

Abstract
Studies of human seizure disorders have revealed that susceptibility to seizures is greatly influenced by genetic factors. In addition to causing epilepsy, genetic factors can suppress seizures and epileptogenesis. Examination of seizure-suppressor genes is challenging in humans. However, such genes are readily identified and analyzed in a Drosophila animal model of epilepsy. In this article, the epilepsy phenotype of Drosophila seizure-sensitive mutants is reviewed. A novel class of genes called seizure-suppressors is described. Mutations defining suppressors revert the “epilepsy” phenotype of neurological mutants. We conclude this review with particular discussion of a seizure-suppressor gene encoding DNA topoisomerase I (top1). Mutations of top1 are especially effective at reverting the seizure-sensitive phenotype of Drosophila epilepsy mutants. In addition, an unexpected class of anti-epileptic drugs has been identified. These are DNA topoisomerase I inhibitors such as camptothecin and its derivatives; several candidates are comparable or perhaps better than traditional anti-epileptic drugs such as valproate at reducing seizures in Drosophila drug-feeding experiments.
1. Introduction
Drosophila has been a model for examining fundamentally important problems in biology, especially developmental biology and neurobiology (Rubin and Lewis, 2000). A lesson from these studies is that findings are generally applicable to other experimental model systems such as Caenorhabditis elegans nematodes and mice due to conservation of fundamental processes and essential gene products (Veraksa et al., 2000; Tickoo and Russell, 2002). An implication from cross-species conservation is that Drosophila has the potential to be a powerful system for modeling human pathologies. This comes, in part, from estimates of 75% of all human disease genes have related sequences in Drosophila (Bier, 2005). Drosophila models have been developed for cancer, cardiac disease, and several neurodegenerative diseases such as Parkinson's disease, Huntington's disease, Alzheimer's disease, and amyotrophic lateral sclerosis (reviewed in Bier and Bodmer, 2004; Bier, 2005; Michno et al., 2005; Vidal and Cagan, 2006). Here we review Drosophila modeling of human seizure disorders.
Human seizure disorders are a significant health concern due to the large number of affected individuals, the potentially devastating ramifications of untreated seizure episodes, and the limitations of antiepileptic drug (AED) options. Seizure-suppressor genes provide a powerful tool for examining seizure disorders and identifying potential AED targets. The major interest in seizure-suppressors is that they may lead to new and significant treatments for human epilepsy. Seizure-suppressor genes could help define targets for unexpected classes of anticonvulsant drugs that are effective new treatments for epilepsy: treatments for intractable syndromes or treatments with reduced side effects. Another possibility is to discover candidate genes that might be used for gene therapy. Among the several questions that arise are: what are seizure-suppressor genes and how might they lead to new therapeutics? What is the entire range of potential gene products that can act as seizure-suppressors? Is this range limited to nervous system-specific gene products, such as signaling molecules or does it include non-nervous system gene products as well? This article focuses on a Drosophila model of epilepsy, illustrating the use of genetic screens to identify seizure-suppressor genes and their potential applications to therapeutics.
2. The utility of Drosophila in studying human seizure disorders
2.1. Animal models of epilepsy
Numerous animal models have been utilized to study epilepsy. Some interesting but uncommon models include baboon, chicken, cat, dog, and Mongolian gerbil (Avoli, 1995; Bertorelli et al., 1995; Menini and Silva-Barrat, 1998; Batini et al., 2004; Lohi et al., 2005). More recently, the model genetic organisms zebrafish and C. elegans have been shown to be valuable in study of seizure disorders (Baraban, 2007). Zebrafish larvae exhibit mammalian-like seizure activity when administered the convulsant drug, pentylenetetrazole (PTZ) (Baraban et al., 2005). PTZ-treated larvae dart around the culture dish, swim in circles, convulse, and then paralyze for several seconds. This behavior is coupled with abnormal brain electrophysiology as recorded using fish electroencephalography, revealing ictal and interictal bursts of neuronal firing during seizure activity. The behavior has been successful in genetic screening for seizure-resistant mutant fish, identifying six such resistant mutants (Baraban, et al., 2007). C. elegans is used to model epilepsy caused by lissencephaly. Worms with a mutated LIS1 gene are more susceptible to PTZ-induced convulsions than normal (Williams et al., 2004). Furthermore, worms depleted for LIS1 pathway components in the worm show genetic interactions that greatly enhance sensitivity to convulsions (Locke, et al., 2006).
Mouse models of epilepsy have been shown to recapitulate many aspects of seizure disorders in humans (Noebels, 2003). Epileptic mice exhibit a variety of spontaneous seizure phenotypes including generalized tonic-clonic seizures and non-convulsive absence seizures. Seizures have an electrophysiological correlate in electrographic recordings from the brains of epileptic mice. In addition to phenotypic similarities, there are genetic similarities between human and mouse epilepsies. Numerous human epilepsy genes cause epileptic phenotypes in mice. Similar to humans, epilepsy genetics in mice frequently follow non-Mendelian, complex inheritance patterns, such as in the case of the EL model of mouse epilepsy (Legare et al., 2000).
In spite of its excellence as a phenotypic and genetic model of human epilepsy, the mouse has some experimental limitations. It is difficult to generate and screen for new mutants. Likewise, it is expensive and labor-intensive to maintain and manipulate large numbers of animals. An alternative model is the fruitfly Drosophila melanogaster. Several features make Drosophila an attractive experimental system. Chemical and transposon-based mutagenesis methods, drug-feeding capabilities and behavioral analyses have large capacity for examining large numbers of experimental animals. Advanced electrophysiology methods and molecular genetic techniques greatly facilitate mutant analysis. Epilepsy mutations, seizure suppressors, and seizure enhancers may be studied in single, double, and triple mutant combinations to gain insights into Mendelian and non-Mendelian inheritance patterns. Many of these capabilities are also available in other genetic model organisms such as C. elegans and zebrafish. Here we focus or recent progress made using the Drosophila model.
