NGBSH, [NGB]=60 μM and [H2O2]=0.15 mM; for h′-NGBs, [NGB]=60 μM and [H2O2]=1 mM. All reactions were performed in 200 mM phosphate buffer, pH 7.5.
| Oxidation (%) | ||||||
|---|---|---|---|---|---|---|
| Protein derivatives | Cys46-SO2H | Cys46-SO3H | Cys55-SO2H | Cys55-SO3H | Cys120-SO2H | Cys120-SO3H |
| NGBS-S | * | * | * | * | 1.7 | 1.5 |
| NGBSH | 0.3 | 0.9 | 0.6 | 1.7 | 2 | 2 |
| p-NGBS-S | * | * | * | * | 2 | 2 |
| p-NGBSH | 2.3 | 4.5 | 2.5 | 7 | 10 | 9 |
| p′-NGBSH | 1 | 2 | 0.5 | 2 | 2 | 1.5 |
| h-NGBS-S | * | * | * | * | 5 | 8 |
| h-NGBSH | 21 | 14 | 4 | 14 | 18 | 16 |
| h′-NGBSH | 0.9 | 4 | 0.7 | 4 | 5 | 6 |
| lpo-NGBS-S | * | * | * | * | 6 | 7 |
| lpo-NGBSH | 22 | 8 | 3 | 7 | 10 | 5 |
*Cys46 and Cys55 are involved in the S–S bond.
Figure 3. Structure of NGB.

Structure of the C46G/C55S/C120S mutant of NGB. The atomic co-ordinates were obtained from [10] and the Protein Data Bank (accession number 1OJ6). The disposition of the side chains of the tyrosine residues (Tyr44, Tyr88, Tyr115 and Tyr137), and the cysteine residues (Cys46, Cys55, and Cys120) present in the wild-type protein are shown.
The modification of the prosthetic group can be detected by direct HPLC-MS/MS analysis of the acidified protein solution, since the haem group was released from the protein. The modified haemin was detected only in the NGB derivatives obtained after reaction with high levels of NO2− and H2O2, i.e. h-NGBS-S and h-NGBSH. In particular, ions with m/z 616 and 661 were identified, corresponding to free haemin and haemin modified with a NO2 group [23,24] respectively. The modification probably occurs at one of the haem vinyl groups, in analogy with the haem nitration observed after treatment of Mb with a large excess of NO2− at pH 5.5 [47]. Integration of the peaks in the EIC (extracted ion current) chromatograms indicated that haemin nitration occurred with a 38% yield in h-NGBS-S, a 14% yield in h-NGBSH, a 25% yield in lpo-NGBS-S and a 27% yield in lpo-NGBSH (Table 2).
The modifications at the cysteine and tyrosine residues were characterized on polypeptide fragments resulting from tryptic digestion of the apoNGB derivatives. The HPLC-ESI-MS/MS analysis showed the presence of many modified peptide fragments (for the molecular masses and m/z values as bicharged ions, see Supplementary Table S1 at http://www.BiochemJ.org/bj/407/bj4070089add.htm). The analysis of the MS/MS spectra using the SEQUEST® algorithm allowed the assignment of the modifications to the nitration of the tyrosine residues to 3-nitrotyrosine [23] and the oxidation of the cysteine residues to sulfinic (R-SO2H) and sulfonic (R-SO3H) acids. As an example of the MS results, the MS/MS spectrum of the peptide from amino acid residue 68 to 94, obtained by the analysis of h-NGBS-S (nitrated at Tyr88), is shown in Figure 4.
Figure 4. CID spectrum of the modified amino acid 68–94 peptide.

MS/MS spectrum of the m/z 1477.6 peak (mass of 2953.3 Da) assigned to the amino acid 68–94 peptide in a double-charged state and with Tyr88 modified to 3-nitrotyrosine. The assignment of the y and b ion series is shown. Above the spectrum, the sequence of the 68–94 peptide, with the modified residue in bold, and the summary of the y and b ions found in the spectrum is shown.
