

Abstract
In cancer cells, an imbalance often exists between histone acetyltransferase (HAT) and histone deacetylase (HDAC) activities, and various drug companies are actively seeking competitive HDAC inhibitors for chemotherapeutic intervention. Cancer cells appear to be more sensitive than nontransformed cells to HDAC inhibitors, which disrupt the cell cycle and induce apoptosis via derepression of genes such as P21 and BAX. However, in the search for potent HDAC inhibitors with cancer therapeutic potential, a tendency exists to overlook or dismiss weak ligands that could prove effective in cancer prevention. Butyrate, diallyl disulfide (DADS), and sulforaphane (SFN) are three dietary agents that exhibit HDAC inhibitory activity in vitro and/or in vivo, and other such dietary agents probably will be discovered that affect HDAC activity. We make the distinction between ‘pharmacologic’ agents that potently derepress gene expression, during therapeutic intervention, and dietary HDAC inhibitors that, as weak ligands, might subtly regulate the expression of genes involved in cell growth and apoptosis. An important issue for future study is to determine the extent to which dietary HDAC inhibitors, by modulating genes such as p21 and Bax, enable normal, nontransformed cells to respond most effectively to external stimuli and toxic insults.
Keywords: histone deacetylase, chromatin remodeling, sulforaphane, garlic, S-allyl compounds, butyrate, chemoprevention
INTRODUCTION
Interest is growing in the posttranslational modifications that comprise the ‘histone code’ and their relationship to cancer development. Histone alterations can include acetylation, methylation, phosphorylation, ubiquitination, and biotinylation [1], and the loss of monoacetylation and trimethylation of histone H4 is a common hallmark of human tumor cells [2]. An imbalance also exists in histone acetyltransferase (HAT) and histone deacetylase (HDAC) activities in cancer cells, and drug companies are working actively in the development of HDAC inhibitors for cancer chemotherapy [3–5]. HDAC inhibitors have the potential to ‘derepress’ genes that affect apoptosis and the cell cycle. For example, compounds such as trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA) activate p21Waf1, cyclins, apoptosis mediators, and transcription factors in cancer cells [3–5].
Histone deacetylase inhibitors disrupt the cell cycle in G2, allowing cells to prematurely enter the M phase, and they also interfere with the mitotic spindle checkpoint [6]. Interestingly, cancer cells are more sensitive than nontransformed cells to apoptosis induction by TSA-related compounds. The mechanistic basis for this sensitivity is unclear, but recent studies have implicated reactive oxygen species, thioredoxin status, and apoptosis mediators such as DR4, DR5, and TRAIL [7,8].
For the reasons alluded to above, HDAC inhibitors provide an attractive avenue for drug development, and considerable attention has focused on potent, high-affinity agents related to TSA and SAHA [3–8]. However, in the search for HDAC inhibitors with cancer therapeutic potential, a tendency exists to overlook or dismiss weak ligands that could prove effective in cancer prevention, including agents present in the human diet.
DIETARY HDAC INHIBITORS
A recent review discussed the cancer chemopreventive properties of three reported dietary HDAC inhibitors [9], namely butyrate, diallyl disulfide (DADS), and sulforaphane (SFN). Computer modeling suggests that these dietary agents have structural features appropriate for HDAC inhibition, namely, (i) a functional group that interacts with the buried zinc atom, (ii) a spacer or linker ‘arm’ that fits within the HDAC pocket, and in some cases (iii) a ‘cap’ group that sits outside the active site [10].
Butyrate contains a short three-carbon spacer attached to a carboxylic acid group, which probably enters into the active site of the enzyme and forms a bidentate ligand with the zinc atom (Figure 1A). HDAC activity is inhibited at high micromolar to low millimolar concentrations in vitro, levels nonetheless achievable in the gastrointestinal tract, where butyrate is the principal oxidative fuel for colonocytes [11,12].
Figure 1.