2.2. Seizure-like activity in adult Drosophila
Electrical shock of sufficient intensity delivered to the brain of Drosophila elicits seizure-like activity (Engel and Wu, 1994; Pavlidis and Tanouye, 1995; Lee and Wu, 2002), similar to all animals with complex nervous systems, including humans. Stimulus is a short wavetrain of high frequency electrical impulses (HF stimulation). Seizure-susceptibility is quantified by seizure threshold: the voltage that HF stimulation becomes an electroconvulsive shock (ECS) and elicits seizure-like activity (Kuebler and Tanouye, 2000). In Drosophila, seizure-like activity can be elicited by HF brain stimulation in all genotypes tested, including seizure-sensitive mutants, wild type, and seizure-suppressor mutants (Kuebler and Tanouye, 2000; Kuebler, et al., 2001). However, seizure-sensitive mutants exhibit seizure thresholds that are much lower than wild-type controls (Kuebler and Tanouye, 2000; Kuebler et al., 2001). Drosophila seizure-like activity is manifest as uncontrolled, abnormal HF neuronal firing that approaches 100 Hz for 3 s. Seizure-like activity appears to be extensive with all neurons thus far examined participating in the seizure. This is about 30 different excitatory motoneurons that apparently transmit glutamate (Kuebler and Tanouye, 2000). Immediately following seizure-like activity is a period of synaptic failure where transmission is interrupted at many central chemical synapses (Fig. 1). Experimentally, motoneurons innervating dorsal longitudinal muscles (DLMs) are monitored to track the occurrence of seizure-like activity and the time course of synaptic failure.
Figure 1. Behavioral and electrophysiological responses of BS mutants to mechanical and electrical stimulation.
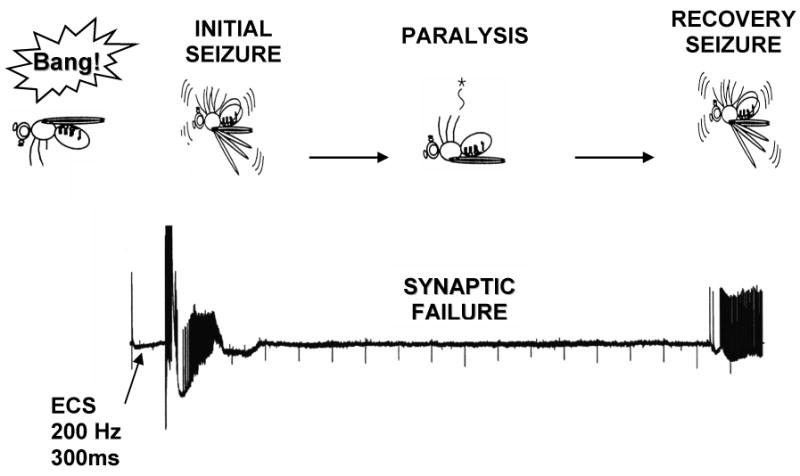
BS flies exhibit a unique behavioral repertoire following a mechanical stimulation (“bang”), such as a tap on the bench-top and vortex on the vortex mixer: they undergo seizure-like behavioral activity (hyperactivity) and subsequent paralysis. The seizure-like behavior is manifested as intense abnormal contraction, wing-flapping, proboscis extension, and leg-shaking; the paralysis is the cessation of physical activity. Upon recovery from paralysis, flies undergo additional bouts of spontaneous seizure-like behavioral activity that vary in intensity depending on the genotype of the fly. The entire cycle from the initial seizure-like behavior to the restoration of normal behavior is termed as the recovery time. This complex behavioral response can be mimicked on an electrophysiological level by administration of a high-frequency ECS (electroconvulsive shock) to the brain of the fly, which results in seizure-like activity followed by synaptic failure. As the fly recovers, a second bout of seizure-like activity is typically seen. Following ECS-induced seizure-like activity, flies exhibit transiently elevated seizure thresholds, corresponding to the behavioral refractory period.
2.3. Drosophila seizure-sensitive mutants
A collection of eleven seizure-sensitive mutants called bang-sensitive (BS) paralytic mutants is the basis for a Drosophila seizure model (Table 1). Seizure-like behaviors are prominent in BS mutants. Following a mechanical shock, such as a tap of the culture vial on the bench top or brief vortex mixing (a ‘bang’), mutants undergo seizure-like behaviors characterized by initial seizure (2 s), temporary paralysis (20-300 s), and recovery seizure (2 s) (Benzer, 1971; Ganetzky and Wu, 1982)(Fig. 1). In addition to mechanical bang stimulation effectiveness, seizure-like behavior in mutants is also elicited by visual stimulation, in particular, stroboscopic light stimulation (R. Hoy, personal communication).
Table 1. List of seizure-sensitive mutants and their gene products.
| Seizure-sensitive Mutant | Gene Product |
|---|---|
| bang senseless (bss1, bss2) | N/A |
| bang sensitive (bas1, bas2) | N/A |
| easily shocked (eas) | ethanolamine kinase (Pavlidis et al., 1994) |
| knockdown (kdn) | citrate synthase (Fergestad et al., 2006) |
| slamdance (sda) | aminopeptidase N (Zhang et al., 2002) |
| technical knockout (tko) | mitochondrial ribosomal protein S12 (Royden et al., 1987) |
| couch potato (cpo) | RNA-binding protein (Glasscock et al., 2005) |
| kazachoc (kcc) | K+ Cl- cotransporter (Hekmat-Scafe et al., 2006) |
| stress-sensitive (sesB) | adenine nucleotide translocase (Zhang et al., 1999a) |
| jitterbug (jbug) | N/A |
| rock-n-roll (rnr) | N/A |
Currently ten different BS mutants have been isolated. The first six mutants comprise the canonical BS strains, characterized by low seizure thresholds (<10 V). The last five BS mutants are non-canonical and exhibit slightly higher seizure thresholds (11-16 V) than the canonical strains.
The canonical BS mutants include bang senseless (bss), bang sensitive (bas), easily shocked (eas), slamdance (sda), technical knockout (tko), and knockdown (kdn). For most of these canonical mutants, the BS behavioral phenotype is completely penetrant and electrophysiological seizure threshold is low, usually below 7 V (Kuebler and Tanouye, 2000). In comparison, normal flies never display a BS behavioral phenotype and have electrophysiological seizure thresholds usually greater than about 35 V (Kuebler and Tanouye, 2000). The molecular basis of bss and bas phenotypes remains unknown. The genes for eas, sda, kdn and tko have been cloned and characterized (table 1). We have settled on three mutants as experimental representatives of the collection. 1) The bss mutant, a severe BS mutant, is behaviorally and electrophysiologically the most sensitive to seizure with a prominent tonic-clonic phenotype. It is also the most difficult BS mutant to suppress genetically and pharmacologically. 2) The eas mutant displays moderate seizure-sensitivity; the gene encodes ethanolamine kinase. 3) The sda mutant displays weak seizure-sensitivity in that it possesses a phenotype that is fully penetrant, but is the easiest to suppress genetically and pharmacologically; the sda gene encodes aminopeptidase.
