

Abstract
The scope of gas phase ion/ion chemistry accessible to mass spectrometry is largely defined by the available tools. Due to the development of novel instrumentation, a wide range of reaction phenomenologies have been noted, many of which have been studied extensively and exploited for analytical applications. This perspective presents the development of mass spectrometry-based instrumentation for the study of the gas phase ion/ion chemistry in which at least one of the reactants is multiply-charged. The instrument evolution is presented within the context of three essential elements required for any ion/ion reaction study: the ionization source(s), the reaction vessel or environment, and the mass analyzer. Ionization source arrangements have included source combinations that allow for reactions between multiply charged ions of one polarity and singly charged ions of opposite polarity, arrangements that enable the study of reactions of multiply charged ions of opposite polarity, and most recently, arrangements that allow for ion formation from more than two ion sources. Gas phase ion/ion reaction studies have been performed at near atmospheric pressure in flow reactor designs and within electrodynamic ion traps operated in the mTorr range. With ion trap as a reaction vessel, ionization and reaction processes can be independently optimized and ion/ion reactions can be implemented within the context of MSn experiments. Spatial separation of the reaction vessel from the mass analyzer allows for the use of any form of mass analysis in conjunction with ion/ion reactions. Time-of-flight mass analysis, for example, has provided significant improvements in mass analysis figures of merit relative to mass filters and ion traps.
Introduction
Prominent among the many contributions made by Graham Cooks to the field of mass spectrometry is the development of novel instrumentation, particularly for tandem mass spectrometry. The development and evaluation of new tools has clearly been a central activity and contributor to the dramatic advances in the capabilities of mass spectrometry over the past several decades. In this perspective, we relate the evolution of instrumentation for research activities focused on the study and use of ion/ion reactions and offer it as one example of many where progress in an area of science is intimately related to and dependent upon the development of instrumentation. It is provided in appreciation for the inspirational example of creativity in instrumentation development [1] provided by Cooks over the years.
Electrospray ionization (ESI) [ 2 ], among its many contributions, enabled the development of macro-molecule ion/ion chemistry as a research area. The multiple-charging phenomenon associated with electrospray makes possible the generation of multiply charged reactant ions. This capability leads to the possibility that at least one ion/ion reaction product can retain charge, which makes such a product directly amenable to study with mass spectrometry. In the past two decades, significant progress has been made in understanding and exploring the gas-phase ion/ion chemistry of high mass multiply charged ions. A variety of potential analytical applications have been developed accordingly. Controlled charge reduction via proton transfer ion/ion reactions, for example, has been shown to be particularly powerful for charge state manipulation. Associated applications have been demonstrated in mixture analysis, especially with the application of the “ion parking” technique [ 3 ] for gas-phase ion concentration and purification [ 4 ], the formation of ions that are not directly produced by ESI for subsequent tandem mass spectrometry studies [5], and the reduction of the product ion charge states to singly and doubly charged species so as to simplify the interpretation of product ion spectra [6]. Ion/ion reactions that allow for multiple proton transfers in a single encounter can lead to the inversion of charge [7] and two such encounters in series can even lead to a net increase in charge for one of the reactants [8,9], which can be desirable for subsequent structural interrogation [10]. The recent demonstration of ion/ion electron transfer to multiply protonated polypeptides that leads to structurally informative fragmentation, a process known as electron transfer dissociation (ETD) [11,12], provides a major new structure determination tool in proteomics. Metal ion transfer [13,14,15] and complex formation [16], while less developed than proton transfer and electron transfer for analytical applications, are also ion/ion reaction phenomenologies that have been examined. The reader is directed to two comprehensive reviews [17,18] of ion/ion chemistry for more detailed information on the reaction phenomenology, fundamental reaction dynamics, and applications. The focus of the current review is to highlight the instrument evolution for gas phase ion/ion chemistry up to the end of 2006.
A fundamental requirement for the study of ion/ion reactions is the ability to form ions of opposite polarity within the context of a single experiment. For this reason, exclusively home-built or extensively modified commercial instrumentation, until very recently, has been used to conduct research in ion/ion chemistry. With the emerging commercial availability of instruments that support ion/ion reactions, it is appropriate to examine the evolution of instrumentation for ion/ion reaction studies. It is convenient to summarize this evolution within the context of the three major components to any ion/ion reaction experiment: ion formation, ion/ion reaction, and mass analysis of the products. The ion formation aspect determines the identities of the reactants and how they are delivered to one another. The next aspect relates to the environment and conditions in which the ion/ion reactions take place. Finally, the form of mass analysis of the products, as with any mass spectrometry experiment, plays an important role in determining the quality of information that can be derived from the experiment, largely through mass measurement accuracy, resolving power, and mass-to-charge range. Therefore, an apparatus suitable for gas phase ion/ion reactions should comprise ionization sources, a reaction vessel (or region) and a mass analyzer, as summarized in Scheme 1.
Scheme 1.

Instrument components for ion/ion reaction studies.
All of the major developments in the exploration of tools for ion/ion reactions can be classified according to one or more of the component aspects indicated in Scheme 1. This perspective, therefore, discusses these developments within the context of the three major components and begins with the ion/ion reaction environment.
I. Reaction Vessel
Perhaps the most important requirement for the study of ion/ion reactions is the provision of an environment that allows for the physical and temporal overlap of oppositely charged ions, preferably at low relative translational energies. Such environments have been created either external or internal to a mass spectrometer. In the former case, ion/ion reactions have taken place at near-atmospheric pressure prior to ion sampling into the vacuum system of a mass spectrometer. In the latter case, reactions have taken place within the confines of an electrodynamic ion trap, typically in the presence of a background gas in the milliTorr range. Instrumentation associated with each of the two categories is described below.
A. Atmospheric Pressure Ion/Ion reactions Followed by Mass Analysis
1. Y-tube/Quadrupole Mass Filter
The first ion/ion reaction studies involving multiply charged ions were described by Ogorzalek-Loo et al. in the early 1990s [19,20]. Ions of opposite polarity were introduced into two arms of a Y-shaped capillary separated by a 60° angle, where they merged in the flow that lead to the inlet of a quadrupole mass filter. A schematic diagram of the apparatus used for this kind of work is shown in Figure 1.
Figure 1.
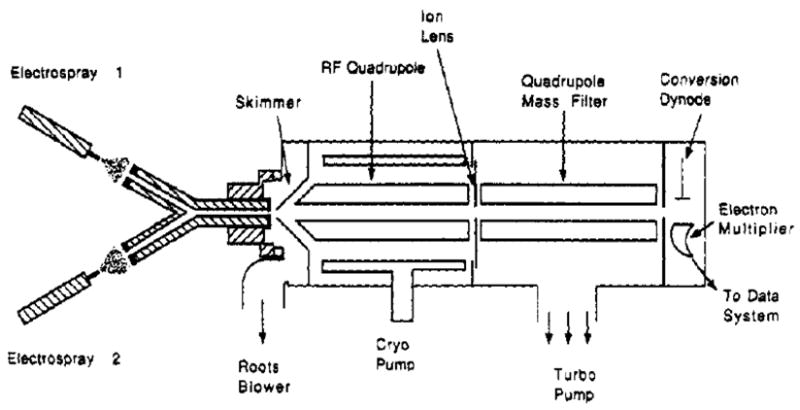
Instrument diagram of the apparatus that incorporates a Y-shaped capillary inlet/flow-reactor for ion/ion reaction studies. Reprinted from reference [20] by permission of Elsevier from “A new approach for the study of gas-phase ion-ion reactions using electrospray ionization”, by Ogorzalek-Loo R. R., Udseth H. R., Smith R. D., Journal of the American Society for Mass Spectrometry, Vol 3, pp 695–705. Copyright 1992 by the American Society for Mass Spectrometry.
The Y-shaped capillary is the novel element of the above apparatus, which functions as a flow reactor at near atmospheric pressure. It provides means for delivering oppositely charged ions to one another, as well as a mixing region for reactions. The residence time in the mixing region has been estimated to be approximately 5×10−4 s. The Y-shape arrangement allows for the use of two spatially separated ion sources to form charged reactants, where three modes of operation have been reported: i) single electrospray (to record data in the absence of ion/ion reactions); ii) dual ESI sources operated at opposite polarity; and iii) ESI with an atmospheric pressure discharge source with all three ion polarity combinations (+/−, −/+ or +/+). ESI droplet desolvation in those studies was facilitated by heated countercurrent gas and/or by heating the capillary. A variety of interesting and potential useful phenomena have been observed for the first time, including proton transfer reactions resulting in reduced charge states and charge inversion. The Y-tube reactor has the advantage of simple implementation of ion/ion reactions on any kind of electrospray mass spectrometer with a relatively high degree of flexibility, in terms of the range of reactant ions amenable to study. On the other hand, it can be difficult to establish well-defined and controlled reaction conditions in the Y-tube reactor. A mixture of ions, solvent vapors, and atmospheric gases were typically present in the Y-tube, which could vary according to the experimental parameters, such as the discharge current, needle inlet spacing and the inlet aperture size. Furthermore, the parameters just mentioned were not completely independent of one another, thereby complicating control of the number of the positive and negative ions delivered to the inlet/reactor, the pressure in the capillary reaction, and the amount of solvent association induced by discharge operation. In addition to complications associated with the front end reactor, the limited mass-to-charge (m/z) range the quadrupole mass filter almost surely prevents detection of many ion/ion reaction products. For these reasons, some of the results described in the publications with the Y-tube setup raised a number of interesting questions that could not be addressed definitively with the instrument in Figure 1.
2. Charge Reduction Electrospray/Time-of-Flight Mass Spectrometry
In 1999, Smith and co-workers reported charge reduction of biopolymer ions formed via electrospray with the use of atmospheric pressure ion/ion reactions [21]. The instrument used in the original study contained three basic components: ESI source, a charge neutralization chamber, as shown in Figure 2, and an orthogonal TOF mass spectrometer [22].
Figure 2.

Schematic diagram of the charge neutralization chamber with α-particle source used for charge reduction of ions formed from ESI, coupled to a time-of-flight mass spectrometer. Reprinted from reference [22] by Scalf et al. with permission from Anal. Chem. 2000, 72, 52–60. Copyright 2000 American Chemical Society.
The analyte species formed by ESI, in the form of ions and charged aerosols, were carried into the neutralization chamber via a flow of gas. The neutralization chamber hosted a disk of 210Po, an α-particle source, which ionized the bath gas to form singly charged ions of both polarities. The subsequent reactions between the singly charged ions derived from the radioactive source and aerosol droplets and/or analyte ions gave rise to charge reduction. In this reactor, the degree of charge reduction occurring in the chamber can be controlled by adjusting the flux of α-particles via shielding the surface area of the 210Po source and the residence time of the analyte ion in the chamber by varying the gas flow speed. The product ions were then directed to an orthogonal TOF mass analyzer with an upper m/z limit of 25,000. Shortly after describing the use of the α-particle-based source, the Smith group developed a corona discharge source for the charge reduction reactions [23]. The configuration of the neutralization chamber was similar to the one shown in Figure 2, except that the 210Po source was replaced by a “point-to-plate” corona discharge source. Control over ion/ion reaction rates was achieved by adjusting the voltage applied to the corona discharge source. An optimized design of the charge reduction chamber for both the corona discharge source and an α-particle source were recently described [24], where an o-ring seal connected the reaction chamber with the inlet of a mass spectrometer, as shown schematically in Figure 3.