The percentage of residue modifications is shown in Tables 2 and 3, and was obtained by comparing the areas of the peaks in the EIC chromatograms that corresponded to the derivatized peptides with those of the corresponding peptides in the starting proteins. The greater efficiency of NGBS-S, in promoting self-nitration of both the haem group and the tyrosine residues by NO2− and H2O2, compared with NGBSH was clearly apparent from the amount of endogenous modifications observed for h-NGBS-S and h-NGBSH (Table 2). This is in agreement with the kinetic parameters for phenol nitration (Table 1), since NGBS-S exhibits a significantly higher catalytic efficiency in terms of kcat/KMnitrite compared with NGBSH.
A complete picture of the cysteine residue derivative formation is provided by the modification of NGBSH under different conditions, resulting in the formation of the h-, p-, h′- and p′-NGBSH derivatives (Table 3). All of the cysteine residues were oxidized to sulfinic and sulfonic acids and the amount of modification induced by NO2− and H2O2 increased with the concentration of the reagents (compare p-NGBSH with h-NGBSH), and was significantly lower in the absence of NO2− (h′-NGBSH compared with p′-NGBSH), indicating that the NGB/NO2−/H2O2 system generates distinctive oxidant species.
The mechanism of protein modification was explored by inducing the formation of the NGB derivatives in the presence of an external catalyst. The enzyme LPO was chosen, since it reacts with H2O2 and NO2− to generate nitrating species with a rate two orders of magnitude larger when compared with NGB [22]. Thus when NGB reacted with NO2− at low H2O2 concentration and in the presence of LPO, the reactive species were generated by the enzyme, making NGB self-promoted derivative formation negligible. This was confirmed by the similarity in the extent of nitration (both at the haem group and the tyrosine residues) obtained for the LPO-promoted modification of NGBS-S and NGBSH (Table 2).
From the analysis of the derivatives lpo-NGBS-S and lpo-NGBSH, it emerged that the IPO-promoted NGB tyrosine residue nitration and cysteine residue oxidation occurred to a lower extent compared with the modifications which are self-promoted by NGB. In contrast, the haem group was the favoured modification site for both lpo-NGBs (Tables 2 and 3). These results indicated that the prosthetic group was the most accessible target for the LPO-generated reactive species, whereas the NGB-generated reactive species exhibited a preference for intramolecular, rather than intermolecular, reactivity.
GdnHCl denaturation assay
The unfolding midpoint [D0] in the presence of GdnHCl for NGBSH, p-NGBSH and h-NGBSH was estimated to be in the range of 4.1–4.2 M, as obtained from the curves of absorbance against denaturant concentration. These values can be compared with those obtained under the same conditions for sperm whale Mb ([D0]=1.46 M [32]), and horse heart Mb ([D0]=1.32 M) (E. Monzani, S. Nicolis, R. Roncone, M. Barbieri, A. Granata and L. Casella, unpublished work). Therefore NGB exhibited a considerably higher stability to GdnHCl than Mbs. Moreover, the derivatization of the protein residues (nitration of tyrosine residues and oxidation of cysteine residues) in the h-NGBSH derivative had very little effect on the stability of the protein. Treatment of NGB with pathophysiological NO2− and H2O2 concentrations, yielding p-NGBSH, induced completely negligible effects on protein stability to GdnHCl denaturation. A similar negligible effect towards denaturation was observed for horse heart Mb after nitration of tyrosine residues in the presence of NO2− and H2O2 ([D0]=1.35 M compared with 1.32 M for native Mb). A more detailed characterization of the unfolding properties of NGB and its derivatives will be carried out separately.
DISCUSSION
In vivo NGB species
Since NGB possesses a bis-histidine six-co-ordinated haem group, the reactivity of the protein towards exogenous ligands is regulated by the dissociation rate of the distal histidine residue. Nevertheless, NGB in its ferrous state has significant reactivity towards small ligands, such as NO, O2 and CO (carbon monoxide). The competition between these external molecules and the histidine residue for the sixth co-ordination site of the FeII centre emerges from the kinetic and thermodynamic constants previously reported [3,4,48]. It follows that, in spite of the high intrinsic affinity of NGB for both O2 and CO, the binding of these ligands is suggested to be slow in vivo [7]. Furthermore, the influence of the internal disulfide bridge on the affinity of ferrous NGBS-S for molecular O2 has been clearly demonstrated [9].
