

Abstract
High-risk HPV-associated anal neoplasms are difficult to treat and biomarkers of malignant progression are needed. A hallmark of carcinogenic progression is genomic instability, which is frequently associated with cell division errors and aneuploidy. The HPV-16 E7 oncoprotein has been previously shown to rapidly induce centriole and centrosome overduplication and to cooperate with HPV-16 E6 in the induction of abnormal multipolar mitoses. Based on this function, it has been suggested that HPV-16 E7 may act as a driving force for chromosomal instability. However, a detailed analysis of centrosome overduplication in primary HPV-associated neoplasms has not been performed so far. Here, we determined the frequency of centrosome overduplication in HPV-associated anal lesions using a recently identified marker for mature maternal centrioles, Cep170. We detected centrosome overduplication in a small but significant fraction of cells. Remarkably, centrosome overduplication, but not aberrant centrosome numbers per se or centrosome accumulation, correlated significantly with the presence of cell division errors. In addition, our experiments revealed that in particular pseudo-bipolar mitoses may play a role in the propagation of chromosomal instability in high-risk HPV-associated tumors. These results provide new insights into the role of centrosome aberrations in cell division errors and encourage further studies on centrosome overduplication as a predictive biomarker of malignant progression in HPV-associated anal lesions.
Introduction
Infection with high-risk HPV types such as HPV-16 is intimately associated with squamous cell carcinomas and pre-cancerous lesions of the anogenital tract (Palefsky, 1995; Palefsky et al., 1991). Men who have sex with men (MSM), in particular those who are seropositive for the human immunodeficiency virus (HIV), but also HIV-positive women, have a high risk for anal HPV infection and the development of high-grade anal intraepithelial lesions (AIN) and anal squamous cell carcinomas (Palefsky, 1998). Such lesions are difficult to treat and surgical therapy is frequently associated with significant post-operative morbidity (Abbasakoor and Boulos, 2005; Berry, Palefsky, and Welton, 2004). Hence, the identification of novel biomarkers to predict malignant progression of AIN would help to improve patient management and quality of life (Palefsky, 2006).
High-grade intraepithelial anal neoplasia has been proposed to represent a pre-cancerous lesion similar to HPV-associated cervical intraepithelial neoplasia (CIN). In CIN, malignant progression has been found to be associated with increasing aneuploidy and centrosome aberrations (Heselmeyer et al., 1997; Kashyap and Das, 1998; Monsonego et al., 1997; Skyldberg et al., 2001; Steinbeck, 1997). Centrosomes are small cytoplasmic organelles that function as microtubule organizing centers in most animal and human cells. They consist of a pair of centrioles, short barrel-shaped microtubule cylinders, which are embedded in pericentriolar material (PCM). During mitosis, centrosomes are involved in the organization and orientation of a bipolar mitotic spindle. Prior to mitosis, the single centrosome duplicates in synchrony with the cell division cycle. In this process, a pre-existing mother centriole serves as a nucleation center for normally only one newly synthesized daughter centriole (Bettencourt-Dias and Glover, 2007; Nigg, 2007; Sluder, 2004; Tsou and Stearns, 2006). The regulatory mechanisms that limit daughter centriole synthesis are poorly characterized but previous studies have shown that certain oncogenic stimuli, including the HPV-16 E7 oncoprotein (see also below), can rapidly disrupt this process (Duensing et al., 2007).
Centrosome aberrations are readily detectable in tumor samples (Lingle et al., 1998), but their use as cancer biomarkers has been hampered by the fact that it was not possible to distinguish between centrosome anomalies that potentially drive chromosomal instability and those that merely arise as a side effect of unrelated cellular insults such as abortive mitoses or incomplete cytokinesis (Duensing and Munger, 2001; Nigg, 2002). This distinction, however, is critical because centrosome aberrations may have fundamentally different consequences dependent on the cellular context in which they occur (Duensing, 2005).
High-risk HPVs encode two oncoproteins, E6 and E7, which function during the viral life cycle by promoting viral genome replication in differentiating keratinocytes. Whereas the HPV E6 oncoprotein inactivates the p53 tumor suppressor, the HPV E7 oncoprotein binds and degrades the retinoblastoma tumor suppressor protein (pRB) as well as the related proteins p107 and p130. HPV E7 is a multifunctional protein and several additional targets have been identified, including the cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p27Kip1, cyclins, histone deacetylases (HDACs) and several transcription regulators (Longworth and Laimins, 2004; Munger and Howley, 2002; zur Hausen, 1996).