As example, the eas BS mutation is described in more detail. Flies carrying eas have a defect in the ethanolamine kinase gene, which interferes with the metabolism of phosphatidylethanolamine, the predominant membrane lipid in Drosophila (Pavlidis et al., 1994). The eas allele that causes the BS behavioral phenotype is easPC80 containing a 2-bp deletion that introduces a frame shift; the resulting truncated protein lacks a kinase domain and is devoid of enzymatic activity (Pavlidis et al., 1994). The BS behavioral phenotype of eas flies is completely penetrant, 100% of flies are paralyzed by mechanical stimulation. The seizure threshold of eas is very low compared to the wild-type (3.4 V, about 10-fold lower than wild-type), making eas a desirable genetic background used to screen for seizure-suppressors. The eas defect may cause increased seizure-susceptibility by affecting the function of integral membrane proteins such as ion channels or by affecting membrane fusion events such as neurotransmitter secretion. A recent study showed that easPC80 also has a mushroom body defect similar to the easala allele, pointing to a new role for eas in developmental mushroom body neuroblast proliferation (Pascual et al., 2005).
Several non-canonical BS mutants have been identified including couch potato (cpo) and kazachoc (kcc). These mutants are “non-canonical” in the sense that, unlike most of the original BS mutants, their BS phenotype is incompletely penetrant and their seizure thresholds tend to be higher (11-16 V), but still below wild-type levels. Despite this weaker BS phenotype, the non-canonical BS mutants appear to model epilepsy well, perhaps better than the canonical BS mutants. They are especially strong enhancers of seizure with prominent effects in genetic interaction. The cpo mutant is a P-element mutation that was isolated in a screen for new BS mutants that utilized sda/+ heterozygotes as a sensitized genetic background (Glasscock and Tanouye, 2005; Zhang et al., 2002). The cpo mutation disrupts an RNA-binding protein leading to a variety of neurological phenotypes including behavioral abnormalities, seizure-susceptibility, and synaptic transmission defects (Glasscock and Tanouye, 2005). The cpo phenotype is reminiscent of the majority of human seizure disorders, which exhibit symptomatic epilepsy as one of a set of complex neurological symptoms. The kcc mutation was identified as a spontaneous mutation present on the second chromosome of a sda-enhancer stock. The mutation renders flies susceptible to epileptic-like seizures due to reduced expression of the K+/Cl− cotransporter gene (Hekmat-Scafe et al., 2006). Drosophila kcc mutations resemble those described for mouse KCC2. Seizure phenotypes are observed in mouse KCC2 knock-down mutants that reduce the normal level of neuronal K+/Cl− cotransporter (Woo et al., 2002; Tornberg et al., 2005). In the more severe knock-down mutant (5% normal KCC2 level), generalized seizures are frequently induced by the mild mechanical stimulation that occurs during handling (Woo et al., 2002).
2.4. Comparisons of seizure-like activity in flies and human seizures
Seizures in flies and humans have several similarities providing support for the utility of this type of investigation. Previous investigations have shown that for Drosophila: (1) all individuals have a seizure threshold; (2) genetic mutations can modulate seizure susceptibility; (3) electroconvulsive shock treatment (ECT) in flies raises the threshold for subsequent seizure-like activity; (4) seizure-like activity spreads through the fly central nervous system (CNS) along particular pathways that are dependent on functional synaptic connections and recent electrical activity; (5) seizure-like activity in flies can be spatially segregated into particular regions of the CNS; (6) Drosophila phenotypes can be ameliorated by the human AEDs sodium valproate, phenytoin, gabapentin, and potassium bromide; and (7) mutations affecting Drosophila sodium channels are excellent seizure suppressors, consistent with the notion that many AEDs are targeting sodium channels (Kuebler and Tanouye, 2000, 2002; Kuebler et al., 2001; Reynolds et al., 2003; Tan et al., 2004).
An especially interesting phenomenon during fly seizure episodes in BS mutants is that upon recovery, the fly is resistant to subsequent BS paralysis for several minutes to over an hour depending on genotype, a time termed the refractory period (Ganetzky and Wu, 1982). This surprising effect is of particular interest because, although mutations causing paralysis were known in the literature, a refractory period for a behavioral defect was unheard of and novel. It has since become clear that all BS mutants have a refractory period (Ganetzky and Wu, 1982), and this has become one of their defining features. Electrophysiologically, flies possess seizure thresholds that are transiently elevated during the reractory period (Pavlidis and Tanouye, 1995; Kuebler and Tanouye, 2000). In humans, refractory period is observed following electroconvulsive shock treatment (ECT) that is used for treatment of epilepsy and depression in the clinic. ECT has numerous anticonvulsant effects, including elevated seizure threshold and decreased seizure duration, which makes it a useful adjunctive therapy in epilepsies that are not amenable to treatment with medication.
3. Genetic suppression of seizure-susceptibility in Drosophila
3.1. Identification of seizure-suppressor mutants
Human seizure disorders represent a pervasive class of disease with unsatisfactory treatment options. In addition, a large number of disparate genes are involved in epileptogenesis. Frequently, there is a lack of obvious functional relationship between mutation and seizure susceptibility, complicating an understanding of epilepsy on a mechanistic level. Seizure-suppressor genes provide a potentially powerful tool for examining seizure disorders and identifying potential AED targets. The general approach to identify suppressors is straightforward: starting with an epilepsy genetic background, second-site suppressor mutations are evaluated for their ability to revert the epilepsy phenotype to wild type. Surprisingly, the basic approach of utilizing second-site suppressor mutations has not been extensively exploited for neurological syndromes.
The most promising aspect of the approach is the apparent abundance of seizure-suppressor mutations and ease of identifying them. Reverse genetics utilizing double mutant combinations, as well as forward genetics screens utilizing a variety of mutagenesis schemes available for Drosophila have all been used productively to isolate seizure-suppressor mutations. Reverse genetics searches focusing on existing Drosophila nervous system excitability mutations have shown that sodium channel (para, mlenapts), potassium channel (Sh), and gap junction mutations (shakB) are all effective seizure-suppressors (Kuebler et al., 2001; Song and Tanouye, 2006). Seizure-suppressor mutations have also been readily isolated in forward genetics mutant screens. Especially interesting are novel and unexpected seizure-suppressor mutations, not obviously affecting electrical signaling. These include DNA topoisomerase I enzyme (top1JS1), a zinc-finger transcription factor escargot (esg), and the meiotic gene mei-P26EG (Song et al., 2007; Hekmat-Scafe et al., 2005; Glasscock et al., 2005). The following is a more detailed description of selected seizure-suppressor mutations. Continued identification of seizure-suppressor mutations through genetic screens will provide a rich source of genetic aberrations that can serve to dissect apart seizure-susceptibility and act as a basis for discovering new AED targets.
