

Abstract
Mitochondria is where the bulk of the cell’s ATP is produced. Mutations occur to genes coding for members of the complexes involved in energy production. Some are a result of damages to nuclear coded genes and others to mitochondrial coded genes. This review describes approaches to bring small molecules, proteins and RNA/DNA into mitochondria. The purpose is to repair damaged genes as well as to interrupt mitochondrial function including energy production, oxygen radical formation and the apoptotic pathway.
Keywords: Mitochondrial DNA, Mitochondrial disease, Translocators, Protein and RNA import, Membrane insertion, Lipophilic cations
1. Introduction
Before embarking on a discussion of macromolecule or drug delivery to mitochondria the uniqueness of the organelle must be considered. Mitochondria are different from other subcellular organelles with respect to their complex two membrane structure. It is hypothesized that mitochondria were originated by endosymbiosis. Research on mitochondrial DNA supports that mitochondria have similarity with α-proteobacteria [1]. The double membrane structure most likely evolved to help preserve some of the essential functions of the organelle. Perhaps the most important function of mitochondria is the syntheses of ATP from ADP and phosphate where the free energy required to perform the reaction (?G0 = + 7.3 Kcal/mole) is derived from energy released during the transfer of electrons through the electron transport system to oxygen. For ATP synthesis to occur, it is necessary that the mitochondria maintain an acidic inner space and an electrochemical potential across the inner membrane. Thus, it is essential that the organelle have the ability to selectively control the permeation of chemicals entering into it. If chemicals could enter freely, then these two features could be destroyed.
During the past few years a number of outstanding review articles have been published that discussed various ways to bring drugs or chemicals into mitochondria [2–6]. This review.article will summarize various unique features of mitochondria and then review how others suggested that those might be exploited to permit compounds to enter it. This article will focus primarily on ways of getting protein and DNA into mitochondria since small molecule entry has been well reviewed by others.
2. Structural features of mitochondria
2.1. Double membrane and matrix space
Mitochondria are composed of a double membrane. Like all membranes, these two are primarily composed of phospholipid bilayers with proteins embedded in them. Since there are two membranes, two aqueous spaces are defined. The inner most is the matrix space while the one between the membranes is simply called the intermembrane space. The outer membrane, like the plasma membrane, has a lipid to protein ratio of 1:1. The outer membrane does not offer a barrier to small molecules. These can simply diffuse through pores in the membrane formed by a membrane spanning protein call porin. Large molecules such as proteins cross the outer membrane by using a unique protein import apparatus. ATP synthesis occurs in the matrix space so it is the inner membrane that provides the bulk of the protection for what enters the organelle. To allow specific compounds to reach the matrix space a number of proteins in the inner membrane function as transporters with each having a specific ligand to move across the membrane. For example, one of these (ATP/ADP carrier) allows ADP to cross the inner membrane while simultaneously transferring out an ATP from the matrix space. Many other such transporter have been described. The composition of inner membrane is different from outer membrane in that it is more proteinacious and it contains an unusual phospholipid, cardiolipin. Other than the metabolite transporters, the inner membrane also has three functionally different proteins; proteins involved in the respiratory chain complex, ATP synthase and protein import machinery.
Within the human mitochondrial matrix space is a small genome of 16569 bp that codes for 13 hydrophobic proteins all of which are involved in electron transfer, along with 22 transfer RNAs and two ribosomal RNAs [7,8]. The matrix space is also the site for major metabolic pathways including the citric acid cycle, urea cycle and fatty acid oxidation. Citric acid cycle is necessary to generate NADH that is the source of electrons for the electron transport system. The terminal acceptor for these electrons is oxygen. During the past decades a number of chemicals have been found that inhibit the transfer of electrons and from these we have learned something about the ability of molecules to enter the matrix space. Cyanide and azide ion both inhibit cytochrome oxidase, thereby blocking the flow of electrons through complex IV and enhance reactive oxygen species (ROS) generation at complex III [9,10]. Carbon monoxide has strong affinity for heme and it binds the heme containing cytochrome oxidase [11]. In contrast, a simple ion such as chloride or sodium can not diffuse into the matrix space, and like phosphate ion, requires a specific translocator on the inner membrane to allow it to get from the cytosol to the matrix space. Calcium ions enter the matrix space but it too requires the action an energy dependant transporter.
Rotenone, a very hydrophobic compound that is isolated from a plant root simply diffuses across the membrane and binds to a protein in complex 1 and inhibits the transfer of electron from NADH to coenzyme Q [12]. Some antibiotic compounds cross the inner membrane and bind to components of the electron transfer system [13,14]. Other hydrophobic molecules, even those carrying a positive charge can enter mitochondria by simply diffusing across the membranes. Rhodamine and JC1 has been used to stain mitochondria. Triphenylphosponium (TPP) ion is another example of a cationic hydrophobic molecule that can cross the mitochondrial membrane. Investigators have shown that drugs attached to TPP can diffuse into mitochondria [15–17]. Thus, hydrophobic molecules have the ability to simply diffuse across the membrane. Ions with delocalized charge can also diffuse across the membrane while ions with a localized charge (i.e. Na+ or Ca2+) require translocators.
2.2. Translocators and transporters
Binding of a drug to virtually any one of the transporters or even porin, the outer membrane protein, could disrupt mitochondria function by preventing an essential compound from entering or leaving. For example, atractyloside binds to the ATP/ADP carrier and affects mitochondria by uncoupling oxidative phosphorylation [14]. To date, not many such drugs have been described.
The vast majorities of mitochondrial proteins are coded by nuclear genes and after synthesis on cytosolic ribosomes are translocated to mitochondria. A receptor-translocator complex exists in mitochondrial membrane that allows these proteins to enter mitochondria [18–20]. Since proteins that are destined for the matrix space have to cross two membranes, there are actually two separate translocators. One is found on each membrane with the one on the outer membrane abbreviated TOM (translocator outer membrane) with the other being TIM (translocator inner membrane). The nuclear encoded proteins that are recognized by the TOM and TIM complexes possess an N-terminal leader sequence [18–22]. Attaching the leader to essentially any protein will enable the protein to be translocated into mitochondria. In fact, fusing the leader to a segment or DNA or RNA will allow those entities to be taken up by mitochondria [23]. More details about using the TOM and TIM complex will be discussed in a subsequent section of the review.
