

Abstract
β-Catenin, a member of the Wnt signaling pathway, is downregulated by glycogen synthase kinase-3β (GSK-3β)-dependent phosphorylation of Ser/Thr residues in the N-terminus of the protein, followed by ubiquitination and proteosomal degradation. In human and rodent cancers, mutations that substitute one of the critical Ser/Thr residues in the GSK-3β region of β-catenin stabilize the protein and activate β-catenin/TCF/LEF target genes. This study examined three oncogenic β-catenin mutants from rat colon tumors containing substitutions adjacent to amino-acid residue Ser33, a key target for phosphorylation by GSK-3β. Compared with wild-type β-catenin (WT), the β-catenin mutants D32G, D32N, and D32Y strongly activated TCF-4-dependent transcription in HEK293 cells, and there was accumulation of β-catenin in the cell lysates. Immunoblotting with phosphospecific antibodies indicated that there was little if any effect on the phosphorylation of Ser37, Thr41 or Ser45; however, the phosphorylation of Ser33 appeared to be affected in the β-catenin mutants. Specifically, antiphospho-β-catenin 33/37/41 antibody identified high, intermediate and low expression levels of phosphorylated β-catenin in cells transfected with D32G, D32N and D32Y, respectively. Experiments with the proteosome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN) revealed ubiquitinated bands on all three mutant β-catenins, as well as on WT β-catenin. The relative order of ubiquitination was WT > D32G > D32N > D32Y, in parallel with findings from the phosphorylation studies. These results are discussed in the context of previous studies, which indicated that amino-acid residue D32 lies within the ubiquitination recognition motif of β-catenin.
Keywords: β-catenin, TCF/LEF, APC, Wnt signaling, colorectal cancer, CTNNB1, human β-catenin gene, Ctnnb1, rat β-catenin gene
Introduction
There is a great deal of current interest in β-catenin, APC and other members of the Wnt signaling pathway and their role in the development of human cancers (Polakis, 2000; Clements et al., 2002; Oving and Clevers, 2002; Lustig et al., 2002). In colorectal cancers, the APC tumor suppressor gene is a common target for mutation (Kinzler and Vogelstein, 1997), but colon tumors with wild-type (WT) APC typically have genetic changes in CTNNB1, the gene for β-catenin (Ilyas et al., 1997; Morin et al., 1997; Sparks et al., 1998). β-Catenin is a cadherin-binding protein involved in cell–cell adhesion (Hirohashi, 1998; Hajra and Fearon, 2002; Conacci-Sorrell et al., 2002), and also functions as a transcriptional activator when complexed in the nucleus with members of the TCF/LEF family of proteins (Polakis, 2000). Control of the cytosolic levels of β-catenin occurs via a multiprotein complex, in which APC, axin/conductin and glycogen synthase kinase-3β (GSK-3β) negatively regulate β-catenin expression (Polakis, 2000). In addition, casein kinase I (CKI) serves as a ‘priming’ kinase for GSK-3β (Hagen and Vidal-Puig, 2002). Phosphorylation of β-catenin has been postulated to occur in a sequential manner (Ser45 → Thr41 → Ser37 → Ser33), although recent evidence suggests that phosphorylation at Ser33, Ser37 or Thr41 can occur in the absence of phosphorylation at Ser45 (Wang et al., 2003). Phosphorylation of these Ser/Thr residues in the N-terminal region of β-catenin targets the protein for ubiquitination and subsequent proteosomal degradation (Aberle et al., 1997; Liu et al., 1999). In primary human colon tumors and colorectal cancer cell lines, mutations in CTNNB1 frequently substitute the critical Ser/Thr residues and stabilize β-catenin (Morin et al., 1997; Ilyas et al., 1997; Sparks et al., 1998), leading to accumulation of β-catenin/TCF complexes in the nucleus and activation of target genes (Oving and Clevers, 2002; Huelsken and Behrens, 2002).
Mutations in β-catenin also have been detected in the tumors from animals treated with chemical carcinogens (Dashwood et al., 1998; Takahashi et al., 1998; Suzui et al., 1999; Blum et al., 2001, 2003). Indeed, there are a number of similarities between human and rat tumors with respect to the β-catenin/Apc pathway. First, as in the human situation (Sparks et al., 1998), colon tumors in the rat contain mutations in Apc or Ctnnb1, but not in both of these genes (Dashwood et al., 1998). Second, these mutations stabilize β-catenin and lead to increased expression of β-catenin in the nucleus of cancer cells (Takahashi et al., 1998). Third, β-catenin/Tcf target genes frequently are overexpressed, including c-myc, c-jun and cyclin D1 (Blum et al., 2001, 2003). Fourth, genetic changes in human CTNNB1 or rat Ctnnb1 typically substitute amino-acids within the N-terminal regulatory domain of β-catenin. Fifth, in both species the spectrum of β-catenin mutations includes direct substitution of critical Ser/Thr residues as well as substitution of amino-acids adjacent to these residues. Interestingly, in carcinogen-induced tumors in the rat, substitution of residues adjacent to Ser33 predominates over substitution of Ser/Thr residues directly. For example, the combined results from studies in which rats were treated with 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP), 2-amino-3-methylimidazo[4,5-f]quinoline (IQ), 1,2-dimethylhydrazine (DMH), azoxymethane or methylazoxymethanol acetate plus 1-hydroxyanthraquinone revealed a total of 59 tumors with β-catenin mutations; nine of these mutations (15%) substituted critical Ser/Thr residues directly, whereas 49 (83%) had genetic changes affecting residues immediately adjacent to Ser33 (Dashwood et al., 1998; Takahashi et al., 1998; Suzui et al., 1999; Blum et al., 2001, 2003).