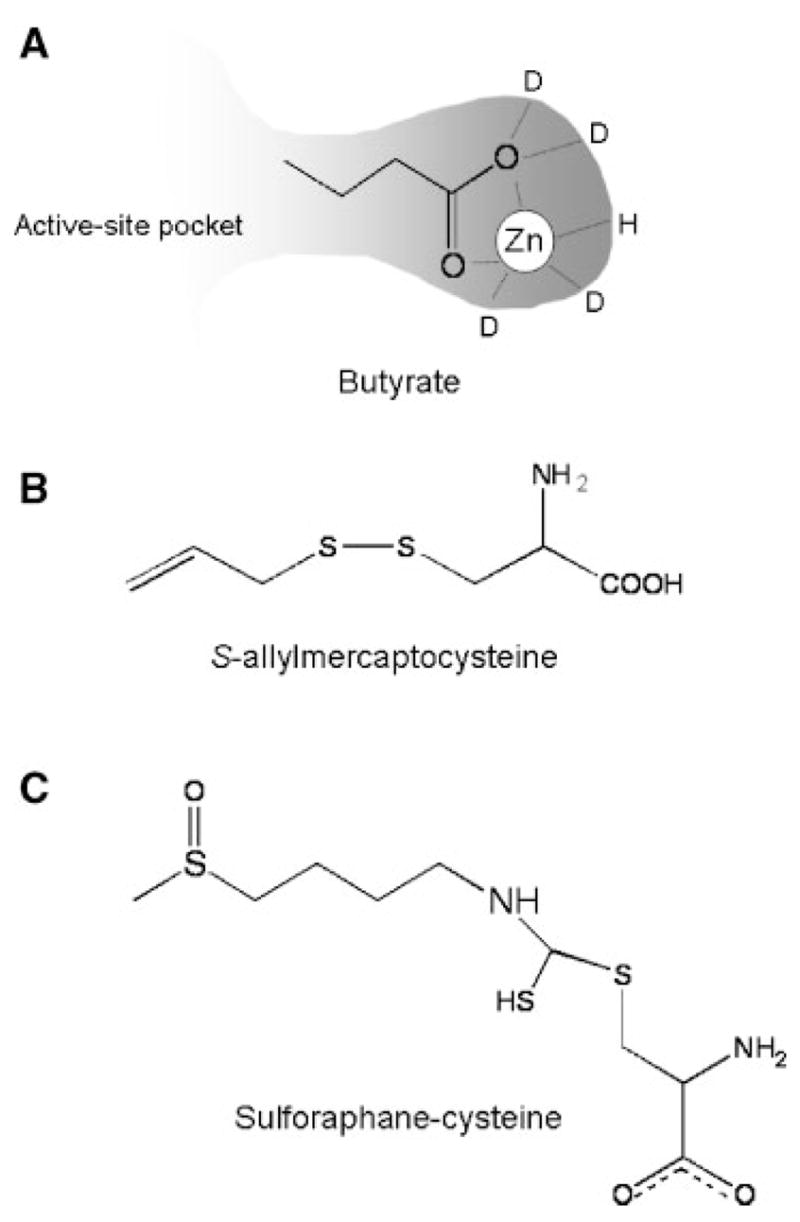
Dietary HDAC Inhibitors. (A) Hypothetical interaction of butyrate with HDAC, showing a presumed bidentate ligand with the active site zinc atom. (B) Structure of S-allylmercaptocysteine, a metabolite of the garlic compound diallyl disulfide, showing the ‘linker’ and carboxylic acid functional group. (C) Orientation of sulforaphane-cysteine, a metabolite of the broccoli phytochemical sulforaphane [15], as modeled in the active site of an HDAC homolog [10].
The garlic compound DADS is metabolized to S-allylmercaptocysteine [13], which resembles butyrate in having a spacer ending with a carboxylic acid functional group (Figure 1B). In human colon cancer cells, 200 μM DADS inhibited HDAC activity significantly and increased acetylated histone status [14].
A similar spacer and carboxylic acid functional group is also present in SFN-cysteine (Figure 1C), which is a metabolite of the broccoli phytochemical SFN. Molecular modeling studies with SFN-cysteine provided good support for complex formation with HDAC [15]. Specifically, the SFN-cysteine α-amino group H-bonded with buried His residues, whereas the SFN-cysteine carboxylate group formed a bidentate ligand with the active site zinc atom. In vitro, SFN-cysteine inhibited HDAC activity significantly in a cell-free system when tested at concentrations in the range 3–15 μM, whereas the SFN parent compound had no effect on HDAC, unless it was incubated with cells to allow for metabolism [15]. Little is known about the concentrations of the parent SFN or its active form(s) in different tissues, or the temporal changes that occur following normal human dietary intake. In a study with four human volunteers who consumed broccoli sprouts [16], peak plasma concentrations were of the order of ~2 μM, using an assay that detected total isothiocyanates and dithiocarbamates.
PREDICTING DIETARY HDAC INHIBITORS
Based on the studies to date, we propose that SFN-cysteine may be the ‘ultimate’ HDAC inhibitor, generated from the parent compound via the mercapturic acid pathway. Indeed, in an in vitro assay for HDAC activity, the SFN parent compound and its metabolite SFN-glutathione (SFN-GSH) had no significant effect, whereas SFN-cysteine and SFN-N-acetylcysteine (SFN-NAC) inhibited HDAC activity significantly [15]. The latter finding is interesting, because it suggests mechanism-based deacetylation by HDAC, with the product (SFN-cysteine) reversibly inhibiting the enzyme.
