

SUMMARY
Deletion of serine 63 from P0 glycoprotein (P0S63del) causes Charcot-Marie-Tooth 1B neuropathy in humans, and P0S63del produces a very similar demyelinating neuropathy in transgenic mice. P0S63del is retained in the endoplasmic reticulum and fails to be incorporated into myelin. Here we report that P0S63del is globally misfolded and Schwann cells mount a consequential canonical unfolded protein response (UPR), that includes expression of the transcription factor CHOP, previously associated with apoptosis in ER-stressed cells. UPR activation and CHOP expression respond dynamically to P0S63del levels and are reversible, but are associated with only limited apoptosis of Schwann cells. Nonetheless, Chop ablation in S63del mice completely rescues their motor deficit and reduces active demyelination two-fold. This is the first indication that signaling through the CHOP arm of the UPR provokes demyelination in inherited neuropathy. In addition, S63del mice provide a unique opportunity to explore how cells can dysfunction yet survive in prolonged ER stress—important for neurodegeneration related to misfolded proteins.
Keywords: Apoptosis, Charcot-Marie-Tooth neuropathy, ER stress, myelin protein zero, P0S63del, Schwann cell, transgene, unfolded protein response
INTRODUCTION
Myelin protein zero (P0) is the major protein produced by myelinating Schwann cells (Kirschner et al., 2004). P0 is a single-pass transmembrane protein, with a large amino-terminal extracellular domain (ECD) and a short carboxy-terminal intracellular tail. The protein is encoded by the Mpz gene and synthesized in the endoplasmic reticulum (ER), where it undergoes N-glycosylation and formation of one intramolecular disulfide bond. In myelin, P0 is present as tetramers that compact the extracellular appositions of myelin wraps via in trans homophilic interactions (D'Urso et al., 1990; Filbin et al., 1990; Shapiro et al., 1996). Accordingly, mice with homozygous disruption of Mpz show uncompaction of myelin in development (Giese et al., 1992) and with heterozygous disruption, altered myelin maintenance in the adult (Martini et al., 1995).
More than 100 mutations of P0, many in the ECD, are associated with hereditary motor and sensory neuropathies (Shy, 2005; Wrabetz et al., 2004). Although the specific pathogenetic mechanisms have not been fully elucidated, dominant inheritance and widely varying phenotypes suggest that many P0 mutations act through gain of abnormal function (Martini et al., 1995; Wrabetz et al., 2006). For example, deletion of serine 63 (S63del) or its mutation to a cysteine (S63C) result in Charcot-Marie-Tooth (CMT) type 1B and Dèjèrine-Sòttas syndrome (DSS) in humans, respectively (Hayasaka et al., 1993; Kulkens et al., 1993). Transgenic mice expressing both mutant and normal P0 confirm that these mutations produce a gain of abnormal function (Wrabetz et al., 2006). The pathomechanisms are likely to differ between the two mutations, for although S63C is incorporated into myelin, producing packing defects, S63del is retained in the ER.
In the ER, membrane proteins are subject to stringent quality control (Ron and Walter, 2007). Accumulation of misfolded proteins in the ER sequesters chaperones like BiP (binding immunoglobulin protein) and activates the unfolded protein response (UPR). As an adaptive response, UPR upregulates transcription of chaperones, temporarily attenuates new translation, and activates protein degradation via the proteasome. However, at high levels of ER stress, UPR signaling contributes to apoptosis (Ron and Walter, 2007). For example, deletion of the gene encoding CHOP (CAATT enhancer-binding protein homologous protein, a transcription factor active in the UPR) protects cells against apoptosis (Marciniak et al., 2004; Zinszner et al., 1998).
Here we report that P0S63del activates a canonical and dose-dependent UPR, including CHOP, and consequentially, demyelination in Schwann cells. ER retention and UPR depend on alteration of the pattern of hydrophobic residues in P0 β-strand C, not changes at residue 63, a mechanism potentially shared by another CMT1B mutation, deletion of phenylalanine 64 (Ikegami et al., 1996). Ablation of Chop in S63del mice reverses behavioural, electrophysiological and morphological abnormalities, indicating the UPR as a novel pathogenetic mechanism in demyelinating peripheral neuropathies. Finally, UPR activation is rapidly reversed by reducing mutant protein levels in mutant nerves, suggesting its dosage as a logical therapeutic target. Given that Schwann cell death follows, not precedes demyelination, we propose that the maladaptive UPR and CHOP in S63del nerves produces a novel cellular dysfunction in Schwann cells.
RESULTS
Protein quality control in S63del nerves
P0S63del causes a demyelinating neuropathy via gain of abnormal function, but does not arrive to the myelin sheath (Wrabetz et al., 2006). To characterize potential toxic mechanisms operative outside of myelin, we surveyed protein quality control in S63del nerves. We considered autophagy (lysosomes), proteasomes, and UPR in S63del nerves. LAMP1 staining was slightly expanded, but did not reveal the lysosomal aggregates that have been reported for PMP22 mutants, where aggresomes are cleared by autophagy (Supp. Figure 1; Fortun et al., 2003). Electron microscopy did not reveal aggregates or swollen ER (Tsang et al., 2007) in S63del Schwann cells (Supp. Figure 2A). Also, EDEM1, a marker of endoplasmic reticulum activated degradation (ERAD), was expressed at normal levels preliminarily suggesting limited ERAD and proteasomal involvement (Supp. Figure 2B).
When expressed in mice of the Mpz-null background, P0S63del is retained in the endoplasmic reticulum (Wrabetz et al., 2006). However, CMT1B patients have both MPZ WT and S63del alleles. Even when P0S63del was co-expressed with P0wt (Figure 5C), immunostaining on teased fibers showed that P0 was still retained in the ER (Figure 1A). These data, taken together with preliminary evidence that the transcription of BiP and CHOP is upregulated in S63del nerves (Wrabetz et al., 2006), suggest that the UPR is an important form of quality control for P0S63del.
Figure 5. Diminished P0S63del levels acutely reverse activation of the UPR.
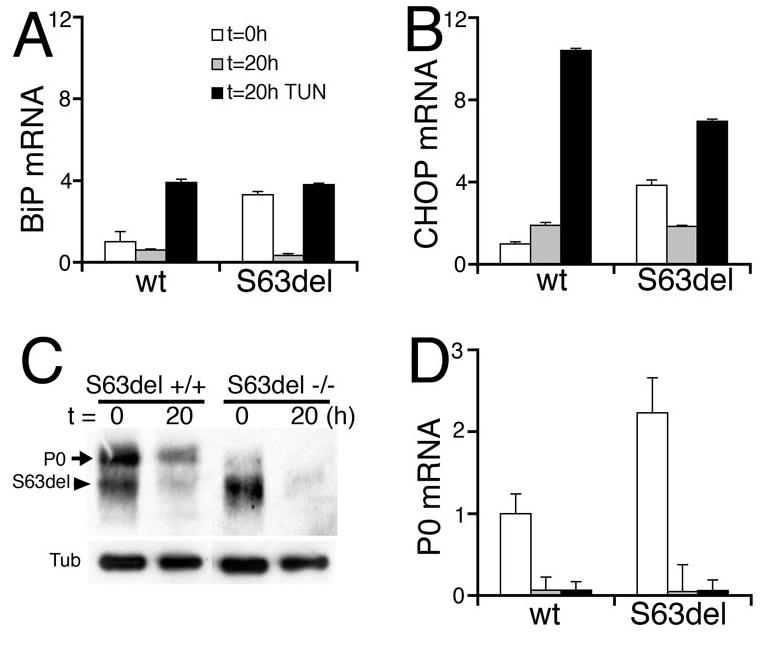
Segments of wt or S63del-L sciatic nerves were snap-frozen either immediately after dissection (t = 0h, white bars) or after incubation ex vivo for 20 hours with (t = 20h TUN, black bars) or without (t = 20h, gray bars) tunicamycin, and analyzed for UPR markers. Levels of mRNA were measured by quantitative RT-PCR (A-B,D) and normalized to PGK1 mRNA. BiP and CHOP mRNAs are induced in S63del mice at t = 0h and remain induced with tunicamycin treatment for twenty hours after axotomy. Instead induction is rapidly reversed without tunicamycin, in parallel with marked reduction of the band containing P0S63del by Western analysis (C, arrowhead), due to the general reduction of P0 mRNA after axotomy (D). (S63del −/−) represents P0 −/− mice transgenic for MpzS63del. β-Tubulin (Tub) is a control for protein loading. Error bars, s.e.m., n = 3 animals.
Figure 1. P0S63del is retained in the ER and induces a dose-dependent UPR.