In the reducing conditions that exist within living cells, NGB would be present in the ferrous NGBSH form, saturated with O2 at approx. 12% [12]. Although the ferric form studied in the present paper may not be the most physiologically relevant form of the protein, the presence of H2O2 and NO2− would rapidly convert ferrous or oxyNGB into the ferric form. Furthermore, metNGB is the product of rapid NO scavenging by oxyNGB, so, in the absence of a metNGB reductase, a system would accumulate metNGB (according to reaction 1) [15].
MetNGB promoted nitration of phenolic compounds
Currently, the only data concerning the behaviour of metNGB with H2O2 indicated an absence of reactivity [19]. The results reported in the present study show an interesting pseudo-enzymatic activity of metNGB in the presence of H2O2 and NO2−. Even if the mechanism of the NGB reaction could be considered to be similar to that of peroxidases, this is in contrast with the absence of activity of metNGB in the oxidation of phenolic compounds observed in the presence of H2O2 alone. In fact, under these conditions, metNGB does not generate the ferryl species compounds I and II, which are the characteristic intermediates of the catalytic cycle of peroxidases. Nevertheless, in analogy with peroxidases, metNGB promotes the nitration of phenolic substrates by activation of NO2− and H2O2. This result is not in contradiction with the absence of the reaction observed between NGB and H2O2, since, besides the peroxidase-like mechanism of nitration through NO2, an alternative mechanism for the nitration of phenolic substrates emerged in our previous studies on peroxidases and Mb in the presence of NO2− and H2O2 [22,23]. This mechanism involved the initial binding of NO2− to the haem iron, followed by the reaction of H2O2 with the co-ordinated NO2−, to produce an iron-bound ONOO− [NGBFeIII-N(O)OO], which acts as the nitrating active species. In the case of peroxidases and Mbs, the ONOO− pathway becomes important only at relatively high NO2− concentrations [22–24], whereas with NGB it is demonstrated that this is the only mechanism active in the nitration of exogenous and endogenous phenolic groups (i.e. tyrosine residues) even in the pathophysiological concentration range of NO2− and H2O2 (Scheme 3).
Scheme 3. Mechanism of metNGB promoted nitration of phenolic compounds.

Schematic representation of the activation of NO2− and H2O2 by the high-spin metNGBs (metNGBS-S or metNGBSH) through the ONOO− pathway, involving the initial binding of NO2− to the haem iron, followed by the reaction with H2O2, to produce an iron-bound ONOO− (NGBFeIII-N(O)OO) nitrating active species. Nitration of phenol occurs at the o-position relative to its hydroxy group. The upper part of the scheme shows the absence of reactivity of metNGB with H2O2.
The ONOO− pathway
The kinetic parameters for HPA nitration reported in Table 1 show the importance of NO2− in discriminating the reactivity of metNGBS-S and metNGBSH. As expected from the influence of the internal S–S bond on the binding rate of exogenous ligands to the iron centre, the kinetic parameters for the two forms of the protein and their dependence on NO2− concentration are significantly different, supporting the hypothesis of direct co-ordination of NO2− to the FeIII centre to promote catalysis. In particular, metNGBS-S exhibits larger values, both for NO2− affinity [extrapolated from kinetic data (1/KMnitrite)] and for reactivity at low NO2− concentrations (kcat/KMnitrite) with respect to metNGBSH, indicating that the interaction of NO2− with the catalytic site regulates the efficiency of nitration at low concentrations of the reagent, in conditions that can occur in vivo. The higher affinity of NO2− for metNGBS-S compared with metNGBSH was confirmed by NO2−-binding studies (Figure 2).
The ONOO− pathway proposed for the nitrating activity of metNGB was examined by the observation of the reaction with 4-hydroxybenzonitrile and phenylacetic acid, which are mechanistic probes of NO2 and ONOO− respectively. The lack of reactivity of hydroxybenzonitrile on one hand, and the presence of several products deriving from nitration and/or hydroxylation of phenylacetic acid on the other hand, is in agreement with the formation of an active species with the chemical properties of ONOO− by metNGB (Scheme 2). It follows that the formation of a protein-bound ONOO− intermediate is a reasonable step for nitration catalysed by metNGB in the presence of NO2− and H2O2, even at rather low concentrations of the reagents.