Figure 3.

Diagram of the charge neutralization chamber with (a) the corona discharge and (b) the α-particle source. Attachment to the mass spectrometer inlet via an o-ring seal greatly improved ion transmission. Reprinted from reference [24] with permission of Elsevier from “Controlling gas-phase reactions for efficient charge reduction electrospray mass spectrometry of intact proteins”, by Frey B. L., Lin Y., Westphall M. S., Smith L. M., Journal of the American Society for Mass Spectrometry, Vol 16, pp 1876–1887. Copyright 2005 by the American Society for Mass Spectrometry.
Ion transfer through the chamber was facilitated by a vacuum-induced air-flow, as compared to the high gas-flow required in the previous designs. The corona discharge source (Figure 3a) was removed from the path of the electrospray ions, thereby reducing interference with the ESI source. The reaction time was fixed by the travel time through the field-free region in the chamber, calculated to be around 10 ms. The control of charge reduction was achieved by adjusting gas flow through the corona discharge, which in turn determined the reagent ion intensity available for reactions. The new design provided an order-of-magnitude increase in ion signals with control over the extent of charge reduction. In addition to the dominant proton transfer reactions, oxidation and nitrate addition were observed as side reactions, which could be reduced by flowing an organic vapor in nitrogen gas through the corona discharge [24]. The α-particle source was reported to have the advantage of producing a higher flux of charge reducing species compared to the corona discharge source. However, the corona discharge source obviated licensing and handling of radio-active materials and the need to replace of the α-particle source after its gradual decay over time.
A common characteristic of the two atmospheric ion/ion reaction strategies discussed above is that mass analysis is physically separated from the ionization and ion/ion reaction regions. The advantages associated with this include simple adaptation of the ion/ion reactors to any mass spectrometer with an ESI interface, independent optimization of the mass analyzer to achieve the best mass analysis performance, no restriction on the reactions imposed by the mass analyzer (e.g., no trapping limit for reactants, as compared to reactions in ion trap instruments (see below)), and the ease of switching reaction polarity. On the other hand, since the ionization and reaction regions are in close proximity, or even integrated, independent control of reactant species and the extent of reaction in the reaction region are limited with these approaches. They do not allow for a true tandem mass spectrometry experiment in which an ion/ion reaction takes place between mass analysis stages.
B. Electrodynamic Ion Traps as Reaction Vessels
The ability of the quadrupole ion trap to store ions of opposite polarity simultaneously was demonstrated nearly three decades ago [25]. Not until 1995, however, was a quadrupole ion trap reported to be used for the study of the reactions of oppositely charged ions [26]. A key advantage of the use of an ion trap as the reaction vessel is its well-known capability for executing MSn experiments [27] as a result of the “tandem-in-time” nature of the device. Furthermore, use of the ion trap for ion/ion reactions facilitates the separation of the ionization and ion/ion reaction steps. These characteristics have been especially useful in the study of ion/ion reactions, particularly for defining ion genealogical relationships. They also enable a wide and growing range of analytical experiments that rely on MSn capabilities. Ion/ion reactions have been implemented both with three-dimensional (3D) ion traps (i.e., those based on conventional Paul traps) and with two-dimensional (2D) ions traps (i.e., those based on linear quadrupole arrays for trapping in x- and y-dimensions and end-plates for trapping in the z-dimension). Developments made with each version of electrodynamic ion trap are discussed briefly in turn.
1. Ion/ion reactions in three-dimensional quadrupole ion traps
In the conventional 3D ion trap, in which a ring-electrode is sandwiched between two end-cap electrodes, ions are stored in each of the three linear dimensions by the radio-frequency (RF) voltage applied between the ring-electrode and the two end-cap electrodes. In the absence of a DC field, ions of opposite polarity are both stored simultaneously at the center of the ion trap. A light bath gas, such as helium, present at roughly 1 mTorr is commonly used in ion trap experiments to translationally cool ions to the center of the ion trap, which is attractive for ion/ion reactions from the standpoint of maximizing spatial overlap and minimizing relative translational energies. Hence, no modification to a 3D ion trap is necessary to enable ion/ion reaction studies, provided means are available to form or admit the reactant ions. However, the use of an ion trap as an ion/ion reaction vessel leads both to constraints and to unique capabilities. For example, the presence of ions of opposite polarity can lead to deviations in expected behavior based on the ion trapping conditions in the absence of oppositely charged ions. When the oppositely charged ions are at low ion densities, the macroscopic motions (i.e., oscillatory frequencies and amplitudes) of the oppositely charged ions are largely independent of one another and are determined by the trapping conditions (e.g., trap radius, RF frequency, RF amplitude, bath gas identity and pressure). However, at some point, ion numbers can become sufficiently large that ion motion can be significantly affected by the presence of oppositely charged ions [28]. In this scenario, predicted ion frequencies based on the operating conditions of the ion trap no longer apply. For this reason, when it is desirable to use the ion trap as the mass analyzer, ions of one polarity are ejected prior to mass analysis of the ions of opposite polarity.
A constraint placed on ion/ion reactions by use of an ion trap as the reaction vessel arises from the limited range of mass-to-charge values that can be stored simultaneously. In the absence of a DC field, the lower limit to mass-to-charge for ion storage is sharply defined by the point at which the qz-value [29] for an ion is 0.908, where
| (1) |
(VRF = RF amplitude, Ω= RF frequency, r0 = ion trap radius, m = mass, z = unit charge, e = electron charge). The upper limit to mass-to-charge for ion storage is less sharply defined but is related to the pseudo-potential trapping well, Dz [29], approximated at qz values below 0.4 by:
| (2) |
Ions with kinetic energies that approach or exceed Dz are not effectively stored. Note that Dz is inversely related to mass-to-charge ratio. Hence, given that Ω and r0 are held constant in most ion trap experiments, the m/z range for mutual ion storage is determined by the value of VRF. Note, however, that the m/z range for mutual ion storage can be expanded if high ion densities of the low m/z reactant can be established such that the strong electric field of the low m/z ions can prevent the escape of the high m/z ions when the ion trapping field is too weak to do so. This technique has been referred to as “trapping by proxy” [30] as the high m/z ions are stored by the electric field of the low m/z ions which are, in turn, stored by the oscillating quadrupolar field.
The constraints imposed by the use of an ion trap as an ion/ion reaction vessel notwithstanding, the ion manipulation capabilities afforded by the ion trap environment provide for unique and useful capabilities. For example, the ion trap allows for ion/ion reactions to be used within the context of an MSn experiment. A major advantage associated with the use of an ion trap as an ion/ion reaction vessel, at least at relatively low ion densities, is the fact that ions assume m/z-dependent frequencies of motion in the oscillating quadrupolar field. This allows for the use of supplementary fields to inhibit selectively ion/ion reaction rates, an approach referred to as ‘ion parking’ [3]. The ion/ion reaction rate of a specific ion can be inhibited by applying a low amplitude (smaller than that used to effect collision-induced dissociation) supplementary radio-frequency signal to the end-cap electrodes at or near to the z-dimension fundamental secular frequency of the ion. This small degree of ion acceleration reduces the spatial overlap of the oppositely charged ions and also increases their relative velocities. The net effect is the inhibition of the ion/ion reaction rate of the ion of interest. This phenomenon allows for the concentration of most of the ion signal initially dispersed among multiple charge states by the ESI process into a single charge state. Single frequency ion parking has been proved to be quite useful in gas-phase concentration and purification of single components of a multi-component mixture for further interrogation via MS/MS [4].
In addition to the single frequency ion parking for ions within a narrow m/z range, ion/ion reaction rates can be inhibited over a broad range, referred as “parallel ion parking”. Parallel ion parking can be implemented in an ion trap in several ways. Grosshans et al. have suggested application of a dipolar DC voltage across the end-cap electrodes [31,32], which leads to a physical separation of cation and anion clouds in the ion trap that is m/z-dependent. In this approach, all ions with m/z ratios above a particular value are parked. Alternatively, a tailored waveform has been used in parallel ion parking as a means to inhibit sequential ion/ion reactions, whereby notches were used to accelerate only product ions differing in m/z from those of the selected reactants [33]. A single frequency ac signal of relatively high amplitude applied in a dipolar fashion on a three-dimensional or linear ion trap can also induce parallel ion parking [34]. The high amplitude gives rise to the acceleration of a range of m/z values compared to the use of the same frequency at much lower amplitudes. Parallel ion parking has been shown to be useful in inhibiting second generation ion/ion reactions in ETD and for the simultaneous concentration of protein ion signals derived from protein mixtures.
2. Ion/ion reactions in linear quadrupole ion traps
With the introduction of linear quadrupole ion trap mass spectrometers [35,36], interest has been directed to the implementation of ion/ion reactions in such devices. Linear ion traps have an inherent efficiency advantage over 3D ion traps with respect to coupling with ion sources, detectors, and other devices. Ions can be admitted and transmitted, for example, along the center axis of the device without having to overcome a large RF barrier, unlike the case with the 3D ion trap. For this reason, the linear ion trap is particularly attractive for implementation as an ion/ion reaction vessel in hybrid instruments. The conventional means for storing ions along the central axis, however, (i.e., the use of static potentials at the ends of the quadrupole array) is not effective for the mutual storage of ions of opposite polarity. As a result, two approaches have been pursued for implementing ion/ion reactions in linear ion traps. One involves adapting the ion trap for mutual ion storage and the other involves storage of one ion polarity while the ions of opposite polarity are passed or transmitted through the stored ion population. The instrumentation developments related to each of these strategies is discussed here.
a. Mutual storage mode ion/ion reactions in linear ion traps
The oscillating RF field of the quadrupole array provides mutual ion polarity storage in the x- and y-dimensions (i.e., in the radial plane) when the voltages applied to pairs of opposing rods are equal in amplitude and 180° out-of-phase (i.e., a condition referred to as ‘balanced RF’). Mutual ion polarity storage, however, is not achieved in the axial or z-dimension when static DC potentials are applied to trapping plates at either of the quadrupole array. Therefore, an additional RF filed is needed to store oppositely charged ions in all dimensions in a linear ion trap. Applying RF voltages directly to the end lenses of a linear ion trap is one straightforward solution to creating an RF barrier at the ends of the array. In 2004, Hunt group demonstrated mutual trapping ion/ion reaction experiments on a modified linear ion trap mass spectrometer (Finnigan LTQ mass spectrometer, Thermo Electron, San Jose, CA) [11]. The major instrument modifications involved superposing secondary RF potentials on the containment lenses of the linear ion trap for axial trapping during the ion/ion reaction period and adding a chemical ionization source for reagent anion formation at the end of the linear ion trap opposite to that used for introducing analyte ions formed via electrospray. The front ESI/rear CI source arrangement allows reactant ions to be introduced into the linear ion trap along the null axis of the RF quadrupole field from the two ends, which results in high ion injection and trapping efficiency. The three segment configuration of the linear ion trap itself permits flexible control of the ion/ion reaction mixing process via adjustment of the DC biases of these elements. As illustrated in Figure 4, the positive and negative ions can be axially segregated or mixed for different purposes during ion injection, ion isolation, reaction initiation, and reaction termination periods.
