

Abstract
There is growing interest in the various mechanisms that regulate chromatin remodeling, including modulation of histone deacetylase (HDAC) activities. Competitive HDAC inhibitors disrupt the cell cycle and/or induce apoptosis via de-repression of genes such as P21 and BAX, and cancer cells appear to be more sensitive than non-transformed cells to trichostatin A and related HDAC inhibitory compounds. This apparent selectivity of action in cancer cells makes HDAC inhibitors an attractive avenue for drug development. However, in the search for potent HDAC inhibitors with cancer therapeutic potential there has been a tendency to overlook or dismiss weak ligands that could prove effective in cancer prevention, including agents present in the human diet. Recent reports have described butyrate, diallyl disulfide and sulforaphane as HDAC inhibitors, and many other dietary agents will be probably discovered to attenuate HDAC activity. Here we discuss ‘pharmacologic’ agents that potently de-repress gene expression (e.g. during therapeutic intervention) versus dietary HDAC inhibitors that, as weak ligands, might subtly regulate the expression of genes involved in cell growth and apoptosis. An important question is the extent to which dietary HDAC inhibitors, and other dietary agents that affect gene expression via chromatin remodeling, modulate the expression of genes such as P21 and BAX so that cells can respond most effectively to external stimuli and toxic insults.
Introduction
Chromatin remodeling—direct versus indirect HDAC inhibition
Considerable attention has focused on the silencing and unsilencing of genes through changes in DNA methylation (1), but such epigenetic modifications in DNA often require prior alterations at the level of the histones. The ‘histone code’ refers to an ever increasing complexity of histone modifications, including acetylation, methylation, phosphorylation, ubiquitination and biotinylation (2). There is growing interest in these post-translational changes and their implications for cancer development. Global loss of monoacetylation and tri-methylation of histone H4 is a common hallmark of human tumor cells (3). A recent commentary (4) also discussed novel protective functions of p53 associated with chromatin remodeling on a global scale, including transcriptional mechanisms that recruit or displace histone deacetylase (HDAC). Direct HDAC inhibitors also can affect changes in gene expression and impact on key regulators of apoptosis and the cell cycle (5–10), such as p21Cip1/Waf1, cyclins (A, E, B1, D1 and D3), apoptosis mediators (CD95, Bax and Bcl-2), transcription factors (GATA-2, c-Myc) and retinoic acid receptors (RAR).
RARs are targets of retinoids, which have been reviewed extensively in terms of their promise and pitfalls for cancer prevention (11,12). One area of particular interest has been that of acute promyelocytic leukemia (APL), because APL patients respond to pharmacologic doses of retinoic acid with disease remission. Molecular studies have focused on the PML/RAR oncogenic transcription factor, and models have been developed to explain the therapeutic mechanisms of retinoids in APL (Figure 1) (http://www.nature.com/nrc/journal/v1/n3/animation/nrc1201-181a_swf_MEDIA1.html, http://www.broad.mit.edu/chemobio/lab_schreiber/anims/animations/trich_retin.html). In brief, RAR/PML binding to the retinoic acid response element (RARE) recruits the CoR/SIN3/HDAC complex and represses transcription (Figure 1A), but agonists such as retinoic acid interact with the RAR and displace CoR/SIN3/HDAC (13), thereby activating gene expression in association with CoA/HAT complexes (Figure 1B). Resistance to retinoic acid treatment can occur in APL, due to the presence of a RAR–PLZF fusion protein and the inability to effectively displace HDAC (14,15). However, therapeutic efficacy in such cases can be improved when retinoids are combined with agents such as trichostatin A or suberoylanilide hydroxamic acid (SAHA), which interact directly with HDAC as competitive inhibitors (16–19) (Figure 1C). The latter compounds have provided valuable insights into the role of specific residues in the HDAC catalytic mechanism and the geometry of the substrate-binding pocket (6). Important structural features for HDAC inhibition appear to be a functional group that interacts with the buried zinc atom, a spacer or linker ‘arm’ that fits within the HDAC pocket, and in many (but not all) inhibitors a ‘cap’ group that sits just outside the active site (6–10).
Fig. 1.