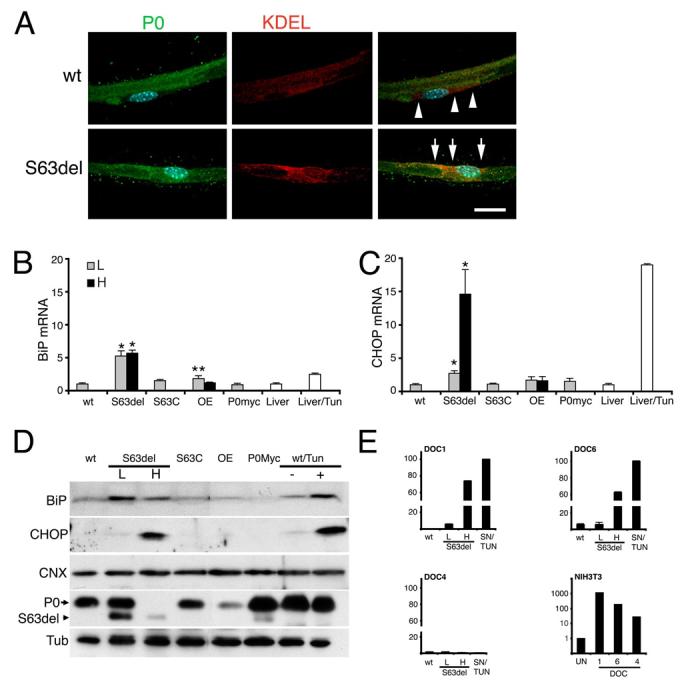
A) Immunofluorescence analysis for P0 or KDEL on teased fibers shows that P0 staining is augmented in the ER of myelinating Schwann cells from P0S63del (arrows), as compared to wt (arrowheads), sciatic nerves. B-C) BiP and CHOP mRNA levels were measured in P28 sciatic nerves by quantitative RT-PCR and normalized to 18S rRNA, wt = 1. L, low, H, high expressor S63del transgenes; OE, P0 wildtype overexpressor; tun, tunicamycin; error bars, s.e.m. for n = individual nerves from 3-5 animals. * p < 0.001, ** p < 0.05 relative to wt by Student's t-test. D) Western analysis for BiP and CHOP was performed on freshly dissected nerves or wt nerve segments cultured overnight with tunicmycin or sham media. Note that CHOP, but not BiP, mRNA and protein levels parallel the ratio of P0S63del (arrowhead) to P0wt (arrow), and the severity of neuropathy (Wrabetz et al., 2006) in S63del mice. Calnexin (CNX) levels do not change. BiP and CHOP are induced upon tunicamycin treatment of wt nerve (wt/Tun +). β-Tubulin (Tub) provides a control for loading. E) DOC1, 6 and 4 mRNA levels are induced by tunicamycin treatment of NIH3T3 cells (note the logarithmic scale), but only DOC1 and 6 are induced in S63del nerves. DOC4 is not induced even by tunicamycin treatment of nerve.
Therefore, we asked whether P0S63del elicited a full UPR, and how the UPR correlated with disease. In mouse, disease severity correlates with MpzS63del mRNA expression (Wrabetz et al., 2006). To examine dose-dependence, we exploited mice expressing S63del mRNA at levels of one (S63del-L) or two (S63del-H) endogenous alleles. BiP and CHOP mRNA levels were increased by 5.2 and 2.7-fold in S63del-L and 5.7 and 14.6-fold in S63del-H sciatic nerves, respectively (Figures 1B and C). Induction of BiP and CHOP mRNA was tissue- and mutation-specific. It was not detected in the brain (data not shown); or in nerves of S63C, P0Myc (pathological model of CMT1B; Previtali et al., 2000) or P0 overexpressor mice (Wrabetz et al., 2000; Yin et al., 2000; Figure 1B). The level of induction was robust, comparable to the levels of BiP and CHOP mRNA acutely induced by tunicamycin (glycosylation inhibitor) in liver (2.5- and 19-fold, Figures 1B and C) or cultured sciatic nerve segments (4- and 10-fold, Figures 5A and B) of normal mice.
Induction of BiP and CHOP in S63del and tunicamycin-treated nerves was confirmed by Western analysis (Figure 1D). As for mRNA levels, CHOP protein induction was higher in S63del-H mice than in S63del-L mice, in parallel with the ratio of mutant P0S63del to P0wt. CHOP activates transcription of the Downstream Of CHOP genes (DOCs) (Wang et al., 1998). Accordingly, DOCs 1 and 6, although not 4, were induced in S63del nerve. Tunicamycin treatment activated DOC4 in NIH3T3 cells, but not in sciatic nerve, raising the possibility that Schwann cells respond differently to CHOP (Figure 1E). Altogether, these data show that BiP, CHOP, and its target genes, are upregulated in S63del nerves to levels comparable to those found when UPR is acutely induced by tunicamycin. Of these, the level of CHOP correlated best with mutant protein level and disease severity.
IRE-1, PERK, and ATF6 pathways are activated in S63del nerves
To characterize the UPR in S63del nerves more fully, we analyzed activation of the three arms of the UPR: IRE-1 and PERK kinases and cleavage of ATF6 transcription factor (Ron and Walter, 2007). Activated IRE-1 non-canonically splices the mRNA of the transcription factor X-box-binding protein 1 (XBP-1), an event specific to UPR (Yoshida et al., 2001). The spliced form of XBP-1 was increased by 2.5, 2.7 and 5-fold in S63del-L, S63del-H and tunicamycin-treated nerves, respectively (Figure 2A). Activated PERK phosphorylates eIF2α, and upregulates translation of the transcription factor ATF4, and transcription of ATF3, CHOP and the oxidoreductin ERO1-Lβ (Jiang et al., 2004; Pagani et al., 2000; Ron and Walter, 2007). Indeed, the levels of eIF2α phosphorylation increased 1.8-fold, and ATF4 3-fold in S63del-L nerves (Figure 2B). Similarly, ATF3 mRNA was induced 2.5-fold in S63del-L, 12.4-fold in S63del-H nerves and 6.1-fold in tunicamycin-treated liver (Figure 2C). Likewise, ERO1-Lβ was induced in S63del-H nerves and tunicamycin-treated liver to a similar extent (Supp. Figure 2C and data not shown). The UPR was activated in Schwann cells, as the transcription factor CHOP, which was undetectable in normal nerves, was localized to the nucleus of S63del Schwann cells (Figure 2D). Finally, dose-dependent ATF6 cleavage was detected in S63del–L and –H nerves (Figure 2E). Thus, P0S63del provokes a dose-dependent activation of IRE-1, PERK, and ATF6 pathways in Schwann cells of S63del nerves, indicating a canonical UPR.
Figure 2. Activation of IRE-1, PERK and ATF6 arms of the UPR in S63del nerves.
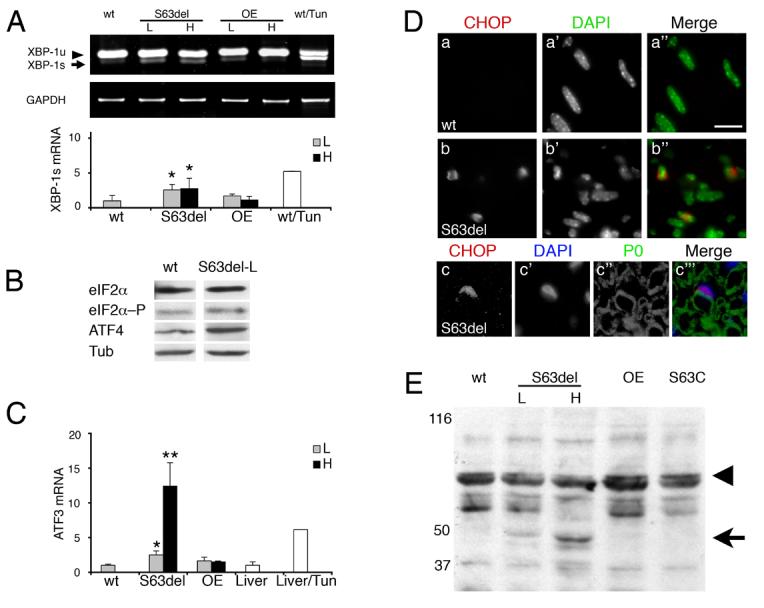
A) IRE-1 catalyzes excision of a 26 nt fragment from XBP-1 mRNA. Semi-quantitative RT-PCR for XBP-1 unspliced (XBP-1u), spliced (XBP-1s), normalized to GAPDH, shows that the spliced XBP-1 is enriched in S63del and tunicamycin-treated nerves. P0wt overexpressor (OE) nerves are included as expression-matched controls for S63del low (L) and high (H). Error bars, s.e.m. for n = 6 individual nerves, *p < 0.05 relative to wt by Student's t-test. B) Levels of eIF2α, eIF2α–P (phosphorylated), and ATF4, were measured by Western analysis. Tubulin (Tub) shows equal loading. C) ATF3 mRNA was measured by quantitative RT-PCR and normalized to phosphoglycerate kinase 1(PGK1) mRNA. ATF3 is induced in a dose-dependent fashion in S63del mice and in tunicamycin-treated livers. Error bars, s.e.m., n = 6, 5, 5, 3, and 3 nerves for wt, S63del-L, S63del-H, OE-L, OE-H, respectively and 3 livers. ** p < 0.01, * p < 0.05 relative to wt by Student's t-test. D) Immunostaining reveals CHOP in elongated, DAPI positive, Schwann cell nuclei in longitudinal sections of S63del nerves at P28 (b–b"), but is undetectable in normal nerves (a–a") Many CHOP-positive Schwann cells contain integral myelin sheaths (c–c"'). Bar = 15 μm in a–b"; 13 μm in c–c"'. E) Western analysis for ATF6 reveals dose-dependent cleavage of the 90kD precursor protein (arrowhead) to the 50kD product (arrow) only in S63del, but not S63C, P0 overexpressor (OE) or wt nerves. Numbers indicate relative molecular weights (Mr).
