

Abstract
Enteric nervous system (ENS) precursors migrate extensively before differentiating to form uni-axonal or multi-axonal neurons. ENS precursor survival, neurite growth, and cell migration are all directed by Ret kinase, but downstream signaling pathways are incompletely understood. We now demonstrate that proteins regulating polarity in other cells including partitioning defective 3 (PAR3), PAR6, protein kinase Cζ (PKCζ), and glycogen synthase kinase 3β (GSK3β) are expressed in developing enteric neurons with a polarized distribution. Blocking PKCζ or GSK3β reduces ENS precursor migration and induces the formation of multi-axonal neurons. Axon elongation also depends on SMURF1 (SMAD specific E3 ubiquitin protein ligase 1), which promotes RhoA degradation and associates with polarity proteins. SMURF1 inhibition, however, does not increase the number of multi-axonal neurons in ENS precursors. These data link cell surface Ret activation with molecular machinery controlling cytoskeletal dynamics and suggest that polymorphisms influencing PKCζ or GSK3β might alter Hirschsprung disease penetrance or expressivity by affecting ENS precursor migration.
Keywords: neuronal polarity, migration, axon specification, partitioning proteins, intracellular signaling, neurite
Introduction
Enteric nervous system (ENS) development requires directed cell migration and precursor differentiation to produce a neuronal network that controls motility and responds to intestinal stimuli (Gershon, 1997; Furness, 2000; Newgreen and Young, 2002a,b). Distinct enteric neuron subtypes differentiate into cells with diverse neurotransmitter expression patterns, electrophysiology, morphology, and function. For >100 years, morphology of enteric neurons has been used as a basis for classification (Dogiel, 1899). This includes “multi-axonal (type II neurons)” and “multi-dendritic, uni-axonal” neurons whose function correlate to some degree with morphology (Brehmer et al., 1999). Because both directed migration and axon specification require cell polarization, we hypothesized that these processes share common molecular mechanisms within the developing ENS.
Polarization is essential for many aspects of development, including asymmetric cell division and establishment of polarized epithelia (Wodarz, 2002; Etienne-Manneville and Hall, 2003a). Recent work has demonstrated that these disparate processes use the same proteins to establish asymmetry and link extracellular signals to the cytoskeleton. This protein complex is organized by PAR (partitioning defective) proteins first discovered in Caenorhabditis elegans mutants that failed to specify anterior/posterior axis (Kemphues et al., 1988). Subsequent work demonstrated that a complex of PAR3/PAR6/atypical protein kinase C (PKC)/cell division cycle 42 (Cdc42) is essential for asymmetric cell division in yeast (Chant, 1999), anterior/posterior polarity in C. elegans (Nance, 2005), neuroblast polarity and axon specification (Shi et al., 2003; Wiggin et al., 2005), astrocyte migration (Etienne-Manneville and Hall, 2003b), and tight junction formation and epithelial to mesenchymal transition (Ozdamar et al., 2005). Furthermore, in astrocytes, Cdc42 activates PKCζ, which directly inhibits glycogen synthase kinase 3β (GSK3β) in a spatially restricted manner to direct motility (Etienne-Manneville and Hall, 2003b). GSK3β is a serine/threonine kinase with many substrates, including microtubule-associated proteins MAP1B, tau, adenomatous polyposis coli, collapsin response mediator protein 2, and the kinesin light chain microtubule motor (Cole et al., 2004; Jope and Johnson, 2004; Quach et al., 2004; Yoshimura et al., 2005). Thus, GSK3β directly influences microtubule growth and transport along microtubules required for axon growth and cell migration. Recent evidence in hippocampal neurons demonstrated that GSK3β also plays a central role in the maintenance and establishment of neuronal polarity (Jiang et al., 2005; Yoshimura et al., 2005, 2006).
We now demonstrate important roles for PKCζ and GSK3β in cell body migration and axon specification in the developing ENS. Furthermore, because neurite growth and cell migration in model systems require localized RhoA inhibition, we tested the hypothesis that, PCKζ and PAR6 interact with the HECT domain E3 ubiquitin ligase SMURF1 (SMAD specific E3 ubiquitin protein ligase 1), shown recently to control RhoA degradation in active cellular protrusions and to regulate cell polarity (Wang et al., 2003). These studies demonstrate that SMURF1 is expressed in ENS precursors in a similar pattern to PAR3, coimmunoprecipitates with a PAR6, GSK3β, and PKCζ, and critically influences neurite growth but does not alter axon–dendrite polarity. These observations have important implications for Hirschsprung disease, a potentially fatal developmental disorder of the ENS in which variable expressivity and partial penetrance is common, and the phenotype often depends on the presence of hypomorphic mutations in several genes.
Materials and Methods
Materials.
The following primary antibodies were used: p75NTR antibody 9651 (1:1000; generous gift from Dr. Moses Chao, New York University Medical Center, New York, NY) (Huber and Chao, 1995), mouse anti-TAU1 (1:100; Chemicon, Pittsburgh, PA), TAUpS396 (1:1000; Invitrogen, Carlsbad, CA), rabbit TuJ1 (1:1000) and mouse TuJ1 (1:100; Covance, Denver, CO), rabbit anti-PAR3 (1:100; BD Biosciences, San Jose, CA), rabbit anti-PAR6 (1:100) and anti-SMURF1 (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-phospho-PKCζ (p-PKCζ) (1:100), and anti-phospho-GSK3β, anti-PKCζ (1:100), and anti-GSK3β (1:100; Cell Signaling Technology, Danvers, MA). The secondary antibodies used were Alexa Fluor 350-, 488-, and 594-conjugated anti-mouse and anti-rabbit antibodies (1:500; Invitrogen). GSK3β inhibitors 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)1H-pyrrole-2,5-dione (SB216763) and 3-[(3-chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrole-2,5-dione (SB415286) were from Tocris Bioscience (Ellisville, MO). Bisindolylmaleimide, myristoylated pseudosubstrate inhibitor selective for PKCζ, and Rho-associated coiled-coil containing protein kinase (ROCK) inhibitors (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide (Y-27632) and (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinolinyl)sulfonyl]homopiperazine (H-1152P) were from Calbiochem (San Diego, CA).
Primary culture of immunoselected ENS precursors.
Enteric neural crest were immunoselected from embryonic day 14.5 (E14.5) Sprague Dawley rat (Charles River Laboratories, Wilmington, MA) small bowel and colon using p75NTR antibody (Wu et al., 1999). Bowel was dissociated with collagenase (1 mg/ml) and dispase (1 mg/ml) to yield a single cell suspension. After p75NTR antibody incubation (9651; 1:1000, 1 h, 4°C) in B27 (Invitrogen) supplemented Neurobasal medium, cells were incubated with goat anti-rabbit coupled paramagnetic beads (1:50, 1 h, 4°C; Miltenyi Biotec, Bergisch Gladbach, Germany) before separating neural crest-derived cells from unselected cells with a positive selection column (MACS separation columns; Miltenyi Biotec). Immunoselected crest-derived cells were plated at a density of 500 cells per well on poly-d-lysine/laminin-coated eight-well chamber slides (Biocoat; Fisher, Pittsburgh, PA) in B27 supplemented Neurobasal medium plus 50 ng/ml glial cell line-derived neurotrophic factor (GDNF). In all experiments, inhibitors were added to the medium 6 h after plating using 5 μm SB216763 and 10 μm SB415286 (GSK3β inhibitors), 50 nm bisindolylmaleimide (PKC inhibitor), 10 μm myristoylated pseudosubstrate inhibitor selective for PKCζ, or 10 μm Y-27632 or 10 nm H-1152P (ROCK inhibitors). Cells were maintained in a humidified environment (5% CO2, 37°C) for 48 h before washing with PBS and fixing in 4% paraformaldehyde (25°C, 20 min). For gene expression experiments, cells were grown in B27 supplemented Neurobasal medium plus 50 ng/ml GDNF for 24, 48, or 72 h. All experiments were performed in triplicate. All analyses were performed blinded.
Immunohistochemistry and image analysis.
After fixation, cells were washed with PBS, blocked with 4% normal donkey serum in TBST (Tris-buffered saline plus 0.1% Triton X-100) (1 h, 37°C), and then incubated in primary antibody (4°C overnight). Antibody binding was visualized with Alexa Fluor 350-, 488-, and 594-conjugated anti-mouse and anti-rabbit secondary antibodies (25°C, 1 h). Images were obtained with an Olympus Optical (Tokyo, Japan) BX60 microscope and an Axiocam digital camera and AxioVision imaging software (Zeiss, Oberkochen, Germany). Photoshop 7.0 (Adobe Systems, San Jose, CA) was used to adjust contrast and brightness so that digital images appear as they did when observed directly through the microscope. Image analysis was performed with NIH ImageJ 1.30.
