

Summary
Hypothalamic-pituitary-adrenocortical (HPA) axis hyperactivity is associated with major depressive disorders, and treatment with classical antidepressants ameliorates not only psychopathological symptoms, but also the dysregulation of the HPA axis. Here, we further elucidated the role of impaired cannabinoid type 1 (CB1) receptor signaling for neuroendocrine and behavioral stress coping in the mouse forced swim test (FST). We demonstrate that the genetic inactivation of CB1 is accompanied by increased plasma corticosterone levels both under basal conditions and at different time points following exposure to the FST. The latter effect could be mimicked in C57BL/6N mice by acute, subchronic and chronic administration of the selective CB1 antagonist SR141716. Further experiments confirmed the specificity of corticosterone-elevating SR141716 actions for CB1 in CB1-deficient mice. Subchronic and chronic pharmacological blockade of CB1, but not its genetic deletion, induced antidepressant-like behavioral responses in the FST that were characterized by decreased floating and/or increased struggling behavior. The antidepressant-like behavioral effects of acute desipramine treatment in the FST were absent in CB1-deficient mice, but the dampening effects of desipramine on FST stress-induced corticosterone secretion were not compromised by CB1-deficiency. However, antidepressant-like behavioral desipramine effects were intact in C57BL/6N mice pre-treated with SR141716, indicating potential developmental deficits in CB1-deficient mice. We conclude that pharmacological blockade of CB1 signaling shares antidepressant-like behavioral effects with desipramine, but reveals opposite effects on HPA axis activity.
Keywords: corticosterone, forced swim test, desipramine, CB1, SR141716, HPA axis, depression
INTRODUCTION
Endocannabinoids act as retrograde messengers in the brain that control the release of several neurotransmitters, including glutamate and GABA, by binding to presynaptic cannabinoid receptor type 1 receptors (CB1) (Marsicano and Lutz, 2006; Chevaleyre et al., 2006). In this manner, the endocannabinoid system functions as a neuromodulatory system to maintain the homeostasis of the brain, which is constantly challenged by physical and psychological stressors. One major neuroendocrine response to stress is the secretion of corticosterone via activation of the hypothalamic-pituitary-adrenocortical (HPA) axis. Expression of CB1 occurs at different levels controlling HPA axis function. These include limbic brain regions such as the hippocampus and amygdala (Marsicano and Lutz, 1999; Mackie, 2005), the paraventricular nucleus of the hypothalamus (PVN) (Cota et al., 2003), the pituitary (Wenger et al., 1999; Pagotto et al., 2001) and the adrenal glands (Galiegue et al., 1995; Buckley et al., 1998), suggesting a multiple role of the endocannabinoid system in the regulation of the hormonal stress response. Indeed, pharmacological blockade of CB1 by the selective antagonist SR141716 (rimonabant) in rodents or inactivation via gene knockout in mice resulted in increased basal and stress-induced ACTH and corticosterone levels (Manzanares et al., 1999; Uriguen et al., 2004; Barna et al., 2004; Haller et al., 2004; Patel et al., 2004; Wade et al., 2006). However, several contradictory results have been reported, in particular with CB1 null mutants (CB1−/−), showing increased stress hormone secretion after novelty stress (Barna et al., 2004; Haller et al., 2004), but not after saline injection (Wenger et al., 2003) or auditory stress (Fride et al., 2005). Similarly, CB1−/− mice were found to have either increased (Barna et al., 2004; Cota et al., 2007), decreased (Uriguen et al., 2004) or similar basal corticosterone levels (Fride et al., 2005; Wade et al., 2006) as compared to wild-type mice. Important factors determining these differences could include the nature of the stressor and the genetic background. Also potential compensatory mechanisms in CB1−/− mice due to the life-long absence of CB1 in these animals have to be considered as recent work from Wade and co-workers suggested (Wade et al., 2006). Thus, in order to establish a general role of endocannabinoid signaling for HPA axis function, it seems essential to substantiate findings in CB1−/− mice with those following pharmacological blockade of CB1 in the respective background strain to exclude potential developmental adaptations.
Increased HPA axis activity is known as a risk factor for depression in humans (Holsboer, 2000; De Kloet et al., 2005). Accordingly, normalization of heightened HPA axis activity seems to be linked to the clinical efficacy of antidepressant treatment (Holsboer, 2000; Ising et al., 2006). CB1-deficient mice display a variety of behavioral and neurovegetative symptoms, which are reminiscent of the melancholic subtype of depression (Hill and Gorzalka, 2005a). These include, among others, hyperactivity of the HPA axis as evidenced by increased CRH expression in the PVN (Cota et al., 2003; Cota et al., 2007), increased corticosterone and ACTH release (Barna et al., 2004; Haller et al., 2004; Cota et al., 2007), attenuated dexamethasone suppression (Cota et al., 2007) and diminished glucocorticoid receptor (GR) expression in the hippocampus (Cota et al., 2007). Accordingly, enhancing endocannabinoid signaling pharmacologically was shown to suppress stress-induced corticosterone secretion (Patel et al., 2004) and to exert antidepressant-like effects in the rat forced swim test (FST), the mouse tail suspension test (TST) and the rat chronic mild stress paradigm (Hill and Gorzalka, 2005b; Gobbi et al., 2005; Bortolato et al., in press). On the other hand, pharmacological blockade of CB1 was also found to exerted antidepressant-like effects in mice in the TST and FST (Shearman et al., 2003; Tzavara et al., 2003; Griebel et al., 2005), which suggested that despite unfavorable neuroendocrine effects, CB1 antagonists could actually have certain antidepressant-like potential (Witkin et al., 2005). This discrepancy between behavioral and neuroendocrine effects of CB1 blockade or deficiency has not yet been thoroughly investigated, particularly not under a chronic treatment schedule, which best relates to the usual long-term application of antidepressants in humans.