The latter hypothesis implies that other antic-arcinogenic isothiocyanates and allyl compounds from cruciferous vegetables and garlic [17–19] might be potential HDAC inhibitors after metabolism via the mercapturic acid pathway, if they have the correct geometry to fit the HDAC pocket. We examined this possibility with a screening test in which human colon cancer cells were treated with various isothiocyanates. The corresponding cell lysates were added to an in vitro HDAC activity assay (Figure 2). HDAC inhibition of the order of ~30% was seen, reproducibly, in cells treated with 15 μM SFN (referred to as ‘SFN(4)’ in Figure 2, due to the spacer length). Isothiocyanates with a similar spacer length as SFN, such as sulforaphene, erucin, and phenylbutyl isothiocyanate exhibited comparable HDAC inhibitory activities, as did compounds with a longer or shorter spacer lengths, such as SFN(6), SFN(9), 6-erucin, and phenethyl isothiocyanate. This is consistent with models showing the HDAC active site occupied with the Cys moiety, and the isothiocyanate ‘cap’ group influencing accessibility to the binding pocket (not shown). Similar findings have been reported for structural analogs of TSA in which the spacer and hydroxamic acid group were retained while substituting the cap group [3–5]. Interestingly, NAC or cysteine alone at 15 μM had no effect on HDAC activity (Figure 2).
Figure 2.
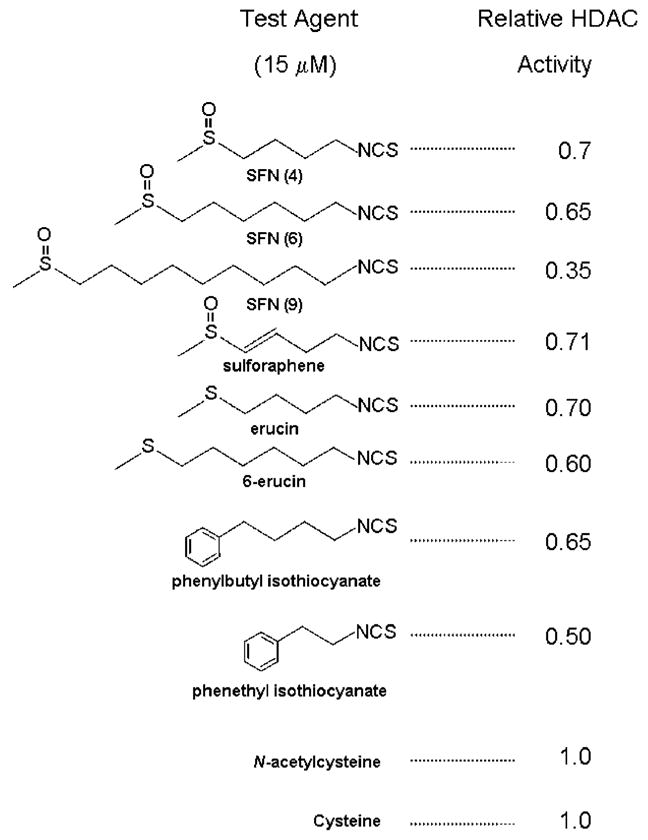
HDAC inhibitory activity of sulforaphane and structurally related isothiocyanates. Human HCT116 colon cancer cells were incubated with the test compounds, and after 48 h the cell lysates were tested in an in vitro HDAC activity assay, using a published procedure [15]. All concentrations were 15 μM.
We draw a distinction here between HDAC inhibition at low doses of SFN (<20 μM) and the effects reported at higher concentrations in vitro. For example, HDAC inhibition would probably play a minor role in cancer cells treated with 40–100 μM SFN; SFN in this dose range generates reactive oxygen species and activates a Bcl-2-associated apoptotic pathway [20,21].
Based on the ‘linker’ model, we speculate about other putative HDAC inhibitors in the human diet of interest. Butyrate is a short-chain fatty acid (Figure 1A), but numerous other dietary fatty acids exist, with their terminal carboxylic acid group positioned adjacent to the zinc atom, which might access the HDAC active site. For example, cis-9,trans-11 and cis-10,trans-12 isomers of the anticarcinogen conjugated linoleic acid might be candidates [22]. Other dietary compounds with the spacer and carboxylic acid functional group include biotin, which is noteworthy due to its role in modifying histone tails as part of the histone code (see above), as well as lipoic acid [23]. Antioxidants such as α-tocopherol, γ-tocopherol, and tocotrienols undergo ω-hydroxylation and β-oxidation, leading to carboxyhydrochroman derivatives with appropriate spacer lengths attached to a carboxylic acid group [24,25]. Recent studies have shown HDAC inhibition by SFN in the colonic mucosa of mice, and suppression of intestinal polyps [26]. Whether other dietary compounds, alone or in combination, will exhibit significant HDAC inhibitory activity in vitro and in vivo, remains to be determined.