Immunoprecipitation studies.
For each immunoprecipitation, p75NTR-positive (p75NTR+) immunoselected cells derived from 50 embryos (E14.5, rat) were maintained in culture for 2 d on fibronectin-coated plastic dishes in B27 supplemented Neurobasal medium plus 50 ng/ml GDNF. Cells were lysed in 1 ml of 100 mm Tris, 0.5% Triton X-100, pH 7.6, plus protease inhibitor cocktail (Complete Mini; Roche, Penzberg, Germany) by repeatedly drawing suspended cells through a 21 gauge needle on ice. Cell extracts were precleared with 20 μl of protein A agarose beads to eliminate nonspecific binding. Supernatants were then incubated for 3 h with SMURF1, PAR6, GSK3β, and PKCζ antibodies at 4°C on a shaker. Twenty microliters of protein A agarose beads was then added, and the mixture was incubated on rotary mixer overnight at 4°C. Beads and the associated immunoprecipitate were collected by centrifugation, washed once in PBS containing 0.5% (v/v) Tween 20, 0.1% (w/v) SDS, and then three times in PBS containing 0.5% (v/v) Tween 20 and 0.5 m NaCl. Pellets were resuspended in SDS-PAGE sample buffer [62.5 mm Tris, pH 6.8, containing 2% (w/v) SDS, 10% (v/v) glycerol, 5% (v/v) β-mercaptoethanol, and 0.001% (w/v) bromophenol blue] and boiled for 5 min. Immunoprecipitates were separated on polyacrylamide gels, and protein immunoblots were probed using antibodies (1:1000) against SMURF1, PAR6, GSK3β, and PKCζ. Control immunoprecipitations were performed by omitting the primary antibody.
Organ culture analysis of neural crest migration.
E11.5 C57BL/6 mouse gut explants containing stomach, small bowel, and colon were pinned to 2% agarose and cultured in 500 μl (DMEM, 10% fetal calf serum, and penicillin/streptomycin) containing GSK3β inhibitors (5 μm SB216763 or 10 μm SB415286) or PKCζ inhibitor (10 μm myristoylated pseudosubstrate inhibitor). After 48 h (37°C, 5% CO2), tissues were fixed (4% paraformaldehyde, 30 min, 25°C) and then processed for whole-mount immunohistochemistry using rabbit polyclonal TuJ1 (1:100) antibody.
GSK3β small interfering RNA studies.
We used a small interfering RNA (siRNA) target sequence (GAACCGAGAGCTCCAGATC) that has been successfully used by several other investigators to silence GSK3β (Yu et al., 2003; Jiang et al., 2005; Yoshimura et al., 2005; Kim et al., 2006). PAGE purified oligonucleotides (IDT, San Diego, CA) for the hairpin siRNA included sites for directional cloning (top strand, 5′ACCGAACCGAGAGCTCCAGATCTTCAAGAGAGATCTGGAGCTCTCGGTTCTTTTTC 3′; bottom strand, 5′TCGAGAAAAAGAACCGAGAGCTCCAGATCTCTCTTGAAGATCTGGAGCTCTCGGTTC 3′) and were annealed before ligation into BbsI and XhoI sites immediately downstream of the U6 promoter in the pBS–hU6 vector. The siRNA expression cassette from pBS–hU6 vector was then subcloned into the FG12 lentiviral vector between the XbaI and XhoI sites, and lentivirus was generated as described previously (Qin et al., 2003). Complementary oligonucleotide pairs for the control siRNA sequence (CAGTCGCGTTTGCGACTGG) were as follows: top strand, 5′ACCGGCA GTCGCGTTTGCGACTGGTTCAAGAGACCAGTCGCAAACGCGACTGTTTTTTCTTTTTC 3′; bottom strand, 5′TCGAGAAAAAGAAAAAACAGTCGCGTTTGCGACTGGTCTCTTGAACCAGTCGCAAACGCGACTGC 3′.
Smurf1 siRNA studies.
The Smurf1 siRNA target sequence (CCGACACTGTGAAAAACAC) (Wang et al., 2003) was cloned into RNAi-Ready pSiren–RetroQ–ZsGreen vector, which expresses a reef coral green fluorescent protein (ZsGreen) (BD Biosciences) by adding BamH1 and EcoR1 sites to enable directional cloning. An Mlu1 site was inserted to facilitate plasmid identification. PAGE purified oligonucleotides (IDT) for the hairpin siRNA were as follows: top strand, 5′gatccGCCGACACTGTGAAAAACACTTCAAGAGAGTGTTTTTCACAGTGTCGGTTTTTTACGCGTG 3′; bottom strand, 5′aattcACGCGTAAAAAACCGACACTGTGAAAAACACTCTCTTGAAGTGTTTTTCACAGTGTCGGCg 3′. Complementary oligonucleotide pairs for the control siRNA sequence (CAGTCGCGTTTGCGACTGG) were as follows: top strand, 5′gatccGCAGTCGCGTTTGCGACTGGTTCAAGAGACCAGTCGCAAACGCGACTGGTTTTTTTACGCGTg3′; bottom strand, 5′aattcACGCGTAAAAAACAGTCGCGTTTGCGACTGGTCTCTTGAACCAGTCGCAAACGCGACTGCg3′.
Smurf1 siRNA virus production.
To produce retrovirus for Smurf1 siRNA, GP2–293 cells (BD Biosciences) (1 × 106) grown on a 60 mm dish were cotransfected 12 h after plating with pSiren–RetroQ–ZsGreen vectors (5 μg; BD Biosciences) and a viral coat protein plasmid (pVSV-G) (5 μg; BD Biosciences) using Fugene 6 (Roche Diagnostics, Indianapolis, IN). Culture medium was replaced 6 h after transfection with 3 ml of new medium [DMEM with glutamine/10% fetal calf serum (Hyclone, Logan, UT)]. Viral particles were harvested from culture medium 72 h after transfection by centrifuging to eliminate cell debris (500 × g, 10 min, 4°C) and then by ultracentrifugation (50,000 × g, 90 min, 4°C). Viral pellet was resuspended in 50 μl of PBS. Viral titer was 4 × 104 pfu/μl.
Viral infection studies.
Enteric neural crest-derived cells from E12.5 CF1 mice were immunoselected from the small bowel and colon as described above and plated on fibronectin (250 μg/ml; Invitrogen) coated four-well Permanox Sonic Seal chamber slides (Nalge Nunc, Woburn, MA). Cells were grown in DMEM plus 10% chick embryo extract (SLI, Bolney, West Sussex, UK). Three hour after plating, cells were infected with virus expressing Smurf1 siRNA, GSK3β siRNA, or control siRNAs by adding 10–20 μl of the viral solution. Medium was changed 12 h after infection. Infection was confirmed by ZsGreen expression in cells infected with Smurf1 siRNA and control virus and by green fluorescent protein (GFP) expression in cells infected with GSK3β siRNA and control virus. After 2 d in culture, cells were trypsinized and replated (10,000 cells per well) on fibronectin (250 μg/ml; Invitrogen) coated four-well Permanox Sonic Seal chamber slides (Nalge Nunc) and grown in DMEM plus 10% chick embryo extract and GDNF (50 ng/ml; PeproTech, Rocky Hill, NJ). For the ROCK inhibitor studies, ROCK inhibitors (10 μm Y-27632 or 10 nm H-1152P) were added at the time of plating. Cells were maintained in a humidified environment (5% CO2, 37°C) for 48 h before washing with PBS and fixing (4% paraformaldehyde, 20 min, 25°C) for immunohistochemical analysis. All experiments were performed in triplicate.
Protein immunoblot to evaluate effectiveness of SB216763 and Smurf1 siRNA.
TAU1 and TAUpS396 (pTAU1) protein immunoblotting was performed on the immunoselected enteric neural crest from E14.5 Sprague Dawley rat. Cells were maintained in culture with or without 5 μm GSK3β inhibitor (SB216763) as described above for 48 h (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). SMURF1 and RhoA protein levels were analyzed in cultured immunoselected enteric neurons that were maintained in culture and infected with control virus or Smurf1 siRNA virus as described above. Cultured cells were lysed with M-per mammalian protein extraction reagent (Pierce, Rockford, IL) supplemented with protease inhibitor cocktail (Roche, Penzberg, Germany) and Halt phosphatase inhibitor cocktail (Pierce). Twenty micrograms of protein were subjected to electrophoresis on 4–12% Tris-glycine SDS-PAGE gradient gels (Invitrogen). Proteins were transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK) before the membranes were probed with the antibodies for TAU1, P-TAU1, SMURF1, and RhoA and developed using Amersham ECL Western blotting detection reagents (GE Healthcare). The films were scanned, and band densities were analyzed using NIH ImageJ 1.30 software.