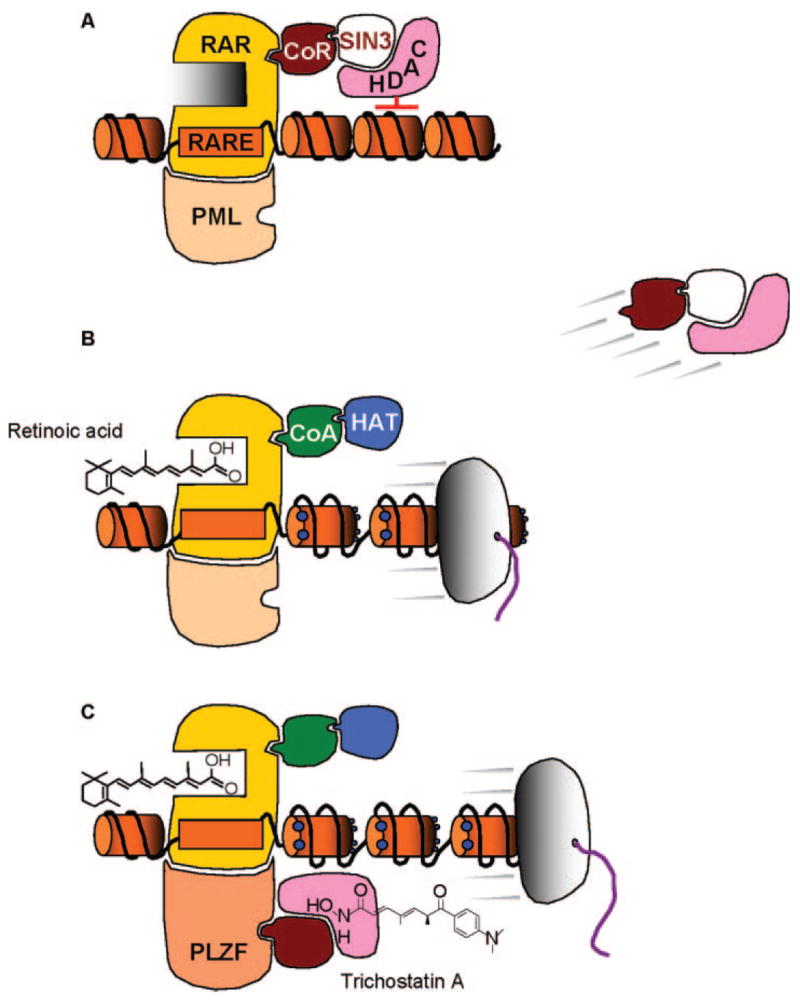
Indirect and direct mechanisms of HDAC modulation, leading to de-repression of silenced genes. (A) Binding of the retinoic acid receptor (RAR) to the retinoic acid response element (RARE), in conjunction with the promyelocytic leukemia (PML) protein, recruits corepressor-Sin3-HDAC (CoR-SIN3-HDAC) complexes. HDAC removes acetyl groups from histones and causes chromatin condensation, leading to gene silencing. (B) Retinoic acid binds to RAR and induces a conformational change in the protein, leading to the release of CoR-SIN3-HDAC and recruitment of co-activator-histone acetyltransferase (CoA-HAT) complexes. HATs, such as p300 and CREB-binding protein, transfer acetyl groups (blue dots) to the histone amino-terminal tails, leading to nucleosomal repulsion, chromatin relaxation and gene transcription. (C) Resistance to retinoic acid treatment, as in the case of the RAR–PLZF (promyelocytic leukemia zinc finger) fusion protein and its associated CoR/HDAC complexes, can be circumvented through the use of competitive inhibitors of HDAC, such as trichostatin A.
HDAC inhibitors have been reported to disrupt the cell cycle in G2, allowing cells to prematurely enter the M phase, as well as interfering directly with the mitotic spindle checkpoint (20). Cell cycle arrest and/or apoptosis is mediated through the de-repression of genes such as P21 and BAX, and cancer cells appear to be more sensitive than non-transformed cells to the actions of HDAC inhibitors. The mechanistic basis for this apparent selectivity of action against cancer cells is far from clear, although recent studies have implicated thioredoxin and intracellular thiol status, the accumulation of reactive oxygen species, and induction of TRAIL (Apo2L, TNFSF10), DR4 and DR5 (21,22).
For the reasons alluded to above, HDAC inhibitors provide an attractive avenue for drug development, and considerable attention has focused on potent, high-affinity agents related to trichostatin A and SAHA (7–10). However, in the search for HDAC inhibitors with cancer therapeutic potential, we believe that there has been a tendency to overlook or dismiss weak ligands that could prove effective in cancer prevention, including agents present in the human diet.