NGB endogenous modifications
The generation and activity of the oxidant species formed by metNGB was examined by identification of the pattern of protein modifications obtained using LPO as an external catalyst, in comparison with metNGB-promoted endogenous modifications (Tables 2 and 3). At the high NO2− concentration (250 mM) used for the preparation of lpo-NGBS-S and lpo-NGBSH derivatives, LPO-promoted nitration proceeded through the formation of the ONOO− active species [22]. The differences in the pattern of residues modified by the LPO-generated ONOO− in comparison with those self-induced by NGB indicated that, in the latter case, ONOO− reacts intramolecularly. Moreover, the ONOO− reactive species are probably responsible for the oxidation of cysteine residues to form the corresponding sulfinic and sulfonic acids. Not only can it promote this type of modifications, as observed in the oxidations of BSA and hMb [24,49], but the high reactivity of this species also accounts for the lower extent of derivatization observed in the h′-NGBSH and p′-NGBSH derivatives (obtained by reaction with H2O2 only) in comparison with the h-NGBSH and p-NGBSH derivatives respectively. The less physiological conditions used in the preparation of the h-NGBSH and h-NGBS-S derivatives, which forced the extent of endogenous modifications, allowed the investigation of the competitive reactivity of the protein residues. Both the presence of three reactive cysteine residues (which may scavenge RNS) in NGBSH, and its poorer catalytic efficiency in the nitration of phenolic compounds compared with NGBS-S, may account for the lower degree of tyrosine residue nitration in h-NGBSH.
From the point of view of the physiological relevance of NGB endogenous modifications, the analysis of p-NGBSH was of particular interest, since this form was obtained from NGBSH (presumed to be the predominant form in vivo [12]) with low concentrations of NO2− and H2O2. Among the protein residues, cysteine residues had the highest reactivity towards the ONOO− active species, and their reaction prevented nitration of tyrosine residues and the haem prosthetic group. It is possible to calculate that approx. 36% of H2O2-oxidizing equivalents introduced were used for oxidation of cysteine residues to sulfinic and sulfonic acids. Oxidation of cysteine residues to sulfinic and sulfonic acid derivatives has been observed [50,51], together with other types of oxidations that can be reversed by reaction with biological thiols [52]. It is worth noting that, among the tyrosine residues, the only one that undergoes nitration in both p-NGBS-S and p-NGBSH is Tyr88, the phenolic hydroxy group of which points toward the large NGB cavity connected to the haem distal site [10]. This may indicate that the ONOO− reactive species endogenously generated at the haem distal site can easily diffuse through the cavity and nitrate Tyr88.
Does human NGB act only as a scavenger?
In the globins, cysteine residues often play specific roles, and this is modulated by the formation of intramolecular or intermolecular disulfide bonds [9]. In the case of NGB, the oxidation state of Cys46 and Cys55 is relevant because it is linked to the peculiar six-co-ordination of the haem. The present study confirms the importance of the internal S–S bond in controlling the reactivity of metNGB towards NO2− and H2O2. It also shows that the protein can be involved in other activities. NGB is thought to act as a scavenger of toxic species generated in vivo under conditions of oxidative stress. But the ability of the metNGB form generated by this activity to activate NO2− and H2O2 means that, besides acting as a scavenger, NGB can be the source of RNS, thus affecting its physiological activity. A significant fraction of the nitrating and oxidizing species (approx. 36% in the conditions used to prepare p-NGBSH) does not escape from the protein, but is consumed in the self-modification of NGB. The easy oxidation of cysteine residues to sulfinic and sulfonic acids may be one way used by NGB to scavenge part of the generated RNS. To gain a better understanding of the physiological relevance of these reactions, it will be important to analyse the state of endogenous modification of samples of the protein isolated after in vivo exposure to oxidative stress conditions.
Online data
Acknowledgments
This work was supported by funds from PRIN (Progetto di Ricerca di Interesse Nazionale) and FIRB (Fondo per gli Investimenti della Ricerca di Base) projects of the Italian MIUR (Ministero dell'Istruzione, dell'Università e della Ricerca Scientifica). The University of Pavia, the European COST D21 Action and CIRCMSB (Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici) are also gratefully acknowledged for support.