3.2 Insights into seizure-suppression mechanisms from mutant analysis
3.2.1 Seizure-suppression by sodium channel mutations
Mutations affecting voltage-gated sodium channels are potent seizure-suppressors and provide validation for the suppressor genetic approach. Reverse genetic experiments were inspired by observations that sodium currents are major targets for several first-line AEDs including phenytoin, carbamezapine, lamotrignine, and valproate (Ragsdale and Avoli, 1998; Catterall, 1999). Two Drosophila sodium channel mutants are paraST76 and mlenapts that affect the channel structural gene and regulate channel expression, respectively (Ramaswami and Tanouye, 1989; Loughney et al., 1989; Kuroda et al., 1991). Both mutants are seizure-resistant mutants showing ECS seizure thresholds that are about 2-3 times higher than for wild type animals (Kuebler et al., 2001). Double mutant combinations between each of the sodium channel mutants and various BS mutants show a reversion of seizure-susceptibility to wild type levels (Kuebler et al., 2001).
The paraST76 and mlenapts mutations exert their effects through a reduction of functional sodium channels (Loughney et al., 1989; Kuroda et al., 1991). This suggests that they inhibit seizures in much the same way as the AEDs phenytoin and carbamazepine (Kuo, 1998; McNamara, 1999). During repetitive firing, these drugs are thought to stabilize sodium channels in the inactive state thereby reducing the number of channels that can be activated. This leads to reduced capacity for HF firing (McLean and MacDonald, 1983; 1986). Indeed, reduced capacity for HF firing has been observed in paraST76 and mlenapts mutants (Nelson and Wyman, 1990; Kuebler et al., 2001).
In a forward genetic screen by mutagenesis using a DNA transposable element, a new para allele paraJS was identified with a transposon insertion in the 3′-UTR (3′ untranslated region) of the para gene (Song and Tanouye, 2007). The paraJS1 mutation is not only exceptionally effective as a seizure suppressor, comparable to paraST76 and mlenapts, but also shows no obvious side-effect phenotypes whether in a wild-type or an eas mutant genetic background. That is, paraJS1 mutant flies have no evidence of sluggish behavior; they show excellent fertility; and they have normal-length life span, unlike other previously identified Drosophila sodium channel mutants. Other behaviors also appear to be normal: grooming, courting, mating, jumping and flying. Identification of the new para allele validates the forward genetic approach, providing evidence that it recapitulates suppressors identified by reverse genetics.
3.2.2. Gap junction mutations suppress seizure-like activity
Electrical synaptic transmission through gap junctions is an important mechanism for synchronizing signaling in the brain, combining together with field effects, chemical synaptic transmission mechanisms, and ionic channel mechanisms in the generation and maintenance of seizures (Carlen et al., 2000). Experimental observations reinforce the notion of gap junction contributions to seizures. Pharmacology that reduces electrical transmission diminishes seizures, and enhanced electrical transmission increases the frequency and severity of seizures (Carlen et al., 2000; Jahromi et al., 2002). For example, carbenoxolone, a gap junction blocker, has been shown to reduce seizures in several animal models of epilepsy (Gajda et al., 2003; Jahromi et al., 2002).
Gap junction proteins in Drosophila are encoded by a family of eight genes called the “innexin” family (Phelan and Starich, 2001). The shakB2 gene encodes a gap junction protein and mutations in this gene have impaired electrical transmission (Krishnan et al., 1993; Phelan et al., 1996). One allele, shakB2, is a loss-of-function mutation in which a T to A substitution inserts a stop codon within the signal sequence (Zhang et al., 1999b). The shakB2 mutant is a seizure-resistant mutant showing an ECS seizure threshold that is about three times higher than for wild type animals (Kuebler et al., 2001). Double mutant combinations between shakB2 and various BS mutants show that it is an effective seizure-suppressor mutation: it completely suppresses seizure-like activity caused by sda, kdn, and jbug mutations. Seizure-like activity caused by eas and tko mutations are partially suppressed by shakB2. Seizure-like activity caused by bas2, bss1 and bss2 mutations are not suppressed by shakB2 (Kuebler et al., 2001; Song and Tanouye, 2006). Based on these interactions with shakB2, it is determined that sda, eas and bss are representative of the entire BS collection. Synapses in the shakB2 mutant have been shown to be impaired in electrical transmission (Thomas and Wyman, 1984; Phelan et al., 1996). Thus, shakB2 is proposed to suppress seizure-like activity by a mechanism similar to that suggested for drugs such as carbenoxolone that block gap junction activity (Szente et al., 2002). Impaired transmission at electrical synapses is thought to interfere with synchronous activation of neuronal populations leading to decreased seizure susceptibility. Observations on shakB2 are generally consistent with this mechanism.
3.2.3 Seizure-suppression by mutation of DNA topoisomerase I
A major interest in seizure-suppressor mutations comes from the potential to develop new epilepsy therapeutics. Novel targets can be discovered for developing drugs that can complement presently available first-line AEDs: new treatments for intractable epilepsy or drugs with reduced side-effects. Also of great interest are genes that may be candidates for use in gene therapy. Gene therapy approaches are not well-developed for treatment of epilepsy and this possibility could initiate from study of seizure-suppressor genes. Genetic screens have identified several unexpected mutations that are surprisingly effective seizure-suppressors. These mutations are unexpected because in each case the gene product is not obviously involved in any type of neuronal signaling. The expectation is that drugs developed from these may lack the deleterious nervous system side-effects commonly associated with AEDs.
New seizure-suppressor mutations include the mei-P26EG mutation. The mei-P26EG suppressor affects an interesting protein known as a ring finger B-box coiled-coil-NHL protein (Glasscock et al., 2005). The suppressor phenotype appears to result from missense mutation of a critical amino acid residue in the NHL protein-protein interaction domain of the protein. The suggestion from this result is that there may be proteins that are interaction partners of mei-P26EG that are additional seizure-suppressors waiting to be identified. The zinc-finger transcription factor esg is another novel seizure-suppressor mutation identified by mutant screening. This is a gain-of-function mutation that suppresses seizure-like activity when the normal product (esg+) is over-expressed ectopically in all nerve cells (Hekmat-Scafe et al., 2005). Gain-of-function mutations such as esg have qualities that make them candidates for gene therapy development.