Previous studies examined the activation of LEF-1-dependent transcription by oncogenic mutants, such as S37A and S45Y, which contained substitutions of critical Ser/Thr residues in the GSK-3β region of β-catenin (Porfiri et al., 1997). However, less information is available for β-catenin mutants containing substitutions adjacent to critical Ser/Thr residues. We sought to examine the functional consequence of substitutions to D32 in β-catenin, since this residue lies immediately adjacent to Ser33, the ‘terminal’ GSK-3β phosphorylation target in β-catenin. Interestingly, in human cancers, substitution of D32 represents the third most common alteration in β-catenin after changes in Ser45 and Thr41 (Nusse, 2003), and substitutions involving D32 occur with a high frequency in carcinogen-induced colon tumors in the rat (Dashwood et al., 1998; Blum et al., 2001, 2003). This provided a means of examining phosphorylation versus ubiquitination events in β-catenin destruction, in the context of previous studies, indicating that D32 lies within the consensus recognition motif for ubiquitination of β-catenin (Aberle et al., 1997). Thus, we tested the hypothesis that substitution of D32 with a bulky aromatic side chain (D32Y) might interfere with phosphorylation of the adjacent Ser33, whereas substitution with a small or basic side chain (D32G, D32N) might have less effect in blocking GSK-3β but might interfere with the subsequent steps involving ubiquitination and proteosomal degradation of β-catenin.
Results
Previous studies examined the activation of LEF-1-dependent transcription by WT and mutant β-catenins S37A and S45Y in human embryonic kidney (HEK) 293 cells, a cell line containing low endogenous levels of cytosolic β-catenin and TCF/LEF (Porfiri et al., 1997). Using a similar assay system, but with TCF-4 (Figure 1a), reporter activities were strongly induced by WT and mutant β-catenins D32G, D32N and D32Y in the presence of TOPflash, which contains perfect TCF/LEF sites (Morin et al., 1997), but not with the corresponding negative control FOPflash. With TOP-flash, β-catenin/TCF-4-dependent reporter activities were comparable at 24 h, but at 48 h they were 3- to 5-fold higher in cells transfected with mutant β-catenins versus WT. At 24 and 48 h post-transfection, respectively, the GSK-3β inhibitor LiCl increased the reporter activity of WT β-catenin eight and two fold compared with WT minus LiCl. In contrast, the proteosome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN) attenuated the reporter activity of WT β-catenin at both time points (Figure 1a).
Figure 1.
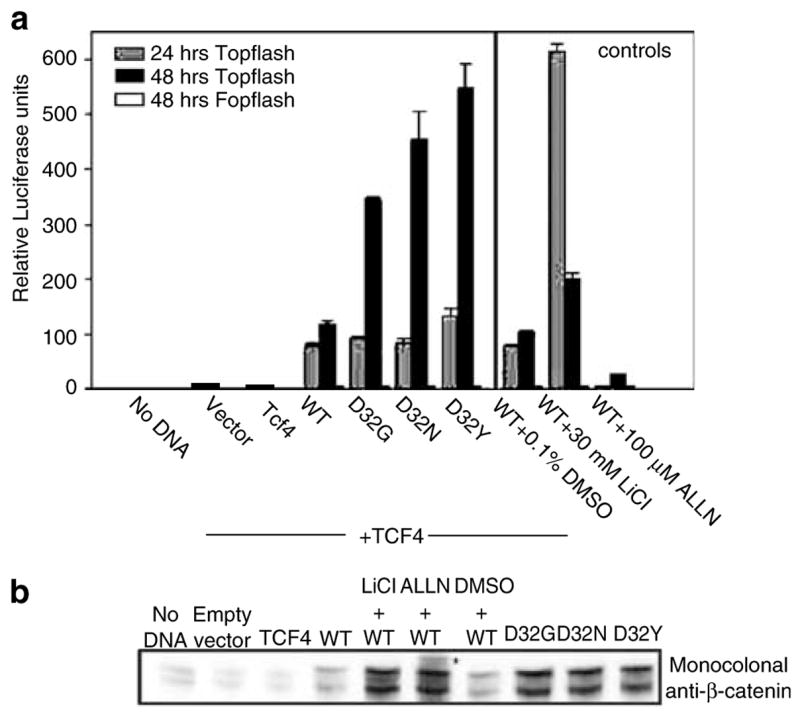
Activation of TCF-4-dependent transcription by WT and mutant β-catenins. (a) A plasmid expressing a TCF/LEF-responsive reporter, TOPflash, or the negative reporter FOPflash (see Morin et al., 1997), was cotransfected into HEK293 cells with empty vector (vector), a plasmid expressing TCF-4 or TCF-4 vector plus a plasmid expressing WT or mutant (D32G, D32N, D32Y) β-catenins. Reporter activation was assessed as luciferase activity in aliquots of cell lysates normalized for protein content. The GSK-3β inhibitor LiCl (30 mM) and the proteosome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN, 100 μM) were included as controls. The data are given as mean ± s.d. of triplicates. (b) Cell lysates obtained 48 h post-transfection were subjected to immuno-blot analysis with monoclonal anti-β-catenin antibody, as well as antibody to β-actin (not shown). The asterisk denotes the position of high molecular weight band(s), detected after treatment with ALLN but not LiCl
Cell lysates from these experiments were subjected to immunoblotting using monoclonal anti-β-catenin antibody (Figure 1b). Equal protein loading was first confirmed, and the same blot was subsequently probed with the primary antibody, see Materials and methods. At 48 h post-transfection, low expression levels were detected for WT β-catenin, whereas high expression levels were detected in cells transfected with D32G, D32N and D32Y. Immunoblotting with antibody specific for the Myc tag in transfected β-catenins also showed similar expression levels for the three β-catenin mutants, 48 h post-transfection (data not presented). Expression levels of WT β-catenin were increased markedly after LiCl and ALLN treatment, but only in the latter case were higher molecular weight bands detected (Figure 1b). Similar results were reported previously (Aberle et al., 1997), and these findings are discussed further below.
The cell lysates obtained 48 h post-transfection were also examined using various phosphospecific antibodies (Figure 2). Initial experiments used antiphospho-β-catenin 33/37/41 antibody, which recognizes β-catenin when it is phosphorylated at one or more of three specific sites, namely Ser33, Ser37 or Thr41. Strong bands were detected in cells transfected with D32G and D32N, but with the same phosphospecific antibody, much weaker bands were detected in cells transfected with D32Y (Figure 2a). Expression levels detected with antiphospho-β-catenin 33/37/41 antibody were, respectively, 6.2-, 5.5- and 1.5-fold higher in cells transfected with D32G, D32N and D32Y versus WT (see the densitometry data in Figure 2a). Using antiphospho-β-catenin 41/45 antibody, high expression levels were detected in cells transfected with D32G or D32N, as well as in cells transfected with D32Y; however, low levels were detected for WT β-catenin (Figure 2b). Finally, α-ABC antibody, which recognizes β-catenin when it is unphosphorylated at Ser37 and/or Thr41, detected low expression levels in lysates from cells transfected with D32G, D32N or D32Y, and no band was seen in cells transfected with WT β-catenin (Figure 2c). With all three phosphospecific antibodies, expression levels of WT β-catenin were increased significantly after treatment with LiCl or ALLN.