References
- 1.Burmester T., Weich B., Reinhardt S., Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt M., Giessl A., Laufs T., Hankeln T., Wolfrum U., Burmester T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J. Biol. Chem. 2003;278:1932–1935. doi: 10.1074/jbc.M209909200. [DOI] [PubMed] [Google Scholar]
- 3.Trent J. T., III, Watts R. A., Hargrove M. S. Human neuroglobin, a hexacoordinate hemoglobin that reversibly binds oxygen. J. Biol. Chem. 2001;276:30106–30110. doi: 10.1074/jbc.C100300200. [DOI] [PubMed] [Google Scholar]
- 4.Uno T., Ryu D., Tsutsumi H., Tomisugi Y., Ishikawa Y., Wilkinson A. J., Sato H., Hayashi T. Residues in the distal heme pocket of neuroglobin. Implications for the multiple ligand binding steps. J. Biol. Chem. 2004;279:5886–5893. doi: 10.1074/jbc.M311748200. [DOI] [PubMed] [Google Scholar]
- 5.Hankeln T., Ebner B., Fuchs C., Gerlach F., Haberkamp M., Laufs T. L., Roesner A., Schmidt M., Weich B., Wystub S., et al. Neuroglobin and cytoglobin in search of their role in the vertebrate globin family. J. Inorg. Biochem. 2005;99:110–119. doi: 10.1016/j.jinorgbio.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Garry D. J., Mammen P. P. A. Neuroprotection and the role of neuroglobin. Lancet. 2003;362:342–343. doi: 10.1016/S0140-6736(03)14055-X. [DOI] [PubMed] [Google Scholar]
- 7.Dewilde S., Kiger L., Burmester T., Hankeln T., Baudin-Creuza V., Aerts T., Marden M. C., Caubergs R., Moens L. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem. 2001;276:38949–38955. doi: 10.1074/jbc.M106438200. [DOI] [PubMed] [Google Scholar]
- 8.Hamdane D., Kiger L., Dewilde S., Green B. N., Pesce A., Uzan J., Burmester T., Hankeln T., Bolognesi M., Moens L., Marden M. C. Coupling of the heme and an internal disulfide bond in human neuroglobin. Micron. 2004;35:59–62. doi: 10.1016/j.micron.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Hamdane D., Kiger L., Dewilde S., Green B. N., Pesce A., Uzan J., Burmester T., Hankeln T., Bolognesi M., Moens L., Marden M. C. The redox state of the cell regulates the ligand binding affinity of human neuroglobin and cytoglobin. J. Biol. Chem. 2003;278:51713–51721. doi: 10.1074/jbc.M309396200. [DOI] [PubMed] [Google Scholar]
- 10.Pesce A., Dewilde S., Nardini M., Moens L., Ascenzi P., Hankeln T., Burmester T., Bolognesi M. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure. 2003;11:1087–1095. doi: 10.1016/s0969-2126(03)00166-7. [DOI] [PubMed] [Google Scholar]
- 11.Smagghe B. J., Sarath G., Ross E., Hilbert J-L., Hargrove M. S. Slow ligand binding kinetics dominate ferrous hexacoordinate hemoglobin reactivities and reveal differences between plants and other species. Biochemistry. 2006;45:561–570. doi: 10.1021/bi051902l. [DOI] [PubMed] [Google Scholar]
- 12.Fago A., Hundahl C., Dewilde S., Gilany K., Moens L., Weber R. E. Allosteric regulation and temperature dependence of oxygen binding in human neuroglobin and cytoglobin. Molecular mechanisms and physiological significance. J. Biol. Chem. 2004;279:44417–44426. doi: 10.1074/jbc.M407126200. [DOI] [PubMed] [Google Scholar]
- 13.Pesce A., Dewilde S., Nardini M., Moens L., Ascenzi P., Hankeln T., Burmester T., Bolognesi M. The human brain hexacoordinated neuroglobin three-dimensional structure. Micron. 2004;35:63–65. doi: 10.1016/j.micron.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Vallone B., Nienhaus K., Matthes A., Brunori M., Nienhaus G. U. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand affinity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:17351–17356. doi: 10.1073/pnas.0407633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunori M., Giuffrè A., Nienhaus K., Nienhaus G. U., Scandurra F. M., Vallone B. Neuroglobin, nitric oxide, and oxygen: functional pathways and conformational changes. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8483–8488. doi: 10.1073/pnas.0408766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y., Jin K., Peel A., Mao X. O., Xie L., Greenberg D. A. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3497–3500. doi: 10.1073/pnas.0637726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y., Jin K., Mao X. O., Zhu Y., Greenberg D. A. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc. Natl. Acad. Sci. U.S.A. 2001;98:15306–15311. doi: 10.1073/pnas.251466698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 19.Herold S., Fago A., Weber R. E., Dewilde S., Moens L. Reactivity studies of the FeIII and FeIINO forms of human neuroglobin reveal a potential role against oxidative stress. J. Biol. Chem. 2004;279:22841–22847. doi: 10.1074/jbc.M313732200. [DOI] [PubMed] [Google Scholar]
- 20.Herold S., Fago A. Reactions of peroxynitrite with globin proteins and their possible physiological role. Comp. Biochem. Physiol. A: Mol. Integr. Physiol. 2005;142:124–129. doi: 10.1016/j.cbpb.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Van der Vliet A., Eiserich J. P., Halliwell B., Cross C. E. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J. Biol. Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 22.Monzani E., Roncone R., Casella L., Galliano M., Koppenol W. H. Mechanistic insight into the peroxidase catalyzed nitration of tyrosine derivatives by nitrite and hydrogen peroxide. Eur. J. Biochem. 2004;271:895–906. doi: 10.1111/j.1432-1033.2004.03992.x. [DOI] [PubMed] [Google Scholar]
- 23.Nicolis S., Monzani E., Roncone R., Gianelli L., Casella L. Metmyoglobin-catalyzed exogenous and endogenous tyrosine nitration by nitrite and hydrogen peroxide. Chem. Eur. J. 2004;10:2281–2290. doi: 10.1002/chem.200304989. [DOI] [PubMed] [Google Scholar]
- 24.Nicolis S., Pennati A., Perani E., Monzani E., Sanangelantoni A. M., Casella L. Easy oxidation and nitration of human myoglobin by nitrite and hydrogen peroxide. Chem. Eur. J. 2006;12:749–757. doi: 10.1002/chem.200500361. [DOI] [PubMed] [Google Scholar]
- 25.Sacksteder C. A., Qian W-J., Knyushko T. V., Wang H., Chin M. H., Lacan G., Melega W. P., Camp D. G., II, Smith R. D., Smith D. J., et al. Endogenously nitrated proteins in mouse brain: links to neurodegenerative disease. Biochemistry. 2006;45:8009–8022. doi: 10.1021/bi060474w. [DOI] [PubMed] [Google Scholar]
- 26.Turko I. V., Murad F. Protein nitration in cardiovascular diseases. Pharmacol. Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 27.Hunt J., Byrns R. E., Ignarro L. J., Gaston B. Condensed expirate nitrite as a home marker for acute asthma. Lancet. 1995;346:1235–1236. doi: 10.1016/s0140-6736(95)92947-9. [DOI] [PubMed] [Google Scholar]
- 28.Torre D., Ferrario G., Speranza F., Orani A., Fiori G. P., Zeroli C. Serum concentrations of nitrite in patients with HIV-1 infection. J. Clin. Pathol. 1996;49:574–576. doi: 10.1136/jcp.49.7.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pryor W. A., Squadrito G. L. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 30.Casella L., De Gioia L., Frontoso Silvestri G., Monzani E., Redaelli C., Roncone R., Santagostini L. Covalently modified microperoxidases as heme-peptide models for peroxidases. J. Inorg. Biochem. 2000;79:31–39. doi: 10.1016/s0162-0134(99)00243-3. [DOI] [PubMed] [Google Scholar]
- 31.Antonini E., Brunori M. Amsterdam: North-Holland Publishing Company; 1971. Hemoglobin and Myoglobin in Their Reactions with Ligands. [Google Scholar]
- 32.Roncone R., Monzani E., Labò S., Sanangelantoni A. M., Casella L. Catalytic activity, stability, unfolding, and degradation pathways of engineered and reconstituted myoglobins. J. Biol. Inorg. Chem. 2005;10:11–24. doi: 10.1007/s00775-004-0606-4. [DOI] [PubMed] [Google Scholar]
- 33.Fuhrhop J. H., Smith K. M. Amsterdam: Elsevier; 1975. Laboratory Methods In Porphyrin And Metalloporphyrin Research. [Google Scholar]