To better characterize the potential benefits or costs of CB1 impairment in terms of antidepressant-like behavioral effects and concomitant hyperactivity of the HPA axis, we investigated the consequences of acute, subchronic and chronic SR141716 treatment in combination with the genetic inactivation of CB1 on behavioral and neuroendocrine measures in the FST, a standard test for assessing antidepressant-like effects in rodents (Cryan and Holmes, 2005), and we evaluated the results in relation to those of desipramine treatment.
METHODS
Animals
Mice were kept under standard conditions with food and water ad libitum. They were housed in groups, either in the animal facility of the University of Texas Southwestern Medical Center under a regular 12 h:12 h light/dark schedule (lights on at 0700 h), or in the animal facility of the Max Planck Institute of Psychiatry under a 12 h:12 h inverted light/dark schedule (lights on at 2100 h). C57BL/6N mice were purchased from Charles River (Germany or USA). Cannabinoid receptor type 1 null-mutant (CB1−/−) mice and their wild-type (CB1+/+) littermates derived from heterozygous breeding pairs, which were backcrossed to the C57BL/6N background for at least 6 generations. They were generated and genotyped as described (Marsicano et al., 2002). Age of tested animals ranged between 2 and 4 months. Female mice were not controlled for their estrus cycle. Initial experiments, where the estrus cycle phase had been determined by vaginal smears, revealed a similar distribution between the different phases in female CB1+/+ and CB1−/− mice (data not shown). Animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Government of Bavaria (Germany) or were approved by the UT Southwestern’s Institutional Animal Care and Use Committee (TX, USA). Two weeks before the experiments, animals were separated and singly housed.
Treatment of blood samples and hormone analysis
Trunk blood was collected in pre-chilled tubes containing EDTA and a protease inhibitor (10 μl aprotinin, Trasylol, Bayer, Germany). Blood samples were centrifuged for 15 min at 2500 g at 4°C. Plasma samples were stored in aliquots at −80°C until assay. Plasma corticosterone concentrations were measured by commercially available RIA kits (MP Biomedicals, Eschwege, Germany), as described elsewhere (Karanth et al., 1997), according to manufacturer’s instructions.
Drugs
SR141716 (NIMH Chemical Synthesis and Drug Supply Program, USA) was dissolved in vehicle solution (1 drop of Tween-80 in 1.5 ml of 2.5% dimethyl sulfoxide in 0.9% saline) and injected IP in a volume of 10 ml/kg body weight. Desipramine hydrochloride (Sigma-Aldrich, Steinheim, Germany) was dissolved in vehicle solution (2% dimethyl sulfoxide in 0.9% saline) and injected IP in a volume of 10 ml/kg body weight.
Forced swim test (FST)
Each mouse was placed into a 5-liter glass beaker (height 23.5 cm; diameter 16.5 cm) containing water up to a height of 15 cm at 25 ± 1°C for 6 min. The water was exchanged after each trial. Floating (immobility) and struggling time were scored over the entire 6 min exposure by pressing preset keys on a computer keyboard, using customized freeware software (EVENTLOG). The resulting two-channel ethogram was further processed by customized software (Winrat Vers. 2.31; Heinz Barthelmes, MPI Munich). A mouse was judged floating when it stopped any movements except those that were necessary to keep its head above water. Vigorous swimming movements involving all 4 limbs of the mouse with the front paws breaking the surface of the water, usually at the walls of the cylinder, were regarded as struggling. Animals’ behavior was analyzed on-line by trained observers who were blind to treatment and genotype. After the FST, animals were placed in their home cages and were left undisturbed until 10 min, 20 min, 30 min or 120 min after the onset of the stressor, when they were killed by decapitation after short isoflurane anesthesia within 45 sec after touching the home cage.
Statistical analysis
Data were analyzed for multiple comparisons using one-, two- or three-way analysis of variance (ANOVA) followed by post-hoc Newman-Keuls Multiple Comparison Test. Homogeneity of variance in independent groups was analyzed using the Levene test and data were subjected to logarithmic or square-root transformation, where required. For two-group comparisons unpaired Student’s t-test was used. Differences were considered statistically significant if p < 0.05. Data are presented as mean ± SEM. Sample sizes are reported in figure legends.
Experiments
All experiments were performed during the second half of the dark phase of the circadian rhythm of the animals under red-light conditions. Each time-point assessed represented an independent batch of male or female animals. Experiments and sample analysis were performed blind to the animals’ treatment or genotype. Experimental graphic charts of the following experiments, depicting the respective time-points of treatment, blood sampling and FST exposure, are shown schematically in the corresponding figures.
Experiment 1: Corticosterone secretion following the pharmacological blockade of CB1
Male C57BL/6N mice (Charles River, USA) were randomly assigned to one out of four treatment groups, which were injected with vehicle or SR141716 (0.5, 2, 10 mg/kg IP) and returned to their home cages. One hour later animals were either killed directly to obtain injection stress-induced control corticosterone levels or exposed to the FST for 6 min and killed 10 min after stressor onset in order to assess the acute effects of SR141716 on immediate FST stress-induced corticosterone levels (compare corresponding Fig. 1). The dose of 10 mg/kg SR141716 was classified as the most potent dose with respect to injection stress-and FST stress-induced corticosterone secretion and was applied in all further experiments.
Figure 1. Acute pharmacological blockade of CB1 dose-dependently increases injection stress- and forced swim test (FST) stress-induced corticosterone secretion.