CONCLUSION
Pharmacologic agents related to TSA and SAHA are available that potently derepress gene expression during therapeutic intervention, but dietary HDAC inhibitors are weak ligands that more subtly regulate genes involved in cell growth and apoptosis. A key issue for future study concerns the extent to which dietary HDAC inhibitors modulate genes such as p21 and Bax so that normal, nontransformed cells can respond most effectively to external stimuli and toxic insults. Combinations of such dietary agents also could prove to be effective when given as a ‘cocktail’ in the clinic, but further work is needed to confirm this possibility. We believe that dietary HDAC inhibitors should not be dismissed simply as weak ligands. Their ability to modulate gene expression through a lifetime of exposure and to impact on cancer chemoprevention warrants further investigation.
Acknowledgments
Research in this laboratory is supported by NIH grants CA65525, CA80176, and CA90890.
Abbreviations
- HDAC
histone deacetylase
- TSA
trichostatin A
- SAHA
suberoylanilide hydroxamic acid
- DADS
diallyl disulfide
- SFN
sulforaphane
References
- 1.Cosgrove MS, Wolberger C. How does the histone code work? Biochem Cell Biol. 2005;83:468–476. doi: 10.1139/o05-137. [DOI] [PubMed] [Google Scholar]
- 2.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 3.Mork CN, Faller DV, Spanjaard RA. A mechanistic approach to anticancer therapy: Targeting the cell cycle with histone deacetylase inhibitors. Curr Pharm Des. 2005;11:1091–1104. doi: 10.2174/1381612053507567. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin F, La Thangue NB. Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 5.Rosato RR, Grant S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol Ther. 2003;2:30–37. doi: 10.4161/cbt.190. [DOI] [PubMed] [Google Scholar]
- 6.Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: Implications for anticancer therapy? EMBO Rep. 2005;6:520–524. doi: 10.1038/sj.embor.7400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ungerstedt JS, Sowa Y, Xu WS, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nebbioso A, Clarke N, Voltz E, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 9.Myzak MC, Dashwood RH. Histone deacetylases as targets for dietary cancer preventive agents: Lessons learned with butyrate, diallyl disulfide, and sulforaphane. Curr Drug Targets. 2004:7. doi: 10.2174/138945006776359467. [DOI] [PubMed] [Google Scholar]
- 10.Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 11.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bach Knudsen KE, Serena A, Canibe N, Juntunen KS. New insight into butyrate metabolism. Proc Nutr Soc. 2003;62:81–86. doi: 10.1079/PNS2002212. [DOI] [PubMed] [Google Scholar]
- 13.Guyonnet D, Berges R, Siess M, et al. Post-initiation modulating effects of allyl sulfides in rat hepatocarcinogenesis. Food Chem Toxicol. 2004;42:1479–1485. doi: 10.1016/j.fct.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Druesne N, Pagniez A, Mayeur C, et al. Diallyl disulfide (DADS) increases histone acetylation and p21Waf1/Cip1 expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 15.Myzak MC, Karplus PA, Chung F-L, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 16.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. 2002;316:43–53. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]
- 17.Bianchini F, Vainio H. Isothiocyanates in cancer prevention. Drug Metab Rev. 2004;36:655–667. doi: 10.1081/dmr-200033468. [DOI] [PubMed] [Google Scholar]
- 18.Keum YS, Jeong WS, Kong AN. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Knowles LM, Milner JA. Possible mechanisms by which allyl sulfides suppress neoplastic cell proliferation. J Nutr. 2001;131:1061s–1066s. doi: 10.1093/jn/131.3.1061S. [DOI] [PubMed] [Google Scholar]
- 20.Shen G, Xu C, Chen C, Hebbar V, Kong AH. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol. 2005;17:1–11. doi: 10.1007/s00280-005-0050-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Singh SV, Srivastava SK, Choi S, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280:19911–19924. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- 22.Pariza MW. Perspectives on the safety and effectiveness of conjugated linoleic acid. Am J Clin Nutr. 2004;79:1132s–1136s. doi: 10.1093/ajcn/79.6.1132S. [DOI] [PubMed] [Google Scholar]
- 23.Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sontag TJ, Parker RS. Cytochrome P450 ω-hydroxylase pathway of tocopherol catabolism. J Biol Chem. 2002;277:25290–25296. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- 25.Birringer M, Pfluger P, Kluth D, Landes N, Brigelius-Flohe R. Identities and differences in the metabolism of tocotrienols and tocopherols in HepG2 cells. J Nutr. 2002;123:3113–3118. doi: 10.1093/jn/131.10.3113. [DOI] [PubMed] [Google Scholar]
- 26.Myzak MC, Dashwood W-M, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apcmin mice. FASEB J. 2006;20:506–508. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]