- 34.Grassetti D. R., Murray J. F. Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Arch. Biochem. Biophys. 1967;119:41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 35.Hogg P. J. Disulfide bonds as switches for protein function. Trends Biochem. Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 36.Du W., Syvitski R., Dewilde S., Moens L., La Mar G. N. Solution 1H NMR characterization of equilibrium heme orientational disorder with functional consequences in mouse neuroglobin. J. Am. Chem. Soc. 2003;125:8080–8081. doi: 10.1021/ja034584r. [DOI] [PubMed] [Google Scholar]
- 37.Nistor S. V., Goovaerts E., Van Doorslaer S., Dewilde S., Moens L. EPR-spectroscopic evidence of a dominant His-FeIII-His coordination in ferric neuroglobin. Chem. Phys. Lett. 2002;361:355–361. [Google Scholar]
- 38.Watanabe Y., Ueno T. Introduction of P450, peroxidase, and catalase activities into myoglobin by site-directed mutagenesis: diverse reactivities of compound I. Bull. Chem. Soc. Jpn. 2003;76:1309–1322. [Google Scholar]
- 39.Roncone R., Monzani E., Nicolis S., Casella L. Engineering and prosthetic group modification of myoglobin: peroxidase activity, chemical stability and unfolding properties. Eur. J. Inorg. Chem. 2004:2203–2213. [Google Scholar]
- 40.Hayashi T., Hitomi Y., Ando T., Mizutani T., Hisaeda Y., Kitagawa S., Ogoshi H. Peroxidase activity of myoglobin is enhanced by chemical mutation of heme-propionates. J. Am. Chem. Soc. 1999;121:7747–7750. [Google Scholar]
- 41.Dunford H. B. New York: Wiley-VCH; 1999. Heme Peroxidases. [Google Scholar]
- 42.Monzani E., Gatti A. L., Profumo A., Casella L., Gullotti M. Oxidation of phenolic compounds by lactoperoxidase. Evidence for the presence of a low-potential compound II during catalytic turnover. Biochemistry. 1997;36:1918–1926. doi: 10.1021/bi961868y. [DOI] [PubMed] [Google Scholar]
- 43.Sampson J. B., Ye Y. Z., Rosen H., Beckman J. S. Myeloperoxidase and horseradish peroxidase catalyze tyrosine nitration in proteins from nitrite and hydrogen peroxide. Arch. Biochem. Biophys. 1998;356:207–213. doi: 10.1006/abbi.1998.0772. [DOI] [PubMed] [Google Scholar]
- 44.Eiserich J. P., Hristova M., Cross C. E., Jones A. D., Freeman B. A., Halliwell B., van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 45.Herold S., Rehmann F. J. Kinetic and mechanistic studies of the reactions of nitrogen monoxide and nitrite with ferryl myoglobin. J. Biol. Inorg. Chem. 2001;6:543–555. doi: 10.1007/s007750100231. [DOI] [PubMed] [Google Scholar]
- 46.Casella L., Monzani E., Roncone R., Nicolis S., Sala A., De Riso A. Formation of reactive nitrogen species at biologic heme centers: a potential mechanism of nitric oxide-dependent toxicity. Environ. Health Perspect. 2002;110:709–711. doi: 10.1289/ehp.02110s5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bondoc L. L., Timkovich R. Structural characterization of nitrimyoglobin. J. Biol. Chem. 1989;264:6134–6145. [PubMed] [Google Scholar]
- 48.Fago A., Mathews A. J., Dewilde S., Moens L., Brittain T. The reactions of neuroglobin with CO: evidence for two forms of the ferrous protein. J. Inorg. Biochem. 2006;100:1339–1343. doi: 10.1016/j.jinorgbio.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 49.Radi R., Beckman J. S., Bush K. M., Freeman B. A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 50.Hamann M., Zhang T., Hendrich S., Thomas J. A. Quantitation of protein sulfinic and sulfonic acid, irreversibly oxidized protein cysteine sites in cellular proteins. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]
- 51.Woo H. A., Jeong W., Chang T.-S., Park K. J., Park S. J., Yang J. S., Rhee S. G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-Cys peroxiredoxins. J. Biol. Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 52.Jacob C., Holme A. L., Fry F. H. The sulfinic acid switch in proteins. Org. Biomol. Chem. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.