Statistical analyses.
Statistical analysis was performed using SigmaStat software (Systat Software, San Jose, CA), and either t test or Mann–Whitney rank sum test were used. Data are presented as mean ± SE. p < 0.05 is considered significant.
Results
Developing enteric neurons express proteins that control polarization in other cell types
E14.5 rat enteric neuron precursors immunoselected with an antibody to p75NTR and maintained in culture were analyzed for the expression of PAR3, PAR6, phospho-PKCζ, and phospho-GSK3β by immunohistochemistry because localization of these proteins influences cell polarity and neurite specification in other cell types. Analyses were limited to cells that had clearly begun to differentiate into neurons based on the expression of neuronal class III β-tubulin (TuJ1+), which is present in all neurites, and Tau1, a marker for axons. At this stage in development, some ENS precursors are still actively proliferating, but others are differentiating into a variety of enteric neuron subtypes (Barlow et al., 2003; Vohra et al., 2006). PAR3, PAR6, p-PKCζ, and p-GSK3β had a similar distribution at each time point. Immunohistochemistry performed 24 h after plating ENS precursors demonstrated that PAR3, PAR6, p-PKCζ, and p-GSK3β were highly expressed throughout the cell body and the longest axon in approximately half of all developing enteric neurons (e.g., for PAR3, this distribution occurred in 48 ± 8% of Tau1 + cells). After 48 h in culture PAR3, p-PKCζ, and p-GSK3β were more highly abundant at the tip of Tau1 + axons than along the axon shaft (79 ± 5% of Tau1 + cells had this distribution for PAR3) (Fig. 1). In contrast, PAR6 was distributed uniformly along the axon shaft and tip (Fig. 1E–H). Each of these proteins was also abundant in the cell body at 24 and 48 h after plating. After 3 d in culture, most of the immunoreactivity of these proteins that control cell polarity localized to the cell body of the enteric neurons (92 ± 3% of Tau1 + cells had this distribution for PAR3; n = 200 neurons analyzed for each time point). These changes in polarity protein localization are similar to those seen in hippocampal neurons and suggest this protein complex determines neurite polarity in the developing ENS.
Figure 1.
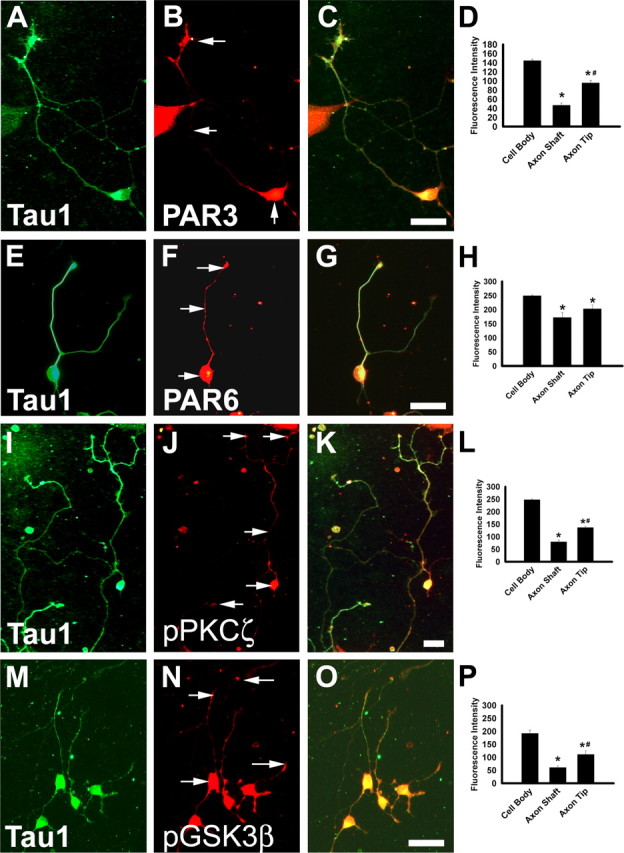
PAR3, PAR6, PKCζ, and GSK3β are produced in developing enteric neurons with a polarized distribution. E14.5 rat immunoselected ENS precursors maintained in culture for 48 h were stained with antibodies that recognize the axonal protein TAU1 (A, E, I, M) or with antibodies to PAR3 (B), PAR6 (F), phosphorylated PKCζ (J), or phosphorylated GSK3β (N). Merged images are shown in C, G, K, and O. Protein localization in the neuron cell body, axon shaft, or axon tip was determined by quantitative analysis of fluorescence intensity using NIH ImageJ 1.30 (D, H, L, P) (n = 50 cells per analysis). Arrows indicate the location of fluorescence intensity measurements. *p < 0.01 compared with cell body. #p < 0.01 compared with axon shaft. Scale bars, 50 μm.
Neuronal polarization requires PKCζ and GSK3β
Although enteric neurons are not a uniform population, rat enteric neuroblasts develop into neurons with a single long axon, or multiple axons over 48 h in culture in reproducible ratios that allow us to investigate the role of PKCζ, GSK3β, and other proteins in axon specification. For example, when immunoselected (i.e., p75NTR expressing) E14.5 rat ENS precursors are cultured under control conditions, 79 ± 4% of TuJ1+ cells have a single Tau1 + axon, 15 ± 3% have two or more axons, and 6 ± 2% have no Tau1 + processes (Fig. 2A–D). To determine whether GSK3β influences enteric neuron polarity, GSK3β activity was blocked throughout the cell using either of two different inhibitors (5 μm SB216763 or 10 μm SB415286). This caused a dramatic increase in the percentage of developing ENS precursors with multiple Tau1 + axons (Fig. 2E–H). Specifically, GSK3β inhibition caused a 3.3-fold increase (p < 0.001) in TuJ1+ cells with multiple axons and a 3.7-fold increase (p < 0.01) in the number of cells with no axons (Fig. 2H). Similarly, infection of ENS precursors with a lentivirus expressing siRNA that efficiently silences GSK3β expression (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) resulted in a 2.5-fold increase (p < 0.001) in the number of enteric neurons with multiple Tau1 + axons and a twofold increase (p < 0.001) in the number of cells with no axon (Fig. 3). Thus, global inhibition of GSK3β increases the percentage of developing enteric neurons with multiple Tau1 + neurites.
Figure 2.

Inhibitors of PKCζ or GSK3β increase the percentage of multi-axonal neurons and the percentage of neurons without axons. E14.5 rat immunoselected ENS precursors were maintained in culture for 48 h (A–C) under control conditions, with a GSK3β inhibitor (SB216763) (E–G), or with a specific PKCζ inhibitor (myristoylated pseudosubstrate inhibitor) (I–K). A, E, I, Enteric neurons were identified with an antibody to neuron-specific β III tubulin (TuJ1). Axons were identified with an antibody to TAU1 (B, F, J). Arrows indicate TAU1-expressing neurites. Merged images are shown in C, G, and K. Cells were classified based on the number of TAU1-expressing neurites present (D, H, L) (n = 125 cells). Similar results were obtained with an alternate GSK3β inhibitor (SB415286). Scale bars, 50 μm.
Figure 3.

GSK3β siRNA increases the percentage of multi-axonal neurons and the percentage of neurons without axons. A–C, E12.5 mouse immunoselected ENS precursors infected with control virus (A–C) or GSK3β siRNA producing virus (D–F) were maintained in culture for 48 h after plating. A, C, D, F, The virus encodes GFP in addition to the siRNA so that infected cells are readily identified. In these experiments, all cells in the dish were infected and all TuJ1+ cells expressed Tau1 +. B, E, Immunohistochemistry was performed using an antibody to TAU1. C, F, Merged images. G, H, Cells were classified based on the number of Tau1+ neurites present (n = 159 cells). Arrows indicate neuron cell bodies. Arrowheads indicate TAU1-expressing neurites. Scale bars, 50 μm.