3. Specific diseases that are of mitochondrial origin
Mitochondria harbors roughly 1000 proteins of which only 13 are mitochondria genome coded. Thus, mitochondrial diseases can arise from defects in both nuclear and mitochondrial genomes [24–28] (Table 1). Mitochondria diseases that arise from the defective nuclear encoded proteins have been recently reviewed by Mackenzie et al [29]. The review discusses pyruvate dehydrogenase deficiency, primary hyperoxaluria type 1, severe alcoholic liver disease and human deafness dystonia syndrome. In pyruvate dehydrogenase deficiency, a point mutation was observed that caused the enzyme-complex to have lower activity in the mitochondrial matrix space when compared to healthy individuals. Primary hyperoxaluria type 1 is the result of two point mutations in the alanine/glyoxylate aminotransferase 1 protein. These mutants are targeted to mitochondria instead of peroxisomes. Human deafness dystonia syndrome is caused by a mutation to the inter membrane space (IMS) protein deafness dystonia peptide 1. Several other mitochondrial diseases occur due to mutation in the nuclear encoded mitochondrial proteins such as those involved in mtDNA maintenance/replications and mtDNA polymerase [30,31].
Table 1.
Diseases resulting from mutations of genes coding for tRNAs and proteins
| mtDNA mutations in the tRNALeu gene | ||
|---|---|---|
| Mutation | Phenotype | |
| A3243G | MEALS | |
| MEALS/MERRF | ||
| PEO | ||
| KSS | ||
| Myopathy | ||
| A3243T | Encephalomyopathy | |
| T3250C | Myopathy with respiratory Failure | |
| A3251G | PEO, myopathy | |
| A3252G | Dementia, diabetes | |
| C3254G | Myopathy, cardiomyopathy | |
| C3256T | PEO, deafness | |
| A3260G | Myopathy, cardiomyopathy | |
| T3271C | MEALS | |
| A3288G | Myopathy | |
| T3291C | MEALS | |
| A3302G | Myopathy with respiratory failure | |
| C3303T | Cardiomyopathy | |
| mtDNA mutation in the tRNALys gene | ||
| Mutation | Phenotype | |
| A8344G | MERRF | |
| MELAS/MERRF | ||
| PEO | ||
| Myopathy | ||
| Leigh’s syndrome | ||
| G8313A | Gastrointestinal dysfunction, dementia, ataxia, deafness, axonal neuropathy | |
| G8328A | Encephalopathy | |
| G8342A | PEO | |
| T8356C | MERRF | |
| G8363A | MERRF | |
| Cardiomyopathy, deafness, ataxia | ||
| A8296G | Hypertrophic cardiomyopathy | |
| mtDNA mutations in other tRNA genes | ||
| Gene | Mutation | Phenotype |
| tRNAPhe | A583G | MELAS |
| tRNAVal | G1606A | Ataxia, dementia, deafness |
| G1642A | MELAS | |
| tRNAIle | A4269G | Encephalopathy |
| T4274C | PEO | |
| T4285C | PEO | |
| A4295G | Cardiomyopathy | |
| tRNATrp | 5537T insert | Ataxia. Leigh’s syndrome |
| G5540A | Ataxia, deafness | |
| tRNAAsp | A5692G | Ataxia |
| G5703A | PEO | |
| tRNACys | A5814G | MELAS |
| tRNASer | 7472C insert | Deafness, ataxia |
| T7512C | MERRF/MELAS | |
| tRNAAsp | G7543A | Infantile encephalopathy |
| tRNAGlu | T14709C | Myopathy and diabetes |
| Mitochondrial oxidative phosphorylation diseases resulting from nuclear gene mutations | ||
| Gene encoding protein respiratory components | Phenotype | |
| Complex I NDUFS1 | Leigh syndrome | |
| Complex I NDUFS2 | Cardiomyopathy- Encephalomyopathy | |
| Complex I NDUFS4 | Leigh syndrome | |
| Complex I NDUFS7 | Leigh syndrome | |
| Complex I NDUFS8 | Leigh syndrome | |
| Complex I NDUFV1 | Leigh syndrome | |
| Complex II SDHA | Leigh syndrome | |
| Complex IV SURF1 | Leigh syndrome | |
| Nuclear genetic disorders of the mitochondrial respiratory chain due to mutations in translation factors: | ||
| Gene | Phenotype/Disease | |
| EFG1 | Leigh syndrome | |
| MRPS16 | Lactic acidosis, dysmophism | |
| EFTu | Leukodystrophy and polymicrogyria | |
| PUS1 | Myopathy and sideroblastic anemia | |
| Nuclear genetic disorders associated with multiple mtDNA deletions or mtDNA depletion: | ||
| Gene | Phenotype/Disease | |
| POLG, POLG2, | Autosomal progressive external ophthalmoplegia | |
| PEO1 and SLC25A4 | ||
| TP | Mitochondrial neurogastrointestinal encephalomyopathy (thymidine phosphorylase deficiency) | |
| POLG and MPV | Alpers–Huttenlocher syndrome | |
| DGUOK | Encephalomyopathy and liver failure | |
The vast majority of diseases of mitochondrial origin are due to defects mainly in the electron transport complexes [32–34]. These are the result of damaged or mutated proteins being incorporated into the complexes. The damaged proteins can be any one of the 13 that are coded by the mitochondrial genome or ones being coded by a nuclear gene since the complexes are composed of subunits of each. A list of the diseases associated with the mitochondrial genome is presented in Figure 1.
Figure 1.
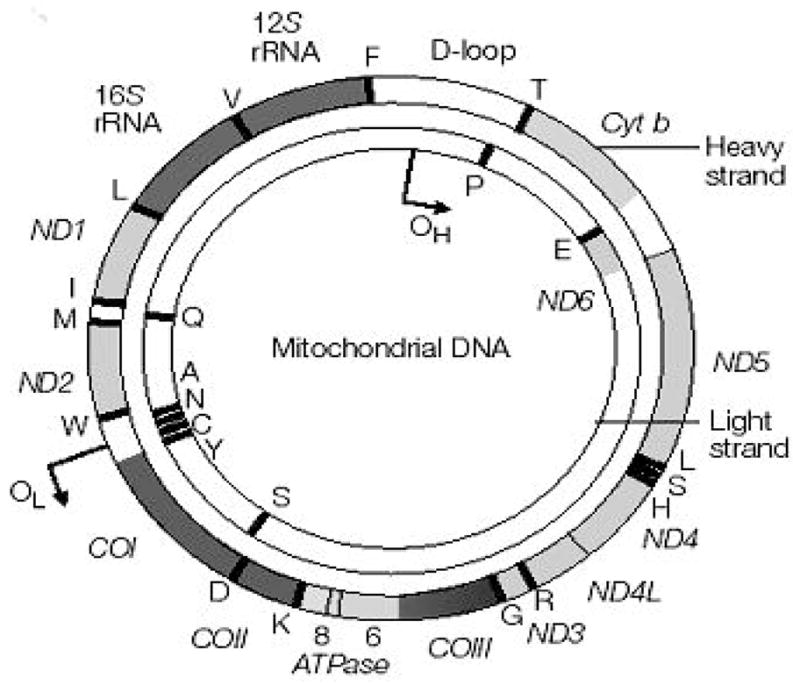
Map of human mitochondrial DNA. Human mitochondrial DNA is 16569 bp long. It codes for 13 polypeptides (ND1-ND6, ND4L, CYT b, COX I-COX III, ATPase6 and ATPase8), 2 rRNAs and 22 tRNAs. The tRNAs are shown as single letter amino acid code. The D loop is a non coding sequence. OH heavy strand replication origin. OL light strand replication origin. The figure was taken from reference 8.
4. Why should drugs be targeted to mitochondria?
In addition to providing energy for the cell through oxidative phosphorylation, mitochondria also serve as a source of carbon compounds for some important pathways. For example, acetyl CoA is produced in mitochondria by the action of the pyruvate dehydrogenase complex. Acetyl CoA is needed for the synthesis of citrate in the matrix space but is also needed to make malonyl CoA in the cytosol to be used in fatty acid biosynthesis. Acetyl CoA can not pass through the membrane but citrate can. Thus, in theory, a drug that interfered with the translocation of citrate that produces acetyl CoA might be useful for weight control.
A recent discovery showed that mitochondria is involved in apoptosis (programmed cell death). It appears that compounds that increase the permeability of the outer membrane can cause cytochrome c to be released; this event is one of the early triggers of the apoptotic pathway. Naturally occurring compounds that are involved are actually proteins of the Bcl-2 family abbreviated as Bax and Bid. These proteins form pores that permits cytochrome c to exit from IMS. On the other hand, Bcl-2 and Bcl-XL inhibit apoptosis [35–38]. Despite intensive investigations, how Bax and Bak form pores in the outer membrane is still not known. The widely accepted theory is that during apoptosis, Bax and Bak change their conformations, oligomerize and form pore in the outer membrane that facilitate the release of cytochrome c. Bcl-2 can inhibit Bax and Bak oligomerization by binding the preformed oligomer [39]. The pro and antiapoptotic proteins, Bax, Bak, Bcl-2 and Bcl-X L all have the ability to form pores in the outer membrane though the shape and ion selectivity of the pores are different [40–43]. The structures of six Bcl-2 family member proteins have been determined [44–46]. The structures of these proteins possess remarkable similarity although their amino acid sequences differ. The structure of Bcl-X L shows that it forms a hydrophobic groove that makes a binding site for proapoptotic protein such as BAD suggesting that the interaction is mainly hydrophobic [47]. This study led the prediction that small molecules that could be used as inhibitors of these proteins. ABT-737 is a potent inhibitor that binds the same hydrophobic region of Bcl-XL and has been used in clinical trials [48].
Voltage dependent anion channel (VDAC) is a highly conserved protein with homology to bacterial porin and it forms an outer membrane pore [49]. VDAC plays a major role in mitochondria associated apoptosis. There are many conflicting theories about its role in apoptosis. It was found that proapoptotic protein Bax was co-immunoprecipitated with VDAC suggesting an interaction between them [50]. Other investigators showed that Bax can favor the high conductance state of VDAC that allowed cytochrome c to be released from the intermembrane space [51]. Rostovtseva et al found the proapoptotic protein tBid, not Bax, binds VDAC and prevents the exchange of ATP/ADP between cytosol and mitochondria leading to mitochondrial swelling resulting in rupture of the mitochondrial membrane. Subsequently, cytochrome c is released into cytoplasm leading to cell death [52]. Studies showed that mitochondria bound hexokinase II also has role in apoptosis [53]. Hexokinase II binds VDAC inhibiting the binding of Bax. Since the binding of Bax to VDAC promotes apoptosis, hexokinase II prevents the cytochrome c release into cytosol and hence prevents apoptosis. Drugs that induce apoptosis would be useful as therapeutic agents.
During the passage of electrons through the electron transport system, oxygen radicals are formed (ROS) which leads to a concept termed oxidative stress [54–56]. Mitochondria destroy the free radicals using vitamin E, ascorbate, coenzyme Q 10, cytochrome c and glutathione or enzymatically with superoxide dismutase, glutathione peroxidase and catalase. If this defense system is compromised free radicals could accumulate in mitochondria. These radicals could cause mtDNA mutation [57], lipid peroxidation [58] and protein oxidation [59]. mtDNA mutations are associated with many physiological complications including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis [26,60]. Antioxidant supplements as well as drugs have been used to help scavenge the free radicals produced in mitochondria [61].
Drugs are necessary for both inhibiting mitochondria in order to kill cancer cells as well as to protect the cells from oxidative damage and to repair defects. Because of the complex nature of the organelle it will be necessary to use many different strategies to get a drug or macromolecule into mitochondrial once it is taken up by the plasma membrane.
5. Strategies used to bring compounds into mitochondria that disrupts its function
5.1. Lipophilic cations
Many of the chemicals that disrupt mitochondrial function take advantage of the hydrophobic nature of the membrane. For example CCCP is a compound used to disrupt the electrochemical potential of the organelle and it simply diffuses across the membrane. The concept of hydrophobic molecules as carriers of drugs into mitochondria was turned into reality when triphenyl phosphonium ion was shown to cross into the matrix space [61,62]. Functional groups could be attached to the phosphorous atom that will be carried into the matrix space and released there. Alternatively, it might be possible to modify the phenyl rings so that drugs could be attached to them. These could then be cleaved from the carrier by enzymes in the matrix space. TPP or a methyl derivative, TPMP have the property of being relatively lipid-soluble, despite their net positive charge. The hydrophobic nature as well as the delocalized positive charge allows for the efficient penetration into mitochondria without requiring a receptor [61].
Oxidative damage to mitochondria is very common in most age-related human diseases including neurodegenerative disorders. This is due to the formation of reactive oxygen species (ROS), produced by the mitochondria [54]. The selective delivery of antioxidants to damaged mitochondria is therefore an effective therapeutic strategy in such human disorders. For example, Murphy’s lab employed VitE and coenzyme Q to treat the cells from patients having Friedreich Ataxia (FA) [61]. The mitochondrial protein frataxin is defective in the FA disease and it contributes to ROS formation in mitochondria. It was observed that VitE and coenzyme Q both were able to rescue the cells from the damage caused by ROS. However, these antioxidants do not have the ability to concentrate in mitochondria. Thus, the amounts needed to afford protection were very high. To overcome the problem, antioxidants were covalently coupled to a triphenylphosphonium cation (Fig. 2), and these compounds were selectively taken up by mitochondria [3,61]. The lipophilic cations easily permeate through the lipid bilayers and subsequently accumulate several hundred-fold within mitochondria because of a large mitochondrial membrane potential (150–170 mV; negative inside). Thus, TPP can be used as a carrier to target antioxidants to mitochondria to rescue cells from oxidative damage.
Figure 2.
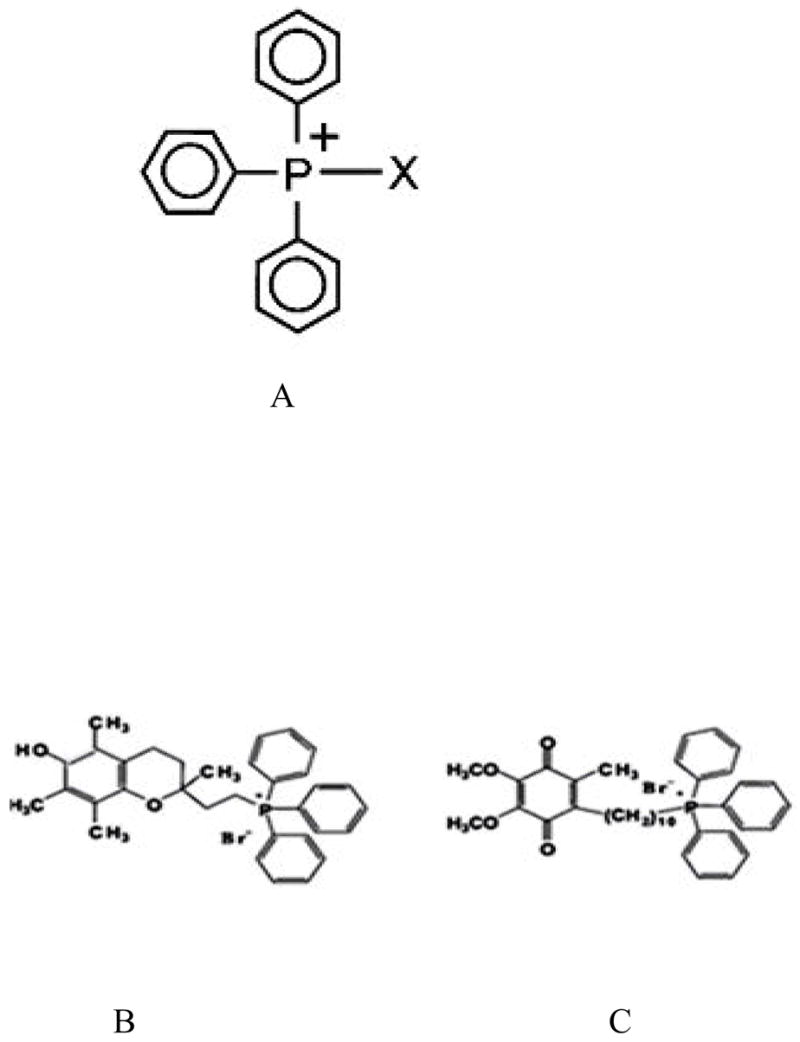
Triphenyl phosphonium ions that can enter mitochondria because of their lipophilic properties. A. Triphenyl phosphonium ion (TPP). Antioxidant moieties (α-tocopherol and ubiquinol) fused to TPP. B. Mito-E2 C. Mito-Q10.
Dequalinium is another delocalized cationic lipid and has the potential to target DNA to mitochondria [5,6]. It binds DNA tightly and can fold into a compact structure. This structure protects the bound DNA from nuclease digestion. Due to its delocalized positive charge it can pass through the plasma membrane by endocytosis. Unlike a nuclear targeting lipid vector, the DNA is not released from the liposome upon contact with the inner side of plasma membrane. Instead, DNA is released only when this liposome interacts with the mitochondrial membrane. The specific components present in the mitochondrial membrane interact with the positively charged liposome and DNA is released at the mitochondrial surface. If DNA is fused to a mitochondrial leader peptide, it will be imported into the matrix space using the mitochondrial import machinery (TOM/TIM). It was shown that dequalinium-DNA can be imported into mitochondrial matrix space when it was incubated with BT20 cells [63].
5.2. Protein-nucleic acid
Shortly after the discovery of the leader sequence, Schatz’s laboratory showed that mitochondria can import single or double stranded DNA whose 5′ end was covalently linked to the C-terminus of a mitochondria precursor protein [64]. The precursor initially employed was the leader sequence from COX IV fused to dihydrofolate reductase. The cysteine residue at the C-terminus of the preprotein was coupled to the 5′ end of 24 nucleotide ssDNA using a bifunctional crosslinker. The protein-DNA adduct was imported into yeast mitochondria. The authors also showed that dsDNA can be imported into isolated mitochondria.
5.3. Peptide-nucleic acid
Peptide nucleic acids (PNAs) are synthetic DNA-like molecules in which the chains of pyrimidine and purine bases are linked by an aminoethyl (pseudopeptide) backbone (Fig. 3) [65,66]. This structure specifically hybridizes to complementary DNA and RNA and is resistance to nuclease and protease attack. As a result, PNAs have the potential to bind damaged mitochondria genome. However, poor permeation through the cell membrane has made it less attractive as a therapeutic agent although some cells like muscle cells are particularly capable of DNA or modified DNA uptake by an unknown mechanism [67].
Figure 3.
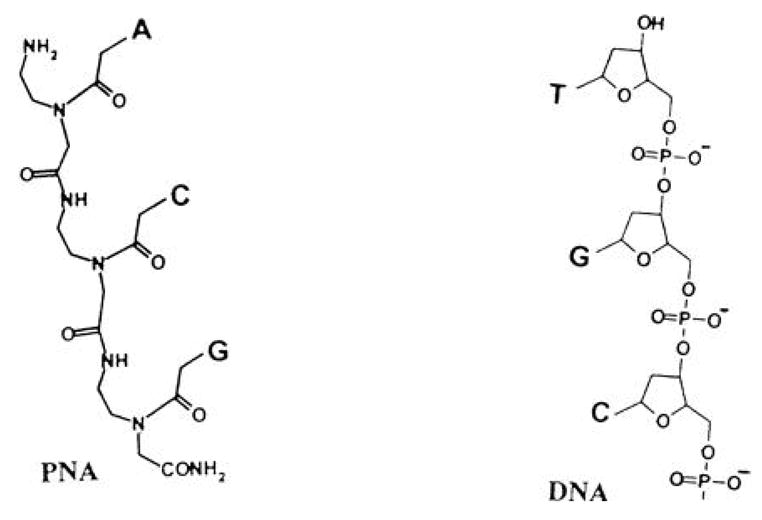
Peptide nucleic acid (PNA) used to bring a DNA-like structure into mitochondria.
Investigators reported that biotinylated PNA was easily incorporated into cells. The mitochondrial leader sequence from COX VIII was fused to the biotinylated PNA, and it was able to localize in mitochondria both in vitro and in vivo [67]. Although the mitochondrial leader peptide was able to carry PNA to mitochondria, the cost of synthesizing the adduct and its low permeability through the plasma membrane has made it unattractive to use. Investigators, instead, used TPP to bring PNA into mitochondria [68]. The investigators applied the PNA-adduct to treat MERRF disease, a disease caused by a point mutation, A8344G tRNALys. The strategy was to study a DNA replication run off assay using two different templates, one from wild type and the other from mutant mtDNA. A PNA 11-mer was conjugated to TPP and was used in the in vitro replication assay. PNA inhibited up to 75% of the mutant DNA replication while no inhibition was found using the wild type DNA template. This PNA-adduct was also employed with cells from MERRF patients that contained both the wild type and mutant mtDNA. The PNA-TPP adduct was found in the mitochondria but the ratio of mutant and wild type DNA was not changed showing that the PNA could not inhibit the replication in intact cells. Though the experiment did not produce the expected results it showed that the PNA-TPP adduct has the potential to enter into mitochondrial matrix when added to a cell line. More work will be needed to make them a useful therapeutic delivery system.
5.4. Proteins and RNA
The leader sequence necessary for protein targeting is removed by a mitochondrial protease after it enters the matrix space. What is of potential interest is that the sequence and size of the leaders vary among the 1000 or so nuclear encoded proteins that enter mitochondria [18]. Further, it has been shown that the leader can bring in virtually any protein as well as segments of RNA or DNA. This means that the leader sequences can be used to bring native or foreign macromolecules into mitochondria.
It has been shown that the leader sequence of the Toho-1, a bacterial signal peptide, can bring GFP to mitochondria [69]. Other laboratories have also shown that bacteria signal peptides can function as a mitochondrial leader [70]. This implies that it could be possible by using a bacterial as well as a mammalian leader peptide to bring any protein into mitochondria. For example, one could use this approach to bring into mitochondria a correct version of a protein coded by damaged gene of either nuclear or mitochondrial origin. Obviously, it would be necessary to find a way to get the correct DNA into the cell, a topic beyond the scope of this review. However, there are well established methods to incorporate a foreign DNA into cells.
5.4.1. Factors to be considered when trying to replace a damaged protein
Coding
One problem when trying to express a mitochondria coded protein in cytosol is that the genetic code for a few amino acids is different in mitochondria than in the nuclear coded genes. ATA codes for methionine in mitochondria but codes for isoleucine in cytosol. TGA codes for tryptophan in mitochondria but it is a stop codon in cytosol. Thus, it will be necessary to change the codons according to the universal nuclear code. Simple PCR reactions can be used to accomplish these changes.
Solubility
All thirteen proteins expressed in mitochondria are very hydrophobic. There is a possibility that if these proteins were expressed in cytosol they might aggregate and precipitate. It is also possible that these very hydrophobic proteins might just bind to any membrane. When these proteins are expressed in mitochondria they are targeted to the inner membrane by a co-translational import mechanism, that is, the protein starts to be incorporated into the inner membrane while it is coming off mitochondrial ribosome [71]. It has been shown that a co-transport/translocation mechanism for protein import from nuclear coded genes also exists [72]. Thus, the hydrophobic nature of allotropically expressed proteins might not be a serious problem if the nuclear coded protein could follow a co-translational import model [73].
Insertion into inner membrane and assembly
Finally, after targeting to the inner mitochondria membrane, it would be necessary to replace a protein that is in embedded in a membrane-bound complex. It is not known precisely how each component assembles into a complex. Most likely the proteins are not free to diffuse in and out of the complex. Thus, it may not possible for a newly imported protein simply to displace the damage protein from an existing complex. Rather, it might be possible to imagine that the new protein can be inserted into the complex during mitochondrial biogenesis.
Several mechanisms exist to insert nuclear coded proteins into the inner membrane (Fig. 4). The carrier proteins such as ATP/ADP carrier protein does not contain any N-terminal-leader sequence but possesses several internal signals necessary for import. The carrier proteins are first recognized by Tom70, an outer membrane protein. In the IMS, they interact with small TIM proteins so that they are not precipitated in the hydrophilic environment of the IMS. Finally, they interact with the TIM22 complex and subsequently are released to inner membrane [74].
Figure 4.

Inner membrane import pathways. The figure shows how the inner membrane proteins are imported and inserted into the inner membrane. The figure was taken from the reference 18.
Some inner membrane proteins contain a leader peptide that is followed by a hydrophobic sequence. The leader peptide first interacts with the TOM complex and then is transferred to the TIM23 complex. The leader peptide crosses the inner membrane and is cleaved by the protease in the matrix space while the hydrophobic signal keeps the protein in the inner membrane. Oxa1p from yeast follows this mechanism [75].
A few preproteins are integrated into the inner membrane by ‘the conservative pathway’. The preproteins contain typical mitochondrial leader sequences that are recognized by TOM and then by TIM23 complex. The proteins are then completely released into matrix space where the leader sequence is removed. The proteins are finally inserted to inner membrane by an export-like system. The export is carried out primarily by Oxa1 complex in the inner membrane. The subunit 9 of F1-F0 ATPase of N. crassa and Oxa1 are examples of proteins using this mechanism [76].
The proteins that are expressed in the mitochondria (cytochrome b of the bc1 complex; Cox1, Cox2, and Cox3 of the cytochrome oxidase; and Atp6, Atp8, and Atp9 of the F0F1-ATPase) are very hydrophobic and are inserted into inner membrane by a co-translational path way. The co-translational import is facilitated by the C-terminus of Oxa1 complex which is extended into matrix that binds the nascent polypeptide chain [18, 71].
The above mentioned mechanisms illustrate various ways of how a protein can be imported into the inner membrane. One or more of these approaches might have to be employed to replace defective proteins in the electron transport complex. Most probably the method which uses the internal signal could not be used since putting an internal signal into a mitochondrial coded protein may disrupt the protein’s function. The other pathways can be used to insert allotopically expressed mitochondria-coded proteins into inner membrane by fusing a signal peptide to the N-terminus to replace a damaged protein.