Especially interesting for AED development is discovery of a new allele of Drosophila DNA topoisomerase I named top1JS1 (called topo I in mammals) (Song and Tanouye, 2007). The top1JS allele was isolated in a seizure-suppressor screen that used the DNA transposable element, P-element, as a mutagen. Suppression was caused by insertion of the P-element in the 5′ untranslated region of the top1 gene: 257 bp upstream of the translation start site. Seizure suppression is due to reduced transcription of the top1 gene: mutant transcription is about 12.5-fold less than normal. The top1 gene is an essential gene: of several known top1 alleles, top1JS is the only viable mutation, while all others are homozygous lethal (Lee et al., 1993; Zhang et al., 2000). The top1JS mutation is a general seizure-suppressor ameliorating phenotypes of sda, eas, and bss (Song and Tanouye, 2007). As example, top1JS suppresses sda seizure-like behaviors and paralysis by about 73%. The threshold for evoking seizures by ECS is raised about 2.5 fold. For eas, behavioral phenotypes are suppressed in 63% of animals and seizure threshold is raised about 3.5 fold. The bss seizure and paralytic behavior are not suppressed by top1JS significantly. That is, most top1JS bss double mutants showed bang-sensitivity. However, there is some indication that top1JS acts to reduce the severity of bss seizure-like behavior, mainly about a two-fold decrease in tonic-clonic-like activity (Song and Tanouye, 2007). Taken together, these results are consistent with top1JS being a general seizure-suppressor: with sda and eas mutants more easily suppressed; and bss mutants more resistant to suppression.
4. Anticonvulsant drug development from seizure-suppressor genes
4.1. Anticonvulsant drug testing in Drosophila
One type of validation of a model for epilepsy therapeutics comes from an assurance that currently available human treatments are effective on Drosophila seizure mutants. Previous discussions in this review compared Drosophila and human seizure disorders behaviorally and electrophysiologically. Especially pertinent are studies showing sodium channel mutants are excellent seizure suppressors because voltage-gated sodium channels are targets of several AEDs (Kuebler et al., 2001; Song and Tanouye, 2007). Further validation for the use of Drosophila in drug development comes from experiments using valproate (Kuebler and Tanouye, 2002). Valproate injected directly into the Drosophila brain is a very potent AED: ECS seizure threshold in bss and sda mutants is immediately elevated to levels comparable to wild-type controls.
There is considerable interest in using Drosophila for high-throughput AED screening (Reynolds et al., 2003; Tan et al., 2004; Stilwell et al., 2006). The most convenient method is to feed seizure-sensitive Drosophila mutants a panel of drugs to be screened and then select for reversion of seizure-like behaviors or paralytic behavior. Promising drugs could then be examined subsequently in more detail by electrophysiological methods, perhaps by direct injection in the brain as for valproate (Kuebler and Tanouye, 2002). AED efficacy in feeding experiments has been by measuring reduction in paralytic recovery time for bss or eas mutants (Reynolds et al., 2004; Tan et al., 2004; Song and Tanouye, 2006). The AEDs phenytoin, gabapentin, potassium bromide, and carbenoxolone have all been deemed effective in Drosophila based on reduction of BS mutant recovery time. Using these criteria, carbamazepine, ethosuximide, and vigabactrin do not appear to be effective AEDs when fed to BS mutant Drosophila (Reynolds et al., 2004). AED drug-feeding experiments have generally not seen dramatic changes in percentages of BS paralysis or dramatic improvements in seizure threshold, although phenytoin may be an exception (Reynolds et al., 2004; Tan et al., 2004; Song and Tanouye, 2006).
4.2. AED development from the top1 seizure-suppressor
4.2.1 An AED target inspired by top1 seizure-suppressor
Identification of the top1JS mutation as a seizure-suppressor has led to consideration of a class of DNA topoisomerase I inhibitors as potentially effective AEDs. These drugs, called top1 (or topo I) inhibitors, have been found to be effective at ameliorating some seizure phenotypes in the Drosophila model (figure 3). DNA topoisomerase I is an essential nuclear enzyme involved in relieving the torsional stress associated with DNA replication, transcription, and chromatin condensation (Champoux, 2001). In this respect, it differs substantially from the targets of presently available AEDs, mainly sodium channels and GABA-related proteins. The top1JS seizure-suppression is consistent with the observation from molecular studies that many syndromes presenting with epilepsy, including human syndromes, mouse knockout mutations, and Drosophila mutations, are not obviously affecting electrical excitability functions (Royden et al., 1987; Pavlidis et al., 1994; Purnamm and McNamara, 1999; McNamara, 1999, Zhang et al., 2002). One might similarly expect that many seizure-suppression mechanisms would exist that are not working via alteration of electrical excitability. It may be possible to gain new insights into mechanisms by which the nervous system can be constructed in ways that reduce seizure sensitivity independent of effects on electrical excitability.
Figure 3. The comparison between genetic interaction and drug feeding.

Drug treatment by CPT phenocopies the genetic interaction between bss and top1JS in reduction of the recovery time. The average recovery time of bss is around 120s (3dp), it is reduced to around 70 s in the double mutant top1JS bss, and 60 s in the CPT-treated bss mutant.
4.2.2 Top1 inhibitors as AEDs suppress Drosophila phenotypes
Top1 inhibitors are the first neurological drug candidates arising from a suppressor genetic approach. Top1 inhibitors have recently drawn considerable pharmacological interest, albeit not as neurological drugs, but as chemotherapeutic drugs in the cancer clinic (Wang et al., 1997; Pommier et al., 1999; Li and Liu, 2000). The top1 inhibitor camptothecin (CPT) is a potent anticancer agent, and its derivatives topotecan and irinotecan have been approved by FDA for treatment of ovarian and colorectal cancer (Mathijssen et al., 2002). Type 1 DNA topoisomerase enzyme is thought to resolve DNA torsional tension by binding DNA and relaxing the helix. After DNA relaxation, top1 enzyme is cleaved and the DNA is religated. CPT top1 inhibitor is thought to work by covalently binding to the top1-DNA complex forming a ternary complex that interferes with religation. This CPT religation interference can lead to apoptosis (Leppard and Champoux, 2005). CPT-induced apoptosis may be essential to its action in cancer therapy and may also contribute to its function as an AED (Song et al., 2007).