Figure 2.
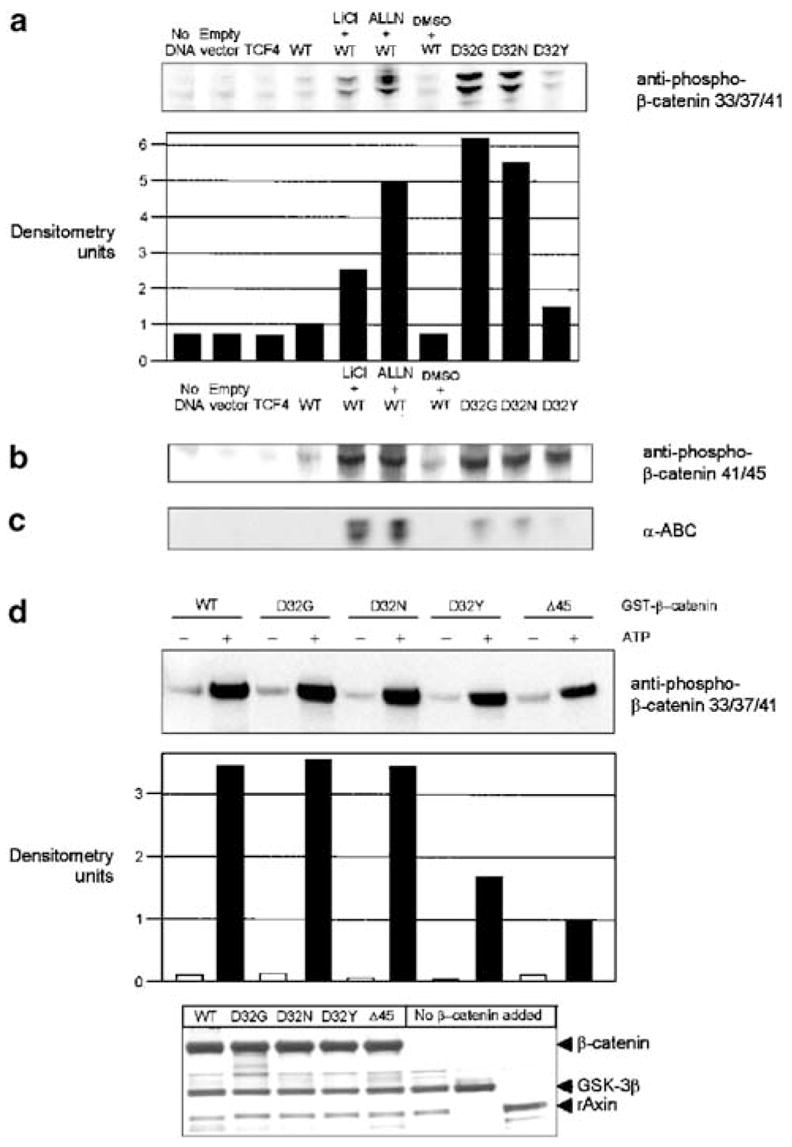
Immunodetection of β-catenin using phosphospecific antibodies. The cell lysates described in Figure 1b were probed with the following antibodies: (a) antiphospho-β-catenin 33/37/41, (b) antiphospho-β-catenin 41/45 and (c) α-ABC (α-active β-catenin, for β-catenin unphosphorylated at Ser45 and Thr41). (d) An in vitro kinase assay was performed with purified GST-tagged rAxin, GSK-3β and WT or mutant β-catenins, in the presence and absence of ATP. Reaction products were analysed by SDS–PAGE with immunodetection using anti-pβ-catenin-33/37/41 antibody. Equal protein loading was confirmed by silver staining (bottom); the last three lanes, respectively, were controls lacking β-catenin but containing GSK-3β plus rAxin, GSK-3β alone or rAxin alone
An in vitro kinase assay was next performed, with purified recombinant GSK-3β, rAxin (Ikeda et al., 1998) and WT or mutant β-catenins, in the presence and absence of ATP (Figure 2d). In immunoblots with antiphospho-β-catenin 33/37/41 antibody, only low background expression levels were detected in the absence of ATP, but there was a strong expression of all phosphorylated β-catenins in the presence of ATP, including D32Y. In addition, Δ45, a β-catenin mutant found in HCT116 human colorectal cancer cells (Morin et al., 1997), was detected with antiphospho-β-catenin 33/37/41 antibody (Figure 2d). The latter finding clearly implies that priming by GSK-3β at Ser45 is not an absolute prerequisite for phosphorylation at other sites in β-catenin, as reported recently (Wang et al., 2003). However, compared with Δ45, the relative efficiency of phosphorylation was > threefold higher for WT, D32G and D32N, and 1.6-fold greater for D32Y (see the densitometry data in Figure 2d). Silver staining after SDS-PAGE confirmed equal loading of all β-catenins in these experiments (Figure 2d, bottom).
Additional experiments were conducted with the proteasome inhibitor ALLN, in cells transfected with WT or mutant β-catenins. Reporter activities were inhibited consistently by 100 μM ALLN (Figure 3a), and total β-catenin expression was increased markedly in the cell lysates (Figure 3b). Interestingly, higher molecular weight bands were detected after ALLN treatment not only for WT β-catenin but also in cells transfected with D32G, D32N or D32Y (see asterisks in Figure 3b). Cell lysates from these experiments were further examined using antiphospho-β-catenin 33/37/41 antibody (Figure 3c); higher expression levels of WT β-catenin were seen in cells treated with ALLN compared with no ALLN. However, the opposite trend was observed in cells transfected with D32G, D32N or D32Y; stronger bands were detected in the absence of ALLN treatment compared with the corresponding + ALLN controls (Figure 3c). Using antiphospho-β-catenin 41/45 and α-ABC antibodies (Figures 3d and e, respectively), β-catenin expression levels were consistently higher in cells treated with 100 μM ALLN compared with the corresponding controls without ALLN, for all β-catenins studied.