Dietary HDAC inhibitors—weak ligands in cancer prevention
A recent review discussed the cancer chemopreventive properties of three reported dietary HDAC inhibitors (23), namely butyrate, diallyl disulfide (DADS) and sulforaphane (SFN). In general, these dietary agents are weak ligands and inhibit HDAC activity at higher concentrations than trichostatin A or SAHA, which are effective in the nanomolar to low micromolar range. A pertinent question, then, concerns the concentrations needed for inhibition of HDAC activity by dietary compounds, and the likelihood that these levels might be achieved under normal physiological conditions.
Butyrate is the smallest known HDAC inhibitor and contains a simple three carbon ‘spacer’ attached to a carboxylic acid group (Figure 2A). Based on the known structural features of the HDAC pocket (6), it is assumed that butyrate fully enters into the active site of the enzyme, and that the carboxylate group of the short-chain fatty acid forms a bidentate ligand with the buried zinc atom. HDAC activity is inhibited at high micromolar to low millimolar concentrations in vitro, levels nonetheless considered to be achievable in the GI tract, where butyrate serves as the principal oxidative fuel for the colonocytes (24–27).
Fig. 2.
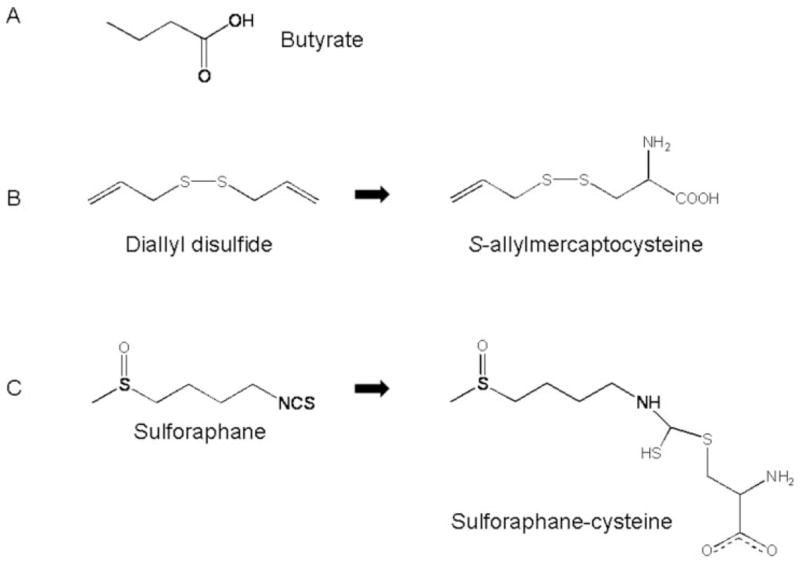
Structures of dietary HDAC inhibitors. (A) The smallest known HDAC inhibitor, butyrate, contains a short three carbon ‘spacer’ attached to a carboxylic acid functional group. (B) The dietary HDAC inhibitor diallyl disulfide, which is present in garlic, can be converted to the metabolite S-allylmercaptocysteine, containing a spacer and a carboxylic acid functional group. (C) Sulforaphane, an isothiocyanate found in broccoli and broccoli sprouts, can be converted to the metabolite SFN–Cys, which has spacer and carboxylic acid functional that fits within the HDAC active site (Figure 3).
A second dietary agent reported to inhibit HDAC activity is the garlic compound DADS (28), which after metabolic conversion to S-allylmercaptocysteine (29) resembles butyrate in having a ‘spacer’ ending with a carboxylic acid functional group (Figure 2B). A similar spacer and carboxylic acid functional group also is present in SFN–cysteine (SFN–Cys) (Figure 2C), a metabolite of the compound SFN, which is found in broccoli and broccoli sprouts. Molecular modeling studies with SFN–Cys provided good support for complex formation with HDAC (Figure 3). Thus, the SFN–Cys a-amino group was positioned to H-bond with buried His residues in the enzyme pocket, and the SFN–Cys carboxylate group formed a bidentate ligand with the active site zinc atom. In vitro, SFN–Cys inhibited HDAC activity significantly in a cell-free system when tested at concentrations in the range 3–15 μM, whereas SFN parent compound had no effect on HDAC unless it was incubated with cells to allow for metabolism (30). Inhibition of the first step in the mercapturic acid pathway (see below) blocked the HDAC inhibition associated with SFN in human colon cancer cells (30), thus establishing the importance of metabolism for HDAC inhibition by SFN. As with most dietary agents, little is known about the concentrations of parent SFN or its active form(s) in different tissues (e.g. colon, prostate), or the temporal changes that occur following normal human dietary intake. In a study with four human volunteers who consumed broccoli sprouts (31), peak plasma concentrations were reported to be in the range 0.94–2.27 μM, based on an assay that detected total isothiocy-anate and dithiocarbamate levels, rather than specific SFN metabolites.