Alteration of β-strand C periodicity in P0 causes ER retention
In S63del nerves, ER retention of P0S63del and UPR activation suggested protein misfolding. Thus, we performed Western analysis for the intramolecular disulfide bond of P0 (Figure 3A). The mobility of P0wt was increased by 3-4 kD under non-reducing conditions, consistent with the presence of a disulfide bond. Non-reduced P0S63del showed a similar shift (arrowhead), but in addition produced a smear, that could represent alternatively folded intermediates. In contrast, another neuropathic mutation at the same residue, P0S63C, that is not retained in the ER, migrated similarly to P0wt.
Figure 3. Alteration of P0 β-strand C polarity causes ER retention and activation of the UPR.
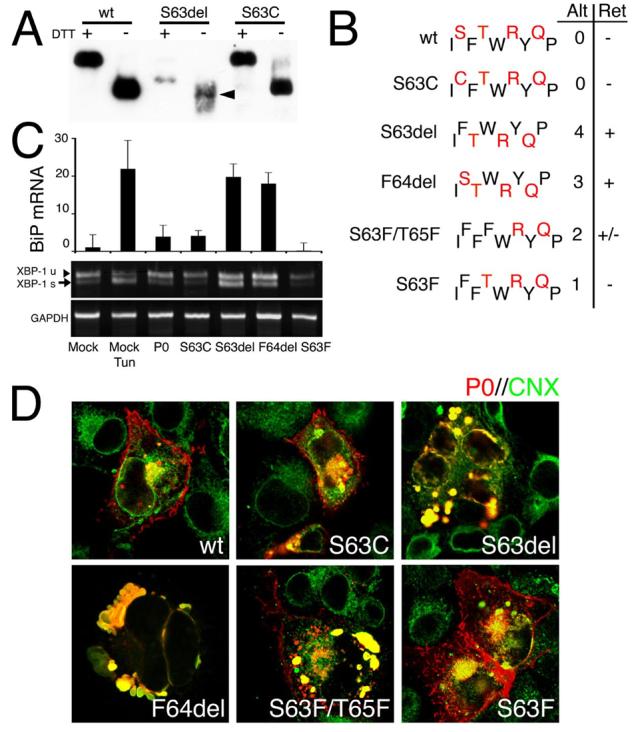
A) Western analysis was performed for P0 under reducing (DTT +) or non-reducing conditions (DTT −) on peripheral nerve lysates from wt, S63del/P0−/− (S63del) or S63C/P0−/− (S63C) mice. Therefore, in mutant mice, only mutant P0 is present. Under non-reducing conditions, wt and mutant P0 show a 3-4 kD shift as expected for the formation of one disulfide bond. Note that non-reduced S63del produces a smear (arrowhead), that might represent protein misfolding. The majority of P0S63del recognized in this analysis is unglycosylated (Wrabetz et al., 2006) and migrates at a lower Mr. In contrast, S63C produces discrete bands with the same mobility as P0wt. B) P0 β-strand C is characterized by an alternating hydrophilic (black) and hydrophobic (red) pattern of residues. In contrast to S63C, deletion of either S63 or F64, or the mutant S63F/T65F and S63F are predicted to alter the orientation of 4, 3, 2 and 1 hydrophobic residues, respectively. The correlation between number of altered (alt) hydrophobic residues and ER retention (Ret +) in transfected cells is shown. COS-7 cells were transfected with constructs encoding various P0wt– or P0mutant–DsRed fusion proteins and analyzed for expression of UPR markers (C) or for trafficking of P0 (D). BiP mRNA was measured by quantitative RT-PCR and normalized to 18S rRNA for transfection efficiency. XBP-1 and GAPDH were measured by semiquantitative RT-PCR. Note that BiP mRNA levels and XBP-1 splicing (XBP-1s, arrow) are enriched in cells expressing either P0S63del or P0F64del mutants, as well as in cells treated with tunicamycin (a representative experiment (of 3) in shown). D) Confocal microscopy of trafficking of P0-DsRed fusion proteins (P0, red) and immunostaining for calnexin (CNX, green) is shown in overlay. P0wt, P0S63C, and P0S63F are delivered to the cell membrane, whereas P0S63del and P0F64del are completely retained in the ER (yellow signal). P0S63F/T65F was mostly retained in the ER, although some was delivered to the plasma membrane in some cells. Bar = 20 μm.
Serine 63 lies in β-strand C, characterized by an alternating hydrophobic and hydrophilic pattern of residues (Figure 3B; Shapiro et al., 1996). We hypothesized that deletion of serine 63 altered the disposition of the 4 hydrophobic residues (FWYP) in β-strand C (Figure 3B, compare wt and S63del). Therefore, we compared the effect of deleting serine 63 to either deletion of phenylalanine 64 (F64del), conversion of both serine 63 and threonine 65 into phenylalanines (S63F/T65F), substitution of serine 63 with phenylalanine (S63F), or substitution of serine 63 with cysteine (S63C), which alter the disposition of 3, 2, 1 or 0 hydrophobic residues, respectively. P0 was fused at its carboxy-terminus in frame with the discosoma red (DsRed) fluorescent protein. Modification of P0 at the carboxy-terminus does not impair targeting of P0 to the plasma membrane in cells (Shames et al., 2003) or to myelin in transgenic sciatic nerve (Pennuto, Fratta, and Wrabetz, unpublished). The chimeras were transiently expressed in COS-7 (Figure 3D) or Chinese hamster ovary (CHO) cells (data not shown) and the cells were immunostained for the ER resident lectin, calnexin (CNX) (Figure 3D). Both P0wt and P0S63C were targeted to the plasma membrane, whereas P0S63del was confined to the ER, consistent with prior analysis of these mutant P0s in transgenic mice (Wrabetz et al., 2006). In addition, transfected P0MycDsRed was trafficked to the cell membrane (data not shown), just as P0Myc arrives to myelin in transgenic sciatic nerve (Previtali et al., 2000), further validating the transfected cells as a model. S63F was trafficked to the cell membrane, whereas F64del and S63F/T65F were completely retained in the ER. Deletion of glutamic acid 71 in the loop connecting β-strands C and C' did not impair protein trafficking (data not shown). S63del and F64del also induced BiP transcription and XBP-1 splicing in COS-7 cells to levels similar to those obtained upon tunicamycin-treatment (Figure 3C). These data suggest that it is not alteration of the side chain at residue 63, but rather a perturbed alignment of hydrophobic residues in β-strand C that results in ER retention and activation of the UPR, providing further support for the notion that P0S63del is globally misfolded.
UPR is dynamic and reversible in sciatic nerve
S63del mice are characterized by hypomyelination, demyelinated fibers and onion bulbs (Wrabetz et al., 2006). A few nascent onion bulbs are detectable at post-natal day 28 (P28) and become evident by 6 months of age. To determine whether the UPR activation preceded or followed the appearance of pathological findings in diseased nerves, we analyzed P0, BiP and CHOP expression across development. In wt and S63del-L mice, BiP and CHOP mRNAs were immediately upregulated during development, mirroring the robust induction of P0 expression which occurs immediately after birth and peaks around P28 (Figure 4), although CHOP rapidly leveled off and was maintained at a plateau suggesting more complicated regulation. Similar results were obtained for S63del-H mice (data not shown). Thus, the robust UPR as measured by peak BiP and CHOP levels is associated with the onset of myelin instability, destruction and failed remyelination marked by onion bulbs.
Figure 4. Induction of the UPR dynamically parallels P0 expression.