The HPV-16 E6 and E7 oncoproteins not only stimulate abnormal centrosome numbers, they have also been instrumental in dissecting distinct mechanisms by which centrosome amplification may arise. The HPV-16 E7 oncoprotein was found to rapidly trigger an excessive production of daughter centrioles thereby rendering cells prone to multipolar mitoses and chromosomal aberrations in a subsequent cell division (Duensing et al., 2001; Duensing et al., 2000). Abnormal centrosome numbers were detected before cells became genomically unstable indicating that they may function as a driving force for chromosomal instability (Duensing et al., 2001). Moreover, it was recently discovered that HPV-16 E7 can promote the concurrent formation of more than one daughter centriole at a single mother, a phenotype that is normally not seen in proliferating cells but that can occur during ciliogenesis (Duensing et al., 2007). In contrast, the HPV-16 E6 oncoprotein was found to provoke an accumulation of centrosomes in cells that were frequently growth-arrested and hence unlikely to generate viable daughter cells and propagate chromosomal instability. Nonetheless, high-risk HPV E6 cooperates with the HPV E7 oncoprotein in the induction of abnormal mitoses and aneuploidy, most likely by relaxing p53 responses (Duensing et al., 2001).
The two basic mechanisms of centrosome amplification, centrosome overduplication and centrosome accumulation, can be discriminated by immunostaining for a novel centrosomal protein, Cep170 (Duensing et al., 2006; Guarguaglini et al., 2005). Cep170 specifically labels mature mother centrioles and therefore allows the distinction between a genuine overduplication of centrosomes in the presence of a single mature mother and centrosome accumulation that usually leads to multiple mature mother centrioles (Guarguaglini et al., 2005). The question whether centrosome overduplication exists in vivo, however, has not been addressed in detail.
Here, we analyzed a total of forty-four benign, pre-malignant or malignant HPV-associated anal lesions by double-immunofluorescence microscopy for γ-tubulin and Cep170 in order to ascertain the frequency of centrosome overduplication. We detected centrosome overduplication in a small but significant number of cells and found that it correlates with the presence of cell division errors. Remarkably, aberrant centrosome numbers per se or centrosome accumulation did not correlate significantly with the presence of cell division errors. We provide evidence that in particular pseudo-bipolar cell divisions may contribute to the propagation of chromosomal instability in human tumors whereas multipolar mitoses may not. Collectively, our results suggest that primary centrosome overduplication increases the risk for cell division errors in high-risk HPV-associated anal neoplasms and encourage further studies into the potential use of centrosome overduplication as a surrogate marker for chromosomal instability and carcinogenic progression.
Results
Centrosome overduplication in high-risk HPV-associated anal lesions
Formalin-fixed, paraffin-embedded specimens from a total of forty-four anal lesions were analyzed (Table 1). All anal squamous cell carcinomas (SCCs; n=14) as well as high-grade squamous intraepithelial lesions (HSILs, anal intraepithelial lesions [AINs] II-III; n=13) (Wright et al., 2002) were tested positive for high-risk HPV types (HPV-16/18 or HPV-31/33). All low-grade squamous intraepithelial lesions (LSILs; condylomas and AIN I; n=6) contained low-risk HPV DNA (HPV-6/11) but also high-risk HPV DNA. Squamous epithelial tissue from hemorrhoidal excision specimens (n=11) was used as control.
Table 1.
Diagnosis, HPV status and summary of centrosome and mitotic aberrations.
| Diagnosis | N= | HPV-16/-18/-31/-33 positive | HPV-6/-11 positive | Mean % of cells with aberrant centrosome numbers | Mean % of cells with centrosome over- duplication | Mean % of cells with centrosome accumulation | Mean % of abnormal metaphases per HPFa | Mean % of abnormal ana-/telophases per HPF |
|---|---|---|---|---|---|---|---|---|
| SCC | 14 | 14 | 1 | 5.8% | 0.7% | 5.2% | 0.4% | 0.04% |
| HSIL | 13 | 13 | 4 | 2.5% | 0.2% | 2.2% | 0.3% | 0.03% |
| LSIL | 6 | 6 | 6 | 4.7% | 0.1% | 4.6% | 0.02% | 0.02% |
| Controlb | 11 | ndc | nd | 1% | 0.03% | 0.9% | 0% | 0% |
SCC, squamous cell carcinoma; HSIL high-grade squamous intraepithelial lesion; LSIL, low-grade squamous intraepithelial lesion.
High-power field.
Squamous epithelial tissue from hemorrhoidal excision specimens.
Not done.
Tissue samples were analyzed by double-immunofluorescence microscopy for γ-tubulin, a PCM protein, and Cep170. Cep170 associates with mature mother centrioles and hence, cells in G1, S or early G2 phase contain a single Cep170-positive centriole. In late G2 phase, the second maternal centriole becomes positive for Cep170 (Guarguaglini et al., 2005; Fig. 1A). Only cells containing aberrant centrosome numbers (more than two centrosomes per cell as detected by γ-tubulin staining) in the presence of a single centrosome positive for Cep170 were counted as centrosome overduplication (Fig. 1A).