Figure 4.
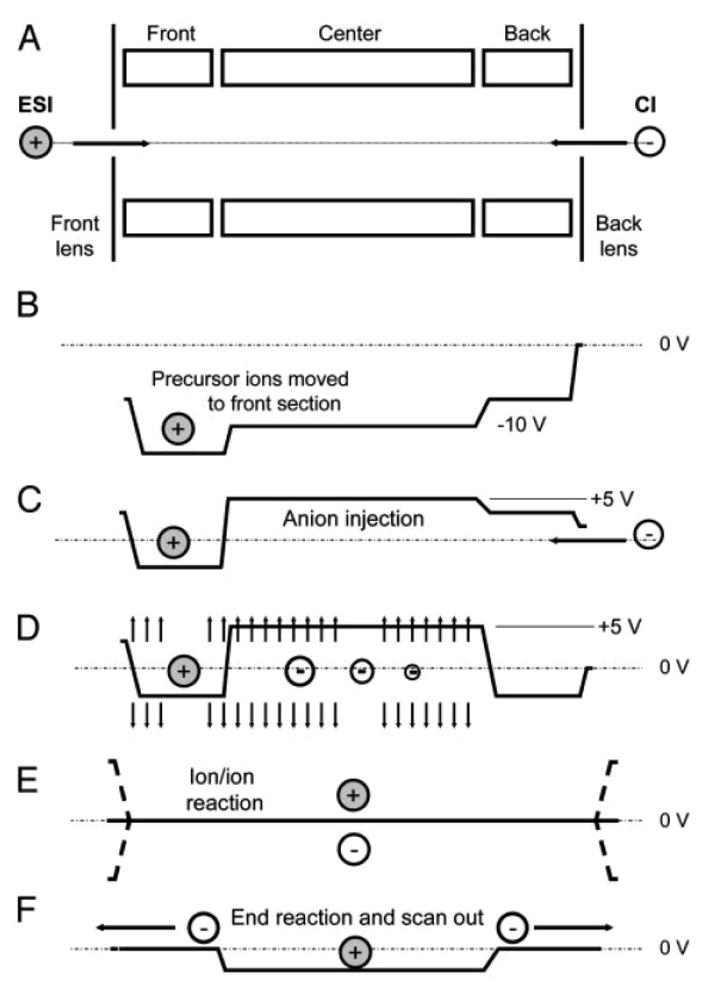
Schematic of steps involving ion/ion reactions in a linear ion trap instrument. Reprinted from reference [11] by Syka et al. with permission from the National Academy of Sciences, USA, copyright 2004.
The initial studies describing ETD have been performed with this apparatus. In more recent studies, the same group has used this instrument setup to demonstrate MSn experiments that combine an ETD step followed by a proton transfer (PT) ion/ion reaction step by adding a second reagent to the CI source to serve as the proton transfer reactant [37]. Whole protein characterization on a chromatographic scale has been demonstrated using this sequential ETD/PT method [38]. The ion/ion reactions of multiply deprotonated peptide ions with xenon radical cations have also been studied using the same instrument setup [39].
An alternative means for creating an RF barrier for mutual ion polarity storage along the z-dimension is to unbalance the RF applied to the quadrupole array [40,41]. When the DC offset of the quadrupole array and the containment lenses are at the same level, subtracting a fraction of RF amplitude applied to one set of opposing rods and increasing by the same amount the RF level on the pair of rods is equivalent to applying the same fraction of RF to the containment lenses. A conceptual depiction is shown in Figure 5, where the degree of unbalance is defined as the percent deviation of the RF amplitude from the average amplitude on both pair of rods. The degree of unbalance can be adjusted by adding and subtracting turns of the inductance coil in the RF circuitry to the two poles of the quadrupole rod sets. Note that in the unbalanced condition, the ions experience an oscillating RF field in the z-dimension only at the ends of the rod set. It is in the fringing regions that the balance between poles is meaningful for trapping in the z-dimension.
Figure 5.
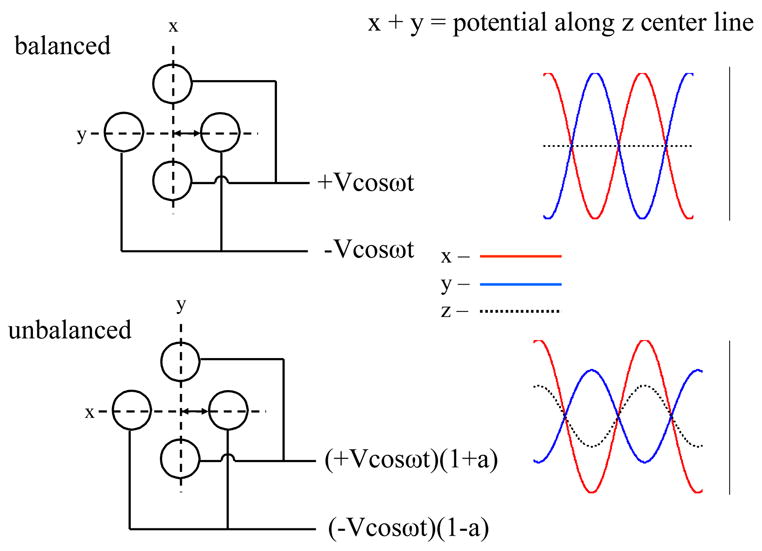
Conceptual depiction of unbalancing the RF amplitudes on the two sets of opposing poles of a quadrupole rod array. V is the RF amplitude applied on the rod, “a” is the degree of unbalance and x, y, z represents the three dimensions. Reprinted from reference [41] with permission of Elsevier from “Mutual storage mode ion/ion reactions in a hybrid linear ion trap”, by Xia Y., Wu J., McLuckey S. A., Londry F. A., Hager J. W., Journal of the American Society for Mass Spectrometry, Vol 16, pp 71–81. Copyright 2005 by the American Society for Mass Spectrometry.
Ion/ion reactions in a linear ion trap operated in an unbalanced RF condition (54%) were demonstrated on a modified hybrid triple quadrupole/linear ion trap mass spectrometer, where an atmospheric sampling glow discharge source was mounted to the side of third quadrupole (Q3) of the assembly for directing negative reagent ions into Q3 between the rods [41]. The schematic diagram of the apparatus is provided in Figure 6. Proton transfer ion/ion reactions were carried out with multiply protonated protein ions formed by ESI and anions derived from glow discharge ionization of perfluro(methyldecaline) (PMD). Many of the key ion/ion proton transfer reaction experiments demonstrated previously with 3D quadruple ion traps were executable in the unbalanced Q3 linear ion trap. However, the presence of RF barriers at the ends of a linear ion trap can compromise other aspects of an MSn experiment if they cannot be created and removed at appropriate points in the sequence of events. For example, high RF barriers at the ends of the array can adversely affect ion injection into the ion trap and can degrades the performance of mass analysis using mass selective axial ejection (MSAE) [ 42 ]. Therefore, subsequent work with this apparatus has employed superposition of RF on containment lenses, as this approach is more readily adapted to software control during the course of an MSn experiment. Mutual storage ion/ion reactions experiments using superposition of RF on the containment lenses were carried out both in Q3, in which RF voltages were applied to the exit lens and IQ3 (see Figure 6), and in Q2, in which the RF voltages were applied to IQ2 and IQ3. The latter experiments required reagent anion accumulation into Q3 from the ASGDI source and subsequent transfer into Q2. In general, ion/ion reaction rates were noted to be roughly one order of magnitude greater in the Q2 experiments, presumably due to the much higher background pressures in Q2 (1-8 mTorr) than in Q3 (5 × 10−5 Torr). Operation at the higher pressure is expected to give rise to better spatial overlap of the oppositely charged ion populations and lower relative velocities.
Figure 6.
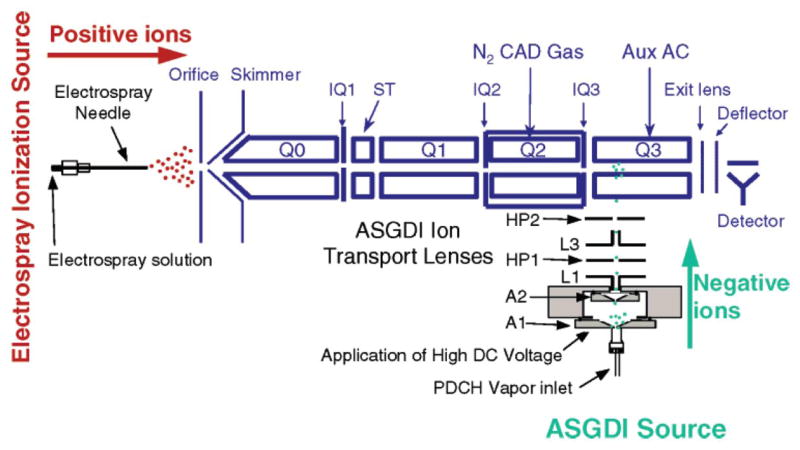
Schematic diagram of a hybrid triple quadrupole/linear ion trap modified by addition of an atmospheric sampling glow discharge ionization source on the side of Q3 linear ion trap for ion/ion reactions. Reprinted from reference [43] by Wu et al. with permission from Anal. Chem. 2004, 76, 5006–5015. Copyright 2004 American Chemical Society.
b. Transmission mode ion/ion reaction in linear ion traps
The linear ion trap also facilitates implementation of ion/ion reaction experiments that do not require mutual ion polarity storage. The relatively high ion injection/transmission efficiencies along the long axis of an RF-only quadrupole array can lead to good spatial overlap with an ion population stored in a linear ion trap using DC potentials applied to the containment lenses. There, in principle, are three ways that transmission mode ion/ion reactions can be effected in a linear ion trap, provided both polarities of ions are introduced in the axial direction, as summarized in Scheme 2. Note that Scheme 2 indicates oppositely charged ions to be injected from opposing ends of a linear ion trap, but that the three transmission mode experiments can also be implemented with ion injection via a common end of a linear ion trap. The first method of transmission mode ion/ion reactions involves the storage of neither ion polarity and relies on reactions taking place between the ions of opposite polarity as they are continuously admitted into the LIT (Method I). This method has yet to be demonstrated. Methods II and III involve storing one ion polarity while ions of the other polarity are continuously admitted into the LIT. Both of these approaches have been demonstrated and evaluated.
Scheme 2.

Three methods for effecting transmission mode ion/ion reaction experiments in a LIT: (I) passing ions of both polarities, (II) positive ion storage/negative ion transmission, (III) positive ion transmission/negative ion storage.
The first example of a transmission mode ion/ion reaction experiment in a linear ion trap was carried out on the triple quadrupole/linear on trap depicted in Figure 6 by Wu et al. in 2004 [43]. In that example, anions were accumulated and stored in Q2 (via initial anion injection into Q3) and positive analyte ions were transmitted through Q2 and collected in Q3 (Method III of Scheme 2). The extent of reaction that a cation can undergo in this approach was determined by the number of anions stored in Q2, Q2 pressure, and the transmission time of the cation through Q2, which is not readily variable over a wide range. Useful proton transfer applications, such as separating ions of different mass but very similar m/z ratios, were demonstrated using this approach. However, formation and admission of reactant ions via ASGDI and radial injection into Q3 proved to be a very inefficient means for admitting ions into Q2 and limited the ability to fully evaluate transmission mode ion/ion reaction experiments for this reason.