Figure 3.
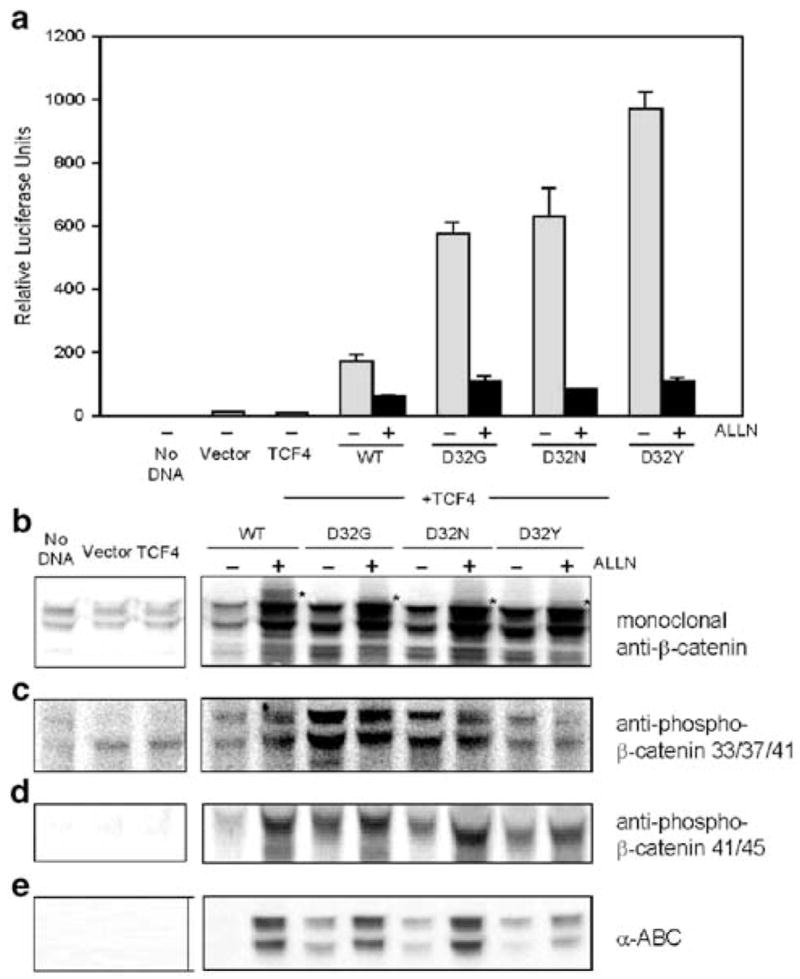
Effect of ALLN on β-catenin/TCF-4-dependent reporter activity and β-catenin expression in HEK293 cells. (a) Reporter activities (TOPflash) were assessed 48 h post-transfection, as described in Figure 1a. Data shown in the figure (mean ± s.d., n =3) are representative findings from three separate experiments conducted in the presence and absence of 100 μM ALLN. The corresponding cell lysates were immunoblotted with antibodies to (b) total β-catenin (monoclonal anti-β-catenin), (c) antiphospho-β-catenin 33/37/41, (d) antiphospho-β-catenin 41/45 and (e) α-ABC. In (b), the asterisk denotes the position of higher molecular band(s), seen after treatment with ALLN
To further optimize the ALLN treatment conditions, dose–response and time–course experiments were performed. Figure 4 shows the results for cells treated with ALLN for 20 h, but similar data were obtained after only 6 h exposure to ALLN (data not presented). In the range 5–50 μM ALLN, a concentration-dependent decrease in reporter activity was observed, except for an unexpected (and reproducible) increase for WT at 5 μM ALLN (Figure 4a). We have no explanation for the latter finding, although it clearly indicates an increase in the pool of transcriptionally active WT β-catenin at low levels of ALLN. In all cases, monoclonal anti-β-catenin antibody detected higher molecular weight bands after treatment with ALLN, and the appearance of these bands coincided with increased expression of cleaved PARP, an indicator of apoptosis (Figures 4a–f). The apparent threshold for increased PARP cleavage was on the order of ~ 10 μM ALLN.
Figure 4.
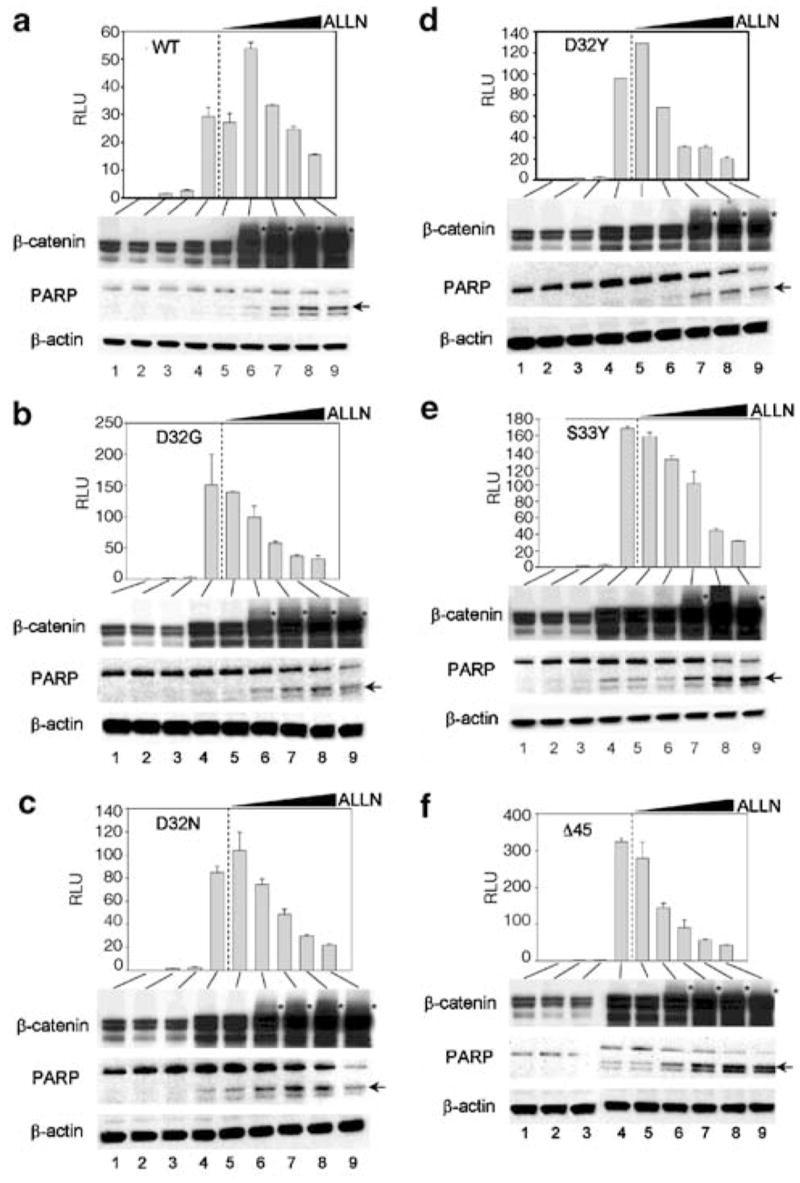
Dose–response for inhibition by ALLN of β-catenin/TCF-4-dependent reporter activity in HEK293 cells, and the expression of β-catenin and poly(ADP-ribose) polymerase (PARP). Cells were transfected with (a) WT, (b) D32G, (c) D32N, (d) D32Y, (e) S33Y or (f) Δ45 β-catenin. TOPflash reporter activities (mean ± s.d., n =3) were determined as described in Figure 3a –lane 1, no DNA; lane 2, empty vector; lane 3, TCF-4; lane 4, TCF-4 plus β-catenin; lanes 5–9, same as lane 4, but with 0 (DMSO vehicle alone), 5, 10, 25 and 50 μM ALLN, respectively. The wedge-shaped symbol indicates increasing concentration of ALLN. The corresponding cell lysates were immunoblotted with antibodies to total β-catenin, PARP and β-actin. An asterisk denotes the position of higher molecular weight band(s) for β-catenin, seen after treatment with ALLN. The arrow indicates cleaved PARP