Figure 1. Centrosome overduplication is present in high-risk HPV-associated anal neoplasms.
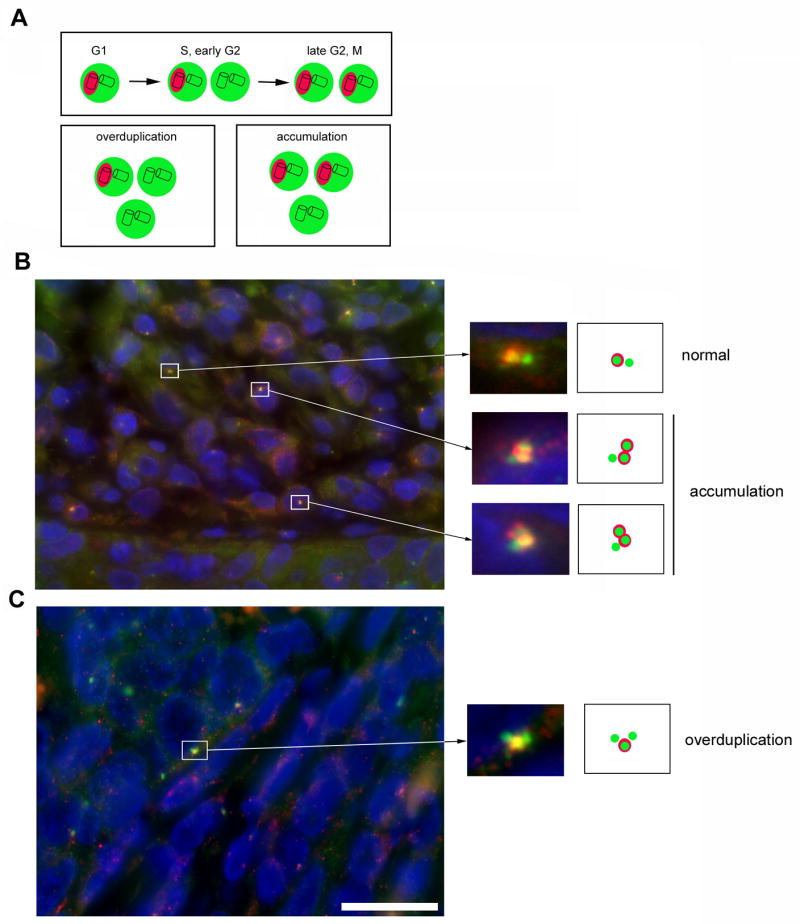
(A) Schematic overview of normal centrosome duplication and pathways that can lead to centrosome amplification. The PCM is in green whereas Cep170-positive centrioles are highlighted in red. Note that during centrosome overduplication in S or early G2 phase of the cell division cycle, only a single centrosome should contain a mature mother centriole that is positive for Cep170.
(B,C) Examples of normal centrosome duplication, centrosome accumulation and centrosome overduplication in two HSILs that contained high-risk HPV DNA. The tissue samples were analyzed by co-immunofluorescence microscopy for the PCM marker γ-tubulin (green) and Cep170 (red). Note the excessive Cep170 staining that may hint to a deregulation of Cep170 expression in HPV-associated neoplasms. Nuclei stained with DAPI. Scale bar indicates 25 μm.
Figures 1B and 1C show examples of normal centrosome content, centrosome overduplication and centrosome accumulation. Because of tissue sectioning, only a fraction of tumor cells shows a detectable centrosome staining. It is noteworthy that many tumors contained cells with enlarged γ-tubulin dots suggesting structural centrosome aberrations (not shown). Cep170 normally localizes to the subdistal appendages of mature maternal centrioles (Guarguaglini et al., 2005). The finding that Cep170 staining was frequently found to be associated with entire centrosomes or even to extend beyond the γ-tubulin signal (Fig. 1B) may hint to a deregulation of its expression in HPV-associated neoplasms.
Abnormal centrosome numbers were detected in 8 out of 11 hemorrhoids (72.7%) and in all HPV-associated anal lesions (100%). The overall number of cells containing supernumerary centrosomes, however, was low in controls (1%; Table 1). Centrosome aberrations were found to be increased to 4.7% of cells in LSILs (4.7-fold; p≤0.001), 2.5% of cells in HSILs (2.5-fold; p≤0.005) and 5.8% of cells in SCCs (5.8-fold; p≤0.005) when compared to benign controls (Fig. 2A).
Figure 2. Numerical centrosome aberrations and centrosome overduplication in benign and malignant anal lesions.