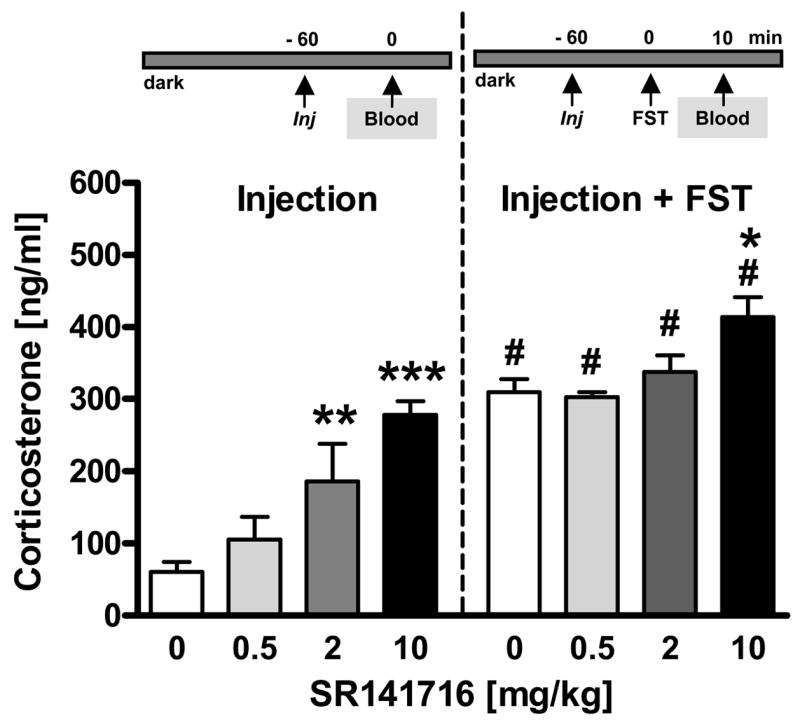
Experiment 1: Male C57BL/6N mice were acutely treated (Injection = Inj) with vehicle (0 mg/kg IP) or SR141716 (0.5, 2, 10 mg/kg IP). One hour later, half of the animals were killed for blood sampling (Blood) without further stressor exposure (Injection), the other half was exposed to the FST and killed 10 min after FST stressor onset (Injection + FST). n = 6–7 per group; *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective vehicle group (0 mg/kg). #p < 0.05 vs. respective injection stress control group (Injection).
Experiment 2: CB1 specificity of SR141716 effects
One hour after injection with vehicle or SR141716 (10 mg/kg IP) male and female CB1+/+ and CB1−/− mice were subjected to the FST and killed 10 min after stressor onset (compare the corresponding Fig. 2).
Figure 2. Acute pharmacological blockade of CB1 increases forced swim test (FST) stress-induced corticosterone secretion in CB1+/+ but not in CB1−/− mice.
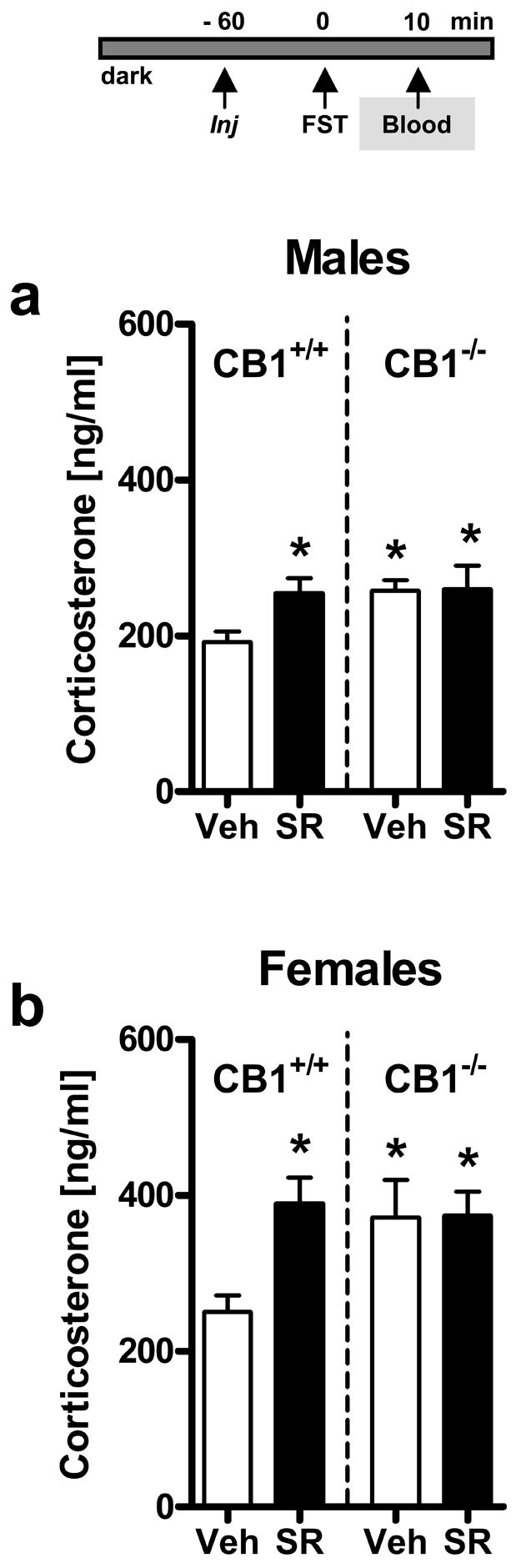
Experiment 2: Male (a) and female (b) CB1 wild-type (CB1+/+) and knockout (CB1−/−) mice (n = 8–16 per group) were treated (Injection = Inj) with vehicle (Veh) or SR141716 (SR; 10 mg/kg IP) 1 h before FST exposure. Mice were killed 10 min after stressor onset, and trunk blood was collected for measurements of plasma corticosterone levels. Note that SR141716 caused a significant increase in corticosterone in CB1+/+ mice without affecting the already elevated corticosterone levels in CB1−/− mice. *p < 0.05 vs. vehicle-treated CB1+/+ mice.
Experiment 3: Time course of FST stress-induced corticosterone secretion in CB1 mutant mice
Because of ethical reasons and because of the shortage of genetically altered CB1+/+ and CB1−/− mice, in Experiment 3 we determined stress hormone levels of animals that had previously undergone repeated forced swimming and whose behavior had been reported in a previous study (Steiner et al., in press). In Experiment 3 male and female CB1+/+ and CB1−/− mice that had been repeatedly exposed to the FST (day 1, FST-1; day 2, FST-2; and day 21, FST-3) were killed 30 min or 120 min after the last stressor onset (FST-3 on day 21; compare corresponding Fig. 3). All time-point groups (basal, 30 min after FST-3 and 120 min after FST-3) represent independent batches of mutant mice, which were bred and exposed to experiments within a time-period of 6 months. They were all housed identically and care was taken to expose all 3 independent batches of mice to identical environmental and experimental conditions.