PKCζ interacts with GSK3β and influences cell polarization in several other contexts (Lin et al., 2000; Etienne-Manneville and Hall, 2001, 2003a; Wang et al., 2003; Higginbotham et al., 2006). To determine whether PKCζ activity is important for neurite specification (axon vs dendrite) in ENS precursors, we used chemical inhibitors to globally block PKCζ activity. Unlike results in hippocampal neurons in which PKC inhibition results in loss of axons (Shi et al., 2003), inhibition of PKC activity in ENS precursors using a general PKC inhibitor (50 nm bisindolylmaleimide) resulted in both a 2.1-fold (p < 0.01) increase in the number of cells with multiple Tau1+ axons and a 4.5-fold (p < 0.001) increase in the number of cells with no axons. Similarly, inhibition of PKCζ using a specific PKCζ inhibitor (10 μm myristoylated pseudosubstrate inhibitor selective for PKCζ) caused a substantial increases in the number of cells with multiple Tau1+ axons (2.3-fold; p < 0.01) or without axons (3.0-fold; p < 0.01) (Fig. 2I–L). To confirm that these effects on axon number in ENS precursors were not influenced by effects of these inhibitors on cell survival, we counted the total number of Ret+ cells per well. None of the inhibitors used had any effect on Ret+ cell number, suggesting that the inhibitors are not inducing cell death (control, 323 ± 30 Ret+ cells per well; SB216763, 318 ± 13 Ret+ cells per well; SB415286, 304 ± 10 Ret+ cells per well; bisindolylmaleimide, 333 ± 38 Ret+ cells per well; PKCζ inhibitor, 355 ± 33 Ret+ cells per well). Thus, global inhibition of either GSK3β or PKCζ activity influences neuronal polarization of cultured enteric neuroblasts, resulting in an increase in multi-axonal cells and an increase in cells without axons.
PKCζ and GSK3β are required for neural crest migration into the distal bowel
Directed migration of ENS precursors also presumably requires polarization of the migrating cells. Because vagal neural crest derivatives that form the ENS have one of the longest migratory routes of any cell in the body during development, we hypothesized that ENS precursor migration would require some of the same molecular machinery used for neurite polarization. To test the hypothesis that PKCζ and GSK3β are important for ENS precursor migration, mouse E11.5 gut was maintained in organ culture for 48 h in the presence of PKCζ or GSK3β inhibitors. At the start of the culture, ENS precursors that migrate from the vagal crest through the bowel to the end of the colon had just reached the ileocecal junction. In the control cultures, 89 ± 3% of the colon was populated by ENS precursors after 48 h in culture. In contrast, treatment with the PKCζ inhibitor reduced colon innervation to 67 ± 4%. GSK3β inhibitors also reduced colon innervation to 54 ± 4% of total colon length. These data confirm an important role for PKCζ and GSK3β in ENS precursor migration (Fig. 4).
Figure 4.

Inhibitors of PKCζ or GSK3β reduce the rate of ENS precursor migration through the distal bowel. E11.5 mouse gut explants containing stomach, small bowel, and colon were cultured for 48 h in control medium (A) or in medium containing GSK3β inhibitor (SB216763) (B) or PKCζ inhibitor (myristoylated pseudosubstrate inhibitor) (C). At the start of the culture, ENS precursors had migrated to the ileocecal junction (ICJ) and are about to enter the colon. D, Quantitative analysis of the percentage of the colon colonized by ENS precursors under different conditions. % Colon colonization = (distance from ICJ to wave front of migrating ENS precursors/distance from ICJ to the end of the colon) × 100. n = 5–8 gut specimens were analyzed under each condition. Similar results were obtained with an alternate GSK3β inhibitor (SB415286). The white bar at the left edge of each image indicates the end of the colon. Arrow in A indicates the leading edge of the wave front of migrating cells. Scale bar, 50 μm.
Polarized distribution of SMURF1 in enteric neurons
Recent work in other systems has suggested that PKCζ-induced changes in cell polarity can be mediated by recruitment of the E3 ubiquitin ligase SMURF1 to cellular protrusions (Wang et al., 2003). SMURF1 then locally degrades RhoA, which in turn regulates actin cytoskeletal dynamics. We therefore hypothesized that SMURF1 might critically regulate neuronal polarity in developing enteric neuroblasts. To test this hypothesis, we first demonstrated by immunohistochemistry that SMURF1 was expressed in ENS precursors (Fig. 5). Remarkably, the immunohistochemical staining pattern for SMURF1 in enteric neuroblasts after 24, 48, or 72 h in culture was very similar to the distribution of PAR3, PKCζ, and GSK3β. Specifically, double-label immunohistochemistry for TAU1 and SMURF1 demonstrated SMURF1 immunoreactivity in the cell body and axonal processes after 24 h in culture (Fig. 5A–C,J). By day 2 in culture, SMURF1 immunoreactivity was more restricted to the cell body and the tip of the growing axon with reduced immunoreactivity in the axon shaft (Fig. 5D–F,K). By the third day in culture, SMURF1 was primarily confined to the cell body of the enteric neurons with very low levels in axons (Fig. 5G–I,L).
Figure 5.
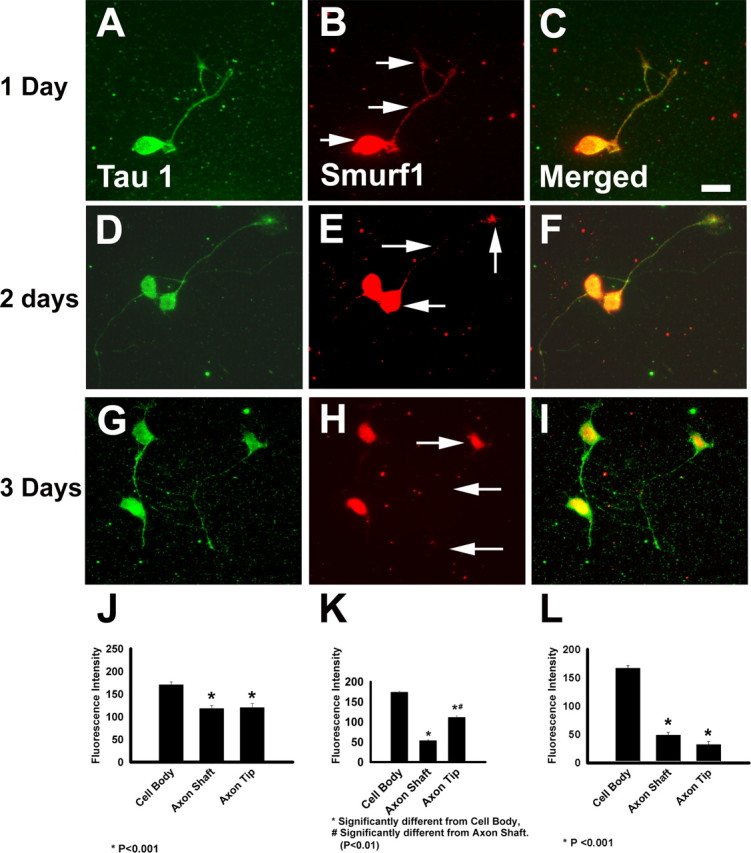
SMURF1 protein is expressed in a polarized distribution in enteric neuron precursors. E14.5 rat immunoselected ENS precursors were maintained in culture for 24, 48, or 72 h before immunohistochemistry with an antibody to TAU1 (A, D, G) or SMURF1 (B, E, H). C, F, I, Merged images. The distribution of SMURF1 immunofluorescence intensity in the cell body, axon shaft, and axon tip was determined after 24 h (J), 48 h (K), or 72 h (L) in culture. Arrows indicate the location of fluorescence intensity measurements. n = 25 cells analyzed for each time point. Scale bar, 50 μm.
SMURF1, PKCζ, and GSK3β associate with PAR6 in enteric neuroblasts
The similar distribution of SMURF1, PKCζ, GSK3β, and PAR3 proteins in developing enteric neuroblasts suggested the possibility that all of these proteins associate with each other in developing ENS precursors. PKCζ, GSK3β, PAR6, and PAR3 are already known to associate in other cell types in which this interaction is important for cell polarization (Lin et al., 2000; Etienne-Manneville and Hall, 2003b). PKCζ and SMURF1 also are known to interact directly in vitro in Mv1Lu and NIH3T3 cells (Wang et al., 2003). To test the hypothesis that SMURF1 associates with other proteins involved in cell polarity in ENS precursors, we performed coimmunoprecipitation experiments and protein immunoblotting (Fig. 6). These analyses confirmed that GSK3β coimmunoprecipitates with both PAR6 and PKCζ from immunoselected ENS precursors and that SMURF1 and PAR6 coimmunoprecipitate with each other. Thus, SMURF1 in ENS precursors associates with the complex of proteins that controls cell polarity.
Figure 6.

PAR6, GSK3β, PKCζ, and SMURF1 proteins coimmunoprecipitate. Cell lysates prepared from p75NTR+ immunoselected cells were immunoprecipitated (IP) using antibodies to SMURF1, PAR6, GSK3β, and PKCζ. Immunoprecipitates were separated by SDS-PAGE, and protein immunoblots were incubated with antibodies against SMURF1, PAR6, GSK3β, and PKCζ as indicated. Blank lanes on the gel were from immunoprecipitations performed with the primary antibody omitted (Ab −ve).