Transmission mode ion/ion reactions in linear ion traps have recently been revisited using a triple quadrupole/linear ion trap instrument with a pulsed dual ion source placed in front of the atmospheric interface [44,45]. This approach to admitting ions into Q2 has overcome the limitations associated with radial injection of ions into Q3 and subsequent transfer into Q2. Proton transfer (charge reduction, charge inversion) and ETD reactions have been conducted in transmission mode via both Methods II and III in the Q2 linear ion trap. When the polarity of the product ions is the same as the transmitted ions, an external device, such as the Q3 linear ion trap, is needed for product ion mass analysis. However, when the detection is for the opposite polarity of ions as the ions in transmission, such an external device is not necessary. The extents of ion/ion reactions via methods II and III were found to be similar when each was conducted under optimized conditions and also comparable to those acquired under mutual storage mode, both in terms of efficiency and information content of the spectra. Ion parking has also been demonstrated with ion/ion reactions implemented in transmission mode [47]. In the case when the parked ions were the stored species, similar performance to that obtained in mutual storage mode was noted. However, lower signals were obtained when the parked ions were the transmitted species, presumably due to poor transmission of the resonantly excited ions through the exit aperture of Q2. Overall, transmission mode ion/ion reactions offer another option for the implementation of ion/ion reactions. An advantage of the transmission mode ion/ion methods is that they do not require measures to be taken to allow for the mutual storage of both ion polarities (i.e., superposition of RF voltages on containment lenses or the use of unbalanced RF). While transmission mode ion/ion reactions have been demonstrated to date only on a hybrid triple quadrupole/LIT instrument, they can, in principle, be used with any type of instrument that employs a quadrupole collision cell. In addition to triple quadrupole instruments, these include, for example, hybrid instruments such as quadrupole/time-of-flight (QTOF), linear ion trap/orbitrap, and linear ion trap/Fourier transform ion cyclotron resonance (FTICR) instruments.
II. Ionization
Many of the instrumentation developments associated with the study of ion/ion reactions have focused on the expansion of ion/ion reaction phenomenology via the exploration of novel reagents and, in some cases, by improving the means by which reactant ions are delivered to one another. The exploration of novel reagents has often involved the use of different types of ionization methods. In the case of the Y-tube studies discussed above, for example, the replacement of one of the ESI emitters with atmospheric pressure chemical ionization expanded the range of species that could be used to react with the analyte ions. It is most straightforward to use atmospheric pressure ionization methods with the approaches that rely on ion/ion reactions at or near atmospheric pressure, however. For this reason, the range of ionization methods that can be interfaced to such reactors in facile fashion is limited. With the ion trap reactors, however, the reaction vessel and ionization methods are largely uncoupled. For this reason, a wider variety of ionization approaches have been explored with ion traps. A summary of the approaches examined to date is given here, first summarizing work with 3D ion traps and then work with 2D ion traps.
A. Ionization approaches with 3D ion traps
The first ion/ion reaction studies in a quadrupole ion trap employed an ESI source to form multiply charged negative ions with ion injection through an end-cap electrode. Singly charge positive ion were formed within the ion trap via electron irradiation of gaseous species admitted into the vacuum system through a 3-mm hole in the ring-electrode [26]. A schematic diagram of this instrument is shown in Figure 7. This approach allowed for the formation of radical cations via electron ionization as well as protonated molecules via ion trap chemical ionization [46]. Both proton transfer [26,47] and electron transfer [48] ion/ion reactions involving multiply charged biopolymer anions were studied with this instrument. The typical experimental sequence involved introduction of multiply charged negative ions (analytes) via an end-cap electrode, ion isolation, in situ formation of singly charged positive ions by gating on the filament to introduce electrons into the ion trap, mutual storage for reaction, and mass analysis using resonance ejection [49]. This combination of ionization approaches was best suited for the study of multiply charged anions with singly charged cations. It was poorly suited, however, for the study of the ion/ion reactions of multiply protonated species due to the inefficient nature of in situ anion formation using a heated filament [50].
Figure 7.
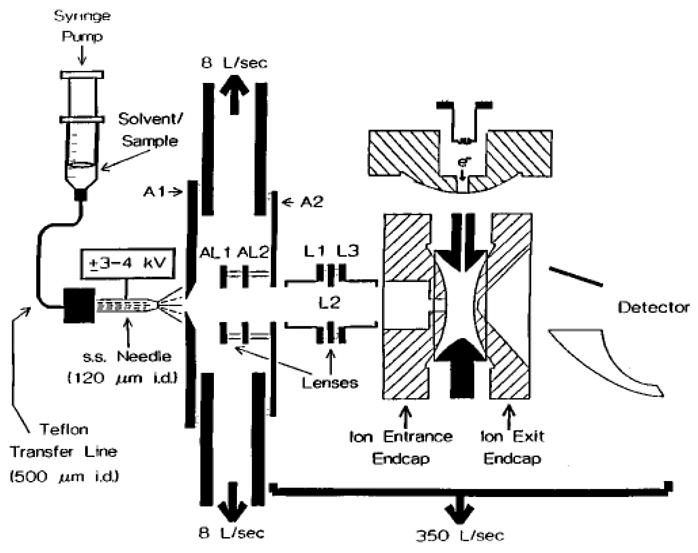
Side-view instrument schematic of a quadrupole ion trap that combines electrospray ionization and in situ ionization effected by injection of electrons through the ring-electrode. Reprinted from reference [17] with permission from John Wiley & Sons Ltd., copyright 1998.
The apparatus shown schematically in Figure 8 was motivated by interest in the study ion/ion reactions between multiply charged positive ions and singly charged negative ions [51]. Compared to the apparatus depicted in Figure 7, the major change of the instrumentation was the replacement of the heated filament by an atmospheric sampling glow discharge ionization (ASGDI) source. The ASGDI source can be used to form either positive or negative reagent ions, but the main motivation for replacing in situ electron ionization with the discharge source is the bright anion beams that can be generated via the discharge for species with relatively high cross-sections for electron capture [52]. With the arrangement shown in Figure 8, each ion source is independently controlled and all the ion species are formed outside the ion trap. The experimental sequence is similar to that for the ESI/heated filament apparatus described earlier, except that a period for glow discharge replaces the electron gated period. A Hitachi M-8000 ion trap mass spectrometer was subsequently modified to provide a similar experimental setup but with significantly improved figures of merit for mass analysis [53], as discussed further in the mass analysis section. Instruments of the geometry of Figure 8 can generate a wide range of reactants for ion/ion chemistry studies. An area of particular emphasis was the study of proton transfer from multiply protonated peptides and proteins to singly charged anions [54]. These studies were facilitated by the ready formation of bright anion beams derived from perfluorocarbons such as perfluro-1,3-dimethylcycolhexane (PDCH). Perfluorocarbon anions proved to be particularly useful reagents for deprotonation of peptides and proteins. Most recently, ETD of multiply protonated peptides in a three-dimensional ion trap with a setup similar to that shown in Figure 8 has been reported [55], with the ASGDI source being used to generate radical anions from species such as sulfur dioxide and nitrobenzene [56].
Figure 8.
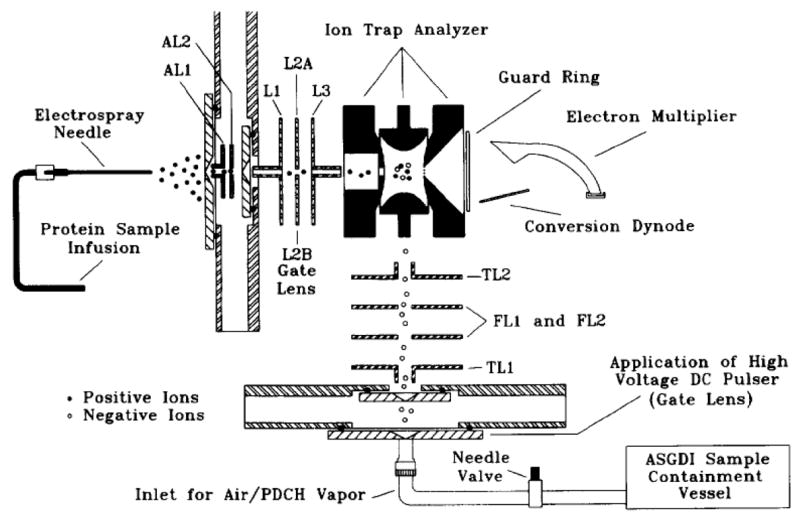
Schematic diagram of a quadrupole ion trap mass analyzer that allows for reactions between ions generated by electrospray ionization (through the end cap electrode) and atmospheric sampling glow discharge ionization (through the ring electrode). Reprinted from reference [51] from Int. J. Mass Spectrom. Ion Processes, Vol 162, Stephenson J. L., Jr., McLuckey S. A., Adaptation of the Paul trap for study of the reaction of multiply charged cations with singly charged anions, pp 89–106. Copyright 1997 with permission from Elsevier.
Glish and co-workers developed an apparatus similar to that shown in Figure 8 to study reactions between Fe+ and FeCO2− ions with oppositely charged peptide and protein ions [57]. The multiply charged peptide/protein ions were formed by ESI and injected into the ion trap via an end-cap electrode, while the singly charged reagent ions were formed using laser desorption/ablation from a stainless steel surface and entered the ion trap via a hole in the ring electrode. Several types of reaction phenomena were observed including charge reduction via electron transfer, adduct formation, and protein/peptide dissociation.
The three instrument geometries just discussed share the use of reagent ion admission via the ring-electrode and in all three cases, the RF voltage used to create the oscillating quadrupolar field was applied to the ring-electrode. The motivation for admitting ions and electrons through the ring-electrode in the first instance was the lack of a straightforward means for admitting ions of both polarities via an end-cap electrode. While significant progress in studying gas phase ion/ion chemistry has been made with these tools, there are significant drawbacks associated with the admitting ions through the ring-electrode. For example, capture of ions admitted through the ring electrode (radial injection) is estimated to be two orders of magnitude less efficient than capture of ions injected via an end-cap electrode (which, in turn, is at least an order of magnitude less efficient than capture of ions admitted axially into a linear ion trap). Furthermore, the ions injected radially experience stronger electric fields than those injected axially, which often leads to significantly greater degrees of fragmentation [53]. These two factors combine to limit the range of species that can be used to react with multiply charged ions injected through the end-cap electrode.