Figure 3. Genetic deletion of CB1 enhances basal and forced swim test (FST) stress-induced corticosterone secretion in a time-dependent manner.

Experiment 3: Male (a) and female (b) CB1 wild-type (CB1+/+) and knockout (CB1−/−) mice (n = 7–13 per group) were either killed under basal conditions (Basal) or were exposed to the FST (FST-3; for details see Methods) and were killed 30 min or 120 min after onset of the stressor for plasma corticosterone measurements. **p < 0.01, ***p < 0.001 vs. respective CB1+/+ group; #p < 0.05 vs. respective basal group.
Two control experiments were performed in order to evaluate whether or not repeated FST exposure leads to an adaptation of the corticosterone response following the stressor. In the first control experiment three groups of male C57BL/6N mice (Charles River, USA) were either once (day 1), twice (day 1 and day 2), or repeatedly (day 1, day 2 and day 21) exposed to the FST and killed 10 min after the last stressor onset, on day 1, day 2 or day 21. In the second control experiment naive male CB1+/+ and CB1−/− mice were exposed to the FST on day 1 and were killed 30 min after stressor onset for assessment of corticosterone and later comparison with corticosterone levels derived from CB1+/+ and CB1−/− mice following repeated FST exposure (Experiment 3).
Experiment 4: Time course of FST stress-induced corticosterone secretion following pharmacological blockade of CB1
Male and female C57BL/6N animals (Charles River, Germany) were exposed twice to the FST on two consecutive days (day 1, FST-1; and day 2, FST-2) and were injected with vehicle or SR141716 (10 mg/kg IP) three times, 2 h before FST-1 on day 1, again 12 h later, and last 2 h before FST-2 on day 2 (compare corresponding Fig. 4), according to an established treatment schedule for the detection of antidepressant-like effects in mice (Wei et al., 2004). Mice were killed 30 or 120 min after the last stressor onset (FST-2, day 2). For baseline stress hormone levels, male and female C57BL/6N mice were repeatedly injected with vehicle or SR141716 according to the same treatment schedule as mentioned above (three injections: once on day 1, again 12 h later, and last on day 2). They were killed 2 h after the last injection on day 2. The time-point of SR141716 injection (2 h before FST, instead of 1 h before FST used in Experiments 1 and 2) was chosen because we wanted to minimize residual effects of injection stress on later blood sampling or later FST exposure. Accordingly, we had recently found that this injection schedule most accurately mimicked behavior of CB1−/− mice in the FST (Steiner et al., in press).
Figure 4. Subchronic pharmacological blockade of CB1 enhances injection stress- and forced swim test (FST) stress-induced corticosterone secretion in a time-dependent manner.

Experiment 4: Male (a) and female (b) C57BL/6N mice (n = 6 per group) were repeatedly treated (Injection = Inj; three times during two days) with vehicle or SR141716 (10 mg/kg IP) and either killed directly 2 h after the last injection (Inj-3) or exposed to the FST (FST-2; for details see Methods) 2 h after the last injection (Inj-3) and killed 30 min or 120 min after onset of the stressor for plasma corticosterone measurements. *p < 0.05, ***p < 0.001 vs. respective vehicle group; #p < 0.05 vs. respective injection stress control group (Injection).
Experiment 5: Behavioral and neuroendocrine effects of desipramine treatment in CB1 mutant mice in response to the FST
Male CB1+/+ and CB1−/− mice were exposed twice to the FST on two consecutive days (day 1, FST-1; day 2, FST-2) and were injected with vehicle or desipramine (20 mg/kg IP) three times, 1 h before FST-1 on day 1, once again 12 h later, and last 1 h before FST-2 on day 2 (compare corresponding Fig. 5), according to an established treatment schedule for detecting antidepressant-like behavioral effects in the FST (Wei et al., 2004). Mice were killed 20 min after the last stressor onset (FST-2 on day 2) because we wanted to investigate desipramine suppressing effects on forced swim stress-induced corticosterone secretion, and these effects had previously been demonstrated for exactly this time-point after FST stress (Conti et al., 2002).
Figure 5. Behavioral and neuroendocrine responses to the forced swim test (FST) in CB1-deficient mice following treatment with desipramine.