SMURF1 is required for neurite formation and elongation in ENS precursors
Because SMURF1 degrades RhoA (Wang et al., 2006) and RhoA inhibits axon growth (Bryan et al., 2005), we initially hypothesized that blocking Smurf1 would reduce neurite length in cultured ENS precursors. To test this hypothesis, E12.5 immunoselected ENS precursors were infected with a retrovirus that produces siRNA to block SMURF1 production (Fig. 7). The virus also produces ZsGreen so that infected cells can be easily identified. ENS precursors were initially maintained in media with chick embryo extract but without additional GDNF for 48 h to allow for siRNA production. Neurospheres formed under these conditions were trypsinized and replated in GDNF-containing medium to encourage neuronal differentiation and neurite growth. Analysis of the infected cells by SMURF1 immunohistochemistry and ZsGreen fluorescence after an additional 48 h in culture demonstrated that Smurf1 siRNA virus-infected cells had essentially no detectable SMURF1 immunoreactivity (Fig. 7D–F), whereas SMURF1 was readily detected in cells infected with a control retrovirus (Fig. 7A–C). The effectiveness of Smurf1 siRNA was also confirmed by protein immunoblot analysis (Fig. 7G), which demonstrated a 90% loss of SMURF1 in infected cells (p < 0.001). Furthermore, Smurf1 siRNA virus-infected TuJ1+ ENS precursors had significantly reduced neurite length compared with cells infected with control virus (Fig. 8A–F,J), and a high percentage of these cells failed to produce neurites during the culture period (Fig. 8K). These data suggested that blocking SMURF1 production significantly impairs neurite formation in ENS precursors.
Figure 7.
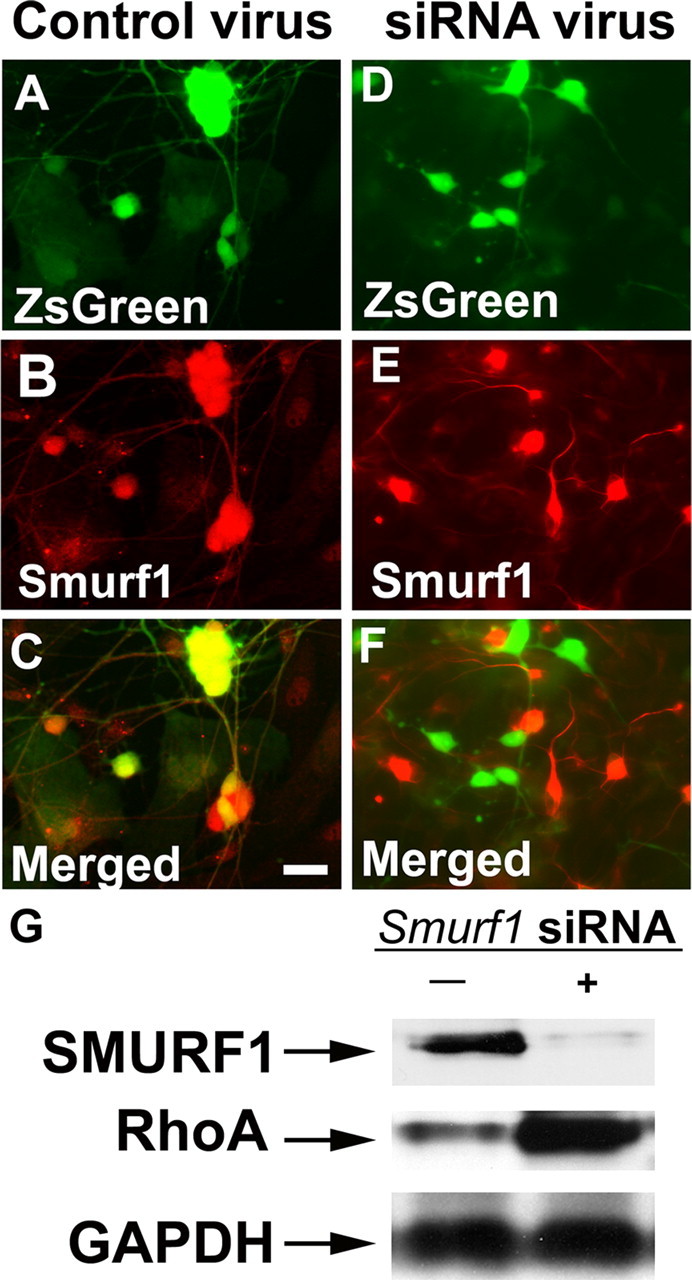
Smurf1 siRNA-expressing virus efficiently reduces SMURF1 protein levels and results in RhoA protein accumulation. E12.5 mouse immunoselected ENS precursors infected with control virus (A–C) or Smurf1 siRNA producing virus (D–F) were maintained in culture for 48 h after plating. A, D, The virus encodes ZsGreen in addition to the siRNA so that infected cells are readily identified. Immunohistochemistry was performed using an antibody to SMURF1 (B, E) and demonstrates that SMURF1 protein is not detected in cells infected with Smurf1 siRNA producing virus. C, F, Merged images. G, Protein immunoblot of cells infected with control virus or Smurf1 siRNA virus. The Smurf1 siRNA efficiently reduces SMURF1 protein levels and causes increased RhoA accumulation.
Figure 8.

Smurf1 siRNA reduces neurite length and increases the percentage of neurons without neurites. E12.5 mouse immunoselected ENS precursors infected with control virus (A–C) or Smurf1 siRNA producing virus (D–F) were maintained in culture for 48 h after plating. A, D, G, ZsGreen expression identifies virus-infected cells. B, E, H, Immunohistochemistry with TuJ1 antibody. C, F, I, Merged images. G–I, Cells expressing Smurf1 siRNA were cultured in the presence of a ROCK inhibitor (10 μm Y-27632). Similar results were obtained with another ROCK inhibitor (10 nm H-1152P). J, K, Quantitative analysis of length for the longest neurite on each cell (n = 132 cells) or of the percentage of cells with no neurites (n = 316 cells). Arrows indicate TuJ1+ neurites. Scale bars, 50 μm. *p < 0.001, significantly different from control. #p < 0.001, significantly different from Smurf1 siRNA treatment group.
Because SMURF1 is known to target RhoA for ubiquitin/proteasome-mediated degradation, we hypothesized that the reduced neurite growth in Smurf1 siRNA-expressing virus-infected cells was attributable to increased RhoA activity. We demonstrated directly that RhoA protein is elevated 2.3-fold in ENS precursors infected with a virus producing Smurf1 siRNA by protein immunoblot analysis (Fig. 7G) (p < 0.001). We then tested the hypothesis that Smurf1 siRNA effects on neurite growth could be reversed by blocking the RhoA action, using an inhibitor of Rho kinase (ROCK), a downstream mediator of RhoA activity important for axon outgrowth in cerebellar granule cells (Bito et al., 2000). These studies demonstrated that the inhibition of neurite growth induced by Smurf1 siRNA was at least partially reversed by ROCK inhibitor (Fig. 8G–K), consistent with the hypothesis that increased RhoA activity limits neurite growth when SMURF1 protein is absent. Interestingly, however, ROCK inhibitor had no effect on neurite length or on the presence of neurites in control siRNA-infected TuJ1+ cells. Together, these results suggest that SMURF1 normally essentially eliminates RhoA/ROCK activity from the tips of growing neurites and that targeting RhoA for degradation is important for neurite growth in ENS precursors.
SMURF1 or ROCK inhibition have different effects on axon specification than PKCζ or GSK3β inhibition
Because PKCζ, GSK3β, and SMURF1 have a similar distribution in ENS precursors and appear to be in the same complex, we had hypothesized that reducing SMURF1 would affect neuronal polarity in the same way as PKCζ or GSK3β inhibition. Specifically, we wanted to determine whether Smurf1 siRNA expression would lead to an increase in the generation of multi-axonal neurons that developed from cultured enteric neuroblasts. To test this hypothesis, immunoselected E12.5 ENS precursors were infected with retroviruses expressing Smurf1 siRNA or a control siRNA and maintained for 48 h in media with chick embryo extract but without added GDNF. Cells were then trypsinized, replated, and cultured in GDNF containing media to induce neurite growth and neuronal differentiation. The number of axons/TuJ1+ cell was evaluated after immunohistochemistry for TAU1. This analysis confirmed that Smurf1 siRNA expression reduced the number of TuJ1+ cells with a single axon and increased the number of neurons without any axons (Fig. 9A,B). In contrast to the results of PKCζ or GSK3β inhibition, however, there was no significant increase in the percentage of TuJ1+ cells with multiple axons despite the fact that blocking PKCζ, GSK3β, or Smurf1 caused comparable reductions in neurite length (Figs. 8J, 9E). Furthermore, treatment of cells infected with Smurf1 siRNA producing virus with a ROCK inhibitor significantly reduced the number of TuJ1+ cells with no axon and increased the percentage of cells with a single axon but had minimal effect on the number on enteric neurons with multiple axons (Fig. 9C,D). These data suggest that, although SMURF1 associates with PAR3, PAR6, PKCζ, and GSK3β in ENS precursors and is important for axon growth, SMURF1 does not influence neuronal polarization or axon specification. Consistent with this finding, Smurf1 inhibition also does not influence the polarized distribution of PAR3 protein in these cells (Fig. 9F).