Based on these findings, cells were treated with 10 μM ALLN for 6 h, and the cell lysates were used in immunoprecipitation experiments. Discrete bands were detected in the region corresponding to ubiquitinated β-catenin in immunoprecipitation experiments with anti-β-catenin antibody followed by immunodetection with antiubiquitin antibody (Figure 5a), or antiubiquitin for immunoprecipitation and anti-β-catenin for immunodetection (data not shown). Because these bands might include endogenous β-catenin, anti-Myc tag antibody was used to pull down the transfected Myc-tagged proteins, and the latter were loaded at equal concentrations in subsequent Western analyses (Figure 5b). Under these conditions, WT β-catenin and the mutants D32G, D32N and D32Y were clearly ubiquitinated (Figure 5c); densitometry measurements of the major ubiquitinated band gave relative levels of expression as follows: WT 1.00, D32G 1.05, D32N 0.81 and D32Y 0.69. As expected, in the Myc pull-down assays the nonubiquitinated 92 kDa form of β-catenin was detected using anti-Myc tag antibody (Figure 5b), but not with antiubiquitin antibody (Figure 5c).
Figure 5.
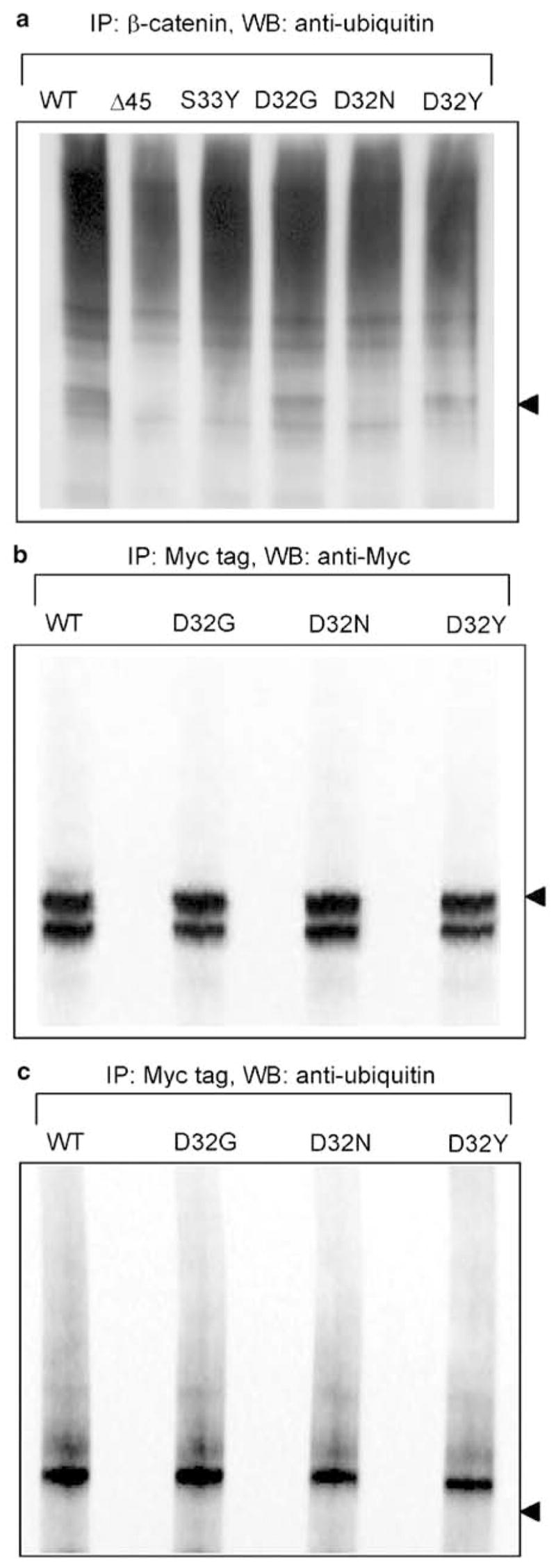
Ubiquitination of WT and mutant β-catenins. (a) HEK293 cells were transfected with WT or mutant β-catenins, as indicated in the figure, and treated for 6 h with 10 μM ALLN. Cell lysates obtained 48 h post-transfection were immunoprecipitated (IP) with antibody to β-catenin, followed by Western blotting with antiubiquitin antibody. Alternatively, anti-Myc tag antibody was used to pull down transfected (Myc-tagged) β-catenins, followed by WB with (b) anti-Myc tag or (c) antiubiquitin antibody. The position of the nonubiquitinated 92 kDa form of β-catenin is indicated with an arrowhead to the right; note the strong expression of these bands in (b) and their absence in (c)
Discussion
Aberle et al. (1997) reported on the appearance of high molecular weight bands in β-catenin following treatment of cells with the proteasome inhibitor ALLN, and stated that these bands coincided with the ubiquitination status of the protein. The ubiquitination recognition motif was identified within the N-terminal GSK-3β region of β-catenin (Aberle et al., 1997). The GSK-3β region is coded by exon 3 in human CTNNB1 and mouse Catnb, or exon 2 of rat Ctnnb1 (Li et al., 2002, 2004), but in these three species an identical ubiquitination recognition motif is present in β-catenin, namely D32-S33-G34-I35-S37 (Aberle et al., 1997).