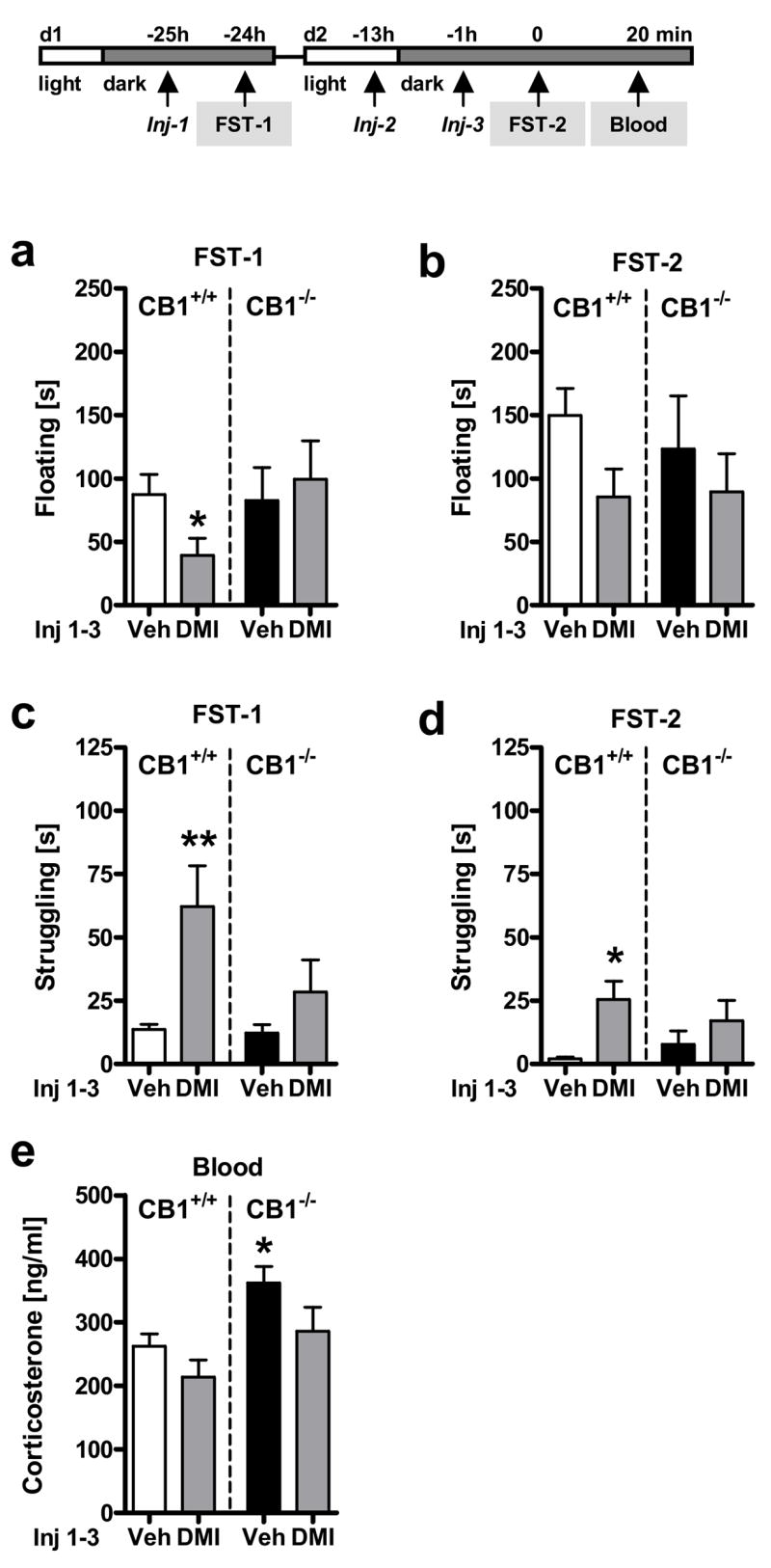
Experiment 5: Male CB1 wild-type (CB1+/+) and knockout (CB1−/−) mice were exposed to the FST on two consecutive days (FST-1, d1; FST-2, d2) and treated (Injection = Inj) three times with either vehicle (Veh) or desipramine (DMI; 20 mg/kg IP) 1 h before the first (Inj-1) and the second FST (Inj-3) and once in between (Inj-2). Floating (a,b) and struggling (c,d) behavior during the entire FST exposure on day 1 (FST-1; a,c) and day 2 (FST-2; b,d). Corticosterone (e) levels 20 min after the onset of the second FST exposure (FST-2). Note that vehicle-treated CB1−/− mice showed a similar behavioral phenotype as vehicle-treated CB1+/+ littermate controls. n = 8–12 per group; *p < 0.05, **p < 0.01 vs. respective vehicle-treated CB1+/+ mice.
Experiment 6: Effects of pharmacological blockade of CB1 on behavioral and neuroendocrine consequences of desipramine treatment in response to the FST
Male C57BL/6N mice (Charles River, Germany) were exposed twice to the FST on two consecutive days (day 1, FST-1; day 2, FST-2) and injected with vehicle or desipramine (20 mg/kg IP) three times, 1 h before FST-1 on day 1, once again 12 h later, and last 1 h before FST-2 on day 2. In addition, mice were pre-treated with vehicle or SR141716 (10 mg/kg; IP) three times, 1 h before each vehicle or desipramine treatment (i.e., 2 h before FST-1 on day 1, once again 12 h later, and last 2 h before FST-2 on day 2) resulting in four treatment groups (Veh-Veh, Veh-DMI, SR-Veh, SR-DMI; compare corresponding Fig. 6). Mice were killed 20 min after the last stressor onset (FST-2 on day 2) in order to determine desipramine suppressing effects on corticosterone secretion (compare Experiment 5).
Figure 6. Lack of interaction between desipramine and SR141716 treatment in C57BL/6N mice.