Figure 9.
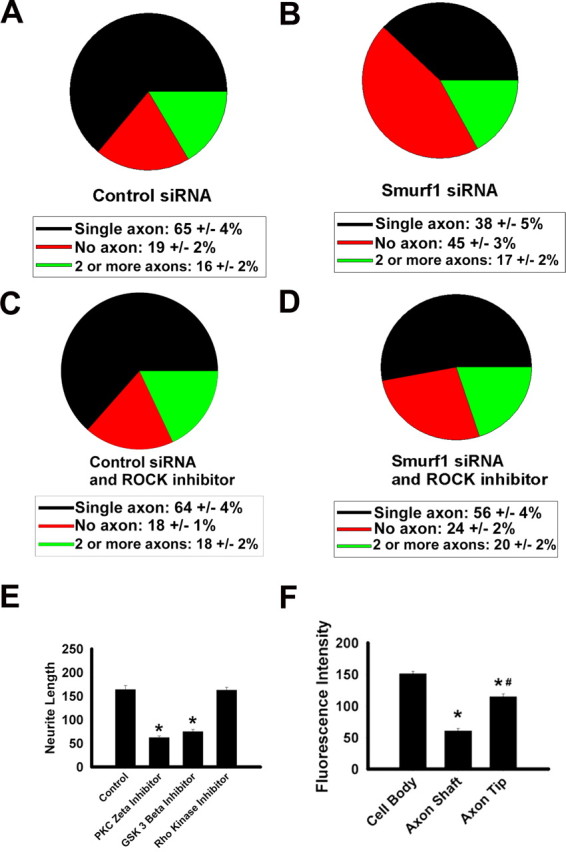
Smurf1 siRNA increases the percentage of cells with no axon but does not increase the percentage of cells with multiple axons. A–D, siRNA-expressing virus-infected E12.5 immunoselected mouse ENS precursors were cultured for 48 h after plating before staining with an antibody to TAU1 and a TuJ1 antibody. C, D, ROCK inhibitor used was Y-27632. A–D, Quantitative analysis of the number of axons per neuron under each condition (n > 289 cells for each graph). E, Quantitative analysis of the length of the longest neurite in each cell (n = 120 cells under each condition). The GSK3β inhibitor used was SB216763, the PKCζ inhibitor used was the myristoylated pseudosubstrate inhibitor, and the ROCK inhibitor used was Y-27632. Similar results were obtained using alternate inhibitors H-1152P (ROCK inhibitor) and SB415286 (GSK3β inhibitor). E, *p < 0.001, significantly different from control. F, Eliminating SMURF1 does not alter PAR3 polarization (n = 34 cells). F, *p < 0.001, significantly different from cell body; #p < 0.001, significantly different from axon shaft.
Discussion
Axon specification is important for the neuronal differentiation. This type of polarized neurite specification is mediated by a protein complex including PKCζ, GSK3β, PAR3, and PAR6 (Wiggin et al., 2005; Yoshimura et al., 2006). Each of these proteins has a demonstrated role in cell polarity in a number of contexts (Wodarz, 2002; Etienne-Manneville and Hall, 2003a). The current study demonstrates that PKCζ, GSK3β, PAR3, and PAR6 are expressed in ENS precursors with protein localization to the axon tip in a pattern resembling that seen in developing hippocampal or dorsal root ganglion cells. Furthermore, globally inhibiting either GSK3β or PKCζ in ENS precursors increases the production of multi-axonal neurons, reduces axon growth, and slows the migration of ENS precursors through the distal bowel. These effects are consistent with a role for GSK3β and PKCζ in ENS precursor polarization.
Recently, SMURF1 has been demonstrated to regulate cell polarity in model cell lines by targeting RhoA for degradation (Wang et al., 2003, 2006; Zhang et al., 2004; Ozdamar et al., 2005; Boyer et al., 2006) and to promote neurite growth in Neuro2A cells by the same mechanism (Bryan et al., 2005). SMURF1 also has other substrates including SMAD proteins activated by bone morphogenetic protein (BMP) signaling (Zhu et al., 1999; Barrios-Rodiles et al., 2005; Alexandrova and Thomsen, 2006), TGFβ and BMP type I receptors (Kavsak et al., 2000; Ebisawa et al., 2001), and Runx2 (runt related transcription factor 2) (Zhao et al., 2003). In the present study, we demonstrate that SMURF1 is expressed in developing mammalian neurons and localized in the cell body and axon tip in a similar distribution to other proteins controlling cell polarity. We also demonstrate that SMURF1 interacts with the polarity complex proteins including PAR6. These data suggested the possibility that SMURF1 and RhoA signaling might be important for ENS precursor polarization. To test this hypothesis, immunoselected ENS precursors were infected with a virus expressing Smurf1 siRNA or control siRNA. Although Smurf1 siRNA-expressing cells had markedly reduced neurite length, there was no increase in axon number in cells with reduced SMURF1 production. This was true even in the presence of ROCK inhibitor that permitted neurite growth from most cells. We further demonstrated that silencing SMURF1 does not influence the polarized distribution of PAR3. Finally, the observation that ROCK inhibitor did not affect neuronal polarity or neurite length in control siRNA-treated cells suggests that, under normal growth conditions, RhoA activity is already low in developing axons because of SMURF1. Thus, although SMURF1 and RhoA/ROCK signaling are important for neurite growth in ENS precursors, there is no clear role for these proteins in ENS precursor polarity.
These findings have important implications for a common potentially fatal human disease in which failure of neural crest-derived precursor migration results in distal colonic aganglionosis. This disorder, called Hirschsprung disease (Skinner, 1996), which affects 1:5000 human infants, has complex genetics with incomplete penetrance and variable expressivity. This occurs because most affected individuals have a combination of hypomorphic mutations that cause aganglionosis (Gabriel et al., 2002; McCallion et al., 2003; Vohra et al., 2006). In particular, reduced signaling by the Ret transmembrane tyrosine kinase is a major risk factor for Hirschsprung disease. For this reason, defining molecular mechanisms through which Ret signaling encourages ENS precursor migration and differentiation is critical for understanding Hirschsprung disease risk factors and may suggest novel methods for preventing or treating this disorder. Furthermore, although enteric neurons have long been appreciated to have complex and diverse patterns of axon growth, almost nothing was previously known about mechanisms that might control neurite specification within the ENS.
Ret is expressed in ENS precursors in which it is the signaling receptor for the GDNF family of ligands (glial cell line-derived neurotrophic factor, neurturin, artemin, and persephin) (Baloh et al., 2000; Manie et al., 2001; Takahashi, 2001; Airaksinen and Saarma, 2002). Substantial work over the past 10 years has demonstrated that GDNF/Ret signaling is important for ENS precursor survival, proliferation, migration, and differentiation (Pichel et al., 1996; Sanchez et al., 1996; Chalazonitis et al., 1998; Enomoto et al., 1998; Hearn et al., 1998; Heuckeroth et al., 1998; Taraviras et al., 1999; Young et al., 2001; Gianino et al., 2003). In fact, gradients of GDNF expression within the bowel are thought to be important for promoting directed ENS precursor migration. For these reasons, Ret signaling pathways have been investigated in many model systems, but mechanisms linking Ret activation to changes in the cytoskeleton needed for directed cell migration or neurite growth are incompletely understood.