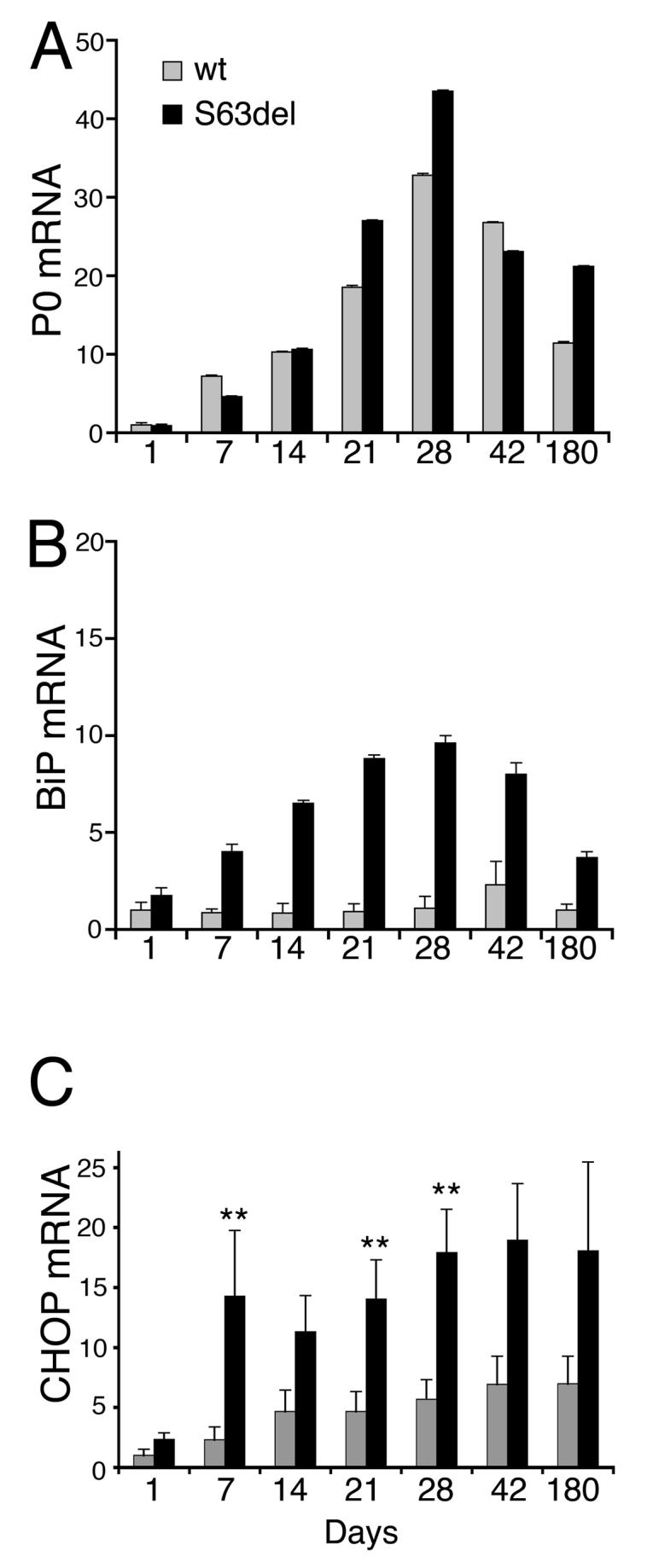
P0 (A), BiP (B) or CHOP (C) mRNA levels in sciatic nerves were measured by quantitative RT-PCR and normalized to 18S rRNA in wt (gray bars) or S63del (black bars) mice. The level of P0 rises in both wt and S63del nerves at P7, with a peak near P28. Only in S63del, but not in wt nerves, BiP rises in parallel, and CHOP rises by P7 to a plateau level that then persists. For P0 and BiP, one representative experiment (animal) of three is shown; error bars, s.e.m.; n = 3. For CHOP the average of 7 experiments (animals) is shown (anchored to P28wt and then expressed relative to P1wt = 1; error bars, s.e.m.; n = 7; ** p < 0.05 relative to wt of the same age by Student's t-test.
The relative amounts of BiP and CHOP differed between S63del-L and S63del-H mice (Figure 1). Given that BiP chaperone helps cells to fold aberrant proteins (Gething, 1999), whereas CHOP may kill overwhelmed cells (Marciniak et al., 2004; Zinszner et al., 1998), the increased level of CHOP in the face of stable amounts of BiP could represent a transition from a cell survival response in S63del-L to an apoptotic reponse in S63del-H. One prediction of this hypothesis would be that in S63del-L nerves the level of BiP is near maximal and can not augmented. Therefore, segments of sciatic nerves were explanted and maintained in culture for 20 hours with or without tunicamycin. At time zero, BiP and CHOP were increased by 3.3 and 3.8-fold in S63del as compared to wt nerves (Figures 5A and B). Surprisingly, 20 hours after axotomy, BiP and CHOP expression decreased to levels similar to wt mice, although parallel treatment with tunicamycin induced BiP/CHOP to 3.9/10.4 and 3.8/6.9-fold in wt and mutant mice, respectively. These data indicate that after axotomy, UPR is almost completely reversed in mutant nerves, and reinduced to similar levels by tunicamycin. Unfortunately, additive effects can not be addressed after axotomy.
One possible explanation was that axotomy of the cultured nerve segments produced acutely decreased mutant P0 (maintainence of Mpz expression depends on axonal signals; Trapp et al., 1988). In fact, 20 hours after axotomy, the level of P0wt was partially reduced (Figure 5C, arrow), whereas P0S63del was almost undetectable, also suggesting that P0S63del is less stable (Figure 5C, arrowhead). Similar results were obtained in sciatic nerve segments from S63del mice deleted of endogenous P0, where only mutant P0 is present (S63del−/−). As expected, this effect was transcriptionally-mediated, as the level of P0 mRNA was very low at 20 hours after axotomy (Figure 5D), and had fallen significantly already by 6 hours after axon cut (data not shown). Thus, the UPR in S63del nerves is continuously activated by sustained expression of mutant protein. Transcriptional induction of BiP and CHOP can be halted by simply discontinuing mutant transgene expression, indicating that the UPR is reversible in vivo.
Ablation of Chop amelioriates neuropathy
Of the UPR-mediators, the level of CHOP expression correlated best with disease severity. To demonstrate a direct role for the CHOP-dependent arm of the UPR in the pathogenesis of demyelination in S63del nerves, we crossed S63del-L and Chop-null mice (Zinszner et al., 1998) and analysed behaviour, neurophysiology and morphology in wt, S63del, Chop-null and S63del/Chop-null littermates. Chop-null mice show no overt neuromuscular defects (Zinszner et al., 1998; see below). Four month-old S63del mice had lost one third to one half of their motor capacity as compared to wt or Chop-null littermates in analysis by accelerating Rotarod (Figure 6A; Wrabetz et al., 2006). Strikingly, S63del/Chop-null mice regained completely normal motor capacity in Rotarod analysis (Figure 6A). This was accompanied by a partial rescue of electrophysiological and morphological abnormalities. In particular, S63del mice manifest reduced nerve conduction velocity (NCV) and increased F-wave latency (Wrabetz et al., 2006). In 6 month old S63del/Chop-null mice, the F-wave latency was significantly improved, even if the NCV did not change. In addition, there were half the number of demyelinated fibers and a trend toward reduction of onion bulbs as compared to S63del mice (Figure 6B and Table). Consistent with unchanged NCV, hypomyelination remained (Figure 6B), as we have documented previously in other transgenic mice that overexpress total P0 (Wrabetz et al., 2000). Finally, morphometric analysis showed no change in the number or size distribution of sciatic nerve axons (Supp. Figure 3). Thus, ablation of Chop rescued demyelination associated with toxic gain of function from P0S63del.
Figure 6. Ablation of Chop restores motor function and reduces demyelination in S63del mice.

A) Rotarod analysis of motor function showed that four month old S63del mice remain for 200 seconds less as compared to wt and Chop-null mice. Instead, S63del/Chop-null mice perform like wt mice. B) Semithin sections stained with toluidine blue are shown from P180 wt, S63del, Chop-null and S63del/Chop-null sciatic nerves. S63del nerves manifest hypomyelination and demyelinated fibers and onion bulbs. Although hypomyelination is not ameliorated in S63del/Chop-null nerves, the number of demyelinating fibers is significantly reduced (Table). Examples of pathological fibers from S63del or S63del/Chop-null nerves are shown magnified below. Arrows indicate demyelinated fibers, stars indicate onion bulbs; and arrowheads indicates remyelinating fibers. Bar = 15 μm in upper panels and 5 μm in the magnified panels below. C) S63del transgene expression was analyzed in rescued nerves by semi-quantitative RT-PCR. Digestion of the amplimer with Dpn II distinguishes wt (326 bp) from transgenic cDNA (244 and 82 bp fragments). D-H) BiP, CHOP and DOC1,6 and 4 expression was analyzed by quantitative RT-PCR and normalized to 18S rRNA. Note that transgene expression and BiP activation remain in S63del/Chop-null nerves, whereas CHOP is undetectable and DOC1 falls below wt levels. DOC6 and 4 are not induced in S63del-L nerves.
Table.
Ablation of Chop reduces demyelination in S63del sciatic nerves.
| Electrophysiology | Morphology | ||||
|---|---|---|---|---|---|
| Genotype | NCV (m/s) | F-wave latency (ms) | G-ratio | % onion bulbs | % dem fibers |
| wt | 39±1.1(15) | 4.8±0.22(15) | 0.67±0.028(6) | 0 | 0 |
| chop−/− | 38±1.2(12) | 4.8±0.13(12) | 0.70±0.010(9) | 0 | 0 |
| S63del | 32±1.7(7) | 6.1±0.45(7) | 0.76±0.015(15) | 0.45±0.110(18) | 0.82±0.095(18) |
| S63del/chop−/− | 31±0.9(10)ns | 5.2±0.15(10)* | 0.74±0.021(11)ns | 0.35±0.103(22)ns | 0.45±0.103(22)** |
All values represent mean ± SEM(n); where n = number of animals, except for G-ratio where n = total microscopic fields from 3-5 animals;
= not significantly different
p < 0.05
p < 0.01 relative to S63del by Student's t-test.