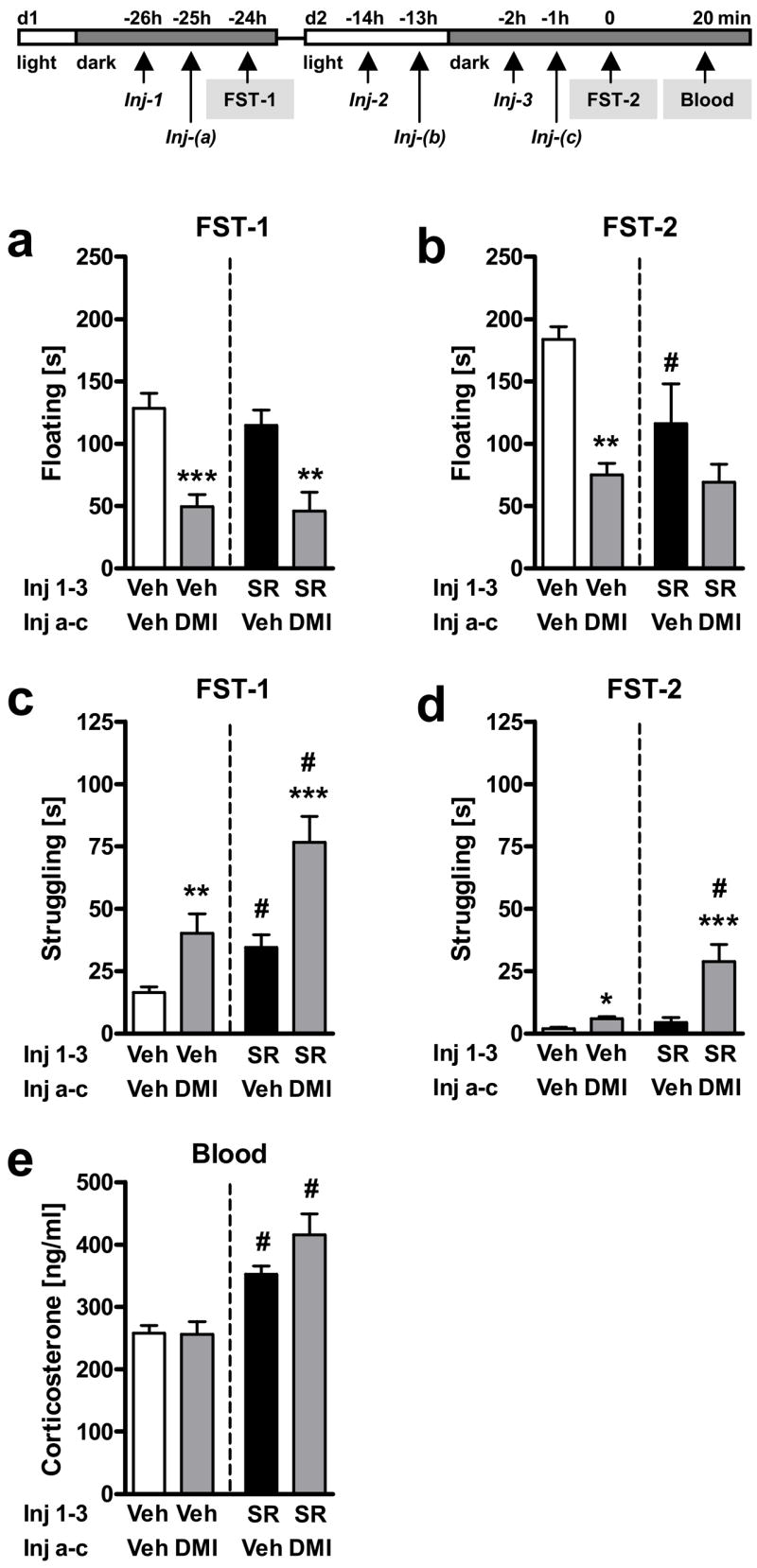
Experiment 6: Male C57BL/6N mice were exposed to the FST on two consecutive days (FST-1, d1; FST-2, d2) and treated three times (Inj-a-c) with either vehicle (Veh) or desipramine (DMI; 20 mg/kg IP), 1 h before the first and the second FST, and once in between. Mice were pre-treated with either vehicle (Veh) or SR141716 (SR; 10 mg/kg IP), 1 h before each desipramine treatment (Inj-1-3). Floating (a,b) and struggling (c,d) behavior during the entire FST exposure on day 1 (FST-1; a,c) and day 2 (FST-2; b,d). Corticosterone (e) levels 20 min after the onset of the second FST exposure (FST-2). n = 10–12 per group; *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective vehicle-treated group (Veh-Veh or SR-Veh); #p < 0.05 vs. respective vehicle pre-treated group (Veh-Veh or Veh-DMI).
Experiment 7: Neuroendocrine consequences of chronic pharmacological blockade of CB1
Male C57BL/6N mice (Charles River, Germany) were chronically pre-treated with vehicle or SR141716 (10 mg/kg; IP) with one injection per day for 10 days (Inj-1-10). Towards the end of that period, at day 10, mice were injected twice (with 12 h in between), in order to maintain the same injection schedule during these last two days as that used for the subchronic SR141716 experiments described in Experiments 4 and 6. Mice were killed 2 h after the last injection (Inj-11) at day 10 for corticosterone assessment and direct comparison with injection stress-induced corticosterone secretion after subchronic SR141716 treatment from Experiment 4. For the last injection both pre-treated groups (vehicle and SR141716) were randomly assigned to one out of two treatment groups, which we acutely challenged with either vehicle or SR141716 (10 mg/kg IP) injection, thus resulting in a total of four groups (Veh-Veh, Veh-SR, SR-Veh, SR-SR; compare corresponding Fig. 7).
Figure 7. Chronic blockade of CB1 by SR141716 leads to reduced responsiveness to corticosterone stimulating effects of an acute SR141716 challenge.
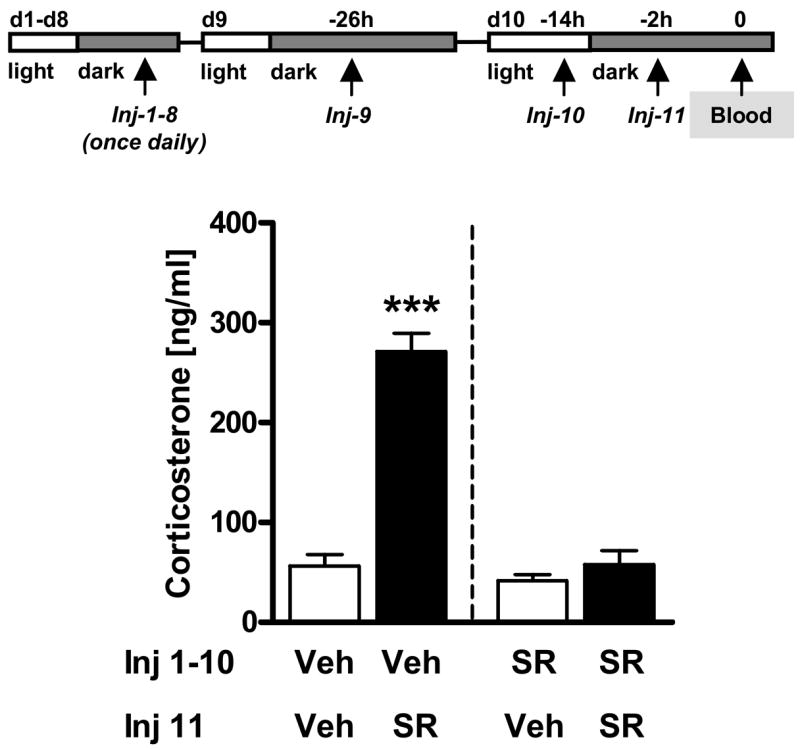
Experiment 7: Male C57BL/6N mice were chronically pre-treated (Injection = Inj) with vehicle (Veh) or SR141716 (SR; 10 mg/kg i.p) for 10 days (d1-10; Inj 1-10). At day 10 (d10), half of the mice of each pre-treatment group (Veh = Vehicle; SR = SR141716) received as last injection (Inj-11) either vehicle or SR141716 (10 mg/kg) resulting in four different groups (Veh-Veh, Veh-SR, SR-Veh, SR-SR; n = 6–7 per group). Animals received one additional pre-treatment injection (Inj-10) 12 h before the last injection in order to comply with the treatment schedule that was previously applied for the subchronic experiments. Two hours after the last injection (Inj-11) animals were killed for corticosterone measurements. ***p < 0.001 vs. all other groups.