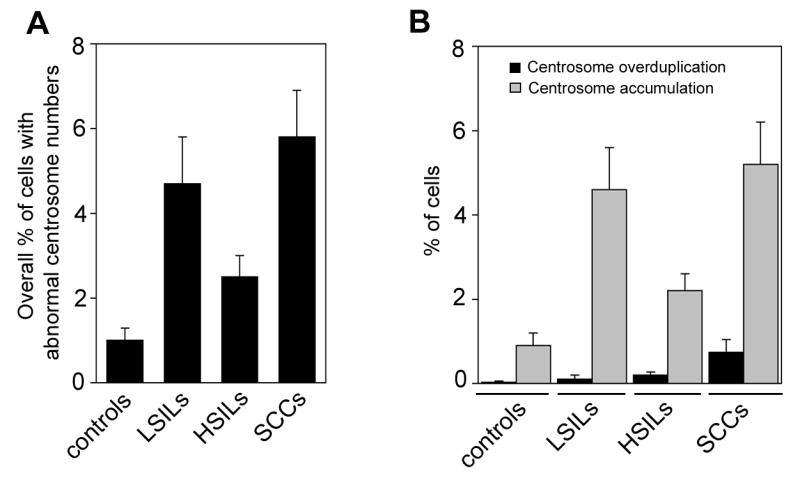
(A,B) Quantification of the overall percentage of cells with abnormal centrosome numbers (more than two per cell, A) or centrosome overduplication (more than two per cell with only one centrosome showing positivity for Cep170, B) in hemorrhoids (controls), LSILs, HSILs or anal SCCs. Each bar represents mean and standard error of results from at least 100 cells from three different areas of each sample.
Centrosome overduplication (more than two centrosomes per cell, only one of them positive for Cep170) was detected in 1 out of 11 controls (9.1%), 1 out 6 LSILs (16.7%), 7 out of 13 HSILs (53.8%) and 6 out of 14 anal SCCs (42.9%; Table 1). The mean percentage of cells with centrosome overduplication was found to increase with malignant grade from 0.03% in controls to 0.1% in LSILs (3.3-fold; p>0.05), 0.2% in HSILs (6.7-fold; p≤0.05) and 0.74% in anal SCCs (24.7-fold; p≤0.05; Fig. 2B).
Centrosome accumulation (more than two centrosomes per cell, at least two of them positive for Cep170) was clearly more frequent than centrosome overduplication and was detected in 8 out of 11 controls (72.7%) and in all HPV-associated anal lesions (100%). The mean percentage of cells with centrosome accumulation was found to increase from 0.9% in controls to 4.6% in LSILs (5.1-fold; p≤0.0005), 2.2% in HSILs (2.4-fold; p≤0.01) and 5.2% in SCCs (5.8-fold; p≤0.005; Table 1 and Fig. 2B).
Taken together, these results show that bona fide centrosome overduplication exists in HPV-associated anal lesions but that it is a relatively rare event. Centrosome aberrations detected in hemorrhoidal control tissue are most likely to be caused by inflammatory and repair processes that can occur together with ploidy changes as it has previously bee described (Lothschutz et al., 2002; Oberringer et al., 1999).
Cell division errors in HPV-associated anal lesions
To explore whether centrosome overduplication can lead to cell division errors as a potential mechanism of aneuploidy, we analyzed all samples for alterations of the normal bipolar metaphase or anaphase spindle pole configuration. In addition to overall frequencies of abnormal meta- or anaphases, we performed a detailed analysis of the type of mitotic defects in metaphase cells in comparison to ana-/telophase cells (Fig. 3).
Figure 3. Cell division errors in high-risk HPV-associated anal lesions.

Examples of normal cell divisions and cell division errors during metaphase (top panels) or ana-/telophase (bottom panels) detected by immunofluorescence microscopy for γ-tubulin. The mitotic figures shown in the inserts are highlighted by arrows. Note that multipolar ana-/telophases were not detected in any sample. Nuclei stained with DAPI. Scale bar indicates 50 μm.
a. Mitotic index
The average frequency of mitotic cells per high-power field (HPF) was found to increase with malignant grade from 0.1% in controls to 0.4% in LSILs (4-fold; p≤0.005), 2.3% in HSILs (23-fold; p≤0.005) and 2.7% in anal SCCs (27-fold; p≤0.0005; Fig. 4A).
Figure 4. Cell division errors increase with malignant grade in HPV-associated anal lesions.