In the present studies, we examined oncogenic D32 mutants of β-catenin from carcinogen-induced rat colon tumors (Dashwood et al., 1998; Blum et al., 2001, 2003). The hypothesis was that D32Y might interfere with phosphorylation of the adjacent Ser33 residue, whereas D32G and D32N might have less effect on phosphorylation, but would nonetheless stabilize β-catenin due to interference with the subsequent ubiquitination and proteosomal degradation steps.
In cells transfected with β-catenins and immuno-blotted with antiphospho-β-catenin 33/37/41 antibody, the relative order of expression appeared to be D32G > D32N > D32Y > WT (see Figures 2a and 3c). There was an apparent inverse association between the protein expression detected at 48 h with antiphospho-β-catenin 33/37/41 antibody and the corresponding reporter activities; thus, with TOPflash the relative order was WT < D32G < D32N < D32Y (Figures 1a and 3a). This trend was also seen in cells transfected with WT or mutant β-catenins but no TCF-4 (data not presented), indicating that the exogenous TCF-4 did not interfere markedly with the phosphorylation events under investigation. Based on these results, and the findings with other phosphospecific antibodies used here, our interpretation is that the substitution D32Y interferes more effectively than D32G or D32N with phosphorylation of the adjacent residue Ser33, and this conclusion was supported by in vitro kinase assays with purified recombinant β-catenins (Figure 2d).
In general, WT β-catenin was observed at low or undetectable levels with phosphospecific antibodies, except after LiCl or ALLN treatment, suggesting rapid phosphorylation, ubiquitination, and proteosomal degradation in HEK293 cells. Phosphorylated β-catenin was detected with phosphospecific antibodies after LiCl treatment (Figures 2a, b); this implies that GSK-3β may not have been inhibited completely under the conditions used, or that an alternative kinase substituted for GSK-3β, such as CKI (Hagen and Vidal-Puig, 2002). Unphosphorylated β-catenin was strongly detected with α-ABC antibody in cells treated with LiCl or ALLN (Figure 2c); our interpretation is that a fraction of the total β-catenin undergoes dephosphorylation, due to the high overall β-catenin levels accumulated in these cells (see Figure 1b). However, this possibility requires further investigation.
Initial experiments with 100 μM ALLN showed increased total β-catenin levels in HEK293 cells but decreased reporter activity (Figures 1–3), indicating an increase in the pool of transcriptionally inactive β-catenin. Proteasome inhibitors such as ALLN represent useful biochemical tools for in vitro studies, but prolonged treatment with such agents leads to disruption of normal cellular functions and eventually to apoptosis. We sought to optimize the ALLN treatment conditions, using PARP cleavage as an indirect measure of apoptosis (Figure 4). An apparent threshold of ~ 10 μM ALLN was observed for inhibition of TCF-4/β-catenin-dependent reporter activity in the absence of apoptosis, and this coincided with the first appearance of β-catenin containing higher molecular weight band(s), indicative of ubiquitination. These data show that the proteosome inhibitor caused accumulation of transcriptionally inactive, ubiquitinated β-catenin, even at concentrations of ALLN that did not produce marked induction of apoptosis. However, more sensitive indicators of apoptosis may be appropriate in future studies.
Ubiquitinated bands were confirmed in cells treated with 10 μM ALLN, using anti-β-catenin or anti-Myc tag antibodies for immunoprecipitation and antiubiquitin antibody for immunodetection (Figure 5). There appeared to be a direct relationship between the level of β-catenin expression detected with antiphospho-β-catenin 33/37/41 antibody (Figure 3c) and the corresponding ubiquitination status of the mutant protein (i.e. D32G > D32N > D32Y). These findings are noteworthy for three reasons. First, they suggest that amino-acid substitution at D32 can negatively impact on the phosphorylation of the adjacent Ser33 residue. This inhibition was most obvious for the bulky side-chain in D32Y, followed by D32N (basic residue) and D32G (small residue). Second, the relative phosphorylation status of β-catenin may dictate the corresponding degree of ubiquitination in D32 mutant proteins. However, we cannot rule out the possibility that, with equivalent levels of phosphorylation, the ubiquitination machinery itself might be inhibited more effectively by the substitution D32Y than by the substitutions in D32N and D32G. Third, substitution at D32 does not appear to preclude ubiquitination of β-catenin, despite the evidence that D32 lies at the start of the ubiquitination recognition motif (Aberle et al., 1997). Other pathways of ubiquitination/degradation of β-catenin remain to be investigated, such as those involving Siah-1 and Hakai (Liu et al., 2001; Matsuzawa and Reed, 2001; Fujita et al., 2002). However, our working hypothesis is that with lower phosphorylation at residues 33/37/41, there is a concomitant decrease in ubiquitination status, and increased stabilization of β-catenin.