The apparatus depicted schematically in Figure 9 has been constructed to address limitations associated ion formation via the ring-electrode. A DC quadrupole is employed to direct sequentially oppositely charged ions into the ion trap via an end-cap electrode [58]. Two ion sources (source A and source B) are situated 90° from the axial dimension of the ion trap and 180° from each other, and the ions from each source are guided in turn through a 90° turn into the center of one end cap electrode. The voltages applied to the DC quadrupole and the lens elements between the ion trap and the DC quadrupole can be switched under computer control to coordinate the accumulation of ions of each polarity in the ion trap. The hardware for each source can be operated as an ASGDI source or as an atmosphere/vacuum interface. This arrangement, therefore, allows for the study of a wide range of ionic reactants through ion source combinations that include ESI/ESI, ESI/ASGDI, and ESI/corona discharge ionization. The improved ion capture efficiency and lesser degree of fragmentation upon ion accumulation also allow for the use of ion sources of moderate to low brightness, such as ESI, and therefore made possible the first ion/ion reaction studies of multiply charged ions of opposite polarity in an ion trap [16,59].
Figure 9.
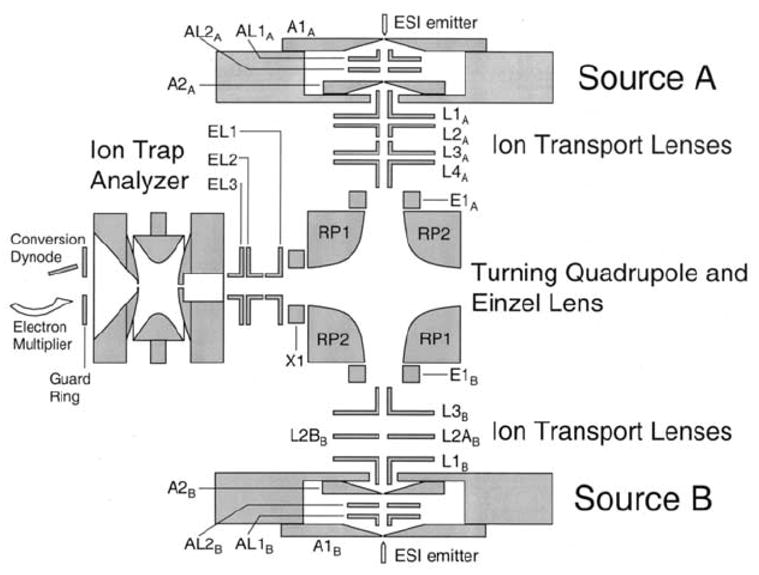
Schematic diagram of the dueling ESI ion trap mass spectrometer. Reprinted from reference [58] with permission of Elsevier from ““Dueling” ESI: instrumentation to study ion/ion reactions of electrospray-generated cations and anions” by Wells J. M., Chrisman P. A., McLuckey S. A., Journal of the American Society for Mass Spectrometry, Vol 13, pp 614–622. Copyright 2002 by the American Society for Mass Spectrometry.
The facility with which electrodynamic ion traps can execute MSn experiments and the high efficiencies associated with ion/ion reactions allows for the use of multiple ion/ion reactions within a single MSn experiment. For some experiments, it is desirable to be able to use more than one type of reagent ion in the “processing” of an analyte ion. In experiments of this kind, it may be desirable to have available more than two ion sources so that each ion source can be individually optimized for the analyte ions and the various reagent ions. A quadrupole ion trap with up to four independent ion sources, as depicted in the schematic diagram of Figure 10, has been constructed to meet this need [60]. This instrument combines features associated with the apparatus shown in Figures 8 and 9. A DC quadrupole is used to direct ions from three independent ion sources to an end-cap electrode, rather than two as in the case of the instrument of Figure 9. As with the apparatus of Figure 8, there is also an ASGDI source mounted orthogonal to the main ion optical axis to inject ions through one ring electrode, as discussed above. This instrument allows remarkable flexibility with respect to the cation/anion combinations that can be used in a single experiment. Novel experiments that employ more than two ion sources have been demonstrated with this device, including the formation multi-unit protein hetero-complexes in the gas-phase [61] and the net increase in the absolute charge of an ion via sequential charge inversion reactions [8,9].
Figure 10.
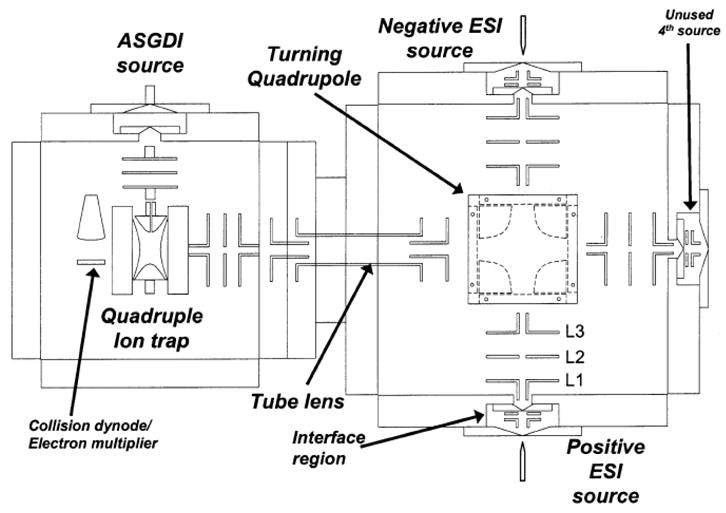
Schematic diagram of multiple-source quadrupole ion trap mass spectrometer. Reprinted from reference [60] by Badman et al. with permission from Anal. Chem. 2002, 74, 6237–6243. Copyright 2002 American Chemical Society.
The most recent description of a three-dimensional ion trap mass spectrometer adapted for ion/ion reactions is that of a commercially available system designed to enable ETD studies (viz., the Bruker HCTultra, also marketed as the Agilent 6340 ETD ion trap). This ion trap system, is comprised of an ESI source for introducing positively charged peptide or protein ions and a chemical ionization source positioned orthogonal to an octopole ion guide leading to an end-cap electrode for injecting radical anions derived from fluoranthene [62,63]. Analyte cations are first accumulated in the ion trap using cation transmission along the octopole ion guide, and the selected precursor is isolated. A gate to transmit negative ions is then opened to allow transmission of anions into the octopole ion guide and on to the entrance end-cap of the ion trap. This approach shares the advantages of the instrument of Figure 9 in that both reagents are injected through an end-cap electrode, thereby avoiding use of the less efficient and more energetic ring-electrode injection.
B. Ionization approaches with linear ion traps
Two of the instrument geometries for admitting oppositely charged ions into linear ion traps have been discussed, i.e., the admission of ions axially from opposite ends of the LIT (see Figure 4) and the injection of one polarity of ions axially and the other radially (see Figure 6). Injection of ions from opposite ends of the LIT, as is done with the LTQ XL linear ion trap marketed by ThermoFinnigan, is facilitated by the use of mass selective radial ejection into detectors on the sides of the quadrupole array for mass analysis [37]. For LITs that use mass selective axial ejection, such as those marketed by ABI and MDS Sciex, a detector is placed adjacent to one of the ends of the quadrupole array. Given the limitations associated with radial injection of ions into ion traps, effort has recently been focused on the development of ion inlets that allow all reactant ions to be admitted through one side of the LIT. This is also desirable for hybrid instruments, even those that employ radial ejection from the LIT, due to limitations in the accessibility of both sides of the LIT as a result of the coupling of the LIT with other analyzers.
For most of the home-built or heavily modified mass spectrometers discussed above for ion/ion reaction studies inside an electrodynamic ion trap, an arrangement of distinct ion sources, each with its own interface, is employed. This maximizes flexibility but from the standpoint of cost and ease of implementation with commercial platforms, it is also desirable to be able to employ a single atmosphere/vacuum interface for all reactant species. Effort has therefore been directed to implementing means for forming and admitting multiple reactant ions through one side of a linear ion trap and via a single vacuum/atmosphere interface. A major objective, however, has been to minimize losses in flexibility, relative to the use of completely independent and distinct ion sources.
The first demonstration of ion/ion reactions in which both reactants are admitted through the same end of the LIT and via a common interface takes advantage of a bipolar ionization source, i.e., a sonic spray ionization (SSI) source, to form both positive and negative ions. SSI is a spray ionization technique first introduced by Hirabayashi et al. in the mid-1990s [64,65], and then adopted and developed further by others [66]. A unique feature associated with SSI is that both positive and negative ions are formed simultaneously. In addition, multiply charged ions can be formed, which is important for ion/ion reaction studies via mass spectrometry. The ion/ion reactions associated with the use of a single SSI source were demonstrated on a triple quadrupole/linear ion trap mass spectrometer in the mutual storage mode, whereby solutions containing both analyte and reagent species are subjected to SSI and the oppositely charged ions are sequentially injected into the Q2 linear ion trap [67]. A variety of potential reagents were examined for the purpose of proton transfer and complex formation reactions. The advantage of using SSI source for ion/ion reactions is that, the SSI source, in principle, can be added to any commercial instrument and be used for most proton-transfer ion/ion reaction experiments demonstrated thus far. The disadvantage, on the other hand, is that analytes and reagents must be mixed in one solution for the simultaneous formation of cationic and anionic reactants. Although no obvious signal suppression was observed in the reported studies, the potential for matrix effects is unavoidable when mixtures of analyte and reagent species are present in solution. Furthermore, the ionization efficiency of sonic spray often does not compare favorably with other forms of spray ionization. Therefore, it is highly desirable to have a dual polarity ionization source that allows for the independent optimization of ionization conditions for analyte and reagent species.
Pulsed dual ESI sources [68] and ESI/APCI sources [69] have been recently described for ion/ion reaction studies that overcome the disadvantages associated with the use of SSI. The home-built dual source is placed directly in front of the atmospheric/vacuum interface of a triple quadrupole/linear ion trap mass spectrometer. A schematic diagram of a pulsed dual electrospray source is shown in Figure 11. The dual ESI/APCI source has a similar arrangement to that of the dual ESI source in which one of the ESI emitters is replaced by an APCI needle. In both cases, alternately pulsed high potentials are applied to each emitter of the dual source, which sequentially generates the oppositely charged ions for subsequent ion/ion reactions. The voltages are pulsed to avoid possible cross-talk between the two emitters if they are both generating ions simultaneously. Minimal compromise in the performance of each ion source due to the placement of the other ion source has been noted. The signals of positive and negative ions upon repeated alternative-pulsing the two emitters has shown good reproducibility such that the combined emitter approach is a robust means for forming both analyte and reactant ions in which each form of ionization can be optimized individually.
Figure 11.