Experiment 8: Behavioral and neuroendocrine consequences of chronic pharmacological blockade of CB1 in response to the FST
Male C57BL/6N mice (Charles River, Germany) were chronically injected with vehicle or SR141716 (10 mg/kg IP) daily for 10 days (Inj-1-11) as described for Experiment 7. Towards the end of that period mice were exposed twice to the FST, on day 9 (FST-1) and again on day 10 (FST-2; compare corresponding Fig. 8). The injection schedule with three injections during these last two days was the same as that used for the subchronic SR141716 experiments described in Experiment 4 and 6. Animals were killed 30 min after the last stressor onset on day 10 (FST-2) in order to allow for a direct comparison with the corticosterone secretion levels after subchronic dosing from Experiment 4, which had shown that SR141716 exerted its strongest corticosterone elevated effect at this time-point after FST stress.
Figure 8. Chronic blockade of CB1 by SR141716 alters behavioral and neuroendocrine responses to the forced swim test (FST).

Experiment 8: Male C57BL/6N mice were exposed to the FST on two consecutive days, on day 9 (d9; FST-1) and day 10 (d10; FST-10) after chronic treatment (Injection = Inj) with vehicle (Veh) or SR141716 (SR; 10 mg/kg i.p) for 10 days (d1-10; Inj 1-11). The injection schedule during the last two days of chronic treatment (d9-10) was kept the same as that previously applied for the subchronic experiments. During days 9 and 10 animals were injected 2 h prior to FST-1 on day 9 (Inj-9), 12 h later (Inj-10), and last 2 h before FST-2 on day 10 (Inj-11). Floating (a) and struggling (b) behavior during the entire FST exposure on day 9 (FST-1) and day 10 (FST-2). Corticosterone (c) levels 30 min after the onset of the second FST exposure (FST-2) on day 10. *p < 0.05, ***p < 0.001 vs. respective vehicle control.
RESULTS
Experiment 1: Pharmacological blockade of CB1 by SR141716 results in increased corticosterone secretion in male C57BL/6N mice
Pharmacological blockade of CB1 with SR141716 (0.5, 2, 10 mg/kg) dose-dependently increased plasma corticosterone levels following injection stress without or with subsequent FST exposure [Treatment: F3,44 = 15, p < 0.001; 2-way ANOVA (Stress, Treatment); Fig. 1]. Post hoc analyses revealed that 2 mg/kg (p < 0.01) and 10 mg/kg (p < 0.001) were effective in elevating injection stress-induced corticosterone levels, whereas only the highest dose of 10 mg/kg was able to further elevate FST stress-induced corticosterone secretion as compared to the respective vehicle-treated group (p < 0.05). As compared to respective injection stress-induced control levels, FST stress itself led to a further increase in corticosterone secretion (Stress: F1,44 = 97.1, p < 0.001) in all groups, independent of treatment (Treatment x Stress: F3,44 = 1.9, p = 0.147).
Experiment 2: Effects of SR141716 on FST stress-induced corticosterone secretion in male and female CB1-deficient mice
In order to determine the specificity of SR141716 for CB1 and to compare pharmacological effects with the effects of genetic deletion of CB1 on stress-induced corticosterone secretion, we injected CB1+/+ and CB1−/− mice with vehicle or the most potent dose of SR141716 (10 mg/kg) before exposure to the FST. Additionally, we conducted the experiment in male and female mice, to assess whether neuroendocrine CB1 effects were sex-dependent. Three-way ANOVA (Genotype, Treatment, Sex) revealed a significant effect of Genotype (F1,88 = 4.27, p < 0.05) reflecting the fact that CB1−/− mice in general showed higher corticosterone secretion than their CB1+/+ littermates (Figs. 2a,b) following FST exposure. These effects were virtually identical to the effects of the pharmacological blockade of CB1 in CB1+/+ mice (Treatment: F1,88 = 5.78, p < 0.05; Figs. 2a,b). A significant Treatment x Genotype interaction (F1,88 = 5.32, p < 0.05) reflects the fact that SR141716 exerted its effects only in CB1+/+ mice, while it had no effect in CB1−/− mice, thus demonstrating the specificity of SR141716 for CB1. Corticosterone elevation was similar in male and female mice, underlined by a non-significant Treatment x Genotype x Sex interaction (F1,88 = 0.78, p = 0.38), but in general, females showed higher corticosterone levels than males (Sex: F1,88 = 24.1, p < 0.001).
Experiment 3: Time course of FST stress-induced corticosterone secretion in male and female CB1 mutant mice
Having confirmed the specificity of SR141716 and the effect of CB1 blockade on the immediate FST stress-induced corticosterone secretion 10 min after onset of the stressor, we also wanted to investigate the effects of genetic inactivation on the time course of FST stress-induced corticosterone secretion. In addition, we also determined basal corticosterone levels in unstressed CB1 mutant mice at the same time of the circadian rhythm at which the stress experiments had been performed.