In summary, the present study has shown that oncogenic mutants of β-catenin found in rat colon tumors, with substitutions at D32, activate TCF-4-dependent transcription and cause overexpression of β-catenin. Through the use of phosphospecific antibodies and the proteosome inhibitor ALLN, D32Y appeared to interfere with phosphorylation of Ser33 to a greater extent than substitutions D32G or D32N. However, none of the substitutions to D32 completely inhibited ubiquitination, despite the fact that D32 lies within the ubiquitination recognition motif in β-catenin. Studies are now in progress on alternative pathways of β-catenin degradation, involving Siah-1 and Hakai.
Materials and methods
Expression constructs
As reported elsewhere (Rubinfeld et al., 1997; Porfiri et al., 1997), fragment switching was used to generate expression vectors for D32G, D32N, D32Y, S33Y, and Δ45 β-catenins, starting with a WT β-catenin cDNA construct that also expressed the Myc epitope tag (a kind gift from Drs Marc van de Wetering and Hans Clevers). The latter construct was subjected to BamHI/XhoI double digestion and the large fragment was purified and ligated to the corresponding BamHI/XhoI double digested insert containing the desired mutation found in carcinogen-induced rat colon or liver tumors (Blum et al., 2001, 2003; Li et al., 2002). To generate procaryotic expression vectors, β-catenin cDNAs were sub-cloned into pGEX-5x-2 (Amersham Pharmacia Biotech) and following transformation into One Shot ® Top 10 competent cell colonies were screened using the supplied primers pGEX5′and pGEX3′. For rAxin, primers were designed to amplify the region 298–506 of rat Axin, using pMALC-2 construct as a template (generous gift from Dr Akira Kikuchi), and the corresponding fragment was subcloned into EcoRI/NotI double digested pGEX-5x-2 expression construct. For GSK-3β cloning, mRNA was isolated from HEK293 cells using the Micro-FastTrack ™ 2.0 mRNA Isolation Kit (Invitrogen), and cDNA was synthesized using the oligo(dT) primer and Thermoscript RT-PCR system (Gibco BRL). GSK-3β forward and reverse primers were designed with EcoRI and NotI restriction sites, respectively, to enable subcloning into pGEX-5x-2, as described above for rAxin. All constructs were confirmed by sequencing in both directions using an ABI Prizm ™ 3.3 sequencer (Center for Gene Research and Biotechnology, Oregon State University).
Cell culture, transient transfections and reporter assays
HEK293 cells were obtained from the American Type Culture Collection (ATCC) and maintained at 37° C under 5% CO2 in minimum essential media (MEM) (Gibco-BRL, Life Technologies) supplemented with 10% horse serum, 2 mM L-glutamine and 1 mM sodium pyruvate. All transfections were performed in triplicate using the Effectene transfection reagent kit (Qiagen), and each experiment was repeated at least three times. HEK293 cells were seeded at a density of 1 × 106 on 60 mm poly-D-lysine-coated plates (Becton Dickinson) and grown to 50–70% confluency. Cells were transfected with 0.5 μg WT or mutant β-catenin DNA, 0.5 μg of TCF-4 construct, and 0.5 μg of TOPflash positive control reporter or the negative control FOPflash. In addition, 0.1 μg of pSV-β-galactosidase vector (Promega) was included as an internal control, and an empty vector was used to give a total of 2 μg DNA in each assay. In some experiments, LiCl was used as an inhibitor of GSK-3β as reported before (Espinosa et al., 2003; Van Gassen et al., 2000). In addition, 6–20 h prior to harvest, ALLN was added as an inhibitor of proteosome-mediated proteolysis; the final concentration was in the range 5–100 μM, as indicated. At the harvest times shown, cells were washed with PBS, lysed for 15 min with Reporter Lysis Buffer (RLB, Promega) and the lysates were cleared by centrifugation at 14 000 r.p.m for 5 min. Luciferase and β-galactosidase activities were determined, respectively, using the Bright–Glo Luciferase (Promega) and Galacto-Star (Tropix) assays in an Orion Microplate Luminometer (Berthold), as described before (Dashwood et al., 2002). Luciferase activity was normalized to β-galactosidase activity and the results were expressed as mean ± s.d. For the data reported here, none of the treatments affected transfection efficiency, based on the results using β-galactosidase as control (data not presented).
SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and immunodetection
Conditions used for SDS–PAGE were described in detail elsewhere (Blum et al., 2001, 2003; Dashwood et al., 2002). In brief, total cell lysates (15–20 μg protein) were subjected to SDS-PAGE using 4–12% Bis-Tris Gels (Novex) and transferred to nitrocellulose membrane (Invitrogen). After staining with Coomassie Brilliant Blue or Amido Black to confirm equal protein loading and transfer, the membrane was blocked with 2% BSA in PBS for 1 h and incubated overnight at 4° C with the indicated primary antibody, followed by secondary antibody conjugated with horseradish peroxidase (Bio-Rad). Detection was by Western Lightning Chemiluminescence Reagent Plus (PE Life Sciences) coupled with image analysis and quantification on an Alphalnnotech photodocumentation system. Four different antibodies were used, which recognize different phosphorylation states of β-catenin: (i) anti-β-catenin monoclonal antibody (Transduction labs), which recognizes total β-catenin regardless of phosphorylation status; (ii) anti-pβ-catenin-41/45 (Cell Signaling Technology), a monoclonal antibody specific for either pThr41, pSer45, or both; (iii) anti-pβ-catenin 33/37/41 (Cell Signaling Technology), a monoclonal antibody that recognizes β-catenin when it is phosphorylated at one of three sites, namely Ser33, Ser37 and/or Thr41; and (iv) α-ABC (α-active β-catenin), a monoclonal antibody that recognizes β-catenin when it is nonphosphorylated at both Ser37 and Thr41 (kindly provided by Dr Hans Clevers). The optimal dilution of each antibody was 1:500, 1:1000, 1:1000 and 1:100, respectively. Immunoblotting with other antibodies was as follows: 1:2000 dilution of monoclonal antiubiquitin (BD Pharmingen), 1:1000 dilution of anti-Myc tag monoclonal antibody (Cell Signaling Technology), 1:1000 dilution of antipoly(ADP-ribose) polymerase (PARP) (BD Biosciences) and 1:5000 dilution of anti-β-actin (Sigma).