NCV, nerve conduction velocity; G-ratio is calculated as axonal diameter/fiber diameter; % dem fibers, % demyelinated fibers.
Rescue was not due to reduced expression of the MpzS63del transgene or reduced activation of the UPR. By RT-PCR analysis, transgenic and endogenous Mpz mRNAs can be distinguished exploiting a polymorphic restriction site (Wrabetz et al., 2000). The amount of MpzS63del mRNA was about 50% of the endogenous Mpz in both S63del and S63del/Chop-null mice at P28 and P180 (Figure 6C; Wrabetz et al., 2006). Consistent with the idea that P0S63del levels determine UPR activation, BiP mRNA levels were similar in S63del and S63del/Chop-null nerves at P28 (Figure 6D), and remained induced 4 to 6-fold in both at P42 and P120 (data not shown). Instead, Chop was undetectable as expected in S63del/Chop-null nerve (Figure 6E). Moreover, DOC1, which is induced in S63del-L nerve was reduced to basal levels in S63del/Chop-null nerve (Figure 6F-H). Therefore, deletion of Chop from S63del mice restored behavioural, electrophysiological and morphological abnormalities despite continued UPR, indicating that the CHOP arm of the UPR is pathogenetic and suggesting that the expression of Chop is a maladaptive response to P0S63del misfolding in myelinating Schwann cells.
Chop activation is associated with delayed apoptosis in S63del nerves
Prolonged ER stress and induced Chop expression can provoke apoptosis (Oyadomari et al., 2002; Rutkowski et al., 2006; Zinszner et al., 1998). In fact, in S63del-L the number of TUNEL positive nuclei was 0.4% at P28 and 0.9% at P180, whereas wt mice and Chop-null mice showed very low levels of apoptosis. S63C, P0-OE mice (dysmyelinating, not demyelinating neuropathies) also showed no increase in apoptosis at P180 (Figure 7A). Ablation of Chop in S63del mice decreased the number of TUNEL positive nuclei, indicating that CHOP causes apoptosis in S63del nerves.
Figure 7. Chop and apoptosis in S63del nerves.

A) TUNEL analysis in longitudinal sections of wt and mutant sciatic nerves at P28 (grey bars), P42 (white bars) and P180 (black bars) reveals that cell death is significantly increased in S63del nerves as compared to wt or other mutant nerves. TUNEL-positive nuclei were usually elongated suggesting they belonged to Schwann cells (data not shown). Ablation of Chop reduces cell death to wt levels in S63del/Chop-null nerves. Note that cell death in S63del nerves at P180 is nearly double that of P28, whereas BiP and Chop expression peak at or before P28 (Figure 4). Error bars, s.e.m., for P28 and P180, n = 27, 5, 14, 5, 6, or 6 animals in wt, Chop-null, S63del, S63del/Chop-null, S63C, or OE (P0 wildtype overexpressor), respectively; for P42 wt and S63del n=3. *p < 0.001 by Student's t-test relative to wt or S63del/Chop-null. B) Cell number per 40x field was estimated by counting DAPI-stained nuclei in longitudinal sections of sciatic nerves. Chop ablation does not alter the nuclei/field in S63del nerves at either P28 or P180. p < *0.002 or &0.05 by Student's t-test relative to wt for n= 9-11 sections (2-3 sections from each of 3-5 animals). C) Cell proliferation was estimated by BrdU incorporation in P28 (grey bars) and P180 (black bars) sciatic nerves from wt and mutant mice. Cell proliferation is increased in S63del mice at P28 and P180 and in S63del/Chop-null mice at P28. Error bars, s.e.m., n = 5, 3, 4, 3, wt, Chop-null, S63del, S63del/Chop-null, respectively. D) Proteolytic cleavage of caspase-12 was analyzed by Western analysis of sciatic nerve lysates from P42 wt, S63del, S63C and P0 OE or P28 wt, Chop-null, S63del or S63del/Chop-null animals. The amount of the 42 kD activated form of caspase-12 (arrowhead) is strongly increased in S63del nerves and remains after Chop ablation. A representative experiment (of 3) is shown. Numbers indicate Mr.
Independently of UPR, demyelination itself also provokes both proliferation and apoptosis in peripheral nerves (Sancho et al., 2001). Therefore, to determine whether apoptosis in S63del nerves was a direct consequence of CHOP signals or secondary to demyelination, we analyzed its timing and whether there was associated proliferation. Even if UPR activation as measured by BiP and Chop expression peaked between P28 and P42, there were only slightly more TUNEL positive nuclei in S63del nerves at P42 as compared to P28, but many more at 6 months old (Figure 7A). This pattern correlates instead very closely to the trend of demyelination (Wrabetz et al., 2006). In addition, in S63del nerves apoptosis was not associated with cell loss, but with an increased number of cells, consistent with supranumerary Schwann cells present in demyelination (Figure 7B). Indeed, cell death was compensated by increased proliferation in S63del nerves at both P28 and P180, typical of demyelination (Figure 7C). We also analyzed for proteolytic cleavage of caspase-12, an event specific to UPR and thought to be associated with cell death (Nakagawa et al., 2000). The 42 kD activated caspase-12 was specifically enriched in S63del nerves (Figure 7D, arrowhead), at P42 when the level of apoptosis was relatively low. Moreover, at P28 ablation of Chop and amelioration of demyelination reduced apoptosis to wt levels in S63del nerves, even if cleaved caspase 12 remained easily detected (Figure 7D). These data provide further evidence that activated caspase 12 is not sufficient to cause cell death in the context of UPR in tissues (Sharma and Gow, 2007). Thus, delayed cell death correlates best with progressive demyelination, suggesting that CHOP induces myelin instability and demyelination, which in turn causes as a secondary consequence, proliferation and cell death.
DISCUSSION
We describe the first hereditary neuropathy associated with UPR. We had previously shown that P0S63del produces demyelinating neuropathy via a dose-dependent, toxic gain of function originating from the ER in transgenic Schwann cells (Wrabetz et al., 2006). Here we show that P0S63del elicits a canonical UPR in Schwann cells. Biochemical and mutation analyses suggest that deletion of S63del alters the conformation of P0 β-strand C, leading to global misfolding and BiP binding to mutant P0. As a consequence, Ire-1, Perk and ATF6 pathways are activated, leading to Chop induction that parallels the severity of neuropathy. Ablation of Chop from S63del mice restores motor function and reduces demyelination, indicating that the CHOP arm of the UPR is pathogenetic in S63del neuropathy. The mechanism, however, is surprising, for whereas cell death is reduced in S63del/Chop-null nerves, the neuropathy phenotype, the timing of cell death and the presence of proliferation indicate that Chop primarily provokes demyelination and only secondarily, cell death. Thus, unlike in oligodendrocytes where Chop is part of the adaptive stress response and ameliorates cell death, in Schwann cells Chop is part of a maladaptive stress response and produces cell-specific dysfunction not primarily related to cell death.
The UPR is specific to S63del expression
The UPR has a physiological role in other cells that must coordinately synthesize high levels of proteins and lipids (Ron and Walter, 2007) and precisely coordinated gene dosage and protein and lipid synthesis is pivotal for normal myelinogenesis (Saher et al., 2005; Suter and Scherer, 2003; Wrabetz et al., 2004). Nevertheless, neither the normal upregulation of myelin protein expression during myelinogenesis, nor the overexpression of normal P0 or other mutant P0s (e.g. P0S63C, P0myc) nor the expression of mutant PMP22Tr-J (Dickson et al., 2002), activated the UPR to significant levels (there was a trend towards increased levels of CHOP mRNA during myelin formation (Figure 4C)). Only P0S63del, retained in the ER, activated UPR significantly.
Loss of β-strand C periodicity causes ER retention and activation of the UPR
We show that ER retention of, and UPR activation by, P0S63del was not due to the lack of S63 itself, but rather to alteration of β-strand C periodicity. S63 is the first residue of β-strand C (Shapiro et al., 1996), characterized by an alterating pattern where the hydrophobic side chains are oriented toward the protein core and the hydrophilic side chains away. Therefore, deletion of serine 63 might alter the orientation of the 4 hydrophobic residues of β-strand C causing protein unfolding, as suggested by the smear produced when S63del is resolved under non-reducing conditions (Figure 3). Accordingly, deletion of F64, a CMT1B mutation in human (Ikegami et al., 1996), was predicted to alter disposition of 3 out of 4 hydrophobic residues. Indeed, F64del was completely retained in the ER and activated the UPR in transfected cells. Instead, S63F, also associated with a CMT1B mutation in human (Blanquet-Grossard et al., 1995), did not alter trafficking or induce an UPR in transfected cells. Clearly, the UPR is only one of several ‘toxic’ gain of function mechanisms associated with Mpz-related neuropathies (Wrabetz et al., 2006).