(A–C) Quantification of overall mitotic cells (A), cells with cell division errors during metaphase (B) or cells with cell division errors during ana-/telophase (C) in hemorrhoids (controls), LSILs, HSILs or anal squamous cell carcinomas (SCCs) per high-power field (HPF). Cell division errors included pseudo-bipolar, tripolar or multipolar configurations. Each bar represents mean and standard error of results from at least 10 HPFs from different areas of each sample.
b. Metaphases
In all 44 samples, a total of 752 metaphases was identified and analyzed. Four-hundred and sixty-two metaphases were non-informative because of loss of spindle poles due to tissue sectioning and excluded from further analyses. Two-hundred and ninety metaphases were assessed for spindle polarity and 184 were found to be bipolar (63.5%). A pseudo-bipolar configuration was detected in 32 of 290 metaphases (11%) and was found only in SCCs (n=9) and HSILs (n=5). We defined pseudo-bipolar meta- or ana-/telophases as cell divisions, which maintain opposing spindle poles but that have more than one γ-tubulin signal on at least one side (Fig. 3). A tripolar configuration was detected in 43 of 290 metaphases (14.8%) and was found only in SCCs (n=9) and HSILs (n=6). A multipolar spindle pole arrangement was detected in 31 of 290 metaphases (10.7%) and was found only in SCCs (n=6) and HSILs (n=4).
When abnormal metaphases were assessed according to the histopathological diagnosis, they were found to be absent in controls (0%), whereas 1 out of 6 LSILs (16.6%), 7 out of 13 HSILs (54%) and all 14 anal SCCs (100%) contained abnormal metaphases.
The average frequency of abnormal metaphases per HPF was found to increase from 0% in controls to 0.02% in LSILs (p>0.05), 0.3% in HSILs (p≤0.05) and 0.4% in anal SCCs (p≤0.005; Table 1 and Fig. 4B).
c. Ana-/telophases
A total of 81 ana-/telophases was assessed in all 44 samples among which 13 were non-informative. Of the remaining 68 ana-/telophases, 54 were bipolar (79.4%), whereas 10 of 68 (14.7%) showed a pseudo-bipolar configuration, which was found in SCCs (n=4), HSILs (n=2) and a LSIL (n=1). A tripolar configuration was detected in 4 of 68 ana-/telophases (5.9%) and was found only in a SCC (n=1) and HSILs (n=2). Remarkably, we did not detect any multipolar ana-/telophases. The underrepresentation of tri- or multipolar anaphases in comparison to metaphases was found to be statistically significant (p≤0.05 and p≤0.005. respectively; chi-square test).
When abnormal ana-/telophases were assessed for each histopathological diagnosis, they were found to be absent in controls (0%), whereas 1 out of 6 LSILs (16.7%), 2 out of 13 HSILs (15.4%) and 4 out of 14 anal SCCs (28.6%) contained abnormal ana-/telophase cells.
The average frequency of abnormal ana-/telophases per HPF increased with malignant grade from 0% in controls to 0.02% in LSILs, 0.03% in HSILs and 0.04% in anal SCCs. Differences between controls and HPV-associated anal lesions did not yield statistical significance (Table 1 and Fig. 4C).
Taken together, our findings indicate that abnormal metaphase or anaphase configurations are tightly associated with high-risk HPV infection and do not occur in benign controls. Moreover, only pseudo-bipolar cell division errors were found at comparable frequencies in metaphase and ana-/telophase cells whereas tripolar cell division errors and in particular multipolar cell division errors were significantly underrepresented in ana-/telophase populations.
Centrosome overduplication correlates with the presence of cell division errors
Lastly, we analyzed all samples for a correlation between the presence of centrosome overduplication and cell division errors (Table 2). Centrosome overduplication was found to coincide with cell division errors in 14 of 44 samples (31.8%; p≤0.01, chi-square test). Of the 14 samples, 9 were anal SCCs and 5 HSILs. No controls or LSILs were found to show a coincidence between centrosome overduplication and cell division errors. No statistically significant correlation was found between overall abnormal centrosome numbers or centrosome accumulation and the presence of cell division errors (p>0.05; chi-square test).
Table 2.
Centrosome overduplication and cell division errors.
| Cell division errors | No cell division errors | Total | |
|---|---|---|---|
| Centrosome overduplication | 14 | 4 | 18 |
| No centrosome overduplication | 9 | 17 | 26 |
|
| |||
| Total | 23 | 21 | 44 |
In conclusion, our results suggest a correlation between centrosome overduplication and cell division errors in anal lesions that may be characteristic for high-grade anal lesions and anal SCCs.
Discussion
The use of centrosome aberrations as surrogate biomarkers for chromosomal instability in cancer has been hindered by the lack of robust assays to detect centrosome overduplication. Centrosome overduplication stands in contrast to centrosome accumulation due to cellular defects that are not directly related to the centrosome duplication cycle such as abortive mitoses or cytokinesis failure, since the latter are associated with a dubious potential to produce viable and genomically unstable progeny (Duensing, 2005). In the present study, we show that (a) detection of centrosome overduplication in formalin-fixed, paraffin-embedded tumor samples is feasible and (b) that centrosome overduplication, although a relatively rare event, correlates significantly with the presence of cell division errors. Our results furthermore suggest that the coincidence of centrosome overduplication and cell division errors is characteristic for high-risk HPV-associated HSILs and anal SCCs.