Purification of GST-tagged proteins and in vitro kinase assays
Escherichia coli strain BL21 was transformed with procaryotic expression plasmids (see above), and 1 μM isopropyl-β-D-thiogalactopyranoside was added to cultures to induce the expression of GST-tagged proteins. Glutathione sepharose ™ 4B beads (Amersham Pharmacia Biotech) were used to isolate GST-tagged proteins, which were quantified using the Bio-Rad Protein Assay (Bio-Rad) and analyzed by SDS-PAGE. The in vitro kinase assay was performed with 400 ng GST-rAxin, 400 ng GST-GSK-3β and 200 ng GST-β-catenin in a total volume of 30 μl, and the reaction was started by adding 3 μl 10X kinase buffer (200 mM Tris-HCl pH 7.5, 100 μM MgCl2, 50 mM DTT and 200 μM ATP). Following incubation at 30° C for 4 h, the reaction was stopped by adding 7.5 μl SDS sample buffer and heating at 75° C for 10 min. Reaction products were analysed by SDS-PAGE with immunodetection using anti-pβ-catenin-33/37/41 antibody.
Immunoprecipitation assays
Cell lysates were incubated for 5 min with 500 μl ice-cold lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml Leupeptin, 20 mM N-ethylmaleimide (Sigma) and protease inhibitor cocktail (Roche Molecular Biochemicals)). Lysate was cleared by centrifugation at 14 000 r.p.m. for 10 min at 4° C, and 400 μg of total protein were used for immunoprecipitation. Lysates were pre-cleared with 2 μl rabbit anti-mouse IgG (Sigma) and 100 μl 5% protein Sepharose CL-4B slurry (Amersham Pharmacea Biotech) for 1 h at 4°C with rotation. Lysates were spun at 13 000 r.p.m. for 5 min. Lysates were incubated at 4° C with 10 μl monoclonal anti-β-catenin for 18 h or 10 μl anti-Myc tag monoclonal antibody (Cell Signaling Technology) for 6 h, followed by 2 μl rabbit anti-mouse IgG for 1 h. Protein A Sepharose CL-4B was added to the immune complex and incubated for 1 h at 4° C with gentle agitation. The Protein A Sepharose CL-4B was recovered by centrifugation at 10 000 r.p.m. and washed 5 times with ice-cold lysis buffer. Proteins were eluted with 30 μl of 3 × SDS buffer and subjected to immunoblot analysis with anti-Myc tag or anti-ubiquitin antibody.
Acknowledgments
Expression constructs for WT β-catenin, TCF-4, TOPflash and FOPflash as well as α-ABC antibody were kindly provided by Dr Marc van de Wetering and Dr Hans Clevers, University Hospital Utrecht, The Netherlands. Dr Akira Kikuchi of Hiroshima University School of Medicine generously supplied a construct for rAxin. We thank Dr Robert Tanguay for use of the gel scanner and associated software. We are grateful for services provided by the Center for Gene Research and Biotechnology, Oregon State University. This work was supported in part by NIH Grants CA65525, CA80176 and CA90890.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum CA, Xu M, Orner GA, Fong AT, Bailey GS, Stoner GD, Horio DT, Dashwood RH. Carcinogenesis. 2001;22:315–320. doi: 10.1093/carcin/22.2.315. [DOI] [PubMed] [Google Scholar]
- Blum CA, Tanaka T, Zhong X, Li Q, Dashwood W-M, Pereira C, Xu M, Dashwood RH. Mol Carcinog. 2003;36:195–203. doi: 10.1002/mc.10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, Groden J, Lowy AM. Cancer Res. 2002;62:3503–3506. [PubMed] [Google Scholar]
- Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. Cancer Res. 1998;58:1127–1129. [PubMed] [Google Scholar]
- Dashwood W-M, Orner GA, Dashwood RH. Biochem Biophys Res Commun. 2002;296:584–588. doi: 10.1016/s0006-291x(02)00914-2. [DOI] [PubMed] [Google Scholar]
- Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. J Biol Chem. 2003;278:32227–32235. doi: 10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Nature Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Hagen T, Vidal-Puig A. Biochem Biophys Res Commun. 2002;294:324–328. doi: 10.1016/S0006-291X(02)00485-0. [DOI] [PubMed] [Google Scholar]
- Hajra KM, Fearon ER. Genes Chromosomes Cancer. 2002;34:255–268. doi: 10.1002/gcc.10083. [DOI] [PubMed] [Google Scholar]
- Hirohashi S. Am J Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J, Behrens J. J Cell Sci. 2002;115:3977–3978. doi: 10.1242/jcs.00089. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. The EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Proc Natl Acad Sci USA. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Li Q, Dixon BM, Al-Fageeh M, Blum CA, Dashwood RH. Gene. 2002;283:255–262. doi: 10.1016/s0378-1119(01)00839-3. [DOI] [PubMed] [Google Scholar]
- Li Q, Dashwood W-M, Zhong X, Al-Fageeh, Dashwood RH. Genomics. 2004;83:231–242. doi: 10.1016/j.ygeno.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. Proc Natl Acad Sci USA. 1999;96:6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N. Mol Cell. 2001;7:927–936. doi: 10.1016/s1097-2765(01)00241-6. [DOI] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa S, Reed JC. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Nusse R. 2003 http://www.stanford.edu/~rnusse/arm/bcatmut.html.
- Oving IM, Clevers HC. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- Polakis P. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Porfiri E, Rubinfeld B, Albert I, Hovanes K, Waterman M, Polakis P. Oncogene. 1997;15:2833–2839. doi: 10.1038/sj.onc.1201462. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri P, Polakis P. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- Suzui M, Ushijima T, Dashwood RH, Yoshimi N, Sugimura T, Mori H, Nagao M. Mol Carcinog. 1999;24:232–237. doi: 10.1002/(sici)1098-2744(199903)24:3<232::aid-mc10>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Fukuda K, Sugimura T, Wakabayashi K. Cancer Res. 1998;58:42–46. [PubMed] [Google Scholar]
- Van Gassen G, De Jonghe C, Nishimura M, Yu G, Kuhn S, St George-Hyslop P, Van Broeckhoven C. Mol Med. 2000;6:570–580. [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Vogelstein B, Kinzler KW. Cancer Res. 2003;63:5234–5235. [PubMed] [Google Scholar]