Fig. 3.
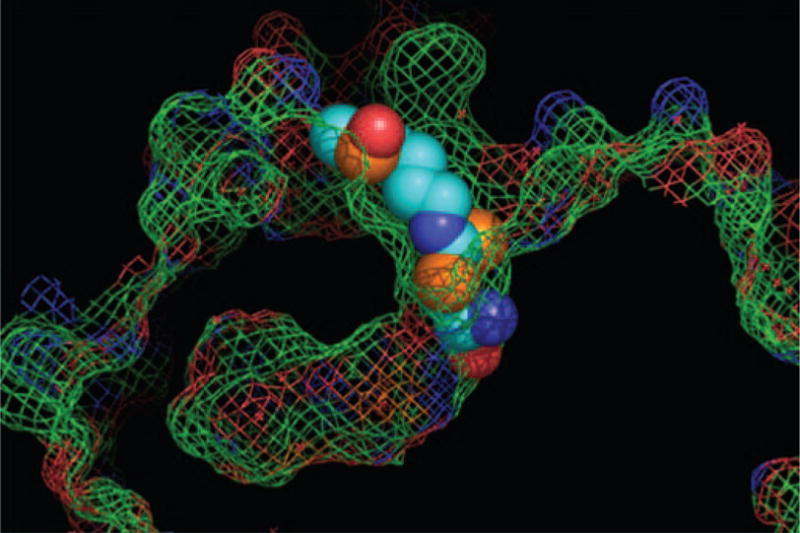
SFN–Cys/HDAC interaction. Molecular modeling studies revealed a plausible interaction for SFN–Cys within the active site of an HDAC, with the carboxylate group of SFN–Cys positioned as a bidentate ligand with the buried zinc atom. See (30) for further details.
The ‘spacer’ theory—predicting dietary HDAC inhibitors
Based on the SFN studies conducted to date, we propose that the ‘ultimate’ HDAC inhibitor is SFN–Cys, generated from the parent compound via the mercapturic acid pathway (Figure 4, lower left). In addition to SFN–Cys formed from SFN–glutathione (SFN–GSH), there may be removal of the acetyl group from SFN–N-acetylcysteine (SFN–NAC) by HDAC, leading to regeneration of SFN–Cys. The latter possibility is interesting, because it represents a mechanism-based (deacetylation) pathway, with the product reversibly inhibiting the enzyme. As mentioned above, direct addition of SFN parent compound to nuclear extracts in vitro had no effect on HDAC activity, and SFN–GSH also had minimal inhibitory activity, whereas SFN–NAC and SFN–Cys attenuated HDAC activity significantly (Figure 4A).
Fig. 4.
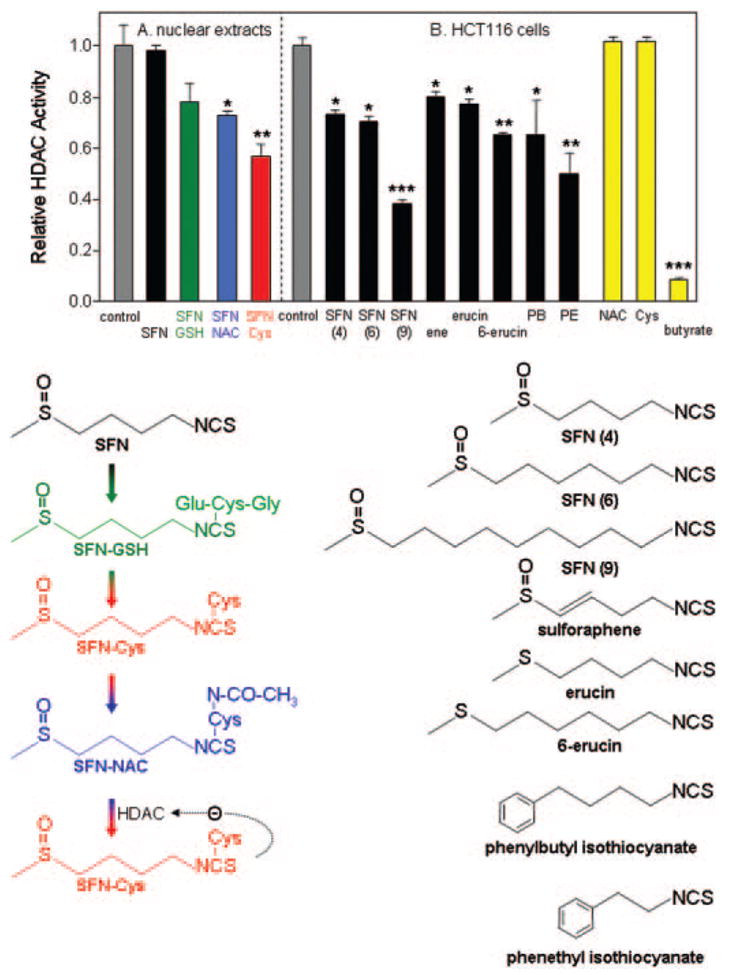
HDAC inhibitory activity of sulforaphane (SFN) and structurally related isothiocyanates. (A) The mercapturic acid pathway converts SFN sequentially to SFN–GSH (SFN–glutathione), SFN–Cys (SFN–cysteine) and SFN–NAC (SFN–N-acetylcysteine). SFN–Cys formed from SFN–GSH, or after deacetylation of SFN–NAC by HDAC, leads to competitive enzyme inhibition, according to the working hypothesis (see text). Direct addition of SFN or SFN–GSH to isolated nuclear extracts in vitro had no significant effect on HDAC activity, whereas SFN–NAC (blue bar) and SFN–Cys (red bar) attenuated HDAC activity significantly. (B) Incubation of SFN with HCT116 human colon cancer cells followed by testing of the cell lysates revealed significant HDAC inhibition, as seen with several other structurally related isothiocyanates. All concentrations were 15 μM, except butyrate which was 1 mM. Data = mean ± SD, n =3. *P < 0.05, **P <0.01, ***P < 0.001, using Student’s t-test versus the corresponding control (gray bar). For details of the HDAC assay and other conditions, see (30).