The amino-acid composition and disposition of P0 β-strand C residues are highly similar to the BiP binding domain in immunoglobulin (Gething, 1999). Under physiological conditions, BiP binds UPR transducers IRE-1, PERK and ATF6, maintaining them inactive (Ron and Walter, 2007). Binding of accumulated misfolded proteins by BiP activates IRE-1 and PERK kinases and ATF6 processing. Therefore, we suggest that alteration of P0S63del β-strand C exposes a hydrophobic surface that promotes BiP binding, thereby activating the UPR in S63del Schwann cells.
UPR directly causes S63del CMT1B neuropathy
CHOP expression correlated well with the level of MpzS63del and disease severity, suggesting a direct role in the pathology. Indeed, genetic ablation of Chop restored motor function. Several observations support that ameliorated demyelination accounts for the rescue. The number of demyelinated fibers was reduced by half, and there was a trend towards reduced numbers of onion bulbs, the hallmark of demyelination in neuropathy. The F-wave latency—a neurophysiological measure of demyelination—returned to near normal. Finally, preterminal sprouting at neuromuscular junctions, typical of demyelination, was rescued in S63del/Chop-null mice (Tinelli, Court and Wrabetz, in preparation; Court et al., in press).
However, Chop ablation did not rescue either hypomyelination or reduced NCV. One possible explanation is that they are caused by P0 overexpression—present in S63del nerves (Figure 4A; Wrabetz et al., 2000). Alternatively, other UPR effectors could alter differentiation (Acosta-Alvear et al., 2007; Tsang et al., 2007) and cause hypomyelination. For example, sustained activation of the MEK/ERK signaling pathway is associated with UPR (Hu et al., 2004), and activated ERK kinases induce Schwann cell de-differentiation (Harrisingh et al., 2004). In addition, ablation of Chop only rescued half of demyelination. The UPR could also produce demyelination independently of CHOP, as it broadly alters both the transcription and stability of specific mRNAs (Hollien and Weissman, 2006; Ron and Walter, 2007), and has global effects on other metastable proteins and their folding with consequential changes in function (Gidalevitz et al., 2006). PMP22 could be considered a metastable protein (Pareek et al., 1997); alterations of PMP22 or its dosage produce the most common hereditary demyelinating neuropathy. These possibilities would all fit with a model of pathogenesis where CHOP produces some demyelination, but other UPR mediators provoke further demyelination, as well as other aspects of the neuropathy.
Although hypomyelination independent of CHOP signaling may explain in part why ablation of Chop rescues rotarod performance but not all of the electrophysiological and morphological aspects of S63del neuropathy, other factors may intervene. Rotarod performance is incompletely sensitive, and we analyzed animals at 4-6 months of age, where demyelination is underway, but axonal damage is limited. In demyelinating neuropathy, disability correlates best with axonal damage, whereas how early myelin alterations produce disability is largely unknown—neuromuscular junction alterations may play a role (Yin et al., 2004).
CHOP and demyelination versus cell death
What role does cell death play in S63del pathogenesis? Under conditions of persistent stress and prolonged activation, the UPR initiates proapoptotic pathways (Rutkowski et al., 2006). Accordingly, we observed an increased number of cells programmed to die in S63del mutant mice. Moreover, CHOP functions as a pro-apoptotic gene both when UPR is activated acutely (Zinszner et al., 1998) and chronically (Oyadomari et al., 2002). However, a series of observations in S63del nerves indicate that UPR and CHOP induce demyelination with secondary cell death and not vice versa. First, mouse models of neuropathy due to death of Schwann cells manifest a different phenotype, congenital hypomyelination (Messing et al., 1992). Instead, S63del nerves show a classic “onion bulb” neuropathy, where myelin is formed normally then becomes unstable and is destroyed. Second, the rise in TUNEL positive nuclei is significantly delayed relative to CHOP induction (compare Figures 7 and 4) and rises after demyelination is detected. Third, there is accompanying proliferation and no decrease in cell numbers. Demyelinating neuropathy from other causes typically manifests cell death and proliferation at levels similar to S63del nerves (Sancho et al., 2001). In contrast, the UPR has been implicated in a number of misfolded protein disorders with primary cell death, including diabetes (Oyadomari et al., 2002), GM1-gangliosidosis (Tessitore et al., 2004), Pelizeaus-Merzbacher disease (Southwood et al., 2002), polyglutamine repeat diseases (Nishitoh et al., 2002) and Alzheimer disease (Hoozemans et al., 2005), where cell death is much more evident (typically 5-20% as measured by % pyknotic, tunel- or caspase 3-positive nuclei; Knapp et al., 1986; McLaughlin et al., 2007; Tessitore et al., 2004; versus less than 1% here). Finally, CHOP is usually detected in non-pyknotic Schwann cell nuclei, associated with normal myelin sheaths (Figure 2), suggesting that CHOP is associated with other early pathogenetic events, not late cell death. This provides further support for the idea that Chop induction in the context of UPR does not guarantee cell death (Rutkowski et al., 2006).
One must be careful not to equate the effects of UPR and CHOP; the UPR comprises a complex group of effectors. Does UPR produce demyelination because the UPR is peculiar in Schwann cells, or because the UPR happens to occur in cells that form myelin? We favor the latter. First, the UPR activation is canonical, CHOP target genes are activated, and P0S63del also activates a UPR in heterologous cells. Instead the UPR, or alterations to its mediators, perturb myelination by both oligodendrocytes (Li et al., 2004; Lin et al., 2005; Richardson et al., 2004; Southwood et al., 2002) and Schwann cells. In contrast, the diverse effects of Chop ablation in the two cells indicates opposing roles. CHOP is adaptive in oligodendrocytes of Rumpshaker mice (Southwood et al., 2002), but maladaptive in Schwann cells of S63del mice. A comparison of CHOP target genes, as well as the relative level of activation of the arms of the UPR, in the two pathological cells could illuminate the differing response.
So how does the CHOP arm of the UPR perturb S63del Schwann cells? It is important to note that the UPR and CHOP are induced during myelin formation, but pathology appears only later in mature myelin (de- not dys-myelination). Given that hereditary demyelinating neuropathies are commonly associated with moderately altered protein dosage (Suter and Scherer, 2003; Wrabetz et al., 2004), an appealing hypothesis is that Chop could target the program of myelin gene expression, perhaps via its stoichiometry. This could occur either transcriptionally—CHOP is a transcription factor that activates a downstream program of gene expression (Marciniak et al., 2004; Wang et al., 1998)—or post-transcriptionally—CHOP target genes regulate dephosphorylation of eIF2α and thereby stress-induced changes in translation of specific mRNAs (Marciniak et al., 2004). It is notable in this regard that subtle changes in translational control may exert dramatic effects on myelin homeostasis in the CNS (Li et al., 2004; Richardson et al., 2004).
Activation of the UPR occurs during myelination and is dynamic and reversible
In S63del nerves, transcriptional induction of BiP and CHOP begins within the first week of age during myelin formation. The observation that the UPR is activated before appearance of pathological symptoms strengthens the idea that the UPR is pathogenetic in S63del nerves. Furthermore, activation of the UPR was dynamically linked to continued expression of MpzS63del. Indeed, when the Mpz mRNA disappeared after axotomy, P0S63del disappeared quickly and the UPR was halted immediately. Given the capacity of Schwann cells to remyelinate, this suggests that pathology could be reversed in vivo. Likewise, disease reversibility in vivo has been reported for in other models of progressive neurodegenerative disorders associated with toxic mutant proteins including Huntington's disease (Yamamoto et al., 2000) and spinocerebellar ataxia type 1 (Zu et al., 2004).
Relevance to neuropathies and other misfolded protein diseases
One third to one half of hereditary neuropathies are due to a myriad of small mutations in more than 20 genes (Suter and Scherer, 2003; Wrabetz et al., 2004). How can we develop unifying treatment strategies? Common therapeutic targets will be key and alterations in protein trafficking and protein quality control are common to many hereditary neuropathies. The UPR due to S63del is pathogenetic, dose-dependent, dynamic and reversible. But elimination of one UPR mediator, Chop, only rescues half of demyelination—other aspects of UPR probably also contribute to pathogenesis. These data provide a rationale for treatment strategies aimed at eliminating directly the mutant protein, thereby converting a more severe toxic neuropathy into a milder neuropathy due to P0 haploinsufficiency. In this light, S63del mice provide the opportunity for proof of principle, by allelic specific RNAi, for example (Wood et al., 2007), that could ameliorate diseases due to toxic protein quality control responses. Finally, S63del mice provide an animal model where misfolded proteins elicit a UPR, causing cellular dysfunction, but not immediate apoptosis. Mechanisms identified in this model may be relevant to the first events of other misfolded protein diseases such as Alzheimer or Parkinson disease, or diabetes.