New data presented, when combined with known Ret signaling pathways, directly link Ret activation at the cell surface to intracellular molecular machinery that regulates the cytoskeleton and suggest a model through which Ret signaling might control neurite outgrowth, cell polarization, and migration of ENS precursors (Fig. 10). This signal begins with localized phosphatidylinositol 3-kinase (PI3) kinase activation, which is also essential for ENS precursor survival, neurite outgrowth, and directed migration (Natarajan et al., 2002; Srinivasan et al., 2005). Because PI3 kinase signaling is required for cell survival, however, PI3 kinase effects on neurite growth or precursor migration are difficult to interpret. Furthermore, previous studies of ENS development ignored the issue of polarization that is required for axon specification and directed migration. These mechanisms have, however, been investigated previously in a number of other cell types (Hirai and Chida, 2003; Arimura and Kaibuchi, 2005; Charest and Firtel, 2006; Yoshimura et al., 2006) in which migration, neurite growth, and specification are controlled by different cell surface receptors. In ENS precursors, localized PI3 kinase activation occurs after Ret phosphorylation at tyrosine 1062 (Hayashi et al., 2000; Takahashi, 2001; Kurokawa et al., 2003). PI3 kinase then phosphorylates phosphatidyl inositol [PI(4,5)P2] to generate PI(3,4,5)P3 [PIP3], which in turn leads to local recruitment to the cell membrane and activation of pleckstrin homology domain proteins, including protein kinase B (also known as AKT), 3′-PI-dependent protein kinase 1 (PDK1), and integrin-linked kinase (Yoganathan et al., 2000). PDK1 phosphorylates and activates PKCζ, whereas PIP3 also stimulates PKCζ autophosphorylation (Hirai and Chida, 2003). In addition, PI3 kinase or PIP3 and PDK1 greatly enhance the activity of GDP/GTP exchange factors (GEF) that activate Rho family GTPases such as Rac1 and Cdc42 (Tian et al., 2002; Innocenti et al., 2003). PI3 kinase activation is also required for the localization of PAR complex proteins, PKCζ, and Cdc42 to the tips of growing axons (Yoshimura et al., 2006). Cdc42 is then thought to activate Rac1 via PAR3 and the Tiam1 (T-cell lymphoma invasion and metastasis 1) GEF (Matsuo et al., 2003; Nishimura et al., 2005). Because Rac1 directly activates PI3 kinase, localized activation of PI3 kinase by receptor tyrosine kinases such as Ret can lead to the formation of a positive feedback loop that facilitates rapid axon extension or directed cell migration. Once this activated complex is enriched at the leading edge of the migrating cell or the tip of the growing axon, changes in actin filament and microtubule dynamics can be mediated by Rho family GTPases and PKCζ.
Figure 10.

Model for how Ret signaling influences ENS precursor cell migration and neurite growth. Proteins highlighted with bold outlining form a positive feedback loop required for cell migration and axon growth. New roles in ENS development were demonstrated for PKCζ, GSK3β, SMURF1, and ROCK through the current studies. GFRα1, Glial cell line derived neurotrophic factor family receptor α1; SHC, Src homology 2 domain containing; GRB2, growth factor receptor-bound protein 2; ILK, integrin-linked kinase; APC, adenomatous polyposis coli; CRMP2, collapsin response mediator protein 2.
In parallel with PI3 kinase/PKCζ-mediated signaling, localized inhibition of GSK3β is also essential for polarized outgrowth of single axons from hippocampal (Shi et al., 2004; Jiang et al., 2005; Yoshimura et al., 2005; Gartner et al., 2006) and dorsal root ganglion (Zhou et al., 2004) neurons and for migration of astrocytes (Etienne-Manneville and Hall, 2003b). This localized GSK3β inhibition appears to be required for adenomatous polyposis coli- and kinesin-mediated transport of PAR3 to the plus ends of rapidly growing microtubules (Shi et al., 2004; Zhou et al., 2004). In contrast to these other neuron types, however, some enteric neurons normally produce multiple axons, suggesting the possibility that reducing PKCζ or GSK3β signaling could be part of the mechanism for producing multi-axonal enteric neuron subtypes in vivo. Interestingly, inhibition of GSK3β could be mediated by AKT, integrin linked kinase (Arevalo and Chao, 2005), or directly by PKCζ (Etienne-Manneville and Hall, 2003b). Thus, the observation that blocking either PKCζ or GSK3β has a similar effect on axon growth and polarization in the ENS is consistent with the hypothesis that local activity of these kinases is essential for normal development. For this reason, global inhibition of kinase activity both increases axon number and slows precursor migration by disrupting normal cell polarity.
Together, these observations provide novel insight into molecular mechanisms critical for formation of the ENS. Specifically, we demonstrate for the first time that proteins controlling neuronal polarity and axon number (PKCζ and GSK3β) are also essential for ENS precursor migration through the bowel. In addition, we find that, although SMURF1 is essential for neurite growth, associates with other proteins controlling polarity, and influences polarity in other cell types, altering SMURF1 or ROCK signaling does not influence axon number in the developing enteric neurons. Finally, although PCKζ and GSK3β activity are not required for survival of ENS precursors, we predict that polymorphisms in GSK3β or PKCζ could alter Hirschsprung disease penetrance or expressivity and contribute to the variable phenotypes observed even within family members that share the same Ret mutation by reducing the migration of ENS precursors through the distal bowel.
Note added in proof.
A recent manuscript demonstrates that Smurf1 is important for neurite growth but not neuronal polarity in hippocampal neurons (Schwamborn et al., 2007). The same authors demonstrate that the related protein Smurf2 regulates neuronal polarity by ubiquitinating and targeting Rap1B for degradation. These observations agree with our findings about the role of Smurf1 in enteric neurons.
Footnotes
R.O.H. was supported by National Institutes of Health (NIH) Grants RO1 DK57038 and RO1 DK6459201, Digestive Disease Research Center Core NIH Grant P30-DK52574, and March of Dimes Grant FY02-182. We thank Dr. Jeffrey Milbrandt and Dr. Louis Muglia for thoughtful comments about this manuscript and Dr. Moses Chao for sharing his p75NTR antibody.
References
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Alexandrova EM, Thomsen GH. Smurf1 regulates neural patterning and folding in Xenopus embryos by antagonizing the BMP/Smad1 pathway. Dev Biol. 2006;299:398–410. doi: 10.1016/j.ydbio.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo JC, Chao MV. Axonal growth: where neurotrophins meet Wnts. Curr Opin Cell Biol. 2005;17:112–115. doi: 10.1016/j.ceb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kaibuchi K. Key regulators in neuronal polarity. Neuron. 2005;48:881–884. doi: 10.1016/j.neuron.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EMJ, Milbrandt J. The GDNF family ligands and receptors—implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron. 2003;40:905–916. doi: 10.1016/s0896-6273(03)00730-x. [DOI] [PubMed] [Google Scholar]
- Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N, Robinson M, Suzuki H, Hayashizaki Y, Jurisica I, Wrana JL. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- Bito H, Furuyashiki T, Ishihara H, Shibasaki Y, Ohashi K, Mizuno K, Maekawa M, Ishizaki T, Narumiya S. A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron. 2000;26:431–441. doi: 10.1016/s0896-6273(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Boyer L, Turchi L, Desnues B, Doye A, Ponzio G, Mege JL, Yamashita M, Zhang YE, Bertoglio J, Flatau G, Boquet P, Lemichez E. CNF1-induced ubiquitylation and proteasome destruction of activated RhoA is impaired in Smurf1−/− cells. Mol Biol Cell. 2006;17:2489–2497. doi: 10.1091/mbc.E05-09-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehmer A, Schrodl F, Neuhuber W. Morphological classifications of enteric neurons—100 years after Dogiel. Anat Embryol (Berl) 1999;200:125–135. doi: 10.1007/s004290050267. [DOI] [PubMed] [Google Scholar]
- Bryan B, Cai Y, Wrighton K, Wu G, Feng XH, Liu M. Ubiquitination of RhoA by Smurf1 promotes neurite outgrowth. FEBS Lett. 2005;579:1015–1019. doi: 10.1016/j.febslet.2004.12.074. [DOI] [PubMed] [Google Scholar]
- Chalazonitis A, Rothman TP, Chen J, Gershon MD. Age-dependent differences in the effects of GDNF and NT-3 on the development of neurons and glia from neural crest-derived precursors immunoselected from the fetal rat gut: expression of GFRα-1 in vitro and in vivo. Dev Biol. 1998;204:385–406. doi: 10.1006/dbio.1998.9090. [DOI] [PubMed] [Google Scholar]
- Chant J. Cell polarity in yeast. Annu Rev Cell Dev Biol. 1999;15:365–391. doi: 10.1146/annurev.cellbio.15.1.365. [DOI] [PubMed] [Google Scholar]