According to the working hypothesis, other reported ‘anti-carcinogenic’ isothiocyanates and allyl compounds from cruciferous vegetables and garlic (32–34) may be potential HDAC inhibitors, if they are metabolized via the mercapturic acid pathway and have the correct geometry to fit the HDAC pocket. We examined this possibility for several isothiocy-anates, using a screening methodology in which HCT116 human colon cancer cells were treated with test compound and the corresponding cell lysates were added to an in vitro HDAC activity assay (Figure 4B). HDAC inhibition was seen, reproducibly, in cells treated with SFN, in contrast to the lack of inhibition obtained upon addition of parent compound to isolated nuclear extracts (compare SFN data in Figure 4A and B). Isothiocyanates with a similar ‘spacer’ length as SFN, such as sulforaphene, erucin and phenylbutyl isothiocyanate exhibited comparable HDAC inhibitory activities, as did compounds with a longer or shorter spacer, such as 6-SFN, 9-SFN, 6-erucin and phenethyl isothiocyanate. This is consistent with molecular modeling studies showing the HDAC active site occupied with the Cys moiety, and the isothiocyanate ‘cap’ group influencing accessibility to the binding pocket (Figure 3). Similar findings have been reported for structural analogs of trichostatin A, in which the spacer and hydroxamic acid group were retained while substituting the cap group (7–10). Interestingly, NAC and cysteine alone had no effect when incubated with HCT116 colon cancer cells at the same concentration of 15 μM, and the cell lysates were used in the HDAC assay (Figure 4B).
We make a distinction here between HDAC inhibition at lower doses of SFN (<20 μM) and the effects reported more typically at higher concentrations. For example, we would expect little if any meaningful contribution of the HDAC mechanism in colon or prostate cancer cells treated with 40–100 μM SFN; SFN in this dose-range has been shown to generate reactive oxygen species, and activates a mitochon-drial/Bcl-2-associated apoptotic pathway that was blocked by 1–10 mM GSH or NAC (35,36). The latter compounds were included ostensibly for their ‘antioxidant’ activities, but other mechanisms might be involved when cells are treated with a ~ 10–100-fold molar excess of GSH or NAC, compared with SFN. In human hepatoma cells, 1 mM NAC protected against Ni-induced histone hypoacetylation (37), but had no apparent effect on histone acetylation in HL-60 human leukemia cells (38).