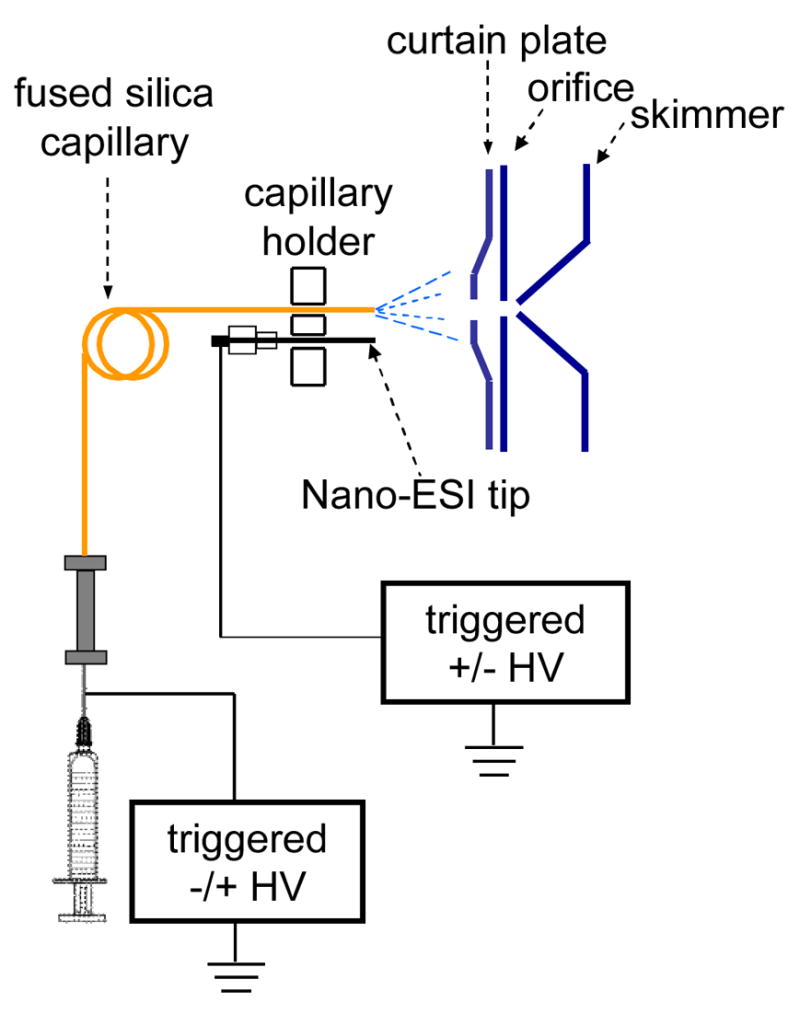
Schematic diagram of the pulsed dual electrospray source coupled with the nano-electrospray ionization interface of a Q TRAP mass spectrometer.
The dual ESI and ESI/APCI sources can both be used for single proton transfer reactions and single electron transfer reactions. ETD experiments with the dual ESI source have been enabled by the recent development of means for forming electron transfer reagents via ESI [70]. However, several ETD reagents that are most readily formed via APCI or CI are more efficient than the ESI derived reagents identified to date. Therefore, at least at this time, the ESI/APCI approach is the more suitable for ETD studies. The alternately pulsed dual ESI source is most suitable in implementation of single and multiple proton-transfer ion/ion reactions. For example, charge inversion reactions, which involve multiple proton transfers, are readily implemented with the dual ESI configuration, but not by the ESI/APCI approach. In this sense, the pulsed dual ESI and ESI/APCI sources are complementary.
The use of pulsed ion sources for the generation of analyte and reagent ions is not necessarily restricted to two ion sources. Provided the necessary power supplies can be gated via the instrument software, it should be possible to incorporate additional emitters. For example, a pulsed triple ionization source, using a common atmospheric/vacuum interface and ion path has been reported [71] to generate different types of ions for sequential ion/ion reaction experiments in a linear ion trap. Compared to the pulsed dual ESI or ESI/APCI source, the pulsed triple ion source is particularly useful for MSn experiments that employ more that one type of reagent ion, such as charge increase via sequential charge inversion reactions [7,8]. The pulsed double or triple ion source holds significant advantages over the SSI source for ion/ion reaction studies in terms of the flexibility to optimize ionization conditions for each ionic species, the wide choice of ion/ion reaction combinations, and superior ionization efficiency. Furthermore, the demonstration of three pulsed ion sources suggests that there is no inherent limit to the number of distinct emitters that might be employed. However, at some point, a static placement of emitters can be expected to lead to significant compromises in sampling efficiency for one or more of the ion sources.
III. Mass analysis for ion/ion reactions
To date, published means for the mass analysis of ion/ion reaction products include a quadrupole mass filter, orthogonal acceleration time-of-flight, and various means for scanning electrodynamic ion traps. Atmospheric pressure ion/ion reaction studies have employed either mass filtering or time-of-flight. In these cases, the ion/ion reaction environment and the mass analyzer are physically de-coupled. The vast majority of ion trap studies, on the other hand, have used the ion trap as both the reaction volume and as the mass analyzer. While the mass analysis figures of merit of an ion trap are modest in some respects, it is convenient to be able to use the reaction vessel as the mass analyzer. Furthermore, the ability of an ion trap to mass analyze high m/z ions is particularly useful for ion/ion reaction studies of ions derived from high mass biomolecules. The upper m/z limit typically supported by most commercial ion trap instruments is 2000–4000. However, ions of much higher m/z ratios can be analyzed with an ion trap by use of, for example, resonance ejection at relatively low qz values [51,72]. In the earliest ion trap ion/ion studies, home-built quadrupole ion trap systems, such as those shown in Figures 7–10, have been controlled by Finnigan ITMS electronics, which are designed for the analysis of ions of m/z below 650. The ion traps in those studies are operated at an angular RF frequency (Ω) =1.1 MHz, ring electrode radius (r0) =1.0 cm, and the amplitude of RF applied to the ring electrode (VRF) =0–7500 V0-p [53]. Singly charged bovine serine albumin ions (~ m/z 66,000) formed via ion/ion deprotonated of multiply charged albumin have been observed in ion traps operated under these conditions via resonance ejection, but with at very low relative abundance. A significant decrease in ion storage efficiency beyond m/z 30,000 is generally observed and is attributed to an increasingly weak trapping field (low Dz, see Equation 2) as the ion m/z increases.
The Hitachi M-8000 quadrupole ion trap mass spectrometer (San Jose, CA), later modified for ion/ion studies to give an instrument similar in geometry to that of Figure 8 [55], is operated at Ω =770 kHz, r0 =0.707 cm, and V =0–8000 Vo-p. With a smaller radius and functioning at a lower angular frequency, the M-8000 provides deeper trapping wells for a given value of VRF than the ITMS systems just mentioned. Ions with m/z values as high as 150,000 have been analyzed via the M-8000 quadrupole ion trap, as illustrated in Figure 13.
Figure 13.
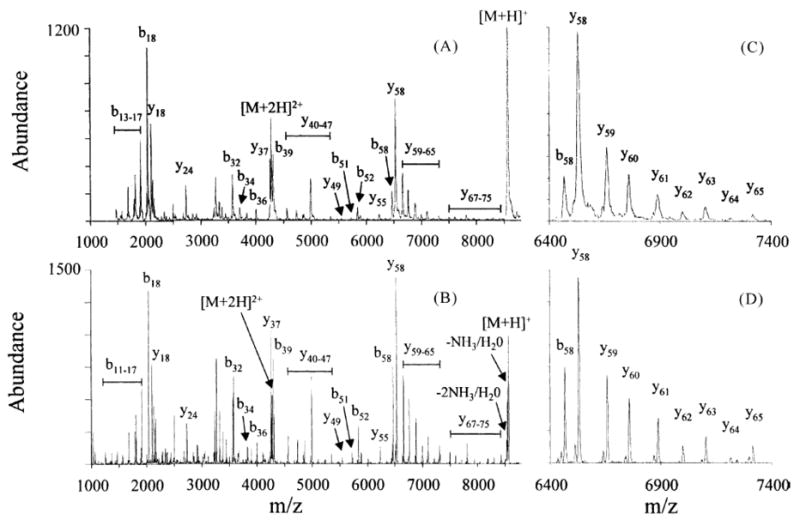
Post ion/ion reaction CID mass spectra of the [M+7H]7+ ion of ubiquitin obtained from (A) Finnigan ITMS and (B) Hitachi model M-8000 mass spectrometers. (C) and (D) show expanded mass-to-charge ratio regions from 6400 to 7400 of the data in A and B, respectively. Reprinted from reference [53] from Int. J. Mass Spectrom., Vol 222, Reid G. E., Wells J. M., Badman E. R., McLuckey S. A., Performance of a quadrupole ion trap mass spectrometer adapted for ion/ion reaction studies, pp 243–258. Copyright 2003 with permission from Elsevier.
A comparison of the mass analysis performance between the ITMS system and the M-8000 system is demonstrated in Figure 13, with experiments involving charge reduction of CID products of [M+7H]7+ ubiquitin ions. The superior mass resolving power of the M-8000 system is apparent. The modified M-8000 quadruople ion trap generally provides mass accuracy of ~200 ppm (external calibration) and a mass resolving power of ~1500 at m/z < 20,000.
The M-8000 quadrupole ion trap mass spectrometer with improved mass analysis performance relative to the ITMS-based system proved to be particularly useful in top-down protein analysis with an ion trap. For example, interpretable MS/MS spectra of proteins up to 25.9 kDa have been obtained on this instrument [6] and modified a priori unknown proteins have been characterized, with the aid of ion/ion proton transfer reactions [73].
Much less information regarding the mass analysis characteristics of linear ion traps at high m/z ratios has appeared, relative to the performance of 3D ion traps. The only published work was collected with the instrument depicted in Figure 6, which is a modified MDS Sciex QTRAP 2000 benchtop instrument, based on the use of mass-selective axial ejection from a low pressure (10−5 Torr range) linear ion trap. An upper m/z limit of at least 33,000 has been observed for the ion/ion reaction products, with resolving power around 1000 for ions with m/z below 20,000. It is premature to draw general conclusions about mass analysis characteristics of MSAE for high m/z ions from the limited studies performed. However, for linear ion traps employing radial ejection, the parameters affecting mass analysis for high m/z ions can be expected to be similar to those for a 3D ion trap.
In most of the ion/ion reaction studies described to date, either the ionization and reaction environment have been closely coupled, as with the atmospheric pressure studies, or the reaction vessel also served as the mass analyzer, as with most of the ion trap experiments. For optimum performance and flexibility, however, it is desirable to be able to de-couple each of the aspects of Scheme 1 as much as possible so that each can be individually and independently optimized. Ion trap-based instruments already largely decouple ionization from the reaction environment and they provide a high degree of experimental flexibility with their MSn capabilities. Therefore, an apparatus comprised an ion trap for ion/ion reactions, coupled with a form of mass analysis with superior mass analysis characteristics relative to those of ion traps, can realize the goal of a tool that largely de-couples all aspects of an ion/ion reaction depicted in Scheme 1. Some of the logical instrument platforms that can be envisioned include hybrid mass spectrometers with a combination of an electrodynamic ion trap with i) a time-of-flight (TOF) mass analyzer, ii) an orbitrap mass analyzer, and iii) a FTICR mass analyzer. A hybrid instrument with TOF mass analysis and ion/ion reaction capabilities has recently been reported based on a modified commercial quadrupole time-of-flight tandem mass spectrometer (QSTAR XL, Applied Biosystems/MDS SCIEX, Concord, ON, Canada) [74]. The instrument consists of three quadrupoles (ion guide Q0, mass filter Q1, collision cell Q2) and a reflectron TOF analyzer with orthogonal injection of ions. A home-built pulsed dual ESI source or a nano-ESI/APCI source has been employed for the formation of reactant ions for proton transfer reactions or ETD reactions. The key hardware modification of the instrument is the superposition of auxiliary RF signals to the containment lenses of the Q2 quadrupole array to enable mutual storage ion/ion reactions. Note that such a modification is not necessary to perform transmission mode ion/ion reactions (methods II and III). The schematic of such an instrument is shown in Figure 14 and an example of an experimental sequence is shown in which DC potentials along the instrument axis at different steps are indicated.