- Charest PG, Firtel RA. Feedback signaling controls leading-edge formation during chemotaxis. Curr Opin Genet Dev. 2006;16:339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogiel AS. Ueber den bau der ganglien in den geflechten des darmes und der gallenblase des menschen und der saugethiere. Arch Anat Physiol Leip Anat Abt Jg. 1899;1899:130–158. [Google Scholar]
- Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EMJ, Milbrandt J. GFRα1 deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cell polarity: Par6, aPKC and cytoskeletal crosstalk. Curr Opin Cell Biol. 2003a;15:67–72. doi: 10.1016/s0955-0674(02)00005-4. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003b;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Furness JB. Types of neurons in the enteric nervous system. J Auton Nerv Syst. 2000;81:87–96. doi: 10.1016/s0165-1838(00)00127-2. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Salomon R, Pelet A, Angrist M, Amiel J, Fornage M, Attie-Bitach T, Olson JM, Hofstra R, Buys C, Steffann J, Munnich A, Lyonnet S, Chakravarti A. Segregation at three loci explains familial and population risk in Hirschsprung disease. Nat Genet. 2002;31:89–93. doi: 10.1038/ng868. [DOI] [PubMed] [Google Scholar]
- Gartner A, Huang X, Hall A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3β) independently of Akt/PKB serine phosphorylation. J Cell Sci. 2006;119:3927–3934. doi: 10.1242/jcs.03159. [DOI] [PubMed] [Google Scholar]
- Gershon M. Genes and lineages in the formation of the enteric nervous system. Curr Opin Neurobiol. 1997;7:101–109. doi: 10.1016/s0959-4388(97)80127-4. [DOI] [PubMed] [Google Scholar]
- Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–2198. doi: 10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kawai K, Kurokawa K, Murakumo Y, Imai T, Funahashi H, Nakao A, Takahashi M. Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene. 2000;19:4469–4475. doi: 10.1038/sj.onc.1203799. [DOI] [PubMed] [Google Scholar]
- Hearn CJ, Murphy M, Newgreen D. GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Dev Biol. 1998;197:93–105. doi: 10.1006/dbio.1998.8876. [DOI] [PubMed] [Google Scholar]
- Heuckeroth RO, Lampe PA, Johnson EMJ, Milbrandt J. Neurturin and GDNF promote proliferation and survival of enteric neuron and glial progenitors in vitro. Dev Biol. 1998;200:116–129. doi: 10.1006/dbio.1998.8955. [DOI] [PubMed] [Google Scholar]
- Higginbotham H, Tanaka T, Brinkman BC, Gleeson JG. GSK3beta and PKCzeta function in centrosome localization and process stabilization during Slit-mediated neuronal repolarization. Mol Cell Neurosci. 2006;32:118–132. doi: 10.1016/j.mcn.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Hirai T, Chida K. Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J Biochem (Tokyo) 2003;133:1–7. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- Huber LJ, Chao MV. Mesenchymal and neuronal cell expression of the p75 neurotrophin receptor gene occur by different mechanisms. Dev Biol. 1995;167:227–238. doi: 10.1006/dbio.1995.1019. [DOI] [PubMed] [Google Scholar]
- Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J Cell Biol. 2003;160:17–23. doi: 10.1083/jcb.200206079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–320. doi: 10.1016/s0092-8674(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Kawai K, Hashimoto M, Ito Y, Takahashi M. Cell signalling and gene expression mediated by RET tyrosine kinase. J Intern Med. 2003;253:627–633. doi: 10.1046/j.1365-2796.2003.01167.x. [DOI] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Manie S, Santoro M, Fusco A, Billaud M. The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet. 2001;17:580–589. doi: 10.1016/s0168-9525(01)02420-9. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Terao M, Nabeshima Y, Hoshino M. Roles of STEF/Tiam1, guanine nucleotide exchange factors for Rac1, in regulation of growth cone morphology. Mol Cell Neurosci. 2003;24:69–81. doi: 10.1016/s1044-7431(03)00122-2. [DOI] [PubMed] [Google Scholar]
- McCallion AS, Stames E, Conlon RA, Chakravarti A. Phenotype variation in two-locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and Ednrb. Proc Natl Acad Sci USA. 2003;100:1826–1831. doi: 10.1073/pnas.0337540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance J. PAR proteins and the establishment of cell polarity during C. elegans development. BioEssays. 2005;27:126–135. doi: 10.1002/bies.20175. [DOI] [PubMed] [Google Scholar]
- Natarajan D, Marcos-Gutierrez C, Pachnis V, De Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–5160. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- Newgreen D, Young HM. Enteric nervous system: development and developmental disturbances—part 1. Pediatr Dev Pathol. 2002a;5:224–247. doi: 10.1007/s10024-001-0142-y. [DOI] [PubMed] [Google Scholar]
- Newgreen D, Young HM. Enteric nervous system: development and developmental disturbances—part 2. Pediatr Dev Pathol. 2002b;5:329–349. doi: 10.1007/s10024-002-0002-4. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Hui SZ, Granholm A-C, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach TT, Duchemin AM, Rogemond V, Aguera M, Honnorat J, Belin MF, Kolattukudy PE. Involvement of collapsin response mediator proteins in the neurite extension induced by neurotrophins in dorsal root ganglion neurons. Mol Cell Neurosci. 2004;25:433–443. doi: 10.1016/j.mcn.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Muller M, Becker AH, Puschel AW. Ubiquitination of the GTPase Rap1B by the ubiquitin ligase Smurf2 is required for the establishment of neuronal polarity. EMBO J. 2007;26:1410–1422. doi: 10.1038/sj.emboj.7601580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Shi SH, Cheng T, Jan LY, Jan YN. APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol. 2004;14:2025–2032. doi: 10.1016/j.cub.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Skinner M. Hirschsprung's disease. Curr Probl Surg. 1996;33:391–461. doi: 10.1016/s0011-3840(96)80009-8. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Anitha M, Mwangi S, Heuckeroth RO. Enteric neuroblasts require the phosphatidylinositol 3-kinase/Akt/Forkhead pathway for GDNF-stimulated survival. Mol Cell Neurosci. 2005;29:107–119. doi: 10.1016/j.mcn.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/s1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- Taraviras S, Marcos-Gutierrez CV, Durbec P, Jani H, Grigoriou M, Sukumaran M, Wang L-C, Hynes M, Raisman G, Pachnis V. Signalling by RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development. 1999;126:2785–2797. doi: 10.1242/dev.126.12.2785. [DOI] [PubMed] [Google Scholar]
- Tian X, Rusanescu G, Hou W, Schaffhausen B, Feig LA. PDK1 mediates growth factor-induced Ral-GEF activation by a kinase-independent mechanism. EMBO J. 2002;21:1327–1338. doi: 10.1093/emboj/21.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohra BPS, Planer W, Armon J, Fu M, Jain S, Heuckeroth R. Reduced entothelin converting enzyme-1 and endothelin-3 mRNA in the developing bowel of male mice may increase expressivity and penetrance of Hirschsprung disease like distal intestinal aganglionosis. Dev Dyn. 2006;236:106–117. doi: 10.1002/dvdy.21028. [DOI] [PubMed] [Google Scholar]
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- Wang HR, Ogunjimi AA, Zhang Y, Ozdamar B, Bose R, Wrana JL. Degradation of RhoA by Smurf1 ubiquitin ligase. Methods Enzymol. 2006;406:437–447. doi: 10.1016/S0076-6879(06)06032-0. [DOI] [PubMed] [Google Scholar]
- Wiggin GR, Fawcett JP, Pawson T. Polarity proteins in axon specification and synaptogenesis. Dev Cell. 2005;8:803–816. doi: 10.1016/j.devcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Wodarz A. Establishing cell polarity in development. Nat Cell Biol. 2002;4:E39–E44. doi: 10.1038/ncb0202-e39. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Chen J-X, Rothman TP, Gershon MD. Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development. 1999;126:1161–1173. doi: 10.1242/dev.126.6.1161. [DOI] [PubMed] [Google Scholar]
- Yoganathan TN, Costello P, Chen X, Jabali M, Yan J, Leung D, Zhang Z, Yee A, Dedhar S, Sanghera J. Integrin-linked kinase (ILK): a “hot” therapeutic target. Biochem Pharmacol. 2000;60:1115–1119. doi: 10.1016/s0006-2952(00)00444-5. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kaibuchi K. Signaling networks in neuronal polarization. J Neurosci. 2006;26:10626–10630. doi: 10.1523/JNEUROSCI.3824-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HM, Hearn CJ, Farlie PG, Canty AJ, Thomas PQ, Newgreen DF. GDNF is a chemoattractant for enteric neural cells. Dev Biol. 2001;229:503–516. doi: 10.1006/dbio.2000.0100. [DOI] [PubMed] [Google Scholar]
- Yu JY, Taylor J, DeRuiter SL, Vojtek AB, Turner DL. Simultaneous inhibition of GSK3alpha and GSK3beta using hairpin siRNA expression vectors. Mol Ther. 2003;7:228–236. doi: 10.1016/s1525-0016(02)00037-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang HR, Wrana JL. Smurf1: a link between cell polarity and ubiquitination. Cell Cycle. 2004;3:391–392. doi: 10.4161/cc.3.4.772. [DOI] [PubMed] [Google Scholar]
- Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J Biol Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]