Although we did not address the contribution of the individual high-risk HPV-encoded oncoproteins to the pathogenesis of centrosome aberrations, previous experiments have shown that centrosome overduplication is predominantly associated with the HPV-16 E7 oncoprotein, whereas the HPV-16 E6 oncoprotein promotes centrosome accumulation (Duensing et al., 2001). Moreover, HPV-16 E7, but not HPV-16 E6, was found to stimulate aberrant centrosome numbers prior to the onset of genomic instability and in diploid cells and hence as a potential driving force of chromosomal instability (Duensing et al., 2001). Despite the differences between the two HPV oncoproteins with respect to the induction of centrosome aberrations, it is highly likely that HPV-16 E6 synergizes with HPV-16 E7 to stimulate cell division errors by attenuating p53-mediated checkpoint responses or pro-apoptotic signaling. This would also provide a possible explanation why the correlation between centrosome overduplication and cell division errors in our study is characteristic of HSIL and anal SCC. In these tumors, HPV DNA may be integrated more frequently resulting in an overexpression of high-risk HPV E6 and E7 oncoproteins and a more profound disruption of host cell checkpoint control. In line with this notion, a link between HPV integration and DNA copy number abnormalities has been detected in HSIL (Gagne et al., 2005).
We detected numerical centrosome aberrations in benign squamous epithelial tissue used as control as well as in LSILs, in the latter group at a frequency that was even higher than in HSILs (Fig. 2A). At the same time, however, LSILs had a lower level of centrosome overduplication and contained more cells with centrosome accumulation. Centrosome accumulation can arise during inflammatory and tissue repair processes (Lothschutz et al., 2002; Oberringer et al., 1999), which are frequent histopathological findings in hemorrhoidal excision specimens. Centrosomes can also accumulate in tetra- or polyploid cells and such ploidy changes, in particular tetraploidy, have been identified in low-grade cervical lesions (Southern, Evans, and Herrington, 1997). It is hence possible that tetraploidization contributes to the relatively high level of centrosome accumulation that we observed in LSILs.
The present results show not only that centrosome overduplication exists in vivo but also provide an additional line of evidence that high-risk HPV-induced centrosome overduplication may function as a driving force for cell division errors. Previous studies have suggested that aneuploidy arises through a tetraploid intermediate (Fujiwara et al., 2005). Such cells are characterized by centrosome accumulation (Duensing, 2005). Although we did not determine the ploidy status in the present study, our finding that centrosome overduplication, but not centrosome accumulation, correlates with cell division errors argues that alternative pathways to aneuploidy may exist without a formation of tetra- or polyploid intermediates.
Multipolar or tripolar metaphases are morphological hallmarks of high-risk HPV associated lesions and their occurrence has been implicated in the development of aneuploidy and carcinogenic progression (Crum et al., 1984; Skyldberg et al., 2001). However, results shown here indicate a significant discrepancy between the frequency of tri- or multipolar metaphases and similar defects at later stages of the cell division process. Similar results were obtained previously using keratinocyte populations that stably expressed HPV-16 oncoproteins (Duensing and Münger, 2002) or in a primary Burkitt’s lymphoma (Duensing et al., 2003). In other words, many of the tripolar and particularly multipolar metaphases seen in tumors may not proceed to completion of cell division and are hence an unlikely source of chromosomally unstable and viable progeny. In contrast, pseudo-bipolar mitotic defects were almost equally represented in metaphase cells and ana-/telophase cells, which suggests that such aberrations are more likely to produce daughter cells. One could envision that such alterations impose more subtle chromosomal changes that may be tolerated even by diploid cells. These findings imply that centrosome aberrations cannot be used as general markers for ongoing chromosomal instability unless accompanied by more detailed analysis of centrosome overduplication as shown here and careful examination of the frequency of cell division errors in both metaphase and ana-/telophase.
In conclusion, our results provide evidence that centrosome overduplication exists in primary high-risk HPV-associated anal lesions and, moreover, that it correlates with the presence of cell division errors. These findings encourage further studies to test the use of centrosome overduplication as a surrogate biomarker of chromosomal instability and carcinogenic progression, for example in anal cytology specimens (Cranston et al., 2007), which may ultimately help to identify patients with anal lesions in need of more intensive monitoring and/or treatment to prevent cancer formation.