Figure 14.

Schematic drawing of a modified quadrupole time-of-flight tandem mass spectrometer (QSTAR XL). The plots below show the potential along the instrument axis at different steps for single ion/ion reaction experiments. Reprinted from reference [74] by Xia et al. with permission from Anal. Chem. 2006, 78, 4146–4154. Copyright 2006 American Chemical Society.
The quadrupole time-of-flight tandem mass spectrometer, abbreviated here as QqTOF, is analogous to the hybrid triple quadrupole/LIT instrument of Figure 6, except that the orthogonal TOF mass analyzer replaces a third linear ion trap (Q3). The TOF provides significant improvements in mass resolving power and mass measurement accuracy. An upper m/z of around 66,000 is observed, which is most likely limited by the poor detection efficiency of singly charged high m/z ions with the relatively low acceleration voltage applied in the TOF section (4 kV). The mass resolving power for the ion/ion products over wide m/z range is about 8000, with mass accuracy around 20 ppm for external calibration and 5 ppm for internal calibration. The post-ion/ion CID spectrum of [M+8H]8+ ubiquitin ions acquired on the modified QqTOF instrument is shown in Figure 15 to provide an indication of the quality of the data. Essentially all the key ion/ion experiments demonstrated with ions traps can be readily implemented, such as proton transfer reactions, ETD reactions, and parallel ion parking for multi-components. The QqTOF mass spectrometer modified with ion/ion capabilities provides a powerful platform for the analysis of the macro-ions in the gas phase and is one example of high performance hybrid instruments that can be envisioned.
Figure 15.

Post ion/ion reaction CID mass spectra of the [M+8H]8+ ion of ubiquitin obtained from the apparatus shown in Figure 14. Reprinted from reference [74] by Xia et al. with permission from Anal. Chem. 2006, 78, 4146–4154. Copyright 2006 American Chemical Society.
Conclusions
Innovation in instrument development has played a fundamental role in the exploration and application of ion/ion reactions involving multiply charged ions. Further opportunities for innovation are apparent in all key aspects of an ion/ion reaction experiment, i.e. approaches for formation and introduction of reactant ions, conditions for conducting reactions, and product ion mass analysis. There are certainly many opportunities to explore the use of other ionization methods for the formation of reactant ions. When the range of gas-phase ions that can be formed via the panoply of ionization methods available to the mass spectrometrist is considered, it is likely that new ion/ion reaction phenomenologies can be discovered. Electrodynamic ion traps are likely to continue to be widely used as ion/ion reaction vessels, particularly as systems based on ion traps that support ion/ion reaction experiments are offered commercially. However, the use of crossed or merged beams for the study of ion/ion reactions could provide information that is difficult or impossible to obtain with the atmospheric pressure and ion trap-based methods. The advent of hybrid instruments that separate the component aspects of an ion/ion reaction experiment makes the development of tools that can reach very high performance standards, in terms of sensitivity, mass resolution, mass accuracy, etc. The recent adaptation of a hybrid quadrupole linear ion trap-orbitrap mass spectrometer for ion/ion reaction studies exemplifies such a trend [75]. Ion/ion reactions might also find use with hybrid instruments capable of providing both mass and ion mobility information. Such developments will underlie improved understanding and use of gas-phase ion/ion reactions and thereby expand the potential of mass spectrometry for the solution of problems in modern science.
Figure 12.
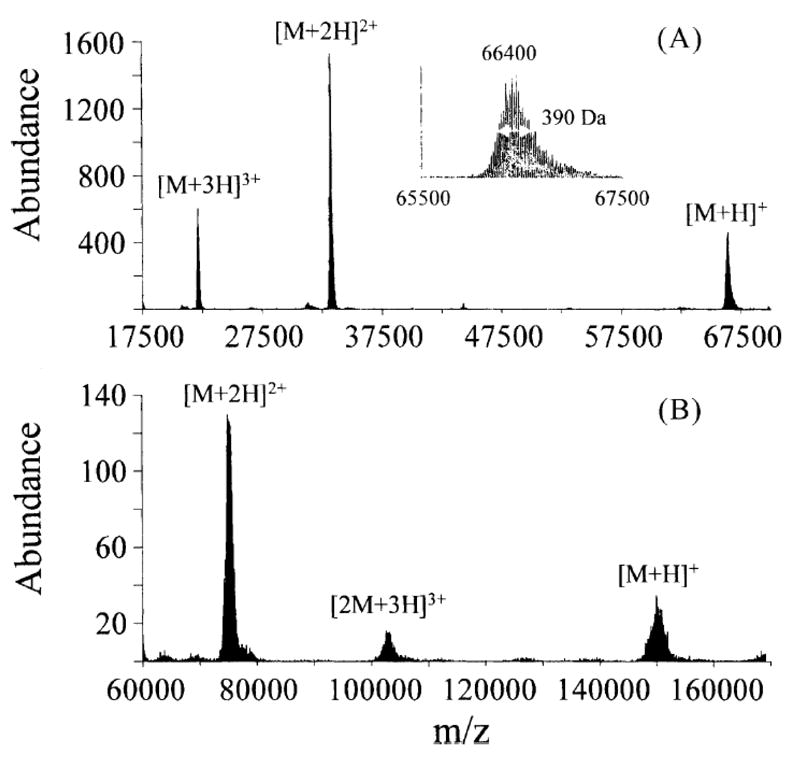
Post ion/ion reaction mass spectrum of (A) bovine serum albumin, and (B) bovine immunoglobulin IgG. Reprinted from reference [53] from Int. J. Mass Spectrom., Vol 222, Reid G. E., Wells J. M., Badman E. R., McLuckey S. A., Performance of a quadrupole ion trap mass spectrometer adapted for ion/ion reaction studies, pp 243–258. Copyright 2003 with permission from Elsevier.
Acknowledgments
The authors acknowledge support from the National Institutes of Health under Grant GM 45372 for much of the ion trap instrument development associated with our work. We also acknowledge Mr. Chris Doerge and Dr. Bob Santini of the Amy Instrumentation Facility for making key contributions to our instrument development work at Purdue. We also acknowledge the support and collaboration of MDS Sciex, and Jim Hager, Frank Londry, and Bruce Thomson, in particular, in our work with hybrid instruments that employ linear ion traps.
Footnotes

Description: Use of ion/ion reactions in electrodynamic ion traps allow ionization and reaction processes to be independently optimized and reactions to be implemented within the context of MSn experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cooks RG. Creativity through instrumentation. Anal Chem. 1985;57:823A–843A. [Google Scholar]
- 2.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass-spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 3.McLuckey SA, Reid GE, Wells JM. Ion parking during ion/ion reactions in electrodynamic ion traps. Anal Chem. 2002;74:336–346. doi: 10.1021/ac0109671. [DOI] [PubMed] [Google Scholar]
- 4.Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. Gas-phase concentration, purification, and identification of whole proteins from complex mixtures. J Am Chem Soc. 2002;124:7353–7362. doi: 10.1021/ja025966k. [DOI] [PubMed] [Google Scholar]
- 5.Stephenson JL, Jr, McLuckey SA. Charge manipulation for improved mass determination of high-mass species and mixture components by electrospray mass spectrometry. J Mass Spectrom. 1998;33:664–672. doi: 10.1002/(SICI)1096-9888(199807)33:7<664::AID-JMS663>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 6.Hogan JM, McLuckey SA. Charge state dependent collision-induced dissociation of native and reduced porcine elastase. J Mass Spectrom. 2003;38:245–256. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 7.He M, Emory JF, McLuckey SA. Reagent anions for charge inversion of polypeptide/protein cations in the gas phase. Anal Chem. 2005;77:3173–3182. doi: 10.1021/ac0482312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He M, McLuckey SA. Two ion/ion charge inversion steps to form a doubly protonated peptide from a singly protonated peptide in the gas phase. J Am Chem Soc. 2003;125:7756–7757. doi: 10.1021/ja0354521. [DOI] [PubMed] [Google Scholar]
- 9.He M, McLuckey SA. Increasing the negative charge of a macroanion in the gas phase via sequential charge inversion reactions. Anal Chem. 2004;76:4189–4192. doi: 10.1021/ac496087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunawardena HP, Emory JF, McLuckey SA. Phosphopeptide anion characterization via sequential charge inversion and electron transfer dissociation. Anal Chem. 2006;78:3788–3793. doi: 10.1021/ac060164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci US A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. Int J Mass Spectrom. 2004;236:33–42. [Google Scholar]
- 13.Newton KA, Amunugama R, McLuckey SA. Gas-phase ion/ion reactions of multiply protonated polypeptides with metal containing anions. J Phys Chem A. 2005;109:3608–3616. doi: 10.1021/jp04416i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton KA, McLuckey SA. Gas-phase peptide/protein cationizing agent switching via ion/ion reactions. J Am Chem Soc. 2003;125:12404–12405. doi: 10.1021/ja036924e. [DOI] [PubMed] [Google Scholar]
- 15.Newton KA, McLuckey SA. Generation and manipulation of sodium cationized peptides in the gas phase. J Am Soc Mass Spectrom. 2004;15:607–615. doi: 10.1016/j.jasms.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Wells JM, Chrisman PA, McLuckey SA. Formation of protein-protein Complexes in vacuo. J Am Chem Soc. 2001;123:12428–12429. doi: 10.1021/ja0170403. [DOI] [PubMed] [Google Scholar]
- 17.McLuckey SA, Stephenson JL., Jr Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Pitteri SJ, McLuckey SA. Recent developments in the ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 2005;24:197–241. doi: 10.1002/mas.20048. [DOI] [PubMed] [Google Scholar]
- 19.Ogorzalek-Loo RR, Udseth HR, Smith RD. Evidence of charge inversion in the reaction of singly charged anions with multiply charged macroions. J Phys Chem. 1991;95:6412–6415. [Google Scholar]
- 20.Ogorzalek-Loo RR, Udseth HR, Smith RD. A new approach for the study of gas-phase ion-ion reactions using electrospray ionization. J Am Soc Mass Spectrom. 1992;3:695–705. doi: 10.1016/1044-0305(92)87082-A. [DOI] [PubMed] [Google Scholar]
- 21.Scalf M, Westphall MS, Krause J, Kaufman SL, Smith LM. Controlling charge states of large ions. Science. 1999;283:194–197. doi: 10.1126/science.283.5399.194. [DOI] [PubMed] [Google Scholar]
- 22.Scalf M, Westphall MS, Smith LM. Charge reduction electrospray mass spectrometry. Anal Chem. 2000;72:52–60. doi: 10.1021/ac990878c. [DOI] [PubMed] [Google Scholar]
- 23.Ebeling DD, Westphall MS, Scalf M, Smith LM. Corona discharge in charge reduction electrospray mass spectrometry. Anal Chem. 2000;72:5158–5161. doi: 10.1021/ac000559h. [DOI] [PubMed] [Google Scholar]
- 24.Frey BL, Lin Y, Westphall MS, Smith LM. Controlling gas-phase reactions for efficient charge reduction electrospray mass spectrometry of intact proteins. J Am Soc Mass Spectrom. 2005;16:1876–1887. doi: 10.1016/j.jasms.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mather RE, Todd JFJ. The quadrupole ion store (quistor).7. Simultaneous positive-negative ion mass-spectrometry. Int J Mass Spectrom Ion Phys. 1980;33:159–165. [Google Scholar]