EXPERIMENTAL PROCEDURES
Animals
All experiments involving animals were performed following experimental protocols approved by the San Raffaele Scientific Institute Animal Care and Use Committee. Transgenic mice expressing either P0 with serine 63 deleted (S63del) or substituted by cysteine (S63C) (Wrabetz et al., 2006), as well as P0 overexpressor (OE), P0-null (Wrabetz et al., 2000), P0-Myc (Previtali et al., 2000), or Chop-null mice (Zinszner et al., 1998) have been previously described. All transgenic lines were maintained on the FVB/N background (Charles River, Calco, Italy). Genotypes were determined as described in the references above. Where indicated, mice were intraperitoneally injected with 1 μg tunicamycin/g body weight in 150 mM dextrose.
Western Analysis
Sciatic nerves from wt and transgenic mice were dissected, immediately frozen in liquid nitrogen and analyzed as previously described (Wrabetz et al., 2000; see supplementary materials for antibodies). Where indicated, sciatic nerves were dissected, and desheathed (to avoid the blood nerve barrier), and incubated with or without 10 μg/ml tunicamycin either overnight or 20 hours in DMEM, 10% fetal calf serum and 100 ng/ml nerve growth factor at 37°C in 5% CO2 humidified atmosphere. Samples were then frozen in liquid nitrogen and processed. To analyze disulfide bond formation, sample buffer lacked 2-mercaptoethanol and either contained or lacked 200 mM dithiothreitol (DTT). Before loading, samples were incubated 10 minutes at room temperature with 0.1M iodoacetamide. All Westerns were repeated at least three times with similar results.
Semiquantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Analysis
Sciatic nerves from wt and transgenic mice were immediately frozen in liquid nitrogen after dissection. Total RNA was prepared with Trizol (Roche Diagnostic GmbH, Germany) and 400 ng of RNA were reverse transcribed using 1 mM dNTPs, 2.5 ng/μl random examers, 40 units RNasin and 20 units AMV Reverse Transcriptase (Promega, WI, U.S.A.). Samples were incubated 10 minutes at room temperature and 1 hour at 42°C. The same volume of RT products was used to amplify glyceraldehyde-3-phosphate dehydrogenase (GAPDH), X-box binding protein (XBP-1) and P0. Digestion of the P0 amplimer for a DpnII polymorphism distinguished wt FVB/N alleles from BALB/C transgenic allele (Wrabetz et al., 2000).
Taqman® Quantitative PCR Analysis
Quantitative PCR was performed as manufacturer's instructions (Taqman®, PE Applied Biosystems Instruments) on an ABI PRISM 7700 sequence detection system (Applied Biosystems Instruments). The relative standard curve method was applied using wt mice as reference. Normalization was performed using either 18S rRNA or phosphoglycerate 1 (PGK1) as reference genes. Target and reference gene PCR amplification was performed in separate tubes with Assays on Demand™ (Applied Biosystems Instruments, see supplementary materials for assay numbers). Total RNA was prepared as described above from normal and mutant mice. To measure BiP and P0 400 ng of RNA were retrotranscribed as described above. For the other genes, 4 μg of total RNA were retrotranscribed using the SuperScriptII RNase H− Reverse Transcriptase (Invitrogen, U.S.A.) as per manufacture's instructions.
DNA constructs
A rat cDNA containing the P0 open reading frame (the kind gift of Marie Filbin, Hunter College, NY, U.S.A.) was cloned into the pBlue-Script vector (Stratagene, La Jolla, CA). The cDNA was amplified by polymerase chain reaction (PCR) with the following oligonucleotides: forward 5'-GGGGAAGCTTATGGCTCCTGGGGCTCC, reverse 5'-GGGGGATCCCGTTTCTTATCCTTGCGAGACTC containing HindIII and BamHI restriction sites (underlined). The reverse primer does not include the stop codon. The HindIII-BamHI fragment was cloned in frame into the corresponding sites of the pDsRed-N1 (Clontech, Palo Alto, CA), generating the pP0-Red vector. Site directed mutagenesis was performed via the QuikChange™ Site-Direct Mutagenesis Kit (Stratagene, La Jolla, USA) as manufacturer's instructions, using as template the pP0-Red plasmid (see supplementary materials for oligonucleotides). All amplimers were confirmed by automated sequence analysis.
Cell Transfection
COS-7 or CHO cells were transfected using a standard calcium phosphate precipitation protocol (Taveggia et al., 1998). Forty eight hours later, the proportion of live transfected cells was determined as a measure of transfection efficiency. Thereafter, total RNA was extracted and 400 ng of total RNA were retro-transcribed as described above. BiP mRNA and 18S rRNA levels were measured by Taqman analysis. The BiP/18S ratio (B) was normalized for transfection efficiency and for any unintended effect of transfection on BiP activation with the weighting calculation: B = [(X*T)+(C*(1−T))]/[(D*R) + (C*(1−R))], where X is the value of BiP/18S induced by the P0mutant, T is the fraction of cells transfected with P0mutantDsRed fusion, C represents the endogenous BiP/18S in untransfected cells arbitrarily set to 1, D is the level of BiP/18S from cells transfected with the sham vector pDsRed, and R is fraction of cells transfected with pDsRed. XBP-1 was analyzed via RT-PCR. Equal amounts of RT products were used to amplify GAPDH and XBP-1. Where indicated, cells were treated for 12 hours with 1μg/ml tunicamycin at 37°C in 5% CO2 humidified atmosphere. In this case, T was considered as 1. Parallel transfections performed with P0mutant-monoDsRed (non-dimerizing) constructs reproduced the intracellular localization and XBP-1 splice results (data not shown).
Immunofluorescence Analysis
Immunofluorescence was performed on cells, sciatic nerves, or teased fibers from sciatic nerves as previously described (Occhi et al., 2005; Pennuto et al., 2003; Wrabetz et al., 2006; see supplementary material for details and antibodies).
Behavioral, Morphological and Electrophysiological Analysis
Motor ability was assessed in 4 month old mice by Rotarod analysis as previously described (Wrabetz et al., 2006). The number of demyelinated axons and onion bulbs were counted blind to genotype in semi-thin sections stained with toluidine blue in images acquired with a 100x objective as described (Quattrini et al., 1996). We analyzed 3500-5000 fibers in 22-40 fields for each genotype in 15 wt, 11 chop-null, 18 S63del and 22 S63del-chop null animals. One of us (A.Q.) was able to distinguish all of the P0S63del from P0S63del/chop-null nerves when blinded to genotype. Semi-automated computer-based morphometry was performed on semi-thin sections of sciatic nerve to determine the G-ratio and the distribution of fiber diameters for myelinated axons as described (Bolis et al., 2005). 6-15 microscopic fields from nerves of 3-5 animals per genotype were analyzed. Electron microscopy was performed as described (Quattrini et al., 1996). Electrophysiology was performed as previously described (Bolino et al., 2004).
Bromodeoxyuridine (BrdU) Incorporation and Terminal Deoxynucleotidyl Transferase-mediated dUTP-biotin nick end labeling (TUNEL) Analysis
Wt and transgenic mice were intraperitoneally injected with 100 μg/g body weight of a 150 mM NaCl solution containing 10 mg/ml BrdU. One hour later, mice were sacrificed and sciatic nerves were embedded in OCT (Miles), and snap frozen in liquid nitrogen. Longitudinal 8 μm cryosections were fixed in cold methanol for 20 minutes at −20°C, washed in PBS and incubated 25 minutes in 2N HCl. After washing in PBS, specimens were incubated 10 minutes in 0.1M Na2B4O7 pH 8.5 and 10 minutes in PBS. Blocking was performed for 30 minutes in 10% goat serum in PBS. Incubation with primary monoclonal anti-BrdU antibody 1:20 (Roche, Switzerland) and secondary goat anti-mouse-FITC 1:50 (Southern Biotech, Birmingham, AL, USA) together with Hoechst 1:1000 was performed in 0.1% bovine serum albumin and 0.1% Triton in PBS. Specimens were then rinsed in 1% BSA and 0.2% Triton in PBS and mounted as described above. TUNEL analysis was performed as previously described (Feltri et al., 2002).