Based on the model proposed, it is interesting to speculate about other putative HDAC inhibitors in the human diet. Ret-inoic acid (Figure 1B) has a cap group, spacer, and carboxylic acid functional group, and might be considered a candidate HDAC inhibitor. However, drug-resistant cases of APL involving the PLZF fusion protein respond to trichostatin A but not retinoic acid (Figure 1C). This might be due, in part, to poor fit between the retinoic acid and the specific HDAC that associates with PLZF, rather than all HDACs per se. Derivatives of retinoic acid are available (e.g. 9-cis, 11-cis, 13-cis and all-trans) with a different degree of flexibility and orientation in the spacer. As far as we are aware, retinoids have not been examined formally for fit in the HDAC pocket and for inhibition of enzyme activity.
As mentioned above, the HDAC inhibitor butyrate is a short-chain fatty acid, but there are numerous other dietary fatty acids, which in theory might gain access to the HDAC active site, with their terminal carboxylic acid group positioned adjacent to the zinc atom. One particularly interesting example is conjugated linoleic acid (CLA), which has anticarcinogenic and antiatherogenic properties, and enhances growth and feed efficiency, through mechanisms that are poorly understood (39). CLA in fact comprises several structurally-related isomers, and the cis-9, trans-11 and cis-10,trans-12 isomers have distinct biological activities, quite different from each other and from the cis-9,cis-12 isomer, linoleic acid (39). Interestingly, c10, t12 CLA decreases Bcl-2 and induces p21, a known HDAC target (39). The shape of the fatty acid tail is strongly influenced by the position and number of double bonds, which might influence access to the HDAC pocket. Other bioactive lipids also have a spacer and –COO− arrangement, such as the prostanoids; although not strictly ‘dietary’, they warrant consideration in terms of possible effects on HDACs.
Molecular modeling studies revealed that the Cys moiety fits well in the HDAC active site when attached to SFN, but no effect was seen on enzyme activity with free Cys (Figure 4B). Free amino acids from dietary sources are unlikely to substitute for Cys, owing to poor fit with the HDAC pocket, and because of the rapid turnover by aminotransferases during normal intermediary metabolism.
Other dietary compounds with the spacer and carboxylic acid functional group include biotin, which is noteworthy since it is used to modify histone tails as part of the histone code (see above), as well as the ‘antioxidant’ lipoic acid (40). Fat-soluble antioxidants such as α-tocopherol, γ-tocopherol and tocotrienols undergo ω-hydroxylation of the side chain and sequential rounds of β-oxidation, leading to carboxyhy-drochroman derivatives with appropriate spacer lengths attached to a carboxylic acid group (41,42). Whether such metabolites would ever reach sufficiently high levels to affect HDAC activity in the nucleus is an open question, but ‘class II’ HDACs also reside in the cytoplasm and might be targets for inhibition (see next section).
Dietary HDAC inhibitors—a double-edged sword?
‘Classical’ HDACs include both class I and class II HDACs that are inhibited by trichostatin A (5–10). Class I HDACs are found exclusively in the nucleus and are expressed in most cell types, whereas class II HDACs shuttle between the nucleus and cytoplasm and appear to have a more restricted tissue distribution. Studies with potent agents related to trichostatin A and trapoxin have clearly established the concept of iso-form-specific HDAC inhibitors (10); however, there is much work to be done to clarify downstream signaling pathways for each HDAC family member and the response to dietary factors, alone as well as in combination, where synergistic or antagonistic effects might be anticipated. Tissue-specific metabolism and the levels of ‘ultimate HDAC inhibitor’, such as SFN–Cys, that might be achieved at the target site, further complicate this issue. In addition, certain dietary agents may increase rather than attenuate HDAC activity, as reported for theophylline in alveolar macrophages from patients with chronic obstructive pulmonary disease (43), and for resveratrol in the activation of human SIRT1 (44). The latter enzyme belongs to the recently discovered NAD+-dependent SIR2 family, designated as class III HDACs, which do not typically respond to trichostatin A. HDAC inhibitors thus have the potential to increase or decrease gene expression, and therapy or prevention based on such mechanisms is still largely a ‘black box’ awaiting further investigation.
Pauling often used the term ‘orthomolecular’ medicine (45,46), which in the context of the present discussion can be viewed as the regulation of genes by the right concentration of a dietary factor in order to maintain optimal cell growth, differentiation, and apoptosis, within a homeostatic norm. By this definition, we distinguish between ‘pharmacologic’ agents that potently de-repress gene expression (e.g. during therapeutic intervention) and dietary HDAC inhibitors that, as weak ligands, more subtly regulate the expression of genes involved in cell growth and apoptosis. An important question is the extent to which dietary HDAC inhibitors, and other dietary agents that affect gene expression in the context of chromatin remodeling [e.g. reversal of DNA hypermethylation by soy isoflavones (47)], modulate genes such as P21 and BAX so that cells can respond most effectively to external stimuli and toxic insults. Studies with dietary HDAC inhibitors are in their infancy, but we believe it is important not to dismiss these agents simply because they are weak ligands; their ability to modulate gene expression in subtle ways and, through a lifetime of exposure, to impact cancer chemoprevention warrants further investigation.
Acknowledgments
We thank Drs Joe Beckman, Mark Leid and Andy Karplus for helpful discussions and critical comments on the manuscript, as well as Jeff Watson, Ganapathy Sarma and Nihal Gooneratne for molecular modeling. Results presented here were from studies supported in part by NIH grants CA65525, CA80176 and CA90890, as well as NIEHS center grant P30 ES00210.
Abbreviations
- APL
acute promyelocytic leukemia
- CLA
conjugated linoleic acid
- HDAC
histone deacetylase
- RAR
retinoic acid receptors
- RARE
retinoic acid response element
- SAHA
suberoylanilide hydroxamic acid
- SFN
sulforaphane
- SFN–Cys
SFN–cysteine
- SFN–GSH
SFN–glutathione
- SFN–NAC
SFN–N-acetylcysteine
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry (Mosc) 2005;70:533–549. doi: 10.1007/s10541-005-0147-7. [DOI] [PubMed] [Google Scholar]
- 2.Cosgrove MS, Wolberger C. How does the histone code work? Biochem Cell Biol. 2005;83:468–476. doi: 10.1139/o05-137. [DOI] [PubMed] [Google Scholar]
- 3.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 4.Allison SJ, Milner J. Remodelling chromatin on a global scale: a novel protective function of p53. Carcinogenesis. 2004;25:1551–1557. doi: 10.1093/carcin/bgh212. [DOI] [PubMed] [Google Scholar]
- 5.de Ruijter AJ, van Gennip AH, Caron HN, Stephan Kemp, Andre BP, Van Kuilenburg. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 7.Mork CN, Faller DV, Spanjaard RA. A mechanistic approach to anticancer therapy: targeting the cell cycle with histone deacetylase inhibitors. Curr Pharm Des. 2005;11:1091–1104. doi: 10.2174/1381612053507567. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin F, La Thangue NB. Histone deacetylase inhibitors open new doors in cancer therapy. Biochem Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 9.Rosato RR, Grant S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol Ther. 2003;2:30–37. doi: 10.4161/cbt.190. [DOI] [PubMed] [Google Scholar]
- 10.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci USA. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 12.Clarke N, Germain P, Altucci L, Gronemeyer H. Retinoids: potential in cancer prevention and therapy. Expert Rev Mol Med. 2004;6:1–23. doi: 10.1017/S1462399404008488. [DOI] [PubMed] [Google Scholar]
- 13.Fazi F, Travaglini L, Carotti D, et al. Retinoic acid targets DNA-methyltransferases and histone deacetylases during APL blast differentiation in vitro and in vivo. Oncogene. 2005;24:1820–1830. doi: 10.1038/sj.onc.1208286. [DOI] [PubMed] [Google Scholar]
- 14.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 15.Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 16.Emionite L, Galmozzi F, Grattarola M, Boccardo F, Vergani L, Toma S. Histone deacetylase inhibitors enhance retinoid response in human breast cancer cell lines. Anticancer Res. 2004;24:4019–4024. [PubMed] [Google Scholar]
- 17.Cote S, Rosenauer A, Bianchini A, Seiter K, Vandewiele J, Nervi C, Miller WH., Jr Response to histone deacetylase inhibition of novel PML/RAR-α mutants detected in retinoic acid-resistant APL cells. Blood. 2002;100:2586–2596. doi: 10.1182/blood-2002-02-0614. [DOI] [PubMed] [Google Scholar]
- 18.He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP, Jr, Rifkind RA, Marks PA, Richon VM, Pandolfi PP. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108:1321–1330. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coffey DC, Kutko MC, Glick RD, Butler LM, Heller G, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001;61:3591–3594. [PubMed] [Google Scholar]
- 20.Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6:520–524. doi: 10.1038/sj.embor.7400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nebbioso A, Clarke N, Voltz E, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 23.Myzak MC, Dashwood RH. Histone deacetylases as targets for dietary cancer preventive agents: Lessons learned with butyrate, diallyl disulfide, and sulforaphane. Current Drug Targets. 2006 doi: 10.2174/138945006776359467. in press. [DOI] [PubMed] [Google Scholar]