In general, FST exposure of CB1+/+ and CB1−/− mice led to a significant rise in corticosterone secretion 30 min after stressor onset, which returned to baseline levels after 120 min [Time: F2,101 = 110.1, p < 0.001; 3-way ANOVA (Genotype, Sex, Time); Figs. 3a,b]. The genetic deletion of CB1 increased corticosterone secretion 30 min after stressor onset as compared to wild-type controls (Genotype: F1,101 = 26.3, p < 0.001; Genotype x Time: F2,101 = 18.1, p < 0.001), similarly to what we previously observed 10 min after FST stressor onset (Fig. 2). Males and females showed similar levels of corticosterone secretion (Sex: F1,101 = 0.006, p = 0.938), independent of genotype (Genotype x Sex: F1,101 = 0.05, p = 0.823) and time point after stressor exposure (Genotype x Time x Sex: F2,101 = 0.568, p = 0.569).
If analyzed separately for male and female mice, post hoc analyses following two-way ANOVA (Genotype, Time) revealed that only male, but not female, CB1−/− mice displayed elevated corticosterone levels as compared to their wild-type littermates under basal conditions (p < 0.01), whereas both male and female CB1−/− mice displayed elevated corticosterone levels 30 min after stressor onset (p < 0.001; Figs. 3a,b), but not 120 min after stressor onset as compared to their CB1+/+ littermates at the respective time point. Furthermore, while both male and female mice showed generally elevated corticosterone levels as compared to basal conditions 30 min after stressor onset (p < 0.001; Figs. 3a,b), only male CB1+/+ and female CB1−/− mice still showed elevated corticosterone levels 120 min after stressor onset as compared to their respective baseline controls (p < 0.05).
As alluded to in the Methods section, above data derived from animals, which have been exposed to the FST already twice before. Therefore, we conducted two control experiments in order to ensure that repeated FST exposure did not confound the data due to occurring habituation of the corticosterone in response to repeated FST exposure. In the first control experiment we could demonstrate that repeated FST exposure of male C57BL/6N mice on day 1 (FST-1), day 2 (FST-2) and day 21 (FST-3) led to an adaptation of floating behavior, but not of corticosterone secretion. Mice floated significantly longer on day 2 (219.8 ± 22.33 s) and day 21 (164.2 ± 24.12 s) than on day 1 (109.4 ± 21.35 s) [Day: F2,24 = 5.9, p < 0.01; one-way ANOVA (Day); n = 8–9 per group]. However, these behavioral adaptations were not paralleled by similar changes in corticosterone secretion assessed 10 min after stressor onset [day 1: 246.3 ± 17.62 ng/ml; day 2: 228.3 ± 9.97 ng/ml; day 21: 206.5 ± 12.95 ng/ml; Day: F2,23 = 2.1, p = 0.15; one-way ANOVA (Day); n = 8 per group]. The second control experiment confirmed that the increased corticosterone levels 30 min after FST stress (FST-3) observed in CB1 mutant mice were not due to a potential deficit in habituation at this later time-point after repeated stressor exposure. Acute FST exposure (FST-1) revealed a significantly stronger increase in corticosterone secretion in CB1−/− mice than in CB1+/+ mice 30 min after FST-1 (265.2 ± 27.56 ng/ml vs. 135.6 ± 11.18 ng/ml; n = 10 per group; t18 = 4.4, p < 0.001; Student’s t-test), which resembles our findings obtained 30 min after repeated FST (FST-3). Furthermore, neither CB1+/+ nor CB1−/− mice from Experiment 3 showed any habituation of the corticosterone response following repeated FST, because the corticosterone response of both CB1+/+ mice (t20 = 1.1, p = 0.29) and CB1−/− mice (t18 = 0.3, p = 0.76) did not differ between plasma taken from mice after acute FST-1 (second control experiment) or repeated FST-3 (Experiment 3).
Experiment 4: Time course of FST stress-induced corticosterone secretion in male C57BL/6N mice subchronically treated with SR141716
To investigate whether our findings of Experiment 3 could be replicated by a pharmacological approach, we applied SR141716 in a subchronic manner (3 consecutive injections) in order to better mimic the CB1 knockout phenotype. In general, FST exposure of C57BL/6N mice led to a significant rise in corticosterone secretion 30 min after FST stressor onset [Time: F2,60 = 106.9, p < 0.001; 3-way ANOVA (Treatment, Sex, Time); Fig. 4], which was more pronounced in females than in males (Sex x Time: F2,60 = 5.6, p = 0.006). Corticosterone levels returned to injection stress-induced control levels 120 min after FST stressor onset. SR141716 treatment caused a general increase of corticosterone secretion both after injection stress and after FST stress (Treatment: F1,60 = 55.5, p < 0.001), which was most pronounced 30 min after FST stressor onset (Treatment x Time: F2,60 = 12.2, p < 0.001), similarly to what we had observed in CB1 mutant mice. Females, independent of treatment, showed lower plasma levels of corticosterone than males in response to injection stress control conditions, but higher levels than males after FST stressor exposure (Sex: F1,60 = 4.3, p = 0.043; Sex x Time: F2,60 = 5.6, p = 0.006).
If analyzed separately for male and female mice, post hoc analyses following two-way ANOVA (Treatment, Time) revealed that male mice treated with SR141716 displayed elevated corticosterone levels as compared to their respective vehicle-treated controls under injection stress control conditions (p < 0.05) as well as 30 min after FST stressor onset (p < 0.001; Fig. 4a), whereas female mice treated with SR141716 only showed significantly elevated corticosterone levels as compared to their respective vehicle-treated controls 30 min after FST stressor onset (p < 0.001; Fig. 4b). 120 min after FST stressor onset corticosterone levels of both male and female mice treated with SR141716 or vehicle returned to levels seen in mice following injection stress only.
Experiment 5: Behavioral but not neuroendocrