

Abstract
Most cardiac Na+ channels open transiently upon membrane depolarization and then are quickly inactivated. However, some channels remain active, carrying the so-called persistent or late Na+ current (INaL) throughout the action potential (AP) plateau. Experimental data and the results of numerical modeling accumulated over the past decade show the emerging importance of this late current component for the function of both normal and failing myocardium. INaL is produced by special gating modes of the cardiac-specific Na+ channel isoform. Heart failure (HF) slows channel gating and increases INaL, but HF-specific Na+ channel isoform underlying these changes has not been found. Na+ channels represent a multi-protein complex and its activity is determined not only by the pore-forming α subunit but also by its auxiliary β subunits, cytoskeleton, calmodulin, regulatory kinases and phosphatases, and trafficking proteins. Disruption of the integrity of this protein complex may lead to alterations of INaL in pathological conditions. Increased INaL and the corresponding Na+ flux in failing myocardium contribute to abnormal repolarization and an increased cell Ca2+ load. Interventions designed to correct INaL rescue normal repolarization and improve Ca2+ handling and contractility of the failing cardiomyocytes. This review considers 1) quantitative integration of INaL into the established electrophysiological and Ca2+ regulatory mechanisms in normal and failing cardiomyocytes and 2) a new therapeutic strategy utilizing a selective inhibition of INaL to target both arrhythmias and impaired contractility in HF.
Keywords: Late sodium current, heart failure, calcium, action potential, numerical model, sodium-calcium exchanger
1. Introduction
Congestive heart failure (HF) is associated with profound abnormalities in both cardiac rhythm and contractile function. Among numerous proteins involved in the cardiac cell alterations in HF, the voltage-gated Na+ channels deserve special consideration, as they seem to be critically involved in abnormal conduction, repolarization, and Ca2+ handling (Bers et al., 2006; Tomaselli and Zipes, 2004). Most Na+ channels open only transiently and are quickly inactivated resulting in the peak transient current, INaT, which determines excitation and conduction. However, some Na+ channels remain active, carrying so-called persistent or late Na+ current (INaL) throughout the action potential (AP) plateau (reviews (Carmeliet, 2006; Noble and Noble, 2006)).
A growing body of evidence accumulated over the last decade shows that INaL provides a major contribution to the AP plateau in ventricular cardiomyocytes (VCs) in a variety of mammalian species including humans (Maltsev et al., 1998a). Single channel studies in human VCs and in heterologously expressed channels show that INaL is produced by the cardiac Na+ channel isoform (Nav1.5), operating in special gating modes (Undrovinas et al., 2002). INaL has a voltage-activated gating similar to INaT and a slow inactivation, which clearly separate INaL from background Na+ currents.
Since late openings of Na+ channel generate both electric current and Na+ influx during the AP plateau, INaL is expected to contribute to at least two known HF cellular mechanisms (depicted in Fig.1): 1) electrophysiological alterations and 2) altered cell Na+ cycling. The latter mechanism is tightly integrated with Ca2+ cycling, as Na+ modulates the Na+/Ca2+ exchanger (NCX) operation (Bers et al., 2006). These anticipated INaL contributions could be amplified at the state of chronic HF that reportedly slows the late Na+ channel gating and increases the whole cell INaL (Maltsev et al., 2007; Maltsev and Undrovinas, 2006; Undrovinas et al., 1999). While mechanisms of the INaL change are mainly unknown, pharmacological properties of INaL remain unchanged in chronic HF and the late channel activity exhibits same gating modes (Maltsev et al., 1998a; Maltsev et al., 2007; Undrovinas et al., 2002; Undrovinas et al., 1999). This raises a possibility that HF alters INaL by affecting Na+ channel environment factors known to modulate Na+ channel gating, such as auxiliary subunits, Ca2+/CaM/CaMKII, cytoskeleton, membrane phospholipid composition , and other scaffolding proteins such as caveolin.
Figure 1.
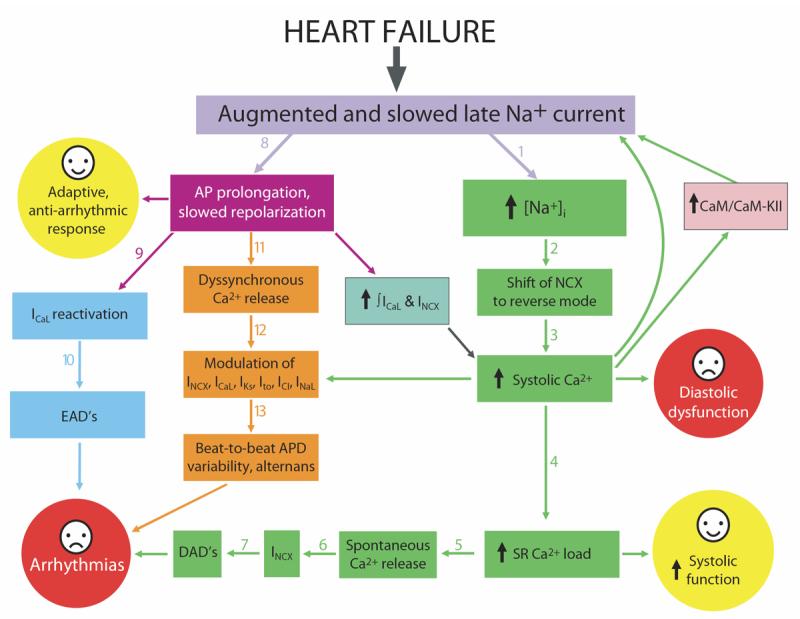
Integration of the late Na+ current into electrophysiological and Ca2+ regulatory mechanism in heart failure (see text for detail).
The importance of INaL contribution into HF mechanisms has been demonstrated in experiments designed to “correct” INaL. It was shown that a partial INaL inhibition and/or acceleration of INaL decay rescue normal repolarization, decrease beat-to-beat AP duration variability, and improve Ca2+ handling and contractility of the failing cells (Maltsev et al., 1998b; Maltsev et al., 2007; Undrovinas et al., 2006; Undrovinas et al., 1999). Thus, INaL has emerged as a novel target for cardioprotection to treat the failing heart (Belardinelli et al., 2006; Maltsev et al., 2001; Noble and Noble, 2006).The focus of this review is to summarize the available data from experiment and numerical modeling of INaL in normal and failing myocardium in order to integrate INaL into the established electrophysiological and Ca2+ regulation mechanisms in normal and failing cardiomyocytes. We believe that such integration will improve our understanding of cardiac cell function under normal and pathological conditions and thus provide a basis for the development of new therapeutic strategies for cardioprotection.
2 The idea and first experimental evidence of a persistent Na+ current from early AP studies
The concept of a persistent (“plateau”) Na+ current in heart cells emerged when the first model of cardiac AP was developed by Denis Noble in 1960-1962 (Noble, 1960; Noble, 1962). At that time, Ca2+ current in cardiac cells (Reuter, 1967) had not yet been discovered, and persistent Na+ current was suggested to explain a relatively long (vs. neuronal) AP plateau duration and cardiac AP shortening when external Na+ is reduced (Weidmann, 1956) (see details in review (Noble, 2006)). Further experimental indication for the existence of a persistent Na+ current and for its contribution to AP duration was found in microelectrode studies in cardiac Purkinje fibers in 1967 (Dudel et al., 1967). It was shown that tetrodotoxin (TTX), as well as many local anesthetic-type antiarrhythmic agents, shortens the cardiac AP (Carmeliet and Saikawa, 1982; Coraboeuf et al., 1979; Davis and Temte, 1969; Dudel et al., 1967; Gliklich and Hoffman, 1978). Coraboeuf and colleagues (Coraboeuf et al., 1979) found that TTX at concentrations lower than 1 μM shortenes AP and induces small hyperpolarization, but does not affect its (dV/dt)max. They concluded that this effect was related to a slow component of Na+ current flowing through a TTX-sensitive Na+ channel. Accordingly, slow Na+ current was believed to be a specific feature of cardiac Purkinje fibers, explaining the longer AP in Purkinje fibers compared to that in the working myocardium.
3. Classification of Na+ currents: the late/persistent Na+ current is different from the “window” current and “background” currents
Further voltage clamp studies have identified several types of single Na+ channel activity and whole cell Na+ currents that could contribute to AP duration. Late openings of Na+ channels were found not only in Purkinje cells but also in myocardial cells. The variety of Na+ channel activities identified so far can be classified (see review (Noble and Noble, 2006)) in terms of the “window” current, the late (persistent) Na+ current i.e. INaL (IpNa), and background Na+ currents.
3.1. The “window” Na+ current
This current was suggested as a theoretical mechanism to explain the persistent Na+ current in cardiac cells (Attwell et al., 1979; Gadsby and Cranefield, 1977). According to the Hodgkin-Huxley formalism (Hodgkin and Huxley, 1952), the “window” current is a non-inactivating component of INaT, resulting from the crossover of its steady-state activation and inactivation curves.
3.2. The late Na+ current, INaL
Voltage clamp studies, however, showed that the concept of the “window” current could not explain some fundamental properties of the late Na+ current, such as its slow inactivation and voltage-dependence.
INaL exhibits slow inactivation
Gintant et al. (Gintant et al., 1984) and Carmeliet (Carmeliet, 1987), using two-microelectrode voltage clamp, demonstrated that INaL in canine and rabbit cardiac Purkinje fibers undergoes a slow inactivation, which deviates from the Hodgkin-Huxley formalism and thus does not support the “window” origin of the current. Furthermore, in single-channel patch-clamp studies, Patlak and Ortiz (Patlak and Ortiz, 1985) discovered bursts of openings of Na+ channels that could underlie the late, slowly inactivated Na+ current in rat VCs. Similar decaying burst activity of Na+ channel was also observed in rabbit Purkinje cells (Zilberter et al., 1994). In addition to bursts, Kiyosue and Arita (Kiyosue and Arita, 1989) identified a sustained channel activity consisting of rare and brief openings in guinea pig VCs. This type of late openings was found to be slowly inactivating in human VCs (Undrovinas et al., 2002)(see section 5 for details). The current produced by the late openings of Na+ channel seemed to play a role in AP plateau not only of Purkinje fibers but also of VCs, because TTX produced 10 to 20 % decrease in AP duration in these cells (Kiyosue and Arita, 1989; Maltsev et al., 1998a). Both TTX- and lidocaine-sensitive slowly inactivating whole-cell inward current in the AP plateau voltage range was reported by Wasserstrom and Salata in dog ventricular myocytes (Wasserstrom and Salata, 1988).
INaL is present within a wide range of voltages similar to that of INaT
The overlap of steady-state activation and inactivation curves occurs within a relatively narrow region of voltages close to INaT activation threshold. However, late openings of the Na+ channel were found at voltages far out of the overlap (e.g. −10 mV, see Fig.5C) (Undrovinas et al., 2002) and significant whole cell INaL is present even at positive voltages (Fig.2B, filled circles) (Maltsev et al., 1998a; Sakmann et al., 2000), where the calculated “window” current is negligible.
Figure 5.

Inactivation of both late scattered mode (A) or burst mode (B) of the late openings of Na+ channel was slowest in failing human cardiomyocytes compared with those from normal human hearts or heterologously expressed Nav1.5. *P<0.05, heart failure vs. normal heart or clone (Mean±SEM). Cell-attached patches: Vh = −140 mV, 24°C. (Reprinted from (Maltsev and Undrovinas, 2006) with permission from European Society of Cardiology). C: recordings of action potentials in failing human cardiomyocytes are shown along with late scattered mode and burst mode openings occurring at −10 mV, i.e. within the voltages of the action potential plateau. Reprinted from (Undrovinas et al., 2002), used with permission from European Society of Cardiology.
Figure 2.

Biophysical properties of the slowly inactivating, late Na+ current (INaL) evaluated by whole cell patch clamp in human ventricular cardiomyocytes (A-E) and human cloned Nav1.5 expressed in tsA201 cells (F). A-B: Late current can be carried either by Na+ or Li+. B: I-V relation for INaL. C: examples of steady-state activation and availability curves, G(Vm) and A(Vp), respectively. D: Examples of original traces illustrating voltage-independent INaL decay. E: slow reactivation of INaL. F: INaL produced by Nav1.5 was assessed as difference current before application of a selective Na+ channel blocker tetrodotoxin (TTX, 30 μM) (20 averaged traces) and after TTX (14 averaged traces). Voltage protocols are shown at the traces. Recording was performed at 24°C. Reprinted from (Maltsev et al., 1998a) (A-E) and (Undrovinas et al., 2002)(F), used with permission.
3.3 Background Na+ currents
A Na+-dependent current, called the Na+ background current (IbNa), was found in rabbit sinoatrial node (Hagiwara et al., 1992) and in guinea pig ventricular and atrial cells (Kiyosue et al., 1993). In contrast to INaL, it is a non-inactivating current with poor cation selectivity and no voltage dependence. The molecular and genetic origins of IbNa remain unknown, but it could result from a leak form of Na+−K+ ATPase (Artigas and Gadsby, 2004) or of NCX (Hilgemann, 2004). This TTX-insensitive IbNa is different from another “background” type Na+ current, which is produced by openings of Na+ channels. A non-inactivating, highly TTX sensitive “background” Na+ channel activity was also recorded within the wide range of membrane potentials in the rabbit Purkinje cells (Zilberter et al., 1994) and in rat cardiomyocytes (Saint et al., 1992). In contrast to slowly inactivating INaL, this type of background Na+ current has much higher sensitivity to TTX and exhibits neither voltage-gated activation, nor voltage-dependent steady-state inactivation; this current persists at potentials ranging from −120 to 0 mV with an almost linear I-V relationship. Experimental hypoxia and metabolic inhibitors, such as cyanide, reportedly augment the highly TTX sensitive background current (Ju et al., 1996). However, studies in human or canine VCs showed neither any TTX-blockable (25 μM) current at membrane potential < −80 mV nor any effect of cyanides, suggesting species-specific differences in the background current (Maltsev et al., 1998a; Maltsev et al., 2007).
A non-inactivating current with almost no voltage-dependency in the range of −60 to −20 mV and no voltage-dependent steady-state inactivation was reported in dog and human myocardium at a non-physiological [Na+]o=5 mM (Valdivia et al., 2005). All these properties are characteristic of a “background” type current rather than INaL. Being saxitoxin (STX) sensitive, it is supposed to be produced by Na+ channel openings; however, the current consists of large, step-like fluctuations (±20 pA, i.e. ∼30% of the current amplitude, see Fig. 3 in (Valdivia et al., 2005)), which are unlikely to originate from individual openings of the STX-sensitive Na+ channels especially at the low [Na+]o. On the other hand, it has been recently demonstrated that low [Na+]o destabilizes inactivation of Na+ channel (Aman and Raman, 2006). It was suggested that occupancy of the channel pore by the permeant ion substantially affects slow inactivation process. Thus, molecular identity, ion selectivity, and gating properties of the ion channel underlying this “background” current remain unknown.
Figure 3.

A: Chronic heart failure slows and increases INaL. A: examples of whole cell INaL recordings in human and dog ventricular cardiomyocytes. B: Idealized INaL and their integrals in normal and failing dog cardiomyocytes of same size (200 pF) calculated using Q10 factors (37°C) and average parameters of INaL density and decay time constant. Larger and slower INaL in failing cardiomyocytes results in substantial increase in total charge (or Na+) transfer by INaL. Gray areas illustrate difference between failing and normal cells. Adapted from (Maltsev et al., 2007), used with permission.
4. Whole-cell INaL in normal and failing human and canine ventricular myocardium
4.1. Biophysical characteristics of INaL
Our patch clamp studies characterized major biophysical and pharmacological characteristics of the whole-cell INaL in human VCs (Maltsev et al., 1998a; Maltsev et al., 2001; Maltsev et al., 2007) (summarized in Fig. 2). Many key properties of INaL turned out to be similar to those of INaT. The voltage dependencies of steady-state activation and availability of INaL are almost identical to those of INaT, and the I-V relationships of these currents almost coincide. Ion selectivity (Li+-permeable and Cs+-impermeable) and pharmacological properties for INaL (see section 6.3 for details) and INaT also proved to be the same. INaL time course measured 200 ms after membrane depolarization is well described by a single exponential decay with a time constant of about 0.5 sec at 24°C. Surprisingly and unlike INaT, the decay time course of INaL does not depend on the membrane potential. Similarly, INaL reactivation is very slow and voltage-independent. Furthermore, inactivation of the major portion of INaT by a short depolarizing prepulse does not affect INaL. This feature indicates that kinetic transitions between different states of the Na+ channels responsible for INaT or INaL are independent. This property of INaL can be exploited for both experimental (separation of these two currents) and theoretical (modeling) purposes.
A slowly inactivating INaL with aforementioned biophysical characteristics is produced by heterologously expressed cardiac Na+ channel isoform main α-subunit Nav1.5 (Fig.2F). It has also been identified in VCs of dogs (Maltsev et al., 2007; Undrovinas et al., 2006; Undrovinas et al., 1999; Zygmunt et al., 2001), guinea pigs (Belardinelli et al., 2006; La et al., 2006; Sakmann et al., 2000), rabbits (Wu et al., 2006) and rats (Chattou et al., 2000).
4.2. Whole cell INaL is larger and slower in chronic HF
The first evidence of importance of INaL in electrophysiological alterations induced by HF was found in 1995 in an experimental dog model of chronic HF (Maltsev et al., 1995). In VCs isolated from these failing hearts, the N-shaped I-V curve for the total current had a significantly increased contribution of a late/persistent, TTX and STX-blockable inward current within the AP plateau range. Experiments in human hearts demonstrated that TTX produces a similar effect on the I-V curve and AP shape of human failing VCs, indicating that INaL is present in humans and thus may have a clinical relevance (Maltsev et al., 1998a). Further patch clamp studies in human and canine hearts have conclusively shown that chronic HF increases INaL density and significantly slows inactivation kinetics of INaL in VCs (Maltsev et al., 2007; Undrovinas et al., 1999) (Fig.3A). Analysis of idealized INaL time course (Fig.3B) in canine normal and failing VCs of the same size (200 pF) shows that 1) absolute INaL difference between normal and failing canine VCs at 37°C is bell-shaped (top panel in Fig. 3B, gray area) with a maximum of 24.5 pA at 90 ms after membrane depolarization, i.e. within the time of AP plateau duration; 2) the relative difference between normal and failing cells increases progressively with membrane depolarization, doubling after 330 ms (Fig. 3B, inset); 3) both absolute and relative differences are much greater at 37°C than at 24°C (Maltsev et al., 2007).
5. Gating of late Na+ channel openings
5.1. Separate Na+ channel gating modes contribute to INaL and INaT
The INaL inactivation can be described in terms of channel gating modes each of which describes a separate set of gating parameters for the same channel type. To our knowledge, the idea and the experimental evidence for gating modes of cardiac Na+ channel were first reported in 1985 by Patlak and Ortiz (Patlak and Ortiz, 1985). In patch-clamped rat VCs, they occasionally observed bursts of 10 or more sequential openings of a single channel that lasted for up to 150 ms. They concluded that “the single channel data cannot be explained by standard models, even those that have two inactivated states or two open states of the channel. Our results suggest that Na+ channels can function in several different ‘modes,’ each with a different inactivation rate.” As INaL lasts hundreds of milliseconds, the numerous short-lived modes (a few milliseconds of membrane depolarization) identified by further patch clamp studies in myocardial cells of a variety of species (mice (Bohle and Benndorf, 1995), guinea pig (Nilius, 1988) and humans (Bohle et al., 2002)) contribute to INaT rather than INaL. More specifically, all 5 gating modes of Na+ channel (F, M1, M2, S, and P-mode) identified by Böhle et al. (Bohle et al., 2002) in human VCs operate within 5 ms of membrane depolarization and thus have no contribution to INaL.
5.2. Gating modes underlying INaL in human VCs: bursts and late scattered openings
Most human cardiac Na+ channels open transiently upon membrane depolarization and then are quickly inactivated (Fig.4A). However, some channels remain active, carrying INaL. In multi-channel cell-attached patches from heterologously expressed Nav1.5 and human VCs, this late activity is arranged in two major gating modes: late scattered mode (LSM) and “burst” mode (Fig 4B,C). The channel openings of the two gating modes have almost identical single channel conductance and the reversal potential (Undrovinas et al., 2002). The latencies of LSM openings reveal voltage-independent ultraslow (hundreds of ms) inactivation, similar to that of whole-cell current decay (Undrovinas et al., 2002). LSM openings render one open voltage-independent state but burst mode openings exhibit one open (voltage-dependent) and two closed states (one voltage-dependent and another voltage-independent) (Undrovinas et al., 2002).
Figure 4.

Examples of Na+ channel activities that underlie peak (“early openings”, panel A) and late (panels B-D) Na+ currents in normal and failing human cardiomyocytes as well as in cardiac channel clone (Nav1.5) expressed in HEK293 cells. Note similarity between modes of late activity in normal hearts, failing hearts, and the clone. Cell-attached patches were held at −130 mV and step clamped to −30 mV at 23°C. Reprinted from (Undrovinas et al., 2002), used with permission from European Society of Cardiology.
5.3 Alteration of late Na+ channel gating in human failing VCs
Since whole cell studies identified slower and larger INaL in the failing VCs (Fig.3)(Maltsev et al., 2007; Undrovinas et al., 1999), one might expect that single channel activity underlying INaL is different in failing cells. However, no qualitative changes in Na+ channel gating were found in failing human VCs (Undrovinas et al., 2002); they exhibit early openings (Fig.4A) and the two modes of late gating (Fig.4D) with the same single channel conductance (Undrovinas et al., 2002) as the late openings of both normal VCs and Nav1.5 (Fig.4B,C). Quantitative characterization and comparison of late Na+ channel gating is rather difficult because assessment of late openings from one and only one channel in the patch is technically impossible. Though it is possible to fabricate a small patch pipette covering a membrane patch with just one Na+ channel (Benndorf, 1988), the probability that it will operate in a late gating mode (either burst mode or LSM) is extremely low. Accordingly, late Na+ channel activity is recorded in relatively large multi-channel (8-15) patches (Maltsev and Undrovinas, 2006; Patlak and Ortiz, 1985; Undrovinas et al., 2002). However, even in such patches late openings of Na+ channel are still insufficient to yield a smooth ensemble average current. Instead, the slow inactivation kinetics of the late channel has been assessed from a decay of the latencies of the channel openings (Undrovinas et al., 2002). This approach has shown a significantly slower inactivation of LSM in failing vs. normal VCs or heterologously expressed Nav1.5 (Fig.5A) (Maltsev and Undrovinas, 2006). As to the burst mode, the total burst length decreased with depolarization and was significantly larger in failing compared to normal myocytes and Nav1.5 (Fig.5B).
6. Molecular identity of INaL in normal and failing ventricular myocardium
6.1. Expression of multiple Na+ channel isoforms in heart
Numerous Na+ channel isoforms have been reported in heart, with Nav1.5 dominating (Goldin et al., 2000). Molecular identity of INaL needs careful consideration especially for failing hearts where gene expression profile changes dramatically. Different transcripts of highly TTX-sensitive neuronal isoforms (Nav1.1, 1.3, 1.6) have been discovered in mouse and dog heart (Nav1.1, 1.2, 1.3) (Haufe et al., 2005; Maier et al., 2002). These studies suggested species-specific expression and protein localization within sarcolemmal compartments. These neuronal Na+ channel isoforms are responsible for 10 to 20% of the Na+ current peak in myocardial and Purkinje cells, respectively (Haufe et al., 2005). Furthermore, four splice variants of SCN5A gene that encodes Nav1.5 have been reported in humans (Tan et al., 2005). These variants are present in human myocardium and differ in their biophysical properties (Tan et al., 2005). Contribution of these isoforms and/or variants in INaL and its modulation by different pathological conditions is not yet understood. The problem of molecular identity of INaL has been approached in many different ways including methods of biophysics, pharmacology, and molecular biology.
6.2. Whole cell and single channel data in human VCs and heterologously expressed Nav1.5
Late Na+ channel openings show similar gating modes and Na+ conductance in normal human cardiomyocytes and in heterologously expressed Nav1.5 (section 5.2). These data indicate the common molecular origin of the late channel openings i.e. they are produced by Nav1.5 in human cardiomyocytes. The heterologously expressed Nav1.5 produces the slowly inactivating whole cell INaL in tsA201 cells (Fig.2F) similar to that observed in human cardiomyocytes (Fig.2A). Studies of late Na+ single-channel activity in failing human cardiomyocytes did not reveal any peculiar Na+ channels, compared to normal hearts or heterologously expressed Nav1.5 (sections 5.2, 5.3). Accordingly, the HF-related alterations in burst and late scattered openings (Fig. 5A,B) that underlie whole-cell INaL changes (Fig.3) (Maltsev et al., 2007) are likely due to a modulation of the same Nav1.5 rather than to an isoform (or a splice variant) switch.
6.3. Pharmacological data in humans and dogs
Difference of 2 amino acids in the Na+ channel pore region accounts for the high affinity of neuronal Na+ channel and low affinity of cardiac Na+ channel for TTX or STX, the feature commonly used to distinguish these Na+ channel isoforms pharmacologically (Heinemann et al., 1992). Dose–response curves for blockade of INaL by TTX and STX reveal only a single-site binding with the half-concentration that is typical for the Nav1.5 in both dog and human failing hearts (IC50 was 1.2 vs. 1.53 μM for TTX and 62 vs. 98 nM for STX comparing dog vs. human cardiomyocytes) (Maltsev et al., 1998a; Maltsev et al., 2007). Further evidence of Nav1.5 origin of INaL is its sensitivity to Cd2+ (IC50=104 μM) (Maltsev et al., 2007), as Cd2+ distinguishes between cardiac and neuronal Na+ channel isoforms (Satin et al., 1992). Furthermore, if INaL did represent the activity of channels with different pharmacological and gating properties, the extent of TTX blockade of INaL would differ at different time points after membrane depolarization, interfering with the decay time course. On the contrary, in both humans and dogs the extent of toxin-induced blockade stayed approximately constant during INaL time course between 200 and 1000 ms. No significant “non-inactivated” or “steady-state” Na+ current blocked by the toxins was observed as of 2 s of membrane depolarization (Maltsev et al., 1998a; Maltsev et al., 2007). These data suggest that within this time frame INaL is mainly determined by the activity of only one type of Na+ channel. Thus, INaL in canine and human VCs has a common molecular origin (i.e. Nav1.5) and, therefore, similar molecular mechanisms could be involved in alteration of INaL in HF in both species.
6.4. siRNA data
Silencing the SCN5A gene responsible for Nav1.5 expression with siRNA decreases INaL by 75% in wide range of membrane potentials (including AP plateau), resulting in a significant reduction of AP duration and variability in dogs with chronic HF (Undrovinas et al., 2005) (Fig.6).
Figure 6.
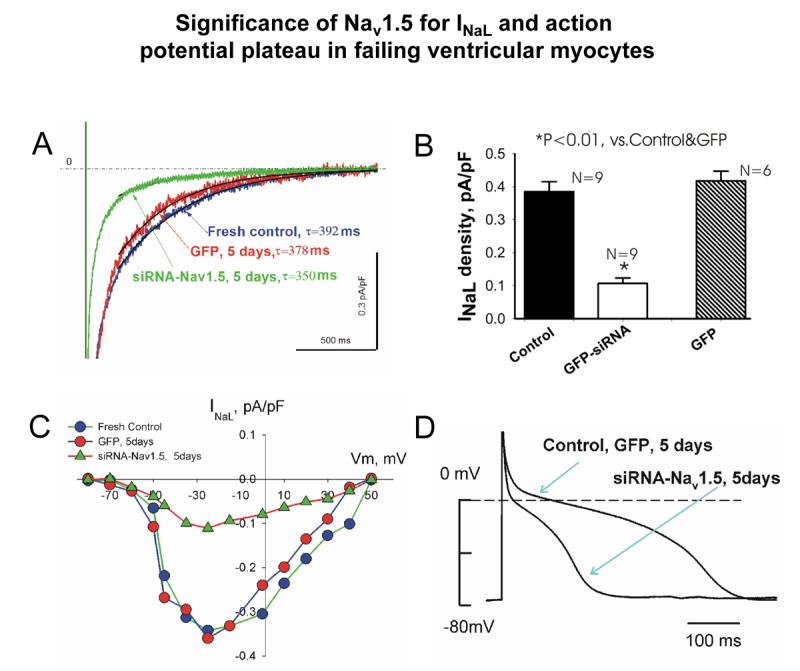
SCN5A gene regulates late sodium current (INaL) and action potential duration in failing heart. Silencing of SCN5A gene expression in ventricular cardiomyocytes of dogs with HF by adenovirally-transferred siRNA causes significant reduction in INaL density (A and B) in wide range of membrane potentials (C) and reduction of action potential duration (D). Shown are typical examples of INaL recordings, current-voltage relationships (whole cell patch clamp, 24°C) and action potential recordings (perforated patch, 35°C, 0.5 Hz pacing rate). Labels: “fresh control”- freshly isolated cells; “GFP” –cultured cells expressing reporter gene GFP, and “siRNA”- cultured cells with silenced SCN5A. Average data for action potential duration: fresh control, 515±51 ms, n=18; GFP control, 479±73 ms, n=8; siRNA, 181±17 ms, n=11; mean±SEM; n is number of cells; * P<0.01, vs. fresh control and GFP, ANOVA. From (Undrovinas et al., 2005).
6.5. Summary for INaL molecular identity
All above findings support the hypothesis that Nav1.5 is a major contributor in INaL in normal and failing myocardium. However, an unknown channel with pharmacological and gating properties similar to Nav1.5 might still be contributing to INaL. The only reported mammalian Na+ channel isoform, other than Nav1.5, that can produce late openings and possesses a low affinity for TTX is Nav1.8 (gene SCN10A) (Akopian et al., 1996). However, up to date mRNA encoding Nav1.8 has not been detected in rat hearts (Akopian et al., 1996), failing human heart tissue, or isolated VCs (Maltsev et al., 2007).
7. Modulation of Na+ channels as a possible mechanism for INaL alterations in pathological conditions
7.1. Na+ channel structure has multiple links to modulatory proteins
The function of the Na+ channel is not fully determined by its protein structure, but depends also on its environment. Na+ channels represent a multi-protein complex comprising not only the main pore-forming α subunit and its auxiliary β subunits, but also components of cytoskeleton, Ca2+-sensitive protein calmodulin, regulatory kinases and phosphatases, trafficking proteins, and extracellular matrix proteins embedded into lipid bilayer plasma membrane. A diagram of Nav1.5 and interacting proteins is shown in Fig.7A. For elegant in-depth reviews on this topic please see (Abriel and Kass, 2005; Meadows and Isom, 2005; Nerbonne and Kass, 2005). Here we highlight only the components that seem to modulate INaL in different pathological conditions, including hypoxia and HF. The III-IV linker (see Fig. 7B) is responsible for Na+ channel inactivation, and mutations in this region disrupt Nav1.5 inactivation causing persistent Na+ current linked to inherited LQT3 syndrome (Bennett et al., 1995). Recently COOH terminal that has binding sites for numerous regulatory proteins, has been implicated in Nav1.5 inactivation (Cormier et al., 2002; Motoike et al., 2004).
Figure 7.

Schematic illustration of Na+ channel as a macromolecular complex. A: The pore forming α subunit of the channel interacts with β-subunits, cytoskeleton and the extracellular matrix (Modified after (Nerbonne and Kass, 2005), used with permission). B: schematic presentation of the α subunit of the cardiac Na+ channel isoform (Nav1.5) with reported sites of interaction with β subunits (restricted only to β1 and β2) and other regulatory proteins.
7.2 Modulation of INaL by β subunits
Many of the mammalian voltage gated Na+ channels are associated with auxiliary β subunits. The β-subunit gene family has four members β1(SCN1B), β2 (SCN2B), β3 (SCN3B) β4 (SCN4B) (see for review (Meadows and Isom, 2005; Nerbonne and Kass, 2005)). All these β-subunits are expressed in rodent hearts and are differently localized to specific subcellular domains and cell types. β1 subunit is non-covalently attached to α subunit, and β2 subunit is covalently linked to α subunit by a disulfide bond (Messner and Catterall, 1986). The protein of these β subunits contains an extracellular amino-terminus, a single transmembrane segment, and an intracellular COOH terminus (Fig. 7B). The extracellular N-terminus of all β subunits contain immunoglobulin domain found in cell adhesion molecules. Particularly, immunoglobulin domain for β2 (and probably for β4) is similar to contactin, whereas for β1 and its splice variant β1A it is similar to myelin Po This unique property allows interactions with variety of signaling molecules and components of the extracellular matrix. With the regard to the intracellular domain, direct interactions of β1 subunit C terminus and ankyrin B in rat brain membranes have been demonstrated, indicating a role of this subunit in Nav localization. Direct interaction between cytoplasmic C terminus domain of Nav1.1 with β1 and β3 has been recently demonstrated (Spampanato et al., 2004). β subunits do not form an ion-conducting pore, but modulate Na+ channel function, Na+ channel protein expression at the plasma membrane (trafficking), and cell adhesion (Meadows and Isom, 2005; Nerbonne and Kass, 2005). More specifically, β1-subunit: 1) is involved in abnormal Na+ channel activity associated with the LQT3 mutation (An et al., 1998), 2) aggravates Na+ channel dysfunction in Brugada syndrome (Makita et al., 2000), 3) modifies the blockade of Na+ channel by fatty acids (Xiao et al., 2000) and lidocaine (Makielski et al., 1999), 4) modulates the trafficking of Nav1.5 (Zhou et al., 2000), and 5) affects the burst mode of the heterologously expressed skeletal muscle Na+ channel isoform in Xenopus oocytes (Chang et al., 1996).
Very few reports are currently available about modulation by β subunits of late openings of the cardiac Na+ channel. Heterologous co-expression of β1 subunit with Nav1.5 in HEK293 cells diminishes a non-inactivating Na+ current (Valdivia et al., 2002). Co-expression of β1 subunit with Nav1.5 in tsA201 cells increases INaL amplitude and significantly slows INaL decay, whereas co-expression of β2 subunit does not affect INaL parameters (Undrovinas and Maltsev, 2001). The potency of β1 subunit to modulate INaL has been confirmed in native cell environment. In normal dog VCs, knocking down of SCN1B by antisense nucleotides significantly accelerated INaL decay (Undrovinas and Maltsev, 2002)(Fig. 8A). In HF, Nav1.5 protein level is downregulated, whereas β1 has remains unchanged, indicating a relatively higher membrane content of β1 (Zicha et al., 2004). This suggests, although indirectly, potential involvement of β1 in the reported INaL alterations in HF. Possible involvement of other β subunits in INaL changes in HF awaits further studies.
Figure 8.

Modulation of INaL by the channel environment elements: auxilliary β1-subunit and β-spectrin-based cytoskeleton. A: In normal dog ventricular myocytes, knocking down of SCN1B by antisense oligonucleotide (β1asOLI) significantly accelerates INaL decay compared to control nonsence oligonucleotide (nsOLI) (Undrovinas and Maltsev, 2002). B: Exposure of the cytoplasmic side of Na+ channel to the specific anti-β-spectrin antibody dramatically enhances activity of late Na+ channel openings culminating in the increased ensemble averaged late current. (rat ventricular myocytes, excised inside-out patch, from Undrovinas, Dubreuil and Makielski, unpublished)
7.3. Modulation of INaL by cytoskeleton
The cytoskeleton is a collection of interacting proteins that forms a framework within the cytoplasm with connections to the membrane that include attachments to integral membrane proteins. This framework maintains cell shape, plasma membrane integrity, and localization of membrane proteins such as ion exchange carriers and ion channels (Fig 7, see for review (Abriel and Kass, 2005; Bennett and Baines, 2001; Meadows and Isom, 2005; Nerbonne and Kass, 2005)).
Ankyrin-B
An adapter protein ankyrin-B, which is expressed in heart, links Nav1.5 to the cytoskeleton. A knockout of ankyrin B in mice affects late Na+ channel openings in cardiomyocytes (Chauhan et al., 2000). Furthermore, mutations in ankyrin B cause the LQT4 syndrome in one French family (Mohler et al., 2003), indicating the importance of channel environment for the channel function. Accordingly, a disruption of any member of this multi-protein complex by pathological conditions may lead to alterations of INaL.
F-actin
Cytochalasin-D, an agent that interferes with F-actin polymerization, slows inactivation of cardiac Na+ channel by inducing bursts of openings. It also affects coupling of steady state activation and availability of Na+ current (Maltsev and Undrovinas, 1996; Undrovinas et al., 1995).
Fodrin
Fodrin (spectrin)-based cytoskeleton, another element of the Na+ channel microenvironment in heart, is a dynamic structure that is altered under a variety of pathological conditions (e.g. ischemia or heart failure (Hein et al., 2000; Heling et al., 2000; Yoshida et al., 1995)). The role of the fodrin-based cytoskeleton in INaL modulation has been confirmed in experiments with anti-β-spectrin antibodies (Fig. 8B) and with the antisense oligonucleotides targeting mRNA's encoding α- and β-fodrin in dog VCs (Undrovinas and Maltsev, 2003).
The fodrin breakdown that occurs in some disease states featuring poor Ca2+ handling can be mediated by the Ca2+ - activated enzyme calpain (Matsumura et al., 2001; Yoshida et al., 1995). Therefore, disturbances of the ankyrin and/or fodrin-based cytoskeleton may affect the Na+ channel inactivation process. Cytoskeletal elements such as ankyrin may bind directly to α and β subunits. The link to β subunits may thus modulate INaL indirectly (as discussed in section 7.2 for β1 subunit).
7.4. Modulation of INaL by lysophosphatidylcholine (LPC)
LPC is the endogenous amphiphilic lipid metabolite that accumulates in ischemic myocardium and represents a major factor causing the electrophysiological alterations that contribute to arrhythmogenesis (Corr et al., 1987; DaTorre et al., 1991). A significant prolongation of QTc interval has been recently found during early transmural ischemia in patients undergoing balloon angioplasty (Kenigsberg et al., 2007). Experimentally, LPC causes depolarization, reduction of the maximal upstroke velocity of AP, sustained abnormal rhythmic activity in Purkinje fibers, and delayed afterpotentials (DADs) in isolated tissue (Arnsdorf and Sawicki, 1981; Corr et al., 1987). The mechanisms of the LPC effects include modifications of Na+ current (Burnashev et al., 1991; Burnashev et al., 1989; Chattou et al., 2000; Shander et al., 1996; Undrovinas et al., 1992). Such modifications account for a reduction of INaT and an emergence of late openings that produce a sustained Na+ current. Interestingly, the late openings caused by LPC form clusters of the synchronized multiple channel openings (Burnashev et al., 1989; Undrovinas et al., 1992); we believe that this functional cooperation of individual channels represents a new fundamental phenomenon and deserves further studies. One of the mechanisms underlying these LPC-induced modifications might be an integration of LPC into the lipid membrane, which would increase the membrane fluidity (Fink and Gross, 1984). This, in turn, enhances motility and interaction of proteins within the membrane. On the other hand, LPC can activate neuromodulation signaling via PKA and PKC (Obata, 2002; Scott et al., 2006), affecting Na+ channel slow inactivation (Chen et al., 2006).
7.5. Modulation of INaL by Ca2+, CaM, and CaM-KII
Ca2+ dependent mechanisms of INaL modulation are discussed in detail later in the review in relation to altered Ca2+ handling in HF VCs (section 9.2).
7.6. Other recently emerged modulators
A novel protein partner of Nav1.5 has been discovered using an yeast 2-hybrid screen (Allouis et al., 2006). This protein, called 14-3-3η, interacts with the cytoplasmic I interdomain of Na+ channel (see Fig. 7B). Although its direct effect on INaL properties has not yet been studied, it was shown that this protein influences the inactivation process by delaying recovery from inactivation. Additionally, Src family tyrosine kinase Fyn has phosphorylation site on Nav1.5A III-IV linker (Ahern et al., 2005), which is known to be responsible for the inactivation. The most recently emerged modulator is a membrane microdomain protein Caveolin-3. This protein was previously linked to Na+ channel trafficking, (Yarbrough et al., 2002) but now is implicated in LQT syndrome (Vatta et al., 2006). Implementation of these new players in INaL modulation in different pathological conditions awaits further studies.
8. Experimental evidence of INaL importance for abnormal repolarization, Ca2+ handling, and contractility in failing myocardium
8.1 Inhibition of INaL normalizes AP duration and beat-to-beat variability and eliminates EADs in HF VCs
AP duration (APD) is extremely frequency dependent in failing VCs (Fig.9A; (Undrovinas et al., 1999)). At low pacing rates of 0.2-0.5 Hz the mean APD is significantly larger than in normal hearts (Fig.9A), and failing cells eventually exhibit EADs (Fig. 5C) (Maltsev et al., 1998a; Maltsev et al., 2007; Undrovinas et al., 1999). In addition to the prolongation, APD also exhibits significant beat-to-beat variability in failing VCs. Although AP prolongation is not evident at physiological frequencies (1Hz and higher), variability of APD in failing cells is substantially larger than in normal cells (compare APD distributions and their SD values in Fig. 9B) (Maltsev et al., 2007; Undrovinas et al., 1999). Increased and slowed INaL (Fig. 3) seems to be an important factor in APD prolongation, variability, and EADs. Partial reduction in the magnitude of INaL caused by specific Na+ channel blockers (TTX,1.5 μM, STX, 100 nM), a new antianginal drug ranolazine (that turned out to be a specific INaL blocker), or injection of external current opposite to INaL during the AP plateau significantly shorten the prolonged APD, decrease beat-to-beat variability of APD, and abolish EADs (Maltsev et al., 1998a; Maltsev et al., 2007; Undrovinas et al., 2006; Undrovinas et al., 1999).
Figure 9.

A, Frequency-dependence of action potential duration in ventricular cardiomyocytes of normal dogs and dogs with chronic heart failure. Note that largest difference occurs at low pacing rates. B: at the low (0.2 Hz) and the physiologic (1 Hz) pacing rates, AP duration in failing myocytes exhibits significant beat-to beat variability (see respective SD values in the APD90 distribution histograms) Adapted from (Undrovinas et al., 1999) used with permission.
8.2. Blockade of INaL improves Ca2+ transient and contractility in HF
The contractile dysfunction in HF is related not only to ongoing loss of functional cardiac units, but also to abnormal function of cardiomyocytes. HF is characterized by systolic dysfunction due to depressed Ca2+ transients (Bers et al., 2006) and by abnormal relaxation of cardiac myocytes (Davies et al., 1995; Gwathmey et al., 1987). At low pacing rates, the prolonged relaxation is associated with the spike-dome configuration of contractile response (Fig.10A). The ratio between amplitudes of the spike and dome phases was suggested to be an index for the severity of HF (Gwathmey et al., 1987). Similar to the shape of abnormal contraction, abnormally prolonged Ca2+ transients have also been observed in both ventricular muscle strips (Gwathmey et al., 1987) and cardiomyocytes isolated from failing hearts (Beuckelmann and Erdmann, 1992; Maltsev et al., 1998b) (Fig.10A). At the higher frequencies, these abnormalities account for the reversal of the force-frequency relationship in failing myocardium, leading to an increase in diastolic [Ca2+]i and diastolic tension (Fig.10B “Control”) (Feldman et al., 1988; Undrovinas et al., 2006). A partial blockade of INaL by STX, TTX or ranolazine greatly improves performance of failing VCs; it abolishes the dome phase of both contraction-relaxation cycle and Ca2+ transient at low pacing rates and prevents the rising diastolic tension and [Ca2+]i at the higher pacing rates in failing VCs (Maltsev et al., 1998b; Undrovinas et al., 2006) (Fig.10).
Figure 10.

A: Examples of effects of a specific Na+ channel blocker saxitoxin (STX) on AP duration, contraction and Ca2+ transient in ventricular cardiomyocytes of dogs with chronic heart failure at a low pacing rate of 0.2 Hz STX reduces AP duration, abolishes “dome” phase of contraction and of Ca2+ transient in failing cells. B: At higher pacing rates a specific INaL blockers ranolazine reduces diastolic tension, and a specific Na+ channel blocker tetrodotoxin (TTX) reduces Ca2+ accumulation (Fluo-4 signals). Adapted from (Maltsev et al., 1998b; Undrovinas et al., 2006), used with permission.
9. Integration of INaL into electrophysiological and Ca2+ mechanisms in HF (Fig.1).
Dramatic improvement of cardiomyocyte function due to inhibition of INaL described above is evidence for a substantial INaL contribution in the failing heart. However, the interpretation of these effects needs extreme care because electrophysiology, contraction, and Ca2+ dynamics in cardiomyocytes are interrelated via multiple feedback mechanisms. The extent of deterioration of cardiomyocyte function and these feedback mechanisms vary greatly with the progression of HF and etiology. Since late openings of Na+ channels generate both an electric current and a Na+ influx, INaL will contribute to at least two established HF mechanisms: 1) electrophysiological alterations (Tomaselli and Zipes, 2004); and 2) altered cell Na+ and Ca2+ cycling (Bers et al., 2006).
9.1 The role of INaL in AP prolongation and EADs
Given the high membrane resistance during the AP plateau, INaL provides a critical contribution to the altered delicate balance of ion currents and thus to the AP duration. The relative contribution of INaL to the AP plateau in failing VCs is amplified by the reduced K+ currents in HF (Tomaselli and Zipes, 2004). Since HF simultaneously increases and slows INaL (sections 4.2 and 5.3), INaL will contribute directly to AP prolongation in HF, thus explaining APD normalization with INaL inhibition described above. Prolonged APD allows more time for ICaL reactivation and thus facilitates EADs (January and Riddle, 1989). Since INaL contributes to AP prolongation, it thus indirectly contributes to EADs (transitions 8-9-10 in Fig.1). Accordingly, inhibiting INaL eliminates EADs likely due to APD shortening.
9.2. INaL and elevated [Na+]i increase Ca2+ entry via NCX and limit depression of systolic function
A major problem in HF is systolic dysfunction, which is associated with smaller Ca2+ transient and sarcoplasmic reticulum (SR) Ca2+ content. The extent of deterioration of systolic function is limited by multiple compensatory mechanisms (listed below), which are indirectly linked to INaL and elevated Na+ (Fig.1).
1) NCX function depends on Na+ and Ca2+ concentrations and membrane voltage. In HF VCs, increased Na+ influx (including that via INaL) shifts the NCX operation from the predominant forward mode to the reverse mode i.e. from Ca2+ efflux to Ca2+ entry (Baartscheer et al., 2003). Elevated Na+ in HF thus limits SR unloading and provides additional Ca2+ influx during the AP (Bers et al., 2006). Interestingly, in addition to preserving SR Ca2+ load, this operational shift in failing human myocardium results in the direct activation of contraction during the terminal phases of the AP via the reverse mode NCX Ca2+ influx (Weisser-Thomas et al., 2003).
2) The vast majority of studies demonstrated that NCX is upregulated in HF (Studer et al., 1994), therefore the above effect could be amplified by the enhanced NCX function.
3) INaL contributes to APD prolongation and thus indirectly prolongs Ca2+ influxes via ICaL and the reverse mode NCX.
4) INaL in HF is positively modulated by intracellular Ca2+ (Maltsev et al., 2002a) which could yield a new possible amplification mechanism of the Ca2+ entry. This creates a positive feedback loop from INaL via NCX to larger cell Ca2+ load, then from the larger Ca2+ load back to INaL. What is known about the mechanisms of Ca2+ modulation of Na+ channels and INaL? Structurally, the carboxyl terminus of Na+ channels has binding sites for Ca2+ itself (Wingo et al., 2004) and for Ca2+-binding protein calmodulin (CaM) that acts as a Ca2+ sensor translating changes in cytoplasmic Ca2+ into cellular responses (Mori et al., 2000). Discovery of these sites inspired multiple studies in heterelogously expressed Na+ channels, including brain, skeletal and cardiac muscle isoforms (Deschenes et al., 2002; Herzog et al., 2003; Kim et al., 2004; Tan et al., 2002; Young and Caldwell, 2005). It was found that some inactivation states of INaT of heterologously expressed cardiac and skeletal Na+ channel isoforms may be modulated directly by Ca2+, CaM and/or via Ca2+ /CaM /CaM-kinase cascade. The most recent study showed that CaMKIIδc enhances INaL and increases [Na]i (Wagner et al., 2006). In normal and especially in failing VCs, elevated [Ca2+]i slows the decay of INaL and increases INaL amplitude and integral (gray area in Fig.11A,B)(Maltsev et al., 2002a). CaM, and CaM-kinase seem to be involved in this modulation because specific antagonists of CaM and CaM-kinase (P209-309 and KN93, respectively) or CaM-KII blocker KN93 significantly accelerate INaL at high [Ca2+]i (Fig.11C) (Maltsev et al., 2002a)
Figure 11.

Modulation of INaL by intracellular Ca2+ and CaM/CaM-Kinase signaling pathway in cardiomyocytes from normal and failing dog hearts. Elevated intracellular Ca2+ concentration up to 1 μM dramatically increases and slowes INaL. A, B: representative traces recorded in cardiomyocytes of failing hearts at “low” and “high” intracellular Ca2+. Gray areas indicate total integrals of INaL that proportionate to Na+ influx by INaL (beginning from 200 ms after depolarization onset depicted by the vertical bar). C: statistical data on INaL decay changes produced by perturbations of Ca2+/CaM/CaM-KII cascade in cardiomyocytes from normal and failing dog hearts. N corresponds to the number of cells tested. Adapted from (Maltsev et al., 2002a), used with permission.
9.3. Adverse effects of increased [Ca2+]i in HF. Role of INaL and NCX in diastolic dysfunction
While the increased cell Ca2+ load limits the depression of systolic function in HF, it also leads to diastolic dysfunction, especially at high rates as described in section 8.2. Relaxation of cardiac myocytes occurs when [Ca2+]i declines, allowing Ca2+ dissociation from the myofilaments. Ca2+ is removed from cytosol, mainly via SERCA, which takes Ca2+ back into the SR, and by NCX operating in forward mode during diastole (Bers, 1991). It is believed that the diastolic dysfunction in HF is mainly due to a reduced SERCA function in HF. At the same time, increased expression and function of NCX in HF tends to offset the deficiency of Ca2+ removal by SERCA (review (Bers et al., 2006)). The contribution of the increased INaL to the Ca2+ removal could be twofold. First, as discussed in section 9.2, INaL and related increase of [Na+]i facilitate Ca2+ influx. Secondly, higher [Na+]i during diastole partially offsets the function of the forward mode NCX and thus worsens the problems of Ca2+ removal from the cytosol and diastolic dysfunction. The improvement of diastolic function by the inhibition of INaL (Fig.10B) can be attributed both to a decrease in Ca2+ load during the AP plateau and to improved removal of Ca2+ by forward mode NCX during diastole. The importance of Na+ influx and of the forward mode NCX function for abnormal Ca2+ handling in HF VCs is illustrated in experiments with temporal substitution of Li+ for Na+ (Fig.12B). These conditions, designed to inhibit NCX function (at least in part, see Fig. 12 legend) and Na+ influx, accelerate Ca2+ transient decay, decrease diastolic [Ca2+]i, and greatly improve overall contractile performance of failing VCs. Since the late current is almost equally carried by Na+ and Li+ (see Fig.2A, B), it is likely preserved under these conditions. This experiment thus illustrates the importance of INaL-related Na+ influx (rather than the INaL-electric current per se) for abnormal Ca2+ handling and cell contraction in failing VCs. A partial blockade of the NCX also improves EC coupling in HF (Hobai et al., 2004) and reduces both EADs and DADs (Nagy et al., 2004; Pogwizd and Bers, 2002), indicating that NCX could be a promising therapeutic target in HF (Shah et al., 2005) (Sipido et al., 2006).
Figure 12.

A: Examples of fluctuations of Ca2+ transient (Fluo 4 signals) observed in ventricular myocytes of a canine chronic HF model at low pacing rates. B: Replacement of external Na+ by Li+ accelerates Ca2+ transients, abolishes Ca2+ oscillations, and decreases diastolic Ca2+ level in canine failing cardiomyocytes at low and high pacing rates, respectively (Undrovinas N., Sabbah H., Undrovinas A., unpublished). To avoid Li+ accumulation in the cells, the Na+-free solution was applied only for a few contraction cycles. Also, a new member NCLX of NCX superfamily has been recently discovered in a variety of tissues including heart (Palty et al., 2004). A distinct feature of NCLX is its ability to slowly transport Li+ in exchange to Ca2+, indicating that Li+ can be no longer considered as an “ideal” NCX blocker. Accordingly, if NCLX is functional in the failing canine myocytes, it could also prevent Li+ accumulation in the experimentation illustrated in panel B.
9.4. Ca2+ overload and increased diastolic Na+: potential importance of INaL for DADs
DADs occur due to spontaneous Ca2+ releases during diastole via the activation of the forward mode of NCX. INaL does not occur at low diastolic potentials in humans and dogs (see Fig. 2B, filled circles), so it cannot have a direct contribution to DADs. However, as discussed above, in HF VCs INaL increases Ca2+ influx leading to SR Ca2+ overload, which, in turn, is critical for initiation of spontaneous Ca2+ release (such as Ca2+ waves) during diastole. INaL involvement in DADs is thus limited to the contribution of the INaL to the Ca2+ overload. On the other hand, high diastolic [Na+]i in HF (Despa et al., 2002) decreases the forward mode NCX current and hence can attenuate the amplitude of DADs. This positive factor of INaL can be counterbalanced by downregulation of IK1 in HF (Kaab et al., 1996), facilitating membrane excitations.
9.5. Role of INaL in dispersion of repolarization: a feedback from abnormal Ca2+ cycling?
The mechanisms of dispersion of repolarization in HF remain unclear. A possible mechanism involves beat-to-beat alternations and/or fluctuations in intracellular Ca2+ cycling transduced to abnormal repolarization by electrogenic feedback mechanisms (review (Wilson et al., 2006), Fig.1). Fluctuations of Ca2+ transient can be observed in failing VCs of a canine chronic HF model (Fig.12A). Listed are three possible indirect contributions of INaL to the beat-to-beat variability problem.
1) The INaL contribution to the abnormal Ca2+ cycling (section 9.2).
2) The INaL dependence on Ca2+/CaM/CaMKII (section 9.2). Thus INaL can serve as one of the electrogenic Ca2+ feedback mechanisms along with other Ca2+ -dependent ion currents, such as ICaL inactivation, IKs, Ito, Ca2+-activated Cl−-current, and NCX.
3) The INaL contributes to AP shape that, in turn, might cause fluctuations of Ca2+ transients. The synchronicity of Ca2+ release depends on the state of phosphorylation of Ca2+ cycling proteins (Song et al., 2001) and on the AP shape (Sah et al., 2002). More specifically, the faster the repolarization, the more synchronous is the Ca2+ release. Failing VCs isolated from the infarct border zone exhibit dyssynchronous SR Ca2+ release from junctional SR (Litwin et al., 2000). Also, SR Ca2+ release is spatially and temporally variable from beat to beat in feline failing VCs (Harris et al., 2005). Since voltage clamped normal and failing feline VCs showed almost the same Ca2+ dynamics, the abnormal AP shape (significant loss of phase 1 AP repolarization) is likely to be the reason for desynchronized Ca2+ release in this feline HF model (Harris et al., 2005). The contribution of INaL could be important for the abnormal early repolarization phase in HF via its increased burst mode (Maltsev and Undrovinas, 2006) (Fig. 5B), especially when Ito is decreased (Tomaselli and Zipes, 2004). Further, late scattered openings of Na+ channel also increase in human HF VCs (Maltsev and Undrovinas, 2006) (Fig. 5A); they persist on the AP plateau (Fig. 5C) and hence tend to prevent further repolarization. Thus late openings of both Na+ gating modes can indirectly (i.e. via the AP shape) contribute to the desynchronized, fluctuating Ca2+ release.
9.6. Summary of INaL role in failing myocardium: friend or foe?
INaL and its Na+ influx can directly and indirectly contribute to several important HF mechanisms related to electrophysiological alterations and ion homeostasis. These contributions could either improve or worsen performance of HF myocardium, i.e. being “friend” or “foe”, respectively (Fig.1). INaL is “friend” as it contributes to 1) APD prolongation as an adaptive and an anti-arrhythmic (anti-re-entry) response (Tomaselli and Zipes, 2004); and 2) Ca2+ entry to limit depression of systolic function. The latter mechanism is an intrinsic, adaptive, digitalis-like effect with all corresponding risks and benefits. Interestingly, a large slow component of the Na+ current decay (burst mode) has been identified in post-myocardial infarction-remodeled myocytes (Huang et al., 2001), i.e. in the transitional period from an infarction to HF. The increased INaL may indeed serve as an initial mechanism of adaptation to match an increased contractility demand for the survived, noninfarcted VCs.
Although APD prolongation can be beneficial in HF, the temporal and spatial dispersion of repolarization that accompanies AP prolongation is critical for arrhythmia and sudden cardiac death (Tomaselli and Zipes, 2004). Additionally, DADs and EADs have critical importance for nonreentrant arrhythmias or triggered activity (recent review (Bers et al., 2006)). Accordingly, INaL is “foe” as it contributes to EADs, DADs, dispersion of repolarization, and diastolic dysfunction.
10. INaL is a novel target for cardioprotection aiming at arrhythmias and Na+-Ca2+ overload. Treatment strategies and requirements for new drugs, targeting Na+ channels
After negative outcomes of Cardiac Arrhythmia Suppression Trial (CAST) (Epstein et al., 1991; Epstein et al., 1993) Class I antiarrhyrhmic drugs (Vaughan Williams, 1984) targeting Na+ channels became close to a “taboo”. The trial tested whether the suppression of ventricular arrhythmias by encainide, flecainide, moricizine after myocardial infarction improves survival. The conclusion was that the “treatment strategies designed solely to suppress these arrhythmias should no longer be followed” (Epstein et al., 1993). However, the discovery of inherited mutations in SCN5A gene that lead to an increased INaL (LQT3 syndrome, see for review (Shah et al., 2005)) and of increased and slowed INaL in acquired, chronic HF (see sections 4.2 and 5.3) gives rise to a revival of Na+ channels as a therapeutic target. These studies suggest that not all Na+ channels must be equally targeted. The emerging paradigm for Na+ channels in HF is that INaT is decreased (Maltsev et al., 2002b; Valdivia et al., 2005; Zicha et al., 2004) but simultaneously INaL is increased (section 4.2). Blockers of INaT are proarrhythmic in HF because they slow conduction, thus worsening conduction problems (review (Shah et al., 2005)) and facilitating development of re-entry. Accordingly, new strategies for treatment must be considered: the new type of “smart” drugs should preferentially block INaL over INaT. This requirement calls for a new classification of Class 1 drugs in the future.
What are the potential benefits of a preferential INaL blockade in HF? Based on the summary of INaL integration into HF mechanisms shown in Fig.1, this could be both an antiarrhythmic effect and an improvement of diastolic function. A preferential blockade of INaL over INaT in failing human VCs was reported for amiodarone (Maltsev et al., 2001), suggesting an explanation of why amiodarone, classified as Class III anti-arrhythmic drug, shows an outstanding efficiency among K+-channel blockers. Table 1 summarizes potencies of various drugs to block INaT and INaL based on their ability to differentially interact with Na+ channel gating states. Based on a new index of the preferential blockade of INaL relative to INaT, the most promising drug is a new antianginal drug ranolazine (Fig.13) (Undrovinas et al., 2006).
Table1.
Interaction of different drugs with resting, inactivated and activated sodium channel responsible for peak transient (INaT) or late (INaL) currents in ventricular cardiomyocytes isolated from failing human and dog hearts: experimental results and theoretical predictions.
| Parameter | Protocol/ drug | Value | |
|---|---|---|---|
| Kd,peak, (μM) |
INaT dose-response, IC50 | Lidocaine | 291 |
| Amiodarone | 87±28 | ||
| Ranolazine | 294 | ||
| Kd,late, (μM) |
INaL dose-response | Lidocaine | 110.6 |
| Amiodarone | 6.7±1.1 | ||
| Ranolazine | 6.5 | ||
| Kdr, (μM) | Calculated from SSI shift for INaL |
Lidocaine | 178 |
| Amiodarone | 14.1±4.2 | ||
| Ranolazine | 7.47 | ||
| Kdi, (μM) | Calculated from SSI shift for INaL |
Lidocaine | 32 |
| Amiodarone | 0.15±0.02 | ||
| Ranolazine | 1.71 | ||
| Kblock, (μM−1s−1) |
Calculated from INaL decay acceleration |
Amiodarone | 0.239 |
Kd,peak Kd,late - half-blocking dose (IC50) was obtained from the single-site binding model fit to dose-response experimental data points for INaT and INaL, respectively, Kdr and Kdi are dissociation constant for the resting and inactivated state obtained from steady-state-inactivation (SSI) shifts in the presence of different drug concentrations (ΔV½ = 6.27*ln((1+[DRUG]/Kdr)/(1+[DRUG]/Kdi)). Kblock, association rate constant for the activated state. Methods used to obtain these values are referred as following protocols: SSI shift corresponds to a mid-point potential shift of Boltzmann function fit to the SSI, Kblock was calculated using a bimolecular model fit to the dose-dependent INaL acceleration assessed by the single-exponential model. Data sources: for amiodarone (Maltsev et al., 2001), lidocaine (Jia et al., 1993), Maltsev&Undrovinas, unpublished, and ranolazine (Undrovinas et al., 2006), accordingly.
Figure 13.

A: Comparison of the potencies of lidocaine, amiodarone and ranolazine to selectively inhibit INaL over the peak transient Na+ current (INaT). Shown are the ratios of IC50, i.e. the drug concentrations causing 50% resting block of the currents. The higher numbers correspond to the higher selectivity to block INaL. B, C: examples of a highly selective blockade of INaL over INaT produced by ranolazine in canine ventricular cardiomyocytes. Reprinted from (Undrovinas et al., 2006), used with permission.
The potential great benefits (preventing arrhythmias and Ca2+ overload) of the preferential INaL blockade can be expected not only in HF but in other cardiac diseases, such as ischemia, in which Na+-Ca2+ overload is a major feature. Since LPC dramatically increases INaL (section 7.4), targeting INaL in ischemia is especially encouraged. Inhibition of INaL by ranolazine reduces Ca2+ overload and left ventricle mechanical dysfunction during ischemia/reperfusion (Fraser et al., 2006). On the other hand, the therapeutic strategy of a preferential INaL blockade should be exercised with care. The balanced and effective therapy targeting INaL should take into account that INaL might be involved in adaptive response (see section 9.6) at different states of cardiac disease, making the question whether INaL is “friend” or “foe” vitally important.
11. Quantitative integration of INaL into electrophysiological and Ca2+ regulatory mechanisms: present state and future perspectives
11.1. INaL simulation using a general function: predictions of INaL role in normal myocardium
The first quantitative integration of INaL into an AP model was done by Sakmann et al. (Sakmann et al., 2000). As described in sections 3.2 and 4.1, a distinctive property of INaL is its voltage-gated activation. In their model, the experimentally obtained I-V relations were fitted by the least-squares method to the general function: I=G(V−E)/(1+exp[V'−V]/k), where I is the current, V is the voltage, Gis the maximum INaL conductance, E is the Na+ equilibrium potential, V' is the voltage at half-maximal activation, and k is the slope factor. These I-V relationships were incorporated into the standard guinea pig ventricular cell model in OXSOFT Heart 4.8. This model predicted the role of INaL in normal myocardium. As discussed in sections 2 and 3, multiple electrophysiology studies demonstrated a significant contribution of INaL in APD. For example, in normal human VCs in the presence of 1.5 μM TTX, APD was reduced by 15 to 20% (Maltsev et al., 1998a). Computer modeling studies fit these data surprisingly well and predicted transmural differences in repolarization based on the INaL gradients across the ventricular wall (Sakmann et al., 2000; Zygmunt et al., 2001). TTX effect on APD duration (% change) in human VCs showed remarkable rate dependence (Maltsev et al., 1998a) indicating that INaL could be involved in the rate-dependent AP adaptation. This mechanism is thought to be related to a slow reactivation of INaL (0.5 s for human VCs (Maltsev et al., 1998a)) resulting in an incomplete recovery from inactivation, INaL reduction, and enhancement of repolarization as rate increases (see review (Carmeliet, 2006)). To test this mechanism in quantitative terms, an AP model will need to integrate the dynamics of INaL reactivation.
11.2. A Markovian chain-based model of late channel openings yielding whole cell INaL
The total Na+ current including INaT and INaL in human VCs has been recently numerically modeled as a set of Markovian kinetic schemes, each of which represents a separate gating mode (Maltsev and Undrovinas, 2006). INaT was described by a single gating mode (Transient Mode, TM) and INaL was described by two gating modes: LSM and the burst mode (BM) (Fig.14A-C). The transition rates for INaL gating modes were estimated from patch clamp single channel data in human VCs.
Figure 14.

A-C: Markov chain kinetic models describing the three major modes of Na+ channel gating in human cardiomyocytes: transient mode (A), late scattered openings (B) and bursts (C), together with simulated traces and transition rates; traces were simulated from 3, 5, and 1 channel(s), respectively. Total simulation times are indicated above the traces. D,E: The model predicts increased and slowed INaL in a failing human ventricular myocyte vs. a normal human myocyte and heterologously expressed Nav1.5 for BM (F) and LSM (G). Inset in panel E shows a substantially larger integral of the LSM current in a failing myocyte vs. a normal myocyte; the box size is 50 pC ×1990 ms. Shown are simulated cumulative activities of 20000 LSM channels or 193 BM channels. Channel numbers were chosen to correspond to a typical human ventricular myocyte. Reprinted from (Maltsev and Undrovinas, 2006) with permission from European Society of Cardiology.
Based on the frequency of occurrence of different modes in patches with known numbers of channels, the populations of channels operating in each gating mode were estimated as follows: 79.8% for TM, 20% for LSM, and 0.2% for BM, yielding an apparent four exponential INa decay (τ=0.4, 4, 50, and 500 ms, at 24°C and −30 mV). In this model, the first two exponentials are produced by two inactivation states of TM, governed by rates c and e in Fig. 14A. The third exponential is related to apparent bursts inactivation, and the forth exponential is determined by inactivation of LSM reopenings governed by rate e in Fig14B. All four predicted components were clearly identified in whole-cell patch clamp recordings in human VCs (Maltsev and Undrovinas, 2006). The model also predicts the voltage and temperature dependence of the late gating modes and the dynamic contributions of each mode into the total Na+ current. The early phase of Na+ current decay at 24°C (<40 ms) involves all three Na+ channel gating modes, the intermediate phase (from 40 to 300 ms) is produced by BM+LSM, although the contribution of BM decreases with depolarization, and ultra-late decay (>300 ms) is determined solely by LSM. This outcome is paradoxical as it means that most INaL is produced by relatively rare scattered openings rather than bursts characterized by abundant channel activity. However, the burst contribution is limited as bursts occur in a much smaller fraction of Na+ channels than LSM (0.2% compared to 20%, respectively) and are inactivated much faster (with an apparent decay time constant of ∼50 ms vs. 500 ms). Furthermore, the contribution from the bursting channels is expected to decrease with depolarization (Fig 5B).
11.3. INaL provides substantial Na+ influx in HF VCs
Two numerical estimates suggest a substantial Na+ influx via INaL (Maltsev and Undrovinas, 2006): 1) INaL-related Na+ influx is similar in magnitude to that of INaT and 2) INaL integral significantly increases in HF. More specifically, a mathematical model based on single-channel data in human VCs described in section 11.2 shows that despite a much smaller channel population operating in LSM compared to TM (20% vs. 79.8%) and a much smaller scale of INaL compared to INaT, LSM and TM channels transfer almost the same amount of Na+ through the plasma membrane during 2 s depolarization. Numerical integration of simulated currents for each mode in a cell with 105 Na+ channels yields electrical charges of 42, 45 and 7 pC for TM, LSM, and BM, respectively. In other words, the INaT peak is about 3 orders of magnitude larger than INaL (50 nA compared to 50 pA), yet its span is about 3 orders of magnitude shorter (2 ms compared to 2 s), resulting in an almost equal total charge transfer.
INaL transfers significantly more Na+ into failing vs. normal VCs (Fig.14E, inset). The total charge transferred by INaL from 10 to 2000 ms is predicted to be 28.5 and 45 pC for normal and failing VCs, respectively, or a ∼58% increase. This model estimate is in line with a 53.6% HF-induced increase of Na+ influx via INaL evaluated from whole cell patch clamp measurements in canine normal and failing VCs (gray area in Fig.3B bottom panel; (Maltsev et al., 2007)). Taking into account that INaT is reduced by 30-40% in HF VCs (Maltsev et al., 2002b; Valdivia et al., 2005; Zicha et al., 2004), the role of INaL in Na+ homeostasis should be even more substantial in the failing cells, in line with the recent finding that [Na]i is significantly increased in failing paced cardiomyocytes (Despa et al., 2002). Modulation of INaL by Ca2+ (section 9.2) is another possibility for a larger Na+ transfer by INaL in HF, especially in cells with high [Ca2+]i.
11.4. Prediction of INaL importance for repolarization, EADs, and DADs
The functionality of integration of INaL into HF mechanisms (Fig.1, labeled by numbers) has been partly numerically tested by Denis Noble and co-authors in their several computer simulation studies, albeit in the models of normal (non-HF) cardiomyocytes. The critical importance of INaL in a hypothetical repolarization failure (transition 8 in Fig.1) has been demonstrated in numerical simulations (Noble and Noble, 2006) using the Sakmann's AP model of guinea pig VC (Sakmann et al., 2000) (described in section 11.1). More specifically, an inhibition of INaL (IpNa in their paper) protects from a repolarization failure produced by a blockade of IKr at an external K+ concentration of 3.5 mM. EADs in failing human VCs can be suppressed by a partial inhibition of INaL with TTX (Maltsev et al., 1998a). This result was reproduced in simulations when EADs were induced in the model by increasing IpNa (Noble, 1999) (transitions 8-9-10 in Fig.1). Other numerical simulations (Noble and Varghese, 1998) performed in a model of atrial cells demonstrated the link of [Na+]i to DADs (interactions 2-3-4-5-6-7 in Fig.1). It was shown that increasing [Na+]i leads to increased Ca2+ loading via NCX. Beyond a threshold (when [Na+]i exceeds 12–15 mM), increased Ca2+ loading triggers repetitive spontaneous release of Ca2+ from the SR. The oscillatory releases, in turn, activate forward mode NCX that produces an inward current and depolarizes cell membrane, resulting in DADs.
11.5. Summary and future perspective
A hypothetical integration of INaL into HF mechanisms, shown in Fig.1, was derived from established HF mechanisms and available data on INaL. It has not been systematically tested in quantitative terms. Quantitative integration of INaL into cell electrophysiological and Ca2+ regulatory mechanisms has been approached so far in non-HF and non-human cardiomyocytes. INaL was approximated with a general function that included only the voltage dependence of INaL activation, but missed the slow inactivation and the recovery from inactivation. These simulations mechanistically describe key phenomena (e.g. AP prolongation, EADs, and DADs) under specific conditions related to INaL and [Na+]i. Since biophysical properties of INaL and late Na+ channel gating have been characterized in detail (sections 4 and 5) and numerically modeled (section 11.2), further studies on INaL integration can consider incorporation of these detailed INaL formulations. Our simulations of INaL demonstrate substantial Na+ flux transferred by INaL that greatly increases in HF (section 11.3). They also describe complex effects of drugs on INaL based on the key parameters of their interactions with Na+ channels (e.g. amiodarone (Maltsev et al., 2001)). Finally, the formulations are based on the data obtained in human VCs (both normal and failing) and thus can be incorporated into human AP models. We believe that the upgraded models will predict whether a particular disease manifestation (such as shown in Fig.1) will wax or wane at a given state of cardiac disease, therapeutic interventions, and genetic factors.
Acknowledgments
We thank Nidas A. Undrovinas for providing data on Ca 2+ transients (Fig.12) and Jeanne Nerbonne for kindly providing a drawing of the Na+ channel macromolecular complex used for Fig.7A.
Sources of funding
This research was supported by grants from the National Institute of Health HL-53819, HL074238, research grant from CV Therapeutics, Palo Alto, CA (AU) , and the Intramural Research Program of the National Institutes of Health, National Institute on Aging (VAM).
References
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–998. doi: 10.1161/01.RES.0000166324.00524.dd. Epub 2005 Apr 2014. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressedy sensory neurones. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Allouis M, Le Bouffant F, Wilders R, Peroz D, Schott JJ, Noireaud J, Le Marec H, Merot J, Escande D, Baro I. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ Res. 2006;98:1538–1546. doi: 10.1161/01.RES.0000229244.97497.2c. Epub 2006 May 1525. [DOI] [PubMed] [Google Scholar]
- Aman TK, Raman IM. Subunit dependence of Na channel open-channel block and slow inactivation in cerebellar neurons. Biophys J. 2006;22:22. doi: 10.1529/biophysj.106.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, Kass RS. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha-and beta1-subunits. Circulation Research. 1998;83:141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- Arnsdorf MF, Sawicki GJ. The effects of lysophosphatidylcholine, a toxic metabolite of ischemia, on the components of cardiac excitability in sheep Purkinje fibers. Circulation Research. 1981;49:16–30. doi: 10.1161/01.res.49.1.16. [DOI] [PubMed] [Google Scholar]
- Artigas P, Gadsby DC. Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. J Gen Physiol. 2004;123:357–376. doi: 10.1085/jgp.200308964. Epub 2004 Mar 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Cohen I, Eisner D, Ohba M, Ojeda C. The steady state STX-sensitive (“window”) sodium current in cardiac Purkinje fibers. Pflügers Arch. 1979;379:137–142. doi: 10.1007/BF00586939. [DOI] [PubMed] [Google Scholar]
- Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–995. doi: 10.1016/s0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92:iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benndorf K. Patch clamp analysis of Na channel gating in mammalian myocardium: reconstruction of double pulse inactivation and voltage dependence of Na currents. Gen Physiol Biophys. 1988;7:353–377. [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- Bers DM. Developments in cardiovascular medicine. Vol. 122. Kluwer Academic Publishers; Dordrecht, Netherlands: 1991. Excitation-contraction coupling and cardiac contractile force. [Google Scholar]
- Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Erdmann E. Ca2+ -currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol. 1992;87:235–243. doi: 10.1007/978-3-642-72474-9_19. [DOI] [PubMed] [Google Scholar]
- Bohle T, Benndorf K. Multimodal action of single Na+ channels in myocardial mouse cells. Biophysical Journal. 1995;68:121–130. doi: 10.1016/S0006-3495(95)80166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohle T, Brandt MC, Lindner M, Beuckelmann DJ. Identification of gating modes in single native Na+ channels from human atrium and ventricle. Circ Res. 2002;91:421–426. doi: 10.1161/01.res.0000033521.38733.ef. [DOI] [PubMed] [Google Scholar]
- Burnashev NA, Undrovinas AI, Fleidervish IA, Makielski JC, Rosenshtraukh LV. Modulation of cardiac sodium channel gating by lysophosphatidylcholine. J Mol Cell Cardiol. 1991;23:23–30. doi: 10.1016/0022-2828(91)90020-m. [DOI] [PubMed] [Google Scholar]
- Burnashev NA, Undrovinas AI, Fleidervish IA, Rosenshtraukh LV. Ischemic poison lysophosphatidylcholine modifies heart sodium channels gating inducing long-lasting bursts of openings. Pflugers Arch. 1989;415:124–126. doi: 10.1007/BF00373151. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Slow inactivation of the sodium current in rabbit cardiac Purkinje fibres. Pflugers Archiv - European Journal of Physiology. 1987;408:18–26. doi: 10.1007/BF00581835. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Action potential duration, rate of stimulation, and intracellular sodium. J Cardiovasc Electrophysiol. 2006;17:S2–S7. doi: 10.1111/j.1540-8167.2006.00378.x. [DOI] [PubMed] [Google Scholar]
- Carmeliet E, Saikawa T. Shortening of the action potential and reduction of pacemaker activity by lidocaine, quinidine, and procainamide in sheep cardiac purkinje fibers. An effect on Na or K currents? Circ Res. 1982;50:257–272. doi: 10.1161/01.res.50.2.257. [DOI] [PubMed] [Google Scholar]
- Chang SY, Satin J, Fozzard HA. Modal behavior of the mu 1 Na+ channel and effects of coexpression of the beta 1-subunit. Biophys J. 1996;70:2581–2592. doi: 10.1016/S0006-3495(96)79829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattou S, Coulombe A, Diacono J, Le Grand B, John G, Feuvray D. Slowly inactivating component of sodium current in ventricular myocytes is decreased by diabetes and partially inhibited by known Na+-H+Exchange blockers. J Mol Cell Cardiol. 2000;32:1181–1192. doi: 10.1006/jmcc.2000.1151. [DOI] [PubMed] [Google Scholar]
- Chauhan VS, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res. 2000;86:441–447. doi: 10.1161/01.res.86.4.441. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol. 1979;236:H561–567. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- Cormier JW, Rivolta I, Tateyama M, Yang AS, Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- Corr PB, Yamada KA, Creer MH, Sharma AD, Sobel BE. Lysophosphoglycerides and ventricular fibrillation early after onset of ischemia. J Mol Cell Cardiol. 1987;19:45–53. doi: 10.1016/s0022-2828(87)80609-0. [DOI] [PubMed] [Google Scholar]
- DaTorre SD, Creer MH, Pogwizd SM, Corr PB. Amphipathic lipid metabolites and their relation to arrhythmogenesis in the ischemic heart. Journal of Molecular & Cellular Cardiology. 1991;23:11–22. doi: 10.1016/0022-2828(91)90019-i. [DOI] [PubMed] [Google Scholar]
- Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- Davis LD, Temte JV. Electrophysiological actions of lidocaine on canine ventricular muscle and Purkinje fibers. Circ Res. 1969;24:639–655. doi: 10.1161/01.res.24.5.639. [DOI] [PubMed] [Google Scholar]
- Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ Res. 2002;90:E49–57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Dudel J, Peper K, Rudel R, Trautwein W. The effect of tetrodotoxin on the membrane current in cardiac muscle (Purkinje fibers) Pflugers Arch Gesamte Physiol Menschen Tiere. 1967;295:213–226. doi: 10.1007/BF01844101. [DOI] [PubMed] [Google Scholar]
- Epstein AE, Bigger JT, Jr., Wyse DG, Romhilt DW, Reynolds-Haertle RA, Hallstrom AP. Events in the Cardiac Arrhythmia Suppression Trial (CAST): mortality in the entire population enrolled. J Am College Cardiol. 1991;18:14–19. doi: 10.1016/s0735-1097(10)80210-4. [DOI] [PubMed] [Google Scholar]
- Epstein AE, Hallstrom AP, Rogers WJ, Liebson PR, Seals AA, Anderson JL, Cohen JD, Capone RJ, Wyse DG. Mortality following ventricular arrhythmia suppression by encainide, flecainide, and moricizine after myocardial infarction. The original design concept of the Cardiac Arrhythmia Suppression Trial (CAST) Jama. 1993;270:2451–2455. [PubMed] [Google Scholar]
- Feldman MD, Gwathmey JK, Phillips P, Schoen F, Morgan JP. Reversal of the force-frequency relationship in working myocardium from patients with end-stage heart failure. J Applied Cardiol. 1988;3:273–283. doi: 10.1016/0022-2828(90)90975-8. [DOI] [PubMed] [Google Scholar]
- Fink KL, Gross RW. Modulation of canine myocardial sarcolemmal membrane fluidity by amphiphilic compounds. Circ Res. 1984;55:585–594. doi: 10.1161/01.res.55.5.585. [DOI] [PubMed] [Google Scholar]
- Fraser H, Belardinelli L, Wang L, Light PE, McVeigh JJ, Clanachan AS. Ranolazine decreases diastolic calcium accumulation caused by ATX-II or ischemia in rat hearts. J Mol Cell Cardiol. 2006;41:1031–1038. doi: 10.1016/j.yjmcc.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Cranefield PF. Two levels of resting potential in cardiac Purkinje fibers. J Gen Physiol. 1977;70:725–746. doi: 10.1085/jgp.70.6.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant GA, Datyner NB, Cohen IS. Slow inactivation of a tetrodotoxin-sensitive current in canine cardiac Purkinje fibers. Biophysical Journal. 1984;45:509–512. doi: 10.1016/S0006-3495(84)84187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliklich JI, Hoffman BF. Sites of action and active forms of lidocaine and some derivatives on cardiac Purkinje fibers. Circ Res. 1978;43:638–651. doi: 10.1161/01.res.43.4.638. [DOI] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- Hagiwara N, Irisawa H, Kasanuki H, Hosoda S. Background current in sino-atrial node cells of the rabbit heart. J Physiol. 1992;448:53–72. doi: 10.1113/jphysiol.1992.sp019029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, Santana LF, Houser SR. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543–550. doi: 10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, Dumaine R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–127. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Terlau H, Imoto K. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflugers Arch. 1992;422:90–92. doi: 10.1007/BF00381519. [DOI] [PubMed] [Google Scholar]
- Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klovekorn WP, Schlepper M, Schaper W, Schaper J. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23:8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW. New insights into the molecular and cellular workings of the cardiac Na+/Ca2+ exchanger. Am J Physiol Cell Physiol. 2004;287:C1167–1172. doi: 10.1152/ajpcell.00288.2004. [DOI] [PubMed] [Google Scholar]
- Hobai IA, Maack C, O'Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–299. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, El-Sherif T, Gidh-Jain M, Qin D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol. 2001;12:218–225. doi: 10.1046/j.1540-8167.2001.00218.x. [DOI] [PubMed] [Google Scholar]
- January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- Jia H, Furukawa T, Singer DH, Sakakibara Y, Eager S, Backer C, Arentzen C, Wasserstrom JA. Characteristics of lidocaine block of sodium channels in single human atrial cells. J Pharmacol ExpTher. 1993;264:1275–1284. [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- Kenigsberg DN, Khanal S, M. K, Krishnan SC. Prolongation of the QTc interval is seen uniformly during early transmural ischemia. J Am Coll Cardiol. 2007 doi: 10.1016/j.jacc.2006.11.035. in press. [DOI] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- Kiyosue T, Arita M. Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ Res. 1989;64:389–397. doi: 10.1161/01.res.64.2.389. [DOI] [PubMed] [Google Scholar]
- Kiyosue T, Spindler AJ, Noble SJ, Noble D. Background inward current in ventricular and atrial cells of the guinea-pig. Proc R Soc Lond B Biol Sci. 1993;252:65–74. doi: 10.1098/rspb.1993.0047. [DOI] [PubMed] [Google Scholar]
- La C, You Y, Zhabyeyev P, Pelzer DJ, McDonald TF. Ultraviolet photoalteration of late Na+ current in guinea-pig ventricular myocytes. J Membr Biol. 2006;210:43–50. doi: 10.1007/s00232-005-0844-6. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski JC, Limberis J, Fan Z, Kyle JW. Intrinsic lidocaine affinity for Na channels expressed in Xenopus oocytes depends on α (hH1 vs. rSkM1) and β1 subunits. Cardiovasc. Res. 1999;42:503–509. doi: 10.1016/s0008-6363(99)00024-3. [DOI] [PubMed] [Google Scholar]
- Makita N, Shirai N, Wandg DW, Sasaki K, George ALJ, Kanno M, Kitabatake A. Cardiac Na+ channel dysfunction in Brugada syndrome is aggravated by β1 - subunit. Circulation. 2000;101:54–60. doi: 10.1161/01.cir.101.1.54. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Lesch M, Undrovinas AI. A non-inactivating inward current in cardiomyocytes of dogs with chronic heart failure. Circulation. 1995;92:I-504. (Abstract) [Google Scholar]
- Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998a;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S, Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Molec Life Sci. 1998b;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: Studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Undrovinas AI. Calmodulin modulates late sodium current in normal and failing myocardium. Circulation. 2002a;106:II-227. [Google Scholar]
- Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002b;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Undrovinas AI. Cytoskeleton controls pool of available sodium channels in the heart. J. Mol. Cell. Cardiol. 1996;28:A162. [Google Scholar]
- Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Saeki E, Otsu K, Morita T, Takeda H, Kuzuya T, Hori M, Kusuoka H. Intracellular calcium level required for calpain activation in a single myocardial cell. J Mol Cell Cardiol. 2001;33:1133–1142. doi: 10.1006/jmcc.2001.1373. [DOI] [PubMed] [Google Scholar]
- Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–458. doi: 10.1016/j.cardiores.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Messner DJ, Catterall WA. The sodium channel from rat brain. Role of the beta 1 and beta 2 subunits in saxitoxin binding. J Biol Chem. 1986;261:211–215. [PubMed] [Google Scholar]
- Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy ZA, Virag L, Toth A, Biliczki P, Acsai K, Banyasz T, Nanasi P, Papp JG, Varro A. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–831. doi: 10.1038/sj.bjp.0706026. Epub 2004 Oct 2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Nilius B. Modal gating behavior of cardiac sodium channels in cell-free membrane patches. Biophysical Journal. 1988;53:857–862. doi: 10.1016/S0006-3495(88)83166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D. Cardiac action and pacemaker potentials based on the Hodgkin-Huxley equations. Nature. 1960;188:495–497. doi: 10.1038/188495b0. [DOI] [PubMed] [Google Scholar]
- Noble D. A modification of the Hodgkin--Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J Physiol. 1962;160:317–352. doi: 10.1113/jphysiol.1962.sp006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D. Reconstruction of cellular mechanisms of genetically based arrhythmias. J Physiol. 1999;518:2P–3P. [Google Scholar]
- Noble D. From the Hodgkin-Huxley axon to the Virtual Heart. J Physiol. 2006 Oct 12; doi: 10.1113/jphysiol.2006.119370. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Varghese A. Modelling of sodium-overload arrhythmias and their suppression. Can J Cardiol. 1998;14:97–100. [PubMed] [Google Scholar]
- Obata T. Adenosine production and its interaction with protection of ischemic and reperfusion injury of the myocardium. Life Sci. 2002;71:2083–2103. doi: 10.1016/s0024-3205(02)01993-8. [DOI] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279:25234–25240. doi: 10.1074/jbc.M401229200. Epub 22004 Apr 25231. [DOI] [PubMed] [Google Scholar]
- Patlak JB, Ortiz M. Slow currents through single sodium channels of the adult rat heart. J Gen Physiol. 1985;86:89–104. doi: 10.1085/jgp.86.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci. 2002;976:454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- Reuter H. The dependence of slow inward current in Purkinje fibres on the extracellular calcium-concentration. J Physiol. 1967;192:479–492. doi: 10.1113/jphysiol.1967.sp008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002;90:165–173. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann BF, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. [DOI] [PubMed] [Google Scholar]
- Satin J, Kyle JW, Chen M, Bell P, Cribbs LL, Fozzard HA, Rogart RB. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- Scott GA, Arioka M, Jacobs SE. Lysophosphatidylcholine mediates melanocyte dendricity through PKCzeta activation. J Invest Dermatol. 2006;5:5. doi: 10.1038/sj.jid.5700567. [DOI] [PubMed] [Google Scholar]
- Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- Shander GS, Undrovinas AI, Makielski JC. Rapid onset of lysophosphatidylcholine-induced modification of whole cell cardiac sodium current kinetics. J Mol Cell Cardiol. 1996;28:743–753. doi: 10.1006/jmcc.1996.0069. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Varro A, Eisner D. Sodium calcium exchange as a target for antiarrhythmic therapy. Handb Exp Pharmacol. 2006;159:199. doi: 10.1007/3-540-29715-4_6. [DOI] [PubMed] [Google Scholar]
- Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. beta-Adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, MacDonald BT, Levin SI, Soltesz I, Benna P, Montalenti E, Isom LL, Goldin AL, Meisler MH. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Tan BH, Valdivia CR, Rok BA, Ye B, Ruwaldt KM, Tester DJ, Ackerman MJ, Makielski JC, Pagel MD, Pu J. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Heart Rhythm. 2005;2:741–747. doi: 10.1016/j.hrthm.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, Anderson ME, Balser JR. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA. Modulation of late cardiac Na+ current by auxiliary β subunits. Pacing Clin Electrophysiol. 2001;24:622. [Google Scholar]
- Undrovinas AI, Maltsev VA. Molecular basis for late Na+ current. Knockdown of Na+ channel subunits in adult cardiomyocytes by antisense oligonucleotides. Biophys J. 2002;82:89a. [Google Scholar]
- Undrovinas AI, Maltsev VA. Spectrin-based cytoskeleton modulates late sodium currents in heart. Biophys. J. 2003;84:25A. [Google Scholar]
- Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Mishra S, Undrovinas NA. Silencing of SCN5A gene by siRNA descrease late sodium current and action potential duration in ventricular cardiomyocytes from dogs with chronic heart failure. Circulation. 2005;112:II-127. [Google Scholar]
- Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. American Journal of Physiology. 1995;269:H203–214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- Valdivia C, Nagatomo T, Makielski J. Late Na currents affected by alpha subunit isoform and beta1 subunit co-expression in HEK293 Cells. J Mol Cell Cardiol. 2002;34:1029. doi: 10.1006/jmcc.2002.2040. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Vaughan Williams EM. A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol. 1984;24:129–147. doi: 10.1002/j.1552-4604.1984.tb01822.x. [DOI] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006;22:22. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Salata JJ. Basis for tetrodotoxin and lidocaine effects on action potentials in dog ventricular myocytes. Am J Physiol. 1988;254:H1157–H1166. doi: 10.1152/ajpheart.1988.254.6.H1157. [DOI] [PubMed] [Google Scholar]
- Weidmann s. Electrophysiologie der Herzmuskelfaser. Huber; Bern: 1956. [Google Scholar]
- Weisser-Thomas J, Piacentino V, 3rd, Gaughan JP, Margulies K, Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–985. doi: 10.1016/s0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Wan X, Rosenbaum DS. Cellular alternans: a mechanism linking calcium cycling proteins to cardiac arrhythmogenesis. Ann N Y Acad Sci. 2006;1080:216–234. doi: 10.1196/annals.1380.018. [DOI] [PubMed] [Google Scholar]
- Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–225. doi: 10.1038/nsmb737. Epub 2004 Feb 2022. [DOI] [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. doi: 10.1124/jpet.105.094862. [DOI] [PubMed] [Google Scholar]
- Xiao YF, Wright SN, Wang GK, Morgan JP, Leaf A. Coexpression with β1-subunit miodifies the kinetics and fatty acid block of hH1α Na+ channels. Am. J. Physiol. 2000;279:H35–H46. doi: 10.1152/ajpheart.2000.279.1.H35. [DOI] [PubMed] [Google Scholar]
- Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T, Kawashima S, Sobue K. Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ Res. 1995;77:603–610. doi: 10.1161/01.res.77.3.603. [DOI] [PubMed] [Google Scholar]
- Young KA, Caldwell JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol. 2005;565:349–370. doi: 10.1113/jphysiol.2004.081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yui J, Hu NN, George ALJ, Murray KT. Activation of proteinkinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 2000;87:33–38. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter YI, Starmer CF, Starobin J, Grant AO. Late Na channels in cardiac cells: the physiological role of background Na channels. Biophys J. 1994;67:153–160. doi: 10.1016/S0006-3495(94)80464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]