Supplementary Material
ACKNOWLEDGMENTS
We thank Desiree' Zambroni for expert technical assistance; Alex Gow, Juan J. Archelos, Lilliana Pedraza, Lucia Notterpek and Roberto Sitia for gifts of antibodies, Steven Scherer for TremblerJ nerves, Ernesto Bongarzone for Twitcher nerves, Rudolf Martini for the Mpz-null mice, and Marie Filbin for the rat P0 cDNA. We also thank Roberto Sitia for insightful discussions. This study was supported by grants from Telethon, Italy (M.L.F., and L.W.); NIH (D.R., DK47119 & ES08681; M.L.F., NS45630; L.W., NS55256); and the Fondazione Mariani, Italy (L.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Blanquet-Grossard F, Pham-Dinh D, Dautigny A, Latour P, Bonnebouche C, Corbillon E, Chazot G, Vandenberghe A. Charcot-Marie-Tooth type 1B neuropathy: third mutation of serine 63 codon in the major peripheral myelin glycoprotein PO gene. Clin. Genet. 1995;48:281–283. doi: 10.1111/j.1399-0004.1995.tb04109.x. [DOI] [PubMed] [Google Scholar]
- Bolino A, Bolis A, Previtali SC, Dina G, Bussini S, Dati G, Amadio S, Del Carro U, Mruk DD, Feltri ML, et al. Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J. Cell Biol. 2004;167:711–721. doi: 10.1083/jcb.200407010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolis A, Coviello S, Bussini S, Dina G, Pardini C, Previtali SC, Malaguti M, Morana P, Del Carro U, Feltri ML, et al. Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J. Neurosci. 2005;25:8567–8577. doi: 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Brophy PJ, Ribcester RR. Remodelling of motor nerve terminals in demyelinating axons of Periaxin-null mice. Glia. doi: 10.1002/glia.20620. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Urso D, Brophy PJ, Staugaitis SM, Gillespie CS, Frey AB, Stempak JG, Colman DR. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron. 1990;4:449–460. doi: 10.1016/0896-6273(90)90057-m. [DOI] [PubMed] [Google Scholar]
- Dickson KM, Bergeron JJ, Shames I, Colby J, Nguyen DT, Chevet E, Thomas DY, Snipes GJ. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9852–9857. doi: 10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, et al. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J. Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin M, Walsh F, Trapp B, Pizzey J, Tennekoon G. Role of myelin Po protein as a homophilic adhesion molecule. Nature. 1990;344:871–872. doi: 10.1038/344871a0. [DOI] [PubMed] [Google Scholar]
- Fortun J, Dunn WA, Jr., Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- Harrisingh MC, Perez-Nadales E, Parkinson DB, Malcolm DS, Mudge AW, Lloyd AC. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. Embo J. 2004;23:3061–3071. doi: 10.1038/sj.emboj.7600309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayasaka K, Himoro M, Sawaishi Y, Nanao K, Takahashi T, Takada G, Nicholson G, Ouvrier R, Tachi N. De novo mutation of the myelin Po gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III) Nat. Genet. 1993;5:266–268. doi: 10.1038/ng1193-266. [DOI] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer's disease. Acta. Neuropathol. (Berl) 2005;110:165–172. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004;279:49420–49429. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Nicholson G, Ikeda H, Ishida A, Johnston H, Wise G, Ouvrier R, Hayasaka K. A novel homozygous mutation of the myelin Po gene producing Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III) Biochem. Biophys. Res. Commun. 1996;222:107–110. doi: 10.1006/bbrc.1996.0705. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner DA, Wrabetz L, Feltri ML. The P0 Gene. In: Lazzarini RA, editor. Myelin Biology and Disorders. Elselvier Academic Press; San Diego: 2004. pp. 523–545. [Google Scholar]
- Knapp PE, Skoff RP, Redstone DW. Oligodendroglial cell death in jimpy mice: an explanation for the myelin deficit. J. Neurosci. 1986;6:2813–2822. doi: 10.1523/JNEUROSCI.06-10-02813.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkens T, Bolhuis PA, Wolterman RA, Kemp S, te Nijenhuis S, Valentijn LJ, Hensels GW, Jennekens FG, de Visser M, Hoogendijk JE, Bass F. Deletion of the serine 34 codon from the major peripheral myelin protein P0 gene in Charcot-Marie-Tooth disease type 1B. Nat. Genet. 1993;5:35–39. doi: 10.1038/ng0993-35. [DOI] [PubMed] [Google Scholar]
- Li W, Wang X, Van Der Knaap MS, Proud CG. Mutations linked to leukoencephalopathy with vanishing white matter impair the function of the eukaryotic initiation factor 2B complex in diverse ways. Mol. Cell. Biol. 2004;24:3295–3306. doi: 10.1128/MCB.24.8.3295-3306.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-{gamma} J. Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat. Genet. 1995;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- McLaughlin M, Karim SA, Montague P, Barrie JA, Kirkham D, Griffiths IR, Edgar JM. Genetic background influences UPR but not PLP processing in the rumpshaker model of PMD/SPG2. Neurochem. Res. 2007;32:167–176. doi: 10.1007/s11064-006-9122-y. [DOI] [PubMed] [Google Scholar]
- Messing A, Behringer RR, Hammang JP, Palmiter RD, Brinster RL, Lemke G. P0 promoter directs expression of reporter and toxin genes to Schwann cells of transgenic mice. Neuron. 1992;8:507–520. doi: 10.1016/0896-6273(92)90279-m. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhi S, Zambroni D, Del Carro U, Amadio S, Sirkowski EE, Scherer SS, Campbell KP, Moore SA, Chen ZL, Strickland S, et al. Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J. Neurosci. 2005;25:9418–9427. doi: 10.1523/JNEUROSCI.2068-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, Cabibbo A, Sitia R. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 2000;275:23685–23692. doi: 10.1074/jbc.M003061200. [DOI] [PubMed] [Google Scholar]
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Bonanomi D, Benfenati F, Valtorta F. Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol. Biol. Cell. 2003;14:4909–4919. doi: 10.1091/mbc.E03-06-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previtali SC, Quattrini A, Fasolini M, Panzeri MC, Villa A, Filbin MT, Li W, Chiu SY, Messing A, Wrabetz L, Feltri ML. Epitope-tagged P(0) glycoprotein causes Charcot-Marie-Tooth-like neuropathy in transgenic mice. J. Cell Biol. 2000;151:1035–1046. doi: 10.1083/jcb.151.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L. β4 integrin and other Schwann cell markers in axonal neuropathy. Glia. 1996;17:294–306. doi: 10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Richardson JP, Mohammad SS, Pavitt GD. Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity. Mol. Cell. Biol. 2004;24:2352–2363. doi: 10.1128/MCB.24.6.2352-2363.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Sancho S, Young P, Suter U. Regulation of Schwann cell proliferation and apoptosis in PMP22- deficient mice and mouse models of Charcot-Marie-Tooth disease type 1A. Brain. 2001;124:2177–2187. doi: 10.1093/brain/124.11.2177. [DOI] [PubMed] [Google Scholar]
- Shames I, Fraser A, Colby J, Orfali W, Snipes GJ. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J. Neuropathol. Exp. Neurol. 2003;62:751–764. doi: 10.1093/jnen/62.7.751. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Doyle JP, Hensley P, Colman D, Hendrickson WA. Crystal structure of the extracellular domain from Po, the major structural protein of peripheral nerve myelin. Neuron. 1996;17:435–449. doi: 10.1016/s0896-6273(00)80176-2. [DOI] [PubMed] [Google Scholar]
- Sharma R, Gow A. Minimal role for caspase 12 in the unfolded protein response in oligodendrocytes in vivo. J. Neurochem. 2007;101:889–897. doi: 10.1111/j.1471-4159.2007.04541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy ME. HMSN Related to MPZ (P0) Mutations. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. W.B. Saunders; Philadelphia: 2005. pp. 1681–1706. [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus- Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Pizzagalli A, Feltri ML, Grinspan JB, Kamholz J, Wrabetz L. MEBA derepresses the proximal myelin basic promoter in oligodendrocytes. J. Biol. Chem. 1998;273:27741–27748. doi: 10.1074/jbc.273.42.27741. [DOI] [PubMed] [Google Scholar]
- Tessitore A, del PMM, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, d'Azzo A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Hauer P, Lemke G. Axonal regulation of myelin protein mRNA levels in actively myelinating Schwann cells. J. Neurosci. 1988;8:3515–3521. doi: 10.1523/JNEUROSCI.08-09-03515.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Cheslett D, Chan WC, So CL, Melhado IG, Chan TW, Kwan KM, Hunziker EB, Yamada Y, et al. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 2007;5:e44. doi: 10.1371/journal.pbio.0050044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. Embo J. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M, Yin H, McClorey G. Modulating the Expression of Disease Genes with RNA-Based Therapy. PLoS Genet. 2007;3:e109. doi: 10.1371/journal.pgen.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, D'Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P, Previtali S, Imperiale D, Zielasek J, Toyka K, et al. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J. Neurosci. 2006;26:2358–2368. doi: 10.1523/JNEUROSCI.3819-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, Feltri M, Quattrini A, Inperiale D, Previtali S, D'Antonio M, Martini R, Yin X, Trapp B, Zhou L, et al. P0 overexpression causes congenital hypomyelination of peripheral nerve. J. Cell Biol. 2000;148:1021–1033. doi: 10.1083/jcb.148.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Kleopa KA, Scherer SS. Inherited Neuropathies: Clinical, Genetic and Biological Features. In: Lazzarini RA, editor. Myelin Biology and Disorders. Elselvier Academic Press; San Diego: 2004. pp. 905–951. [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yin A, Kidd G, Wrabetz L, Feltri M, Messing A, Trapp B. Schwann cell myelination requires timely and precise targeting of P0 protein. J. Cell Biol. 2000;148:1009–1020. doi: 10.1083/jcb.148.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Pioro EP, McDonough J, Dutta R, Feltri ML, Wrabetz L, Messing A, Wyatt RM, Balice-Gordon RJ, Trapp BD. Dysmyelinated lower motor neurons retract and regenerate dysfunctional synaptic terminals. J. Neurosci. 2004;24:3890–3898. doi: 10.1523/JNEUROSCI.4617-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.