Mitochondrial membrane insertion of allotopically expressed proteins have been reported. ATPase6 and ATPase8 were expressed in cytosol and were imported into mitochondria with the use of a mitochondrial leader peptide. Nagley et al showed that a construct containing a leader peptide from ATPase9 fused to ATPase6 subunit from F1-F0 ATPase synthase expressed in cytosol were targeted to mitochondria [77]. This chimeric protein eventually replaced the defective ATPase6 component in the complex. Later Manfredi et al and Zullo et al separately showed that ATPase6 subunit can be expressed in cytosol and be brought to mitochondria [78, 79]. More work will have to be done to determine how general were these findings.
5.4.2. Import of restriction enzymes to be used for mitochondrial gene therapy
mtDNA mutations could produce a new site for a restriction enzyme. The strategy to destroy the mtDNA was to import a unique restriction endonuclease by fusing a leader peptide to it. NARP disease results from the point mutation in mtDNA (T8399G) which results in impair electron transport. The point mutation creates a new SmaI-XmaI site which is not present in wild type mtDNA. A mitochondrial leader sequence was fused to the Sma1 endonuclease and was imported into mitochondria [80]. The protein selectively degraded the mutant mtDNA but not the wt mtDNA suggesting its potential therapeutic use.
5.4.3. Import of tRNA into mitochondria
Human mitochondria code all the tRNAs inside the matrix and hence have no tRNA import machinery. Plants, protozoa and fungi, however, have well defined tRNA import machinery in their mitochondrial membrane [81–84]. It has been shown that human cytoplasmic tRNALys(UUU) is imported into the mitochondria of T. brucei [85], L. tropica. [86] and isolated Leishmania mitochondria [86]. The imported tRNA was able to function properly. This shows that human tRNA can utilize the fungal tRNA import machinery to be imported into mitochondria. Kolesnikova et al also showed that yeast tRNALys could be imported into human mitochondria, although human mitochondria do not possess tRNA import machinery and was able to rescue mitochondrial function suffering from MERRF disease [87]. Very recently, investigators showed that human mitochondria could import tRNA after introducing Leishmania RNA import complex (RIC) into the mitochondria of human cells [89]. More importantly, imported tRNALys into human mitochondria rescued the function of mitochondria possessing defective tRNALys. This study shows that a defective tRNA in human mitochondria can be replaced by a cytosolic tRNA using fungal RIC [88].
5.4.4. Leaders as inhibitors of mitochondrial protein import
Peptides corresponding to a leader sequence typically were found not be good inhibitors of protein import. This was an unexpected finding since it is the leader alone that interacts with TOM 20, a major component of the import apparatus [18]. It is possible that during import of a precursor protein, a heat shock-like protein is needed to anchor the precursor protein to the receptor complex [89]. Should that be the case, then on would need more than just a small peptide to act as an inhibitor.
Finding an inhibitor, be it peptide or an organic compound, for protein import could lead to cell death if newly synthesized proteins could not be brought into mitochondria. An alternative use could be to prevent proteins such as hexokinase II from binding to porin, an event found in cancer cells [90]. In some advanced cancers an isozyme of hexokinase associates with the outer membrane by binding to porin. Thus, when ATP exits mitochondria it becomes the co-substrate for the kinase and allows the cancerous cell to utilize glucose more efficiently than does a non-cancerous cell. To date, few, if any, peptides or compounds have been developed to either prevent import from occurring or to inhibit the binding of proteins to mitochondria.
6. Future
Diseases related to mitochondrial function are often the result of mutations that lead to changes in the proteins involved in the electron transport system. To correct these, some form of gene therapy will have to be employed. Different problems will exist depending upon whether the mutation was to a nuclear or mitochondria gene. We understand how to bring a nuclear coded gene product to the mitochondria. In contrast, there are only a few examples of correctly inserting a mitochondria gene product after the protein is allotropically expressed. More work will have to be done in this area before it can be determined whether or not it will be feasible to replace all or most of the damaged mitochondrial coded gene products.
Small molecules have been shown to affect mitochondria function. Use of triphenyl phosphonium ions has made it possible to bring many different molecules into mitochondria. The range of compounds should be expanded to include ones that can induce apoptosis in cancer cells as well as to inhibit (or activate) damaged proteins that were a result of mutated genes. More compounds are needed to protect the cell from the damaged caused by oxidative stress. Various techniques are available to allow one to bring virtually any type compound, be it a macromolecule or a small organic molecule, into the organelle. It will be necessary to show that the compound is truly functional once it is imported. Unfortunately, due to the complex nature of mitochondria, no one technique will enable all molecules to be imported.
Acknowledgments
This work was supported in part by National Institute of Health grants AA10795, AA05812 and GM 53269. This is journal paper number xxxxx from the Purdue University Agriculture Experiment Station.
Abbreviations
- mtDNA
mitochondrial DNA
- PNA
peptide nucleic acid
- TOM
translocase outer membrane
- TIM
translocase inner membrane
- TPP
triphenyl phosphonium ion
- IMS
inter membrane space
- VDAC
voltage dependent anionic channel
- MERRF
Myoclonic epilepsy and ragged-red fibres
- NARP
Neurogenic weakness, ataxia and retinitis pigmentosa
- MELAS
Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes
- KSS
Kearns–Sayre syndrome
- PEO
Progressive external ophthalmoplegia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gray MW, Lang BF, Burger G. Mitochondria of protests. Annu Rev Genet. 2004;38:477–524. doi: 10.1146/annurev.genet.37.110801.142526. [DOI] [PubMed] [Google Scholar]
- 2.Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 3.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 4.Yamada Y, Akita H, Kogure K, Kamiya H, Harashima H. Mitochondrial drug delivery and mitochondrial disease therapy - An approach to liposome-based delivery targeted to mitochondria. Mitochondrion. 2007;7:63–71. doi: 10.1016/j.mito.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Weissig V, Boddapati SV, Cheng SM, D’Souza GG. Liposomes and liposome-like vesicles for drug and DNA delivery to mitochondria. J Liposome Res. 2006;16:249–264. doi: 10.1080/08982100600851169. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 7.McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease--its impact, etiology, and pathology. Curr Top Dev Biol. 2007;77:113–155. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- 8.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer G. Current issues in the chemistry of cytochrome c oxidase. J Bioenerg Biomembr. 1993;25:145–151. doi: 10.1007/BF00762856. [DOI] [PubMed] [Google Scholar]
- 10.Villani G, Attardi G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic Biol Med. 2000;29:202–210. doi: 10.1016/s0891-5849(00)00303-8. [DOI] [PubMed] [Google Scholar]
- 11.Haab P. The effect of carbon monoxide on respiration. Experientia. 1990;46:1202–1206. doi: 10.1007/BF01936937. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 13.Duewelhenke N, Krut O, Eysel P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob Agents Chemother. 2007;51:54–63. doi: 10.1128/AAC.00729-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malekova L, Kominkova V, Ferko M, Stefanik P, Krizanova O, Ziegelhoffer A, Szewczyk A, Ondrias K. Bongkrekic acid and atractyloside inhibits chloride channels from mitochondrial membranes of rat heart. Biochim Biophys Acta. 2007;1767:31–44. doi: 10.1016/j.bbabio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut LA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 16.James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, Murphy MP. Interaction of the mitochondria-targeted antioxidant mitoq with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- 17.Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RA, Murphy MP. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J Biol Chem. 2005;280:24113–24126. doi: 10.1074/jbc.M501148200. [DOI] [PubMed] [Google Scholar]
- 18.Neupert W, Herrmann JM. Translocation of Proteins into Mitochondria. Annu Rev Biochem. 2007 doi: 10.1146/annurev.biochem.76.052705.163409. in press. [DOI] [PubMed] [Google Scholar]
- 19.Mokranjac D, Neupert W. Protein import into mitochondria. Biochem Soc Trans. 2005;33:1019–1023. doi: 10.1042/BST20051019. [DOI] [PubMed] [Google Scholar]
- 20.Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. J Biol Chem. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- 21.Dolezal V, Likic J, Tachezy T. Lithgow, Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–318. doi: 10.1126/science.1127895. [DOI] [PubMed] [Google Scholar]
- 22.Koehler CM. New developments in mitochondrial assembly. Annu Rev Cell Dev Biol. 2004;20:309–335. doi: 10.1146/annurev.cellbio.20.010403.105057. [DOI] [PubMed] [Google Scholar]
- 23.Seibel M, Bachmann C, Schmiedel J, Wilken N, Wilde F, Reichmann H, Isaya G, Seibel P, Pfeiler D. Processing of artificial peptide-DNA-conjugates by the mitochondrial intermediate peptidase (MIP) Biol Chem. 1999;380:961–967. doi: 10.1515/BC.1999.119. [DOI] [PubMed] [Google Scholar]
- 24.Howell N, Elson JL, Chinnery PF, Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet. 2005;21:583–586. doi: 10.1016/j.tig.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Kang D, Hamasaki N. Mitochondrial disease: maintenance of mitochondrial genome and molecular diagnostics. Adv Clin Chem. 2006;42:217–254. doi: 10.1016/s0065-2423(06)42006-0. [DOI] [PubMed] [Google Scholar]
- 26.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 27.Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 28.Chan DC. Mitochondria: dynamic organelles in disease, aging. and development Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 29.Mackenzi JA, Payne RM. Mitochondrial protein import and human health and disease. Biochim Biophys Acta. 2006;1772:509–523. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kollberg G, Moslemi AR, Darin N, Nennesmo I, Bjarnadottir I, Uvebrant P, Holme E, Melberg A, Tulinius M, Oldfors A. POLG1 mutations associated with progressive encephalopathy in childhood. J Neuropathol Exp Neurol. 2006;65:758–768. doi: 10.1097/01.jnen.0000229987.17548.6e. [DOI] [PubMed] [Google Scholar]
- 31.Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarzi E, Brown MD, Lebon S, Chretien D, Munnich A, Rotig A, Procaccio V. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet A. 2007;143:33–41. doi: 10.1002/ajmg.a.31565. [DOI] [PubMed] [Google Scholar]
- 33.Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:1103–1110. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- 34.DiMauro S. Mitochondrial myopathies. Curr Opin Rheumatol. 2006;18:636–641. doi: 10.1097/01.bor.0000245729.17759.f2. [DOI] [PubMed] [Google Scholar]
- 35.Matsuyama S, Reed JC. Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ. 2000;7:1155–1165. doi: 10.1038/sj.cdd.4400779. [DOI] [PubMed] [Google Scholar]
- 36.Galluzzi L, Larochette N, Zamzami N, Kroemer G. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene. 2006;25:4812–4830. doi: 10.1038/sj.onc.1209598. [DOI] [PubMed] [Google Scholar]
- 37.Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol. 2006;18:685–689. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 39.Ruffolo SC, Shore GC. BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem. 2003;278:25039–25045. doi: 10.1074/jbc.M302930200. [DOI] [PubMed] [Google Scholar]
- 40.Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 41.Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 42.Schendel SL, Montal M, Reed JC. Bcl-2 family proteins as ion-channels. Cell Death Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- 43.Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, Reed JC. Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem. 1999;274:21932–21936. doi: 10.1074/jbc.274.31.21932. [DOI] [PubMed] [Google Scholar]
- 44.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S, Ng SL, Fesik SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 45.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 46.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci U S A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 48.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 49.O’Rourke B. Mitochondrial Ion Channels. Annual Review of Physiology. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capano M, Crompton M. Biphasic translocation of Bax to mitochondria. Biochem J. 2002;367:169–178. doi: 10.1042/BJ20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci USA. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem. 2004;279:13575–13583. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- 53.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 54.Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 55.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 56.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 57.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 58.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 59.Stadtman ER, Oliver CN. Metal-catalyzed oxidation of proteins. Physiological consequences. J Biol Chem. 1991;266:2005–2008. [PubMed] [Google Scholar]