Materials and Methods
Immunofluorescence microscopy
Formalin-fixed, paraffin-embedded tissue sections were baked overnight at 65°C and deparaffinized thrice in xylene for 10 min each. Sections were then dehydrated twice in 100% ethanol for 10 min each. Sections were rehydrated in a graded ethanol series (90%, 70%, 50%) for 5 min each followed by washing in dH2O twice for 2 min each. All steps were carried out at room temperature (RT). Slides were then microwaved in 0.01 M citrate buffer (pH 6.0) thrice for 10 min each. Slides were allowed to cool down to RT followed by washing in dH2O for 1 min and phosphate buffered saline (PBS) twice for 1 min each. Sections were blocked with 10% normal donkey serum (Jackson Immunoresearch) for 30 min at RT, washed in PBS and incubated with an anti-□-tubulin monoclonal antibody (GTU-88; Sigma) at a 1:500 dilution for two days at 4°C. Slides were then incubated at 37°C for at least 1 h followed by incubation with a FITC-conjugated donkey-anti-mouse secondary antibody at a 1:100 dilution (Jackson Immunoresearch) for at least 2 h at 37°C. Slides were washed twice in PBS for 15 min followed by incubation with a rabbit anti-Cep170 antibody (kindly provided by Erich A. Nigg, Max Planck Institute of Biochemistry, Martinsried, Germany) at a 1:1000 dilution overnight at 4°C. Slides were warmed up to 37°C for at least 1 h followed by incubation with a Rhodamine Red anti-rabbit secondary antibody (Jackson Immunoresearch) at a 1:1000 dilution for 2 h at 37°C. Slides were then washed twice in PBS for 15 min, stained with DAPI (Vector Laboratories) and analyzed using an Olympus AX70 epifluorescence microscope. All sections were scored independently by two trained pathologists (A.D. and S.D.). At least three different areas in histomorphologically abnormal regions were scored (these included all epithelial layers in cases of SIL). Three different areas containing squamous epithelial tissue were scored for hemorrhoidal excision specimens.
HPV testing
The presence of high-risk HPV types (HPV-16/18 or HPV-31/33) or low-risk HPV types (HPV-6/11) was determined using the Rembrandt/PanPath in situ hybridization kit (Invitrogen) according to manufacturer’s recommendations.
Statistical analysis
Statistical significance was calculated using Student’s t test for independent samples or the chi-square test. P values ≤0.05 were considered to be statistically significant.
Acknowledgments
We are grateful to Erich A. Nigg (Max Planck Institute of Biochemistry, Martinsried, Germany) and Giulia Guarguaglini (University of Rome “La Sapienza”, Rome, Italy) for sharing the Cep170 antibody. We would like to thank Ross Cranston for critical reading of the manuscript and Peter Veldkamp for helpful suggestions. This work was supported by the GLBT Fund for Health and Wellness of The Pittsburgh Foundation, NIH/NCI grant R01 CA112598 and a Research Scholar Grant from the American Cancer Society (to S.D.).
References
- Abbasakoor F, Boulos PB. Anal intraepithelial neoplasia. Br J Surg. 2005;92(3):277–90. doi: 10.1002/bjs.4967. [DOI] [PubMed] [Google Scholar]
- Berry JM, Palefsky JM, Welton ML. Anal cancer and its precursors in HIV-positive patients: perspectives and management. Surg Oncol Clin N Am. 2004;13(2):355–73. doi: 10.1016/j.soc.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol. 2007;8(6):451–63. doi: 10.1038/nrm2180. [DOI] [PubMed] [Google Scholar]
- Cranston RD, Hart SD, Gornbein JA, Hirschowitz SL, Cortina G, Moe AA. The prevalence, and predictive value, of abnormal anal cytology to diagnose anal dysplasia in a population of HIV-positive men who have sex with men. Int J STD AIDS. 2007;18(2):77–80. doi: 10.1258/095646207779949772. [DOI] [PubMed] [Google Scholar]
- Crum CP, Ikenberg H, Richart RM, Gissman L. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310(14):880–3. doi: 10.1056/NEJM198404053101403. [DOI] [PubMed] [Google Scholar]
- Duensing A, Ghanem L, Steinman RA, Liu Y, Duensing S. p21(Waf1/Cip1) deficiency stimulates centriole overduplication. Cell Cycle. 2006;5(24):2899–902. doi: 10.4161/cc.5.24.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007 doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S. A tentative classification of centrosome abnormalities in cancer. Cell Biol Int. 2005;29:352–359. doi: 10.1016/j.cellbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61(6):2356–60. [PubMed] [Google Scholar]
- Duensing S, Lee BH, Dal Cin P, Münger K. Excessive centrosome abnormalities without ongoing numerical chromosome instability in a Burkitt’s lymphoma. Mol Cancer. 2003;2:30. doi: 10.1186/1476-4598-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97(18):10002–7. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. Centrosome abnormalities, genomic instability and carcinogenic progression. Biochim Biophys Acta. 2001;2(8):M81–8. doi: 10.1016/s0304-419x(00)00025-1. [DOI] [PubMed] [Google Scholar]
- Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437(7061):1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Gagne SE, Jensen R, Polvi A, Da Costa M, Ginzinger D, Efird JT, Holly EA, Darragh T, Palefsky JM. High-resolution analysis of genomic alterations and human papillomavirus integration in anal intraepithelial neoplasia. J Acquir Immune Defic Syndr. 2005;40(2):182–9. doi: 10.1097/01.qai.0000179460.61987.33. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. The Forkhead-associated Domain Protein Cep170 Interacts with Polo-like Kinase 1 and Serves as a Marker for Mature Centrioles. Mol Biol Cell. 2005;16(3):1095–107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heselmeyer K, Macville M, Schrock E, Blegen H, Hellstrom AC, Shah K, Auer G, Ried T. Advanced-stage cervical carcinomas are defined by a recurrent pattern of chromosomal aberrations revealing high genetic instability and a consistent gain of chromosome arm 3q. Genes Chromosomes Cancer. 1997;19(4):233–40. [PubMed] [Google Scholar]
- Kashyap V, Das BC. DNA aneuploidy and infection of human papillomavirus type 16 in preneoplastic lesions of the uterine cervix: correlation with progression to malignancy. Cancer Lett. 1998;123:47–52. doi: 10.1016/s0304-3835(97)00396-0. [DOI] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A. 1998;95(6):2950–5. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68(2):362–72. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothschutz D, Jennewein M, Pahl S, Lausberg HF, Eichler A, Mutschler W, Hanselmann RG, Oberringer M. Polyploidization and centrosome hyperamplification in inflammatory bronchi. Inflamm Res. 2002;51(8):416–22. doi: 10.1007/pl00000323. [DOI] [PubMed] [Google Scholar]
- Monsonego J, Valensi P, Zerat L, Clavel C, Birembaut P. Simultaneous effects of aneuploidy and oncogenic human papillomavirus on histological grade of cervical intraepithelial neoplasia. Br J Obstet Gynecol. 1997;104:723–727. doi: 10.1111/j.1471-0528.1997.tb11984.x. [DOI] [PubMed] [Google Scholar]
- Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nature Rev Cancer. 2002;2:1–11. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17(5):215–21. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Oberringer M, Lothschutz D, Jennewein M, Koschnick M, Mutschler W, Hanselmann RG. Centrosome multiplication accompanies a transient clustering of polyploid cells during tissue repair. Mol Cell Biol Res Commun. 1999;2(3):190–6. doi: 10.1006/mcbr.1999.0172. [DOI] [PubMed] [Google Scholar]
- Palefsky J. Human papillomavirus-associated malignancies in HIV-positive men and women. Curr Opin Oncol. 1995;7(5):437–41. doi: 10.1097/00001622-199509000-00009. [DOI] [PubMed] [Google Scholar]
- Palefsky J. Biology of HPV in HIV infection. Adv Dent Res. 2006;19(1):99–105. doi: 10.1177/154407370601900120. [DOI] [PubMed] [Google Scholar]
- Palefsky JM. Human papillomavirus infection and anogenital neoplasia in human immunodeficiency virus-positive men and women. J Natl Cancer Inst Monogr. 1998;23:15–20. doi: 10.1093/oxfordjournals.jncimonographs.a024166. [DOI] [PubMed] [Google Scholar]
- Palefsky JM, Holly EA, Gonzales J, Berline J, Ahn DK, Greenspan JS. Detection of human papillomavirus DNA in anal intraepithelial neoplasia and anal cancer. Cancer Res. 1991;51(3):1014–9. [PubMed] [Google Scholar]
- Skyldberg B, Fujioka K, Hellstrom AC, Sylven L, Moberger B, Auer G. Human papillomavirus infection, centrosome aberration, and genetic stability in cervical lesions. Mod Pathol. 2001;14(4):279–84. doi: 10.1038/modpathol.3880303. [DOI] [PubMed] [Google Scholar]
- Sluder G. Centrosome duplication and its regulation in the higher animal cell. In: Nigg EA, editor. Centrosomes in Development and Disease. Wiley-VCH, Weinheim; Germany: 2004. pp. 167–189. [Google Scholar]
- Southern SA, Evans MF, Herrington CS. Basal cell tetrasomy in low-grade cervical squamous intraepithelial lesions infected with high-risk human papillomaviruses. Cancer Res. 1997;57(19):4210–3. [PubMed] [Google Scholar]
- Steinbeck RG. Proliferation and DNA aneuploidy in mild dysplasia imply early steps of cervical carcinogenesis. Acta Oncol. 1997;36:3–12. doi: 10.3109/02841869709100723. [DOI] [PubMed] [Google Scholar]
- Tsou MF, Stearns T. Controlling centrosome number: licenses and blocks. Curr Opin Cell Biol. 2006;18(1):74–8. doi: 10.1016/j.ceb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Wright TC, Jr, Cox JT, Massad LS, Twiggs LB, Wilkinson EJ. 2001 Consensus Guidelines for the management of women with cervical cytological abnormalities. Jama. 2002;287(16):2120–9. doi: 10.1001/jama.287.16.2120. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288(2):F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]