- 24.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bach Knudsen KE, Serena A, Canibe N, Juntunen KS. New insight into butyrate metabolism. Proc Nutr Soc. 2003;62:81–86. doi: 10.1079/PNS2002212. [DOI] [PubMed] [Google Scholar]
- 26.McIntosh GH, Royal PJ, Pointing G. Wheat aleurone flour increases cecal β-glucuronidase activity and butyrate concentration and reduces colon adenoma burden in azoxymethane-treated rats. J Nutr. 2001;131:127–131. doi: 10.1093/jn/131.1.127. [DOI] [PubMed] [Google Scholar]
- 27.Bach Knudsen KE, Serena A, Kjaer AK, Jorgensen H, Engberg R. Rye bread enhances the production and plasma concentration of butyrate but not the plasma concentrations of glucose and insulin. J Nutr. 2005;135:1696–1704. doi: 10.1093/jn/135.7.1696. [DOI] [PubMed] [Google Scholar]
- 28.Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duee PH, Martle P, Chaumontet C. Diallyl disulfide (DADS) increases histone acetylation and p21waf1/cip1 expression in human colon cancer cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 29.Guyonnet D, Berges R, Siess MH, Pinnert MF, Chagnon MC, Suschetet M, Le Bon AM. Post-initiation modulating effects of allyl sulfides in rat hepatocarcinogenesis. Food Chem Toxicol. 2004;42:1479–1485. doi: 10.1016/j.fct.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 31.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. 2002;316:43–53. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]
- 32.Bianchini F, Vainio H. Isothiocyanates in cancer prevention. Drug Metab Rev. 2004;36:655–667. doi: 10.1081/dmr-200033468. [DOI] [PubMed] [Google Scholar]
- 33.Keum YS, Jeong WS, Kong AN. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 34.Knowles LM, Milner JA. Possible mechanisms by which allyl sulfides suppress neoplastic cell proliferation. J Nutr. 2001;131:1061s–1066s. doi: 10.1093/jn/131.3.1061S. [DOI] [PubMed] [Google Scholar]
- 35.Shen G, Xu C, Chen C, Hebbar V, Kong AH. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol. 2005 Sep 17;:1–11. doi: 10.1007/s00280-005-0050-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 36.Singh SV, Srivastava SK, Choi S, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280:19911–19924. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- 37.Kang J, Zhang Y, Chen Y, Chen H, Lin C, Wang Q, Ou Y. Nickel-induced histone hypoacetylation: the role of reactive oxygen species. Toxicol Sci. 2003;74:279–286. doi: 10.1093/toxsci/kfg137. [DOI] [PubMed] [Google Scholar]
- 38.Kang J, Chen J, Zhang D, Da W, Ou Y. Synergistic killing of human leukemia cells by antioxidants and trichostatin A. Cancer Chemother Pharmacol. 2004;54:537–545. doi: 10.1007/s00280-004-0845-7. [DOI] [PubMed] [Google Scholar]
- 39.Pariza MW. Perspectives on the safety and effectiveness of conjugated linoleic acid. Am J Clin Nutr. 2004;79:1132s–1136s. doi: 10.1093/ajcn/79.6.1132S. [DOI] [PubMed] [Google Scholar]
- 40.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sontag TJ, Parker RS. Cytochrome P450 ω-hydroxylase pathway of tocopherol catabolism. J Biol Chem. 2002;277:25290–25296. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- 42.Birringer M, Pfluger P, Kluth D, Landes N, Brigelius-Flohe R. Identities and differences in the metabolism of tocotrienols and tocopherols in HepG2 cells. J Nutr. 2002;123:3113–3118. doi: 10.1093/jn/131.10.3113. [DOI] [PubMed] [Google Scholar]
- 43.Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–695. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 45.Pauling L. Orthomolecular psychiatry. Varying the concentrations of substances normally present in the human body may control mental disease. Science. 1968;160:265–271. doi: 10.1126/science.160.3825.265. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez MJ, Miranda-Massari JR, Mora EM, Guzman A, Riordan NH, Riordan HD, Casciari JJ, Jackson JA, Roman-Franco A. Orthomolecular oncology review: ascorbic acid and cancer 25 years later. Integr Cancer Ther. 2005;4:32–44. doi: 10.1177/1534735404273861. [DOI] [PubMed] [Google Scholar]
- 47.Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res. 2005;11:7033–7041. doi: 10.1158/1078-0432.CCR-05-0406. [DOI] [PubMed] [Google Scholar]