CPT, apigenin, and kaempferol are plant phytochemicals that inhibit top1 activity (Pommier et al., 1998; 1999; Snyder and Gillies, 2002; Boege et al., 1996). These top1 inhibitors have been shown to ameliorate phenotypes in BS mutants indicating that they have actions similar to AEDs in the Drosophila epilepsy model. For example, bang-resistant behavior is observed in a small number of eas mutants (3.5% of animals) that are reared in culture medium containing apigenin (drug-rearing experiments) (Song et al., 2007). Although a fairly modest reversion, nonetheless this is better than for most other AEDs thus far examined by feeding experiments, including valproate, and potassium bromide (Kuebler and Tanouye, 2002; Tan et al., 2004), although may be less effective than phenytoin (Reynolds et al., 2004). Top1 inhibitors reduce the paralytic recovery time of BS mutants in drug-rearing experiments and in short-term drug feeding experiments, resembling the effects of AEDs in the Drosophila model (Song et al., 2007; Kuebler and Tanouye, 2002; Tan et al., 2004; Reynolds et al., 2004). For example, bss flies fed CPT in short-term drug feeding recover from paralysis about two-thirds faster than control flies (Song et al., 2007). In addition, tonic-clonic-like activity is nearly completely suppressed. Electrophysiological recordings corroborate the behavioral results. Drug treatment causes a modest albeit significant increase in seizure threshold. Synaptic failure time is greatly decreased (Song et al., 2007). A comparison of drug feeding experiments in the Drosophila suggests that, in general, top1 inhibitors may be a less effective anti-epileptic agent compared to phenytoin, but may be better than valproate, potassium bromide, and carbenoxolone (Kuebler and Tanouye, 2002; Reynolds et al., 2004; Tan et al., 2004; Song and Tanouye, 2006). In addition, top1 inhibition appears to be tolerated much better than valproate with considerably less toxicity. Further experiments in mammalian animal model systems will provide insights into the pharmacological perspectives to evaluate top1 as an anticonvulsant compared to currently available AEDs on the market.
5. Conclusions
A major obstacle in neurological drug discovery comes about because of the limitation of organism-based drug screening that is presently a fairly low throughput assay. For example, screening and testing of drugs for epilepsy can be primarily dependent on laborious tests of rodents utilizing convulsants such as pentylenetetrazol or picrotoxin to induce seizure (White et al., 2002; Yang and Frankel, 2004). Here, we report on an alternative approach that relies on Drosophila seizure-suppressor genes to identify potential AED targets. This approach has, thus far, proven to be surprisingly productive mainly due to the ease with which novel seizure-suppressor mutations have been identified. Using this approach, a new, novel class of potentially effective AEDs may have been discovered, top1 inhibitors including CPT and related drugs. This is a promising first try using the power of the Drosophila seizure-suppressor gene approach. The hope is that continued identification of seizure-suppressor genes will lead to even more promising AED candidates. If further tests confirm the use of top1 inhibitors and other promising AED candidates in the neurology clinic, we suggest Drosophila will have a major impact on directly improving the human condition through advances of medical treatments in nervous system and other debilitating disorders.
Figure 2. Seizure-like activity in intact sda and wild type CS flies.

The BS mutant sda fly is more susceptible to seizure-like activity than the wild type CS fly and therefore has a much lower seizure threshold. (A) Seizure-like activity is elicited in a sda fly by a high frequency stimulus of low strength (8 V) and displayed at a high sweep speed. The HF stimulus (labeled HF) is a short wavetrain (0.5 ms pulses at 200 Hz for 300 ms) of electrical stimuli delivered to the brain. Recording is from a muscle fiber (DLM, dorsal longitudinal muscle) and reflects the activity of the single DLM motoneuron that innervates it. The seizure-like activity is widespread as similar firing can be found in recordings from seven different muscle groups in the fly following HF stimulation (Kuebler and Tanouye, 2000). (B) A low-voltage HF stimulus of 8 V fails to elicit seizure-like activity in a wild type CS fly because the stimulus is below the seizure threshold. Following the HF stimulus artifact, there is no seizure-like activity observed in this recording displayed at a high sweep speed. Note also that there is no period of synaptic failure and single-pulse stimulation of the GF (0.5 Hz) continues to evoke DLM potentials. Two such effective single-pulse stimuli are depicted in this trace; each was effective in evoking a DLM potential. (C) Seizure-like activity is elicited in a wild type CS fly by a high voltage HF stimulus (32 V) which is above the threshold for seizure. Vertical calibration bar is 20 mV, horizontal calibration bar is 300 ms (figure modified from Kuebler and Tanouye, 2002, figure 1).
Table 2. List of seizure-suppressor mutants and their gene products.
| Seizure-suppressor Mutant | Gene Product |
|---|---|
| paralytic (paraST76, paraJS) | Sodium channel |
| maleless no-action-potential temperature sensitive (mlenapts) | RNA helicase like protein |
| Shaker (sh) | Potassium channel |
| Shaking B (shakB2) | Gap junction protein |
| escargot (esg) | Zinc-figure transcription factor |
| meiosis defect (mei-p26EG) | Ring finger B-box coiled-coil-NHL protein |
| top1(top1JS) | DNA topoisomerase I |
Acknowledgments
This work was supported by an NIH research grant and an Epilepsy Foundation grant to M.T.
Abbreviations
- AED
antiepileptic drug
- BS
bang-sensitive Drosophila mutant
- CNS
central nervous system
- CPT
camptothecin
- DLM
dorsal longitudinal muscle
- ECS
electroconvulsive shock
- ECT
electroconvulsive shock treatment
- HF
high-frequency
- PSI
peripherally-synapsing interneuron
- top1
DNA topoisomerase I enzyme
- TUNEL
terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avoli M. Feline generalized penicillin epilepsy. Ital J Neurol Sci. 1995;16:79–82. doi: 10.1007/BF02229078. [DOI] [PubMed] [Google Scholar]
- Baraban SC. Emerging epilepsy models: insights from mice, flies, worms, and fish. Curr Opin Neurol. 2007;20:164–168. doi: 10.1097/WCO.0b013e328042bae0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Dinday MT, Castro PA, Chege S, Guyenet S, Taylor MR. A large-scale mutagenesis screen to identify seizure-resistant zebrafish. Epilepsia. 2007;48:1151–1157. doi: 10.1111/j.1528-1167.2007.01075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Taylor MR, Castro PA, Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005;131:759–768. doi: 10.1016/j.neuroscience.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Batini C, Teillet MA, Naquet R. An avian model of genetic reflex epilepsy. Arch Ital Biol. 2004;142:297–312. [PubMed] [Google Scholar]