- 26.Herron WJ, Goeringer DE, McLuckey SA. Ion-ion reactions in the gas-phase: proton transfer reactions of protonated pyridine with multiply-charged oligonucleotide anions. J Am Soc Mass Spectrom. 1995;6:529–532. doi: 10.1016/1044-0305(95)00199-N. [DOI] [PubMed] [Google Scholar]
- 27.Louris JN, Brodbelt-Lustig JS, Cooks RG, Glish GL, Vanberkel GJ, Mcluckey SA. Ion isolation and sequential stages of mass-spectrometry in a quadrupole ion trap mass-spectrometer. Int J Mass Spectrom Ion Processes. 1990;96:117–137. [Google Scholar]
- 28.Stephenson JL, Jr, McLuckey SA. Anion effects on storage and resonance ejection of high mass-to-charge cations in quadrupole ion trap mass spectrometry. Anal Chem. 1997;69:3760–3766. [Google Scholar]
- 29.March RE, Londry FA. Theory of quadrupole mass spectrometry in practical aspects of ion trap mass spectrometry. In: March RE, Todd JFJ, editors. Chapter 2. Vol. 1. CRC Press; Boca Raton: 1995. [Google Scholar]
- 30.McLuckey SA, Wu J, Bundy JL, Stephenson JL, Jr, Hurst GB. Oligonucleotide mixture analysis via electrospray and ion/ion reactions in a quadrupole ion trap. Anal Chem. 2002;74:976–984. doi: 10.1021/ac011015y. [DOI] [PubMed] [Google Scholar]
- 31.Grosshans PB, Ostrander CM, Walla CA. US Patent 6,570, -15 B1. 2003. [Google Scholar]
- 32.Grosshans PB, Ostrander CM, Walla CA. US Patent 6,674, -067 B2. 2004. [Google Scholar]
- 33.Chrisman PA, Pitteri SJ, McLuckey SA. Parallel ion parking: improving conversion of parents to first-generation products in electron transfer dissociation. Anal Chem. 2005;77:3411–3414. doi: 10.1021/ac0503613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chrisman PA, Pitteri SJ, McLuckey SA. Parallel ion parking of protein mixtures. Anal Chem. 2006;78:310–316. doi: 10.1021/ac0515778. [DOI] [PubMed] [Google Scholar]
- 35.Hager JW. A new linear ion trap mass spectrometer. Rapid Commun Mass Spectrom. 2002;16:512–526. doi: 10.1002/rcm.1020. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz JC, Senko MW, Syka JEP. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13:659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 37.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci US A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi A, Bai DL, Geer LY, Shabanowitz J, Hunt DF. Analysis of intact proteins on a chromatographic time scale by electron transfer dissociation tandem mass spectrometry. Int J Mass Spectrom. 2007;259:197–203. doi: 10.1016/j.ijms.2006.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. Electron transfer dissociation of peptide anions. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Hager JW, Londry FA. US Patent, 2005/0263697 A1. 2005. [Google Scholar]
- 41.Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW. Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 42.Londry FA, Hager JW. Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Hager JW, Xia Y, Londry FA, McLuckey SA. Positive ion transmission mode ion/ion reactions in a hybrid linear ion trap. Anal Chem. 2004;76:5006–5015. doi: 10.1021/ac049359m. [DOI] [PubMed] [Google Scholar]
- 44.Liang X, Hager JW, McLuckey SA. Transmission mode ion/ion electron transfer dissociation in a linear ion trap. Anal Chem. 2007;79:3363–3370. doi: 10.1021/ac062295q. [DOI] [PubMed] [Google Scholar]
- 45.Liang X, McLuckey SA. Transmission mode ion/ion proton transfer reactions in a linear ion trap. J Am Soc Mass Spectrom. 2007;18:882–890. doi: 10.1016/j.jasms.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creaser CS. In: Practical aspects of ion trap mass spectrometry, volume iii. March RE, Todd JFJ, editors. CRC Press; Boca Raton: 1995. pp. 239–253. [Google Scholar]
- 47.Herron WJ, Goeringer DE, McLuckey SA. Product ion charge state determination via ion/ion proton transfer reactions. Anal Chem. 1996;68:257–262. doi: 10.1021/ac950895b. [DOI] [PubMed] [Google Scholar]
- 48.Herron WJ, Goeringer DE, McLuckey SA. Gas-phase electron-transfer reactions from multiply-charged anions to rare-gas cations. J Am Chem Soc. 1995;117:11555–11562. [Google Scholar]
- 49.Kaiser RE, Jr, Cooks RG, Stafford GC, Syka JEP, Hemberger PH. Operation of a quadrupole ion trap mass-spectrometer to achieve high mass charge ratios. Int J Mass Spectrom Ion Processes. 1991;106:79–115. [Google Scholar]
- 50.Berberich DW, Yost RA. Negative chemical-ionization in quadrupole ion trap mass spectrometry - effects of applied voltages and reaction-times. J Am Soc Mass Spectrom. 1994;5:757–764. doi: 10.1016/1044-0305(94)80008-1. [DOI] [PubMed] [Google Scholar]
- 51.Stephenson JL, Jr, McLuckey SA. Adaptation of the Paul trap for study of the reaction of multiply charged cations with singly charged anions. Int J Mass Spectrom Ion Processes. 1997;162:89–106. [Google Scholar]
- 52.McLuckey SA, Glish GL, Asano KG, Grant BC. Atmospheric sampling glow discharge ionization source for the analysis of trace organics in ambient air. Anal Chem. 1988;60:2220–2228. [Google Scholar]
- 53.Reid GE, Wells JM, Badman ER, McLuckey SA. Performance of a quadrupole ion trap mass spectrometer adapted for ion/ion reaction studies. Int J Mass Spectrom. 2003;222:243–258. [Google Scholar]
- 54.Stephenson JL, Jr, McLuckey SA. Ion/ion reactions in the gas-phase: proton transfer reactions involving multiply-charged proteins. J Am Chem Soc. 1996;118:7390–7397. [Google Scholar]
- 55.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Electron transfer ion/ion reactions in a three-dimensional quadrupole ion trap: Reactions of doubly and triply protonated peptides with SO2•−. Anal Chem. 2005;77:1831–1839. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. Complementary structural information from a tryptic N-linked glycopeptide via electrontransfer ion/ion reactions and collision-induced dissociation. J Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Payne AH, Glish GL. Gas-phase ion/ion interactions between peptides or proteins and iron ions in a quadrupole ion trap. Int J Mass Sepctrom. 2001;204:47–54. [Google Scholar]
- 58.Wells JM, Chrisman PA, McLuckey SA. “Dueling” ESI: Instrumentation to study ion/ion reactions of electrospray-generated cations and anions. J Am Soc Mass Spectrom. 2002;13:614–622. doi: 10.1016/S1044-0305(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 59.Wells JM, Chrisman PA, McLuckey SA. Formation and characterization of protein-protein complexes in vacuo. J Am Chem Soc. 2003;125:7238–7249. doi: 10.1021/ja035051l. [DOI] [PubMed] [Google Scholar]
- 60.Badman ER, Chrisman PA, McLuckey SA. A quadrupole ion trap mass spectrometer with three independent ion sources for the study of gas-phase ion/ion reactions. Anal Chem. 2002;74:6237–6243. doi: 10.1021/ac026020w. [DOI] [PubMed] [Google Scholar]
- 61.Gunawardena HP, McLuckey SA. Synthesis of multi-unit protein hetero-complexes in the gas phase via ion-ion chemistry. J Mass Spectrom. 2004;39:630–638. doi: 10.1002/jms.629. [DOI] [PubMed] [Google Scholar]
- 62.Brekenfeld A, Ledertheil T, Lubeck M, Baessmann C, Hartmer R. Combination of ergodic and non-ergodic ion activation, new approcah for PTM-peptide identification. Proceedings of the 53rd ASMS Conference; San Antonio, TX. 2005. [Google Scholar]
- 63.Tang N, Goodley PC, Horn D, Miller C, Hoerth P, Mohsin S. Improving protein identification using a combination of collision induced dissociation and electron transfer dissociation MS/MS. Proceedings of the 54th ASMS Conference; Seattle, WA. 2006. [Google Scholar]
- 64.Hirabayashi A, Sakairi M, Koizumi H. Sonic spray mass-spectrometry. Anal Chem. 1995;67:2878–2882. doi: 10.1021/ac00113a023. [DOI] [PubMed] [Google Scholar]
- 65.Hirabayashi A, Sakairi M, Koizumi H. Sonic spray ionization method for atmospheric-pressure ionization mass-spectrometry. Anal Chem. 1994;66:4557–4559. [Google Scholar]
- 66.Takats Z, Nanita SC, Cooks RG, Schlosser G, Vekey K. Amino acid clusters formed by sonic spray ionization. Anal Chem. 2003;75:1514–1523. doi: 10.1021/ac0260793. [DOI] [PubMed] [Google Scholar]
- 67.Xia Y, Liang X, McLuckey SA. Sonic spray as a dual polarity ion source for ion/ion reactions. Anal Chem. 2005;77:3683–3689. doi: 10.1021/ac0481811. [DOI] [PubMed] [Google Scholar]
- 68.Xia Y, Liang X, McLuckey SA. Pusled dual electrospray ionization for ion/ion reactions. J Am Soc Mass Spectrom. 2005;16:1750–1756. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 69.Liang XR, Xia Y, McLuckey SA. Alternately pulsed nano-electrospray ionization/atmospheric pressure chemical ionization for ion/ion reactions in an electrodynamic ion trap. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang T, Emory JF, O’Hair RAJ, McLuckey SA. Electron transfer reagent anion formation via electrospray ionization and collision-induced dissociation. Anal Chem. 2006;78:7387–7391. doi: 10.1021/ac061409v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liang X, Han H, Xia Y, McLuckey SA. A pulsed triple ionization source for sequential ion/ion reactions in an electrodynamic ion trap. J Am Soc Mass Spectrom. 2007;18:369–376. doi: 10.1016/j.jasms.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Kaiser RE, Jr, Cooks RG, Moss J, Hemberger PH. Mass range extension in a quadrupole ion trap mass spectrometer. Rapid Commun Mass Spectrom. 1989;3:50–53. [Google Scholar]
- 73.Amunugama R, Hogan JM, Newton KA, McLuckey SA. Whole protein dissociation in a quadrupole ion trap: Identification of an a priori unknown modified protein. Anal Chem. 2004;76:720–727. doi: 10.1021/ac034900k. [DOI] [PubMed] [Google Scholar]
- 74.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang X, Londry FA, Yang MJ, McLuckey SA. Implementation of ion/ion reactions in a quadrupole/time-of-flight tandem mass spectrometer. Anal Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of electron-transfer dissociation on a hybrid linear ion trap-orbitrap mass spectrometer. Anal Chem. 2007;79:3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]