- 60.Trifunovic A. Mitochondrial DNA and ageing. Biochim Biophys Acta. 2006;1757:611–617. doi: 10.1016/j.bbabio.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 62.Rottenberg H. Membrane potential and surface potential in mitochondria: uptake and binding of lipophilic cations. J Membr Biol. 1984;81:127–138. doi: 10.1007/BF01868977. [DOI] [PubMed] [Google Scholar]
- 63.D’Souza GG, Rammohan R, Cheng SM, Torchilin VP, Weissig V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J Control Release. 2003;92:189–197. doi: 10.1016/s0168-3659(03)00297-9. [DOI] [PubMed] [Google Scholar]
- 64.Vestweber D, Schatz G. DNA-protein conjugates can enter mitochondria via the protein import pathway. Nature. 1989;338:170–172. doi: 10.1038/338170a0. [DOI] [PubMed] [Google Scholar]
- 65.Corradini R, Sforza S, Tedeschi T, Totsingan F, Marchelli R. Peptide nucleic acids with a structurally biased backbone: effects of conformational constraints and stereochemistry. Curr Top Med Chem. 2007;7:681–694. doi: 10.2174/156802607780487759. [DOI] [PubMed] [Google Scholar]
- 66.Wolf Y, Pritz S, Abes S, Bienert M, Lebleu B, Oehlke J. Structural requirements for cellular uptake and antisense activity of peptide nucleic acids conjugated with various peptides. Biochemistry. 2006;45:14944–1454. doi: 10.1021/bi0606896. [DOI] [PubMed] [Google Scholar]
- 67.Chinnery PF, Taylor RW, Diekert K, Lill R, Turnbull DM, Lightowlers RN. Peptide nucleic acid delivery to human mitochondria. Gene Ther. 1999;6:1919–1928. doi: 10.1038/sj.gt.3301061. [DOI] [PubMed] [Google Scholar]
- 68.Muratovska A, Lightowlers RN, Taylor RW, Turnbull DM, Smith RA, Wilce JA, Martin SW, Murphy MP. Targeting peptide nucleic acid (PNA) oligomers to mitochondria within cells by conjugation to lipophilic cations: implications for mitochondrial DNA replication, expression and disease. Nucleic Acids Res. 2001;29:1852–1863. doi: 10.1093/nar/29.9.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mukhopadhyay A, Ni L, Yang CS, Weiner H. Bacterial signal peptide recognizes HeLa cell mitochondrial import receptors and functions as a mitochondrial leader sequence. Cell Mol Life Sci. 2005;62:1890–1899. doi: 10.1007/s00018-005-5178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagai T, Abe A, Sasakawa C. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J Biol Chem. 2005;280:2998–3011. doi: 10.1074/jbc.M411550200. [DOI] [PubMed] [Google Scholar]
- 71.Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM. Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J. 2003;22:6448–6457. doi: 10.1093/emboj/cdg623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mukhopadhyay A, Ni L, Weiner H. A co-translational model to explain the in vivo import of proteins into HeLa cell mitochondria. Biochem J. 2004;382:385–392. doi: 10.1042/BJ20040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukhopadhyay A, Zullo SJ, Weiner H. Factors that might affect the allotopic replacement of a damaged mitochondrial DNA-encoded protein. Rejuvenation Res. 2006;9:182–190. doi: 10.1089/rej.2006.9.182. [DOI] [PubMed] [Google Scholar]
- 74.Truscot KN, Wiedemann N, Rehling P, Muller H, Meisinger C, Pfanner N, Guiard B. Mitochondrial import of the ADP/ATP carrier: the essential TIM complex of the intermembrane space is required for precursor release from the TOM complex. Mol Cell Biol. 2002;22:7780–7789. doi: 10.1128/MCB.22.22.7780-7789.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herrmann JM, Neupert W, Stuart RA. Insertion into the mitochondrial inner membrane of a polytopic protein, the nuclear-encoded Oxa1p. EMBO J. 1997;16:2217–2226. doi: 10.1093/emboj/16.9.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rojo EE, Stuart RA, Neupert W. Conservative sorting of F0-ATPase subunit 9: export from matrix requires delta pH across inner membrane and matrix ATP. EMBO J. 1995;14:3445–3451. doi: 10.1002/j.1460-2075.1995.tb07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nagley P, Farrell LB, Gearing DP, Nero D, Meltzer S, Devenish RJ. Assembly of functional proton-translocating ATPase complex in yeast mitochondria with cytoplasmically synthesized subunit 8, a polypeptide normally encoded within the organelle. Proc Natl Acad Sci. 1988;85:2091–2095. doi: 10.1073/pnas.85.7.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–399. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- 79.Zullo SJ, Parks WT, Chloupkova M, Wei B, Weiner H, Fenton WA, Eisenstadt JM, Merril CR. Stable transformation of CHO Cells and human NARP cybrids confers oligomycin resistance (oli(r)) following transfer of a mitochondrial DNA-encoded oli(r) ATPase6 gene to the nuclear genome: a model system for mtDNA gene therapy. Rejuvenation Res. 2005;8:18–28. doi: 10.1089/rej.2005.8.18. [DOI] [PubMed] [Google Scholar]
- 80.Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10:3093–3039. doi: 10.1093/hmg/10.26.3093. [DOI] [PubMed] [Google Scholar]
- 81.Entelis NS, Kolesnikova OA, Martin RP, Tarassov IA. RNA delivery into mitochondria. Adv Drug Deliv Rev. 2001;49:199–215. doi: 10.1016/s0169-409x(01)00135-1. [DOI] [PubMed] [Google Scholar]
- 82.Small I, Marechal-Drouard L, Masson J, Pelletier G, Cosset A, Weil JH, Dietrich A. In vivo import of a normal or mutagenized heterologous transfer RNA into the mitochondria of transgenic plants: towards novel ways of influencing mitochondrial gene expression. EMBO J. 1992;11:1291–1296. doi: 10.1002/j.1460-2075.1992.tb05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Delage L, Dietrich A, Cosset A, Marechal-Drouard L. In vitro import of a nuclearly encoded tRNA into mitochondria of Solanum tuberosum. Mol Cell Biol. 2003;23:4000–4012. doi: 10.1128/MCB.23.11.4000-4012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delage L, Duchene AM, Zaepfel M, Marechal-Drouard L. The anticodon and the D-domain sequences are essential determinants for plant cytosolic tRNAVal import into mitochondria. Plant J. 2003;34:623–633. doi: 10.1046/j.1365-313x.2003.01752.x. [DOI] [PubMed] [Google Scholar]
- 85.Hauser R, Schneider A. tRNAs are imported into mitochondria of Trypanosoma brucei independently of their genomic context and genetic origin. EMBO J. 1995;14:4212–4220. doi: 10.1002/j.1460-2075.1995.tb00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goswami S, Chatterjee S, Bhattacharyya SN, Basu S, Adhya S. Allosteric regulation of tRNA import: interactions between tRNA domains at the inner membrane of Leishmania mitochondria. Nucleic Acids Res. 2003;31:5552–5559. doi: 10.1093/nar/gkg773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kolesnikova OA, Entelis NS, Jacquin-Becker C, Goltzene F, Chrzanowska-Lightowlers ZM, Lightowlers RN, Martin RP, Tarassov I I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum Mol Genet. 2004;13:2519–2534. doi: 10.1093/hmg/ddh267. [DOI] [PubMed] [Google Scholar]
- 88.Mahata B, Mukherjee S, Mishra S, Bandyopadhyay A, Adhya S. Functional delivery of a cytosolic tRNA into mutant mitochondria of human cells. Science. 2006;314:471–474. doi: 10.1126/science.1129754. [DOI] [PubMed] [Google Scholar]
- 89.Voos W. A new connection: chaperones meet a mitochondrial receptor. Mol Cell. 2003;11:1–3. doi: 10.1016/s1097-2765(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 90.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]