- Benzer S. From the gene to behavior. J Am Med Assoc. 1971;218:1015–1022. [PubMed] [Google Scholar]
- Bertorelli R, Adami M, Ongini E. The Mongolian gerbil in experimental epilepsy. Ital J Neurol Sci. 1995;16:101–106. doi: 10.1007/BF02229081. [DOI] [PubMed] [Google Scholar]
- Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genetics. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- Bier E, Bodmer R. Drosophila, an emerging model for cardiac disease. Gene. 2004;342:1–11. doi: 10.1016/j.gene.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Boege F, Straub T, Kehr A, Boesenberg C, Christiansen K, Anderson A, Jacob F, Kohrle J. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J Biol Chem. 1996;271:2262–2270. doi: 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- Carlen PL, Frances S, Zhang L, Naus C, Kushnir M, Velazquez JL. The role of gap junctions in seizures. Brain Res Rev. 2000;32:235–241. doi: 10.1016/s0165-0173(99)00084-3. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv Neurol. 1999;79:441–456. [PubMed] [Google Scholar]
- Champoux JJ. DNA topoisomerases: structure, function and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Engel JE, Wu CF. Altered mechanoreceptor response in Drosophila bang-sensitive mutants. J Comp Physiol A. 1994;175:267–278. doi: 10.1007/BF00192986. [DOI] [PubMed] [Google Scholar]
- Fergestad T, Bostwick B, Ganetzky B. Metabolic disruption in Drosophila bang-sensitive seizure mutants. Genetics. 2006;173:1357–1364. doi: 10.1534/genetics.106.057463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajda Z, Gyengesi E, Hermesz E, Ali KS, Szente M. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia. 2003;44:1596–1600. doi: 10.1111/j.0013-9580.2003.25803.x. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Wu CF. Indirect suppression involving behavioral mutants with altered nerve excitability in Drosophila melanogaster. Genetics. 1982;100:597–614. doi: 10.1093/genetics/100.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Tanouye MA. Drosophila couch potato mutants exhibit complex neurological abnormalities including epilepsy phenotypes. Genetics. 2005;169:2137–2149. doi: 10.1534/genetics.104.028357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Singhania A, Tanouye MA. The mei-P26 gene encodes a RING finger B-box coiled-coil-NHL protein that regulates seizure susceptibility in Drosophilia. Genetics. 2005;170:1677–1689. doi: 10.1534/genetics.105.043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Dang KN, Tanouye MA. Seizure suppression by gain-of-function escargot mutations. Genetics. 2005;169:1477–1493. doi: 10.1534/genetics.104.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl-cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi SS, Wentlandt K, Piran S, Carlen P. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol. 2002;88:1893–1902. doi: 10.1152/jn.2002.88.4.1893. [DOI] [PubMed] [Google Scholar]
- Krishnan SN, Frei E, Swain G, Wyman RJ. Passover, a gene required for synaptic connectivity in the giant fiber system of Drosophila. Cell. 1993;73:967–977. doi: 10.1016/0092-8674(93)90274-t. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Modifications of seizure susceptibility in Drosophila. J Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Zhang HG, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Anticonvulsant valproate reduces seizure-susceptibility in mutant Drosophila. Brain Res. 2002;958:36–42. doi: 10.1016/s0006-8993(02)03431-5. [DOI] [PubMed] [Google Scholar]
- Kuo CC. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal sodium channels. Mol Pharmacol. 1998;54:712–721. [PubMed] [Google Scholar]
- Kuroda MI, Kernan MJ, Kreber R, Ganetzky B, Baker BS. The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell. 1991;66:935–947. doi: 10.1016/0092-8674(91)90439-6. [DOI] [PubMed] [Google Scholar]
- Lee J, Wu CF. Electroconvulsive seizure behavior in Drosophila: analysis of the physiological repertoire underlying a stereotyped action pattern in bang-sensitive mutants. J Neurosci. 2002;22:11065–11079. doi: 10.1523/JNEUROSCI.22-24-11065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Brown SD, Chen A, Hsieh TS. DNA topoisomerase I is essential in Drosophila melanogaster. Proc Natl Acad Sci USA. 1993;90:6656–6660. doi: 10.1073/pnas.90.14.6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legare ME, Bartlett FS, II, Frankel WN. A major effect QTL determined by multiple genes in epileptic EL mice. Genome Res. 2000;10:42–48. [PMC free article] [PubMed] [Google Scholar]
- Leppard JB, Champoux JJ. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma. 2005;114:75–85. doi: 10.1007/s00412-005-0345-5. [DOI] [PubMed] [Google Scholar]
- Li T, Liu L. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2000;41:53–77. doi: 10.1146/annurev.pharmtox.41.1.53. [DOI] [PubMed] [Google Scholar]
- Lock CJ, Williams SN, Schwarz EM, Caldwell GA, Caldwell KA. Genetic interactions among cortical malformation genes that influence susceptibility to convulsions in C. elegans. 2006 doi: 10.1016/j.brainres.2006.08.067. [DOI] [PubMed] [Google Scholar]
- Lohi H, Young EJ, Fitzmaurice SN, Rusbridge C, Chan EM, Vervoort M, Turnbull J, Zhao XC, Ianzano L, Paterson AD, Sutter NB, Ostrander EA, André C, Shelton GD, Ackerley CA, Scherer SW, Minassian BA. Expanded repeat in canine epilepsy. Science. 2005;307:81. doi: 10.1126/science.1102832. [DOI] [PubMed] [Google Scholar]
- Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- Mathijssen RH, Loos WJ, Verweij J, Sparreboom A. Pharmacology of topoisomerase I inhibitors irinotecan (CPT-11) and topotecan. Curr Cancer Drug Targets. 2002;2:103–123. doi: 10.2174/1568009023333890. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399(Suppl):A15–A22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- McLean MJ, MacDonald RL. Multiple actions of phenytoin on mouse spinal cord neurons in cell culture. J Pharmacol Exp Ther. 1983;227:779–789. [PubMed] [Google Scholar]
- McLean MJ, Macdonald RL. Sodium valproate, but not ethosuximide, produces use- and voltage-dependent limitation of high frequency repetitive firing of action potentials of mouse central neurons in cell culture. J Pharmacol Exp Ther. 1986;237:1001–1011. [PubMed] [Google Scholar]
- Menini C, Silva-Barrat C. The photosensitive epilepsy of the baboon. A model of generalized reflex epilepsy. Adv Neurol. 1998;75:29–47. [PubMed] [Google Scholar]
- Michno K, van de Hoef D, Wu H, Boulianne GL. Modeling age-related diseases in Drosophila: can this fly? Curr Topics Dev Biol. 2005;71:199–223. doi: 10.1016/S0070-2153(05)71006-1. [DOI] [PubMed] [Google Scholar]
- Nelson JC, Wyman RJ. Examination of paralysis in Drosophila temperature-sensitive paralytic mutations affecting sodium channels; a proposed mechanism of paralysis. J Neurobiol. 1990;21:453–469. doi: 10.1002/neu.480210307. [DOI] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Pascual A, Chaminade M, Preat T. Ethanolamine kinase controls neuroblast divisions in Drosophila mushroom bodies. Dev Biol. 2005;280:177–186. doi: 10.1016/j.ydbio.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Tanouye MA. Seizures and failures in the giant fiber pathway of Drosophila bang-sensitive paralytic mutants. J Neurosci. 1995;15:5810–5819. doi: 10.1523/JNEUROSCI.15-08-05810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Nakagawa M, Wilkin MB, Moffat KG, O'Kane CJ, Davies JA, Bacon JP. Mutations in shaking-B prevent electrical synapse formation in the Drosophila giant fiber system. J Neurosci. 1996;16:1101–1113. doi: 10.1523/JNEUROSCI.16-03-01101.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim Biophys Acta. 1998;1400:83–105. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Update. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- Purnam RS, McNamara JO. Seizure disorders in mutant mice: relevance to human epilepsies. Curr Opin Neurobiol. 1999;9:281–287. doi: 10.1016/s0959-4388(99)80041-5. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, Avoli M. Sodium channels as molecular targets for antiepileptic drugs. Brain Res Rev. 1998;26:16–28. doi: 10.1016/s0165-0173(97)00054-4. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Tanouye MA. Two sodium channel genes in Drosophila: implications for channel diversity. Proc Natl Acad Sci USA. 1989;86:2079–2082. doi: 10.1073/pnas.86.6.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ER, Stauffer EA, Feeney L, Rojahn E, Jacobs B, McKeever C. Treatment with the antiepileptic drugs phenytoin and gabapentin ameliorates seizure and paralysis of Drosophila bang-sensitive mutants. J Neurobiol. 2003;58:503–513. doi: 10.1002/neu.10297. [DOI] [PubMed] [Google Scholar]
- Royden CS, Pirrotta V, Jan LY. The tko locus, site of a behavioral mutation in D. melanogaster, codes for a protein homologous to prokaryotic ribosomal protein S12. Cell. 1987;51:165–173. doi: 10.1016/0092-8674(87)90144-9. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Lewis EB. A brief history of Drosophila's contributions to genome research. Science. 2000;287:2216–2218. doi: 10.1126/science.287.5461.2216. [DOI] [PubMed] [Google Scholar]
- Snyder RD, Gillies PJ. Evaluation of the clastogenic, DNA intercalative, and topoisomerase II-interactive properties of bioflavonoids in Chinese hamster V79 cells. Envir Molec Mutagenesis. 2002;40:266–276. doi: 10.1002/em.10121. [DOI] [PubMed] [Google Scholar]
- Song J, Tanouye MA. Seizure suppression by shakB2, a gap junction mutation in Drosophila. J Neurophysiol. 2006;95:627–635. doi: 10.1152/jn.01059.2004. [DOI] [PubMed] [Google Scholar]
- Song J, Hu J, Tanouye M. Seizure suppression by top1 mutations in Drosophila. J Neurosci. 2007;27:2927–2937. doi: 10.1523/JNEUROSCI.3944-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Tanouye M. A role for para sodium channel gene 3′ UTR in the modification of Drosophila seizure susceptibility. Devel Neurobiol. 2007 doi: 10.1002/dneu.20519. in press. [DOI] [PubMed] [Google Scholar]
- Stilwell GE, Saraswati S, Littleton JT, Chouinard SW. Development of a Drosophila seizure model for in vivo high-throughput drug screening. Eur J Neurosci. 2006;24:2211–2222. doi: 10.1111/j.1460-9568.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- Szente M, Gajda Z, Said Ali K, Hermesz E. Involvement of electrical coupling in the in vivo ictal epileptiform activity induced by 4-aminopyridine in the neocortex. Neurosci. 2002;115:1067–1078. doi: 10.1016/s0306-4522(02)00533-x. [DOI] [PubMed] [Google Scholar]
- Tan JS, Lin F, Tanouye MA. Potassium bromide, an anticonvulsant, is effective at alleviating seizures in the Drosophila bang-sensitive mutant bang senseless. Brain Res. 2004;1020:45–52. doi: 10.1016/j.brainres.2004.05.111. [DOI] [PubMed] [Google Scholar]
- Thomas JB, Wyman RJ. Mutations altering synaptic connectivity between identified neurons in Drosophila melanogaster. J Neurosci. 1984;4:530–538. doi: 10.1523/JNEUROSCI.04-02-00530.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickoo S, Russell S. Drosophila melanogaster as a model system for drug discovery, and pathway screening. Curr Opin Pharmacol. 2002;2:555–560. doi: 10.1016/s1471-4892(02)00206-0. [DOI] [PubMed] [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Veraksa A, Del Campo M, McGinnis W. Developmental patterning genes, and their conserved functions: from model organisms to humans. Mol Genet Metab. 2000;69:85–100. doi: 10.1006/mgme.2000.2963. [DOI] [PubMed] [Google Scholar]
- Vidal M, Cagan RL. Drosophila models for cancer research. Curr Opin Genetics Dev. 2006;16:10–16. doi: 10.1016/j.gde.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Wang HK, Morris-Natschke SL, Lee KH. Recent advances in discovery and development of topoisomerase inhibitors as antitumor agents. Medicinal Res Rev. 1997;17:367–425. doi: 10.1002/(sici)1098-1128(199707)17:4<367::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- White SH, Woodhead JH, Wilcox KS, Kupferberg HJ, Wolf HH. Discovery and preclinical development of anitepileptic drugs. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. Lippincott Williams & Wilkins; 2002. pp. 36–48. [Google Scholar]
- Williams SN, Locke CJ, Braden AL, Caldwell KA, Caldwell GA. Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in Caenorhabditis elegans. Hum Mol Gen. 2004;13:2043–2059. doi: 10.1093/hmg/ddh209. [DOI] [PubMed] [Google Scholar]
- Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, Steller H, Meier P. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4:445–450. doi: 10.1038/ncb799. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Yang Y, Frankel WN. Genetic approaches to studying mouse models of human seizure disorders. Adv Exptl Med Biol. 2004;548:1–11. doi: 10.1007/978-1-4757-6376-8_1. [DOI] [PubMed] [Google Scholar]
- Zhang CX, Chen AD, Gettel NJ, Hsieh TS. Essential functions of DNA Topoisomerase I in Drosophila melanogaster. Dev Biol. 2000;222:27–40. doi: 10.1006/dbio.2000.9704. [DOI] [PubMed] [Google Scholar]
- Zhang HG, Tan J, Reynolds E, Kuebler D, Faulhaber S, Tanouye MA. The Drosophila slamdance gene: a mutation in an aminopeptidase can cause seizure, paralysis and neuronal failure. Genetics. 2002;162:1283–1299. doi: 10.1093/genetics/162.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Roote J, Brogna S, Davis AW, Barbash DA, Nash D, Ashburner M. Stress sensitive B encodes an adenine nucleotide translocase in Drosophila melanogaster. Genetics. 1999a;153:891–903. doi: 10.1093/genetics/153.2.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Curtin KD, Sun YA, Wyman RJ. Nested transcripts of gap junction gene have distinct expression patterns. J Neurobiol. 1999b;40:288–301. doi: 10.1002/(sici)1097-4695(19990905)40:3<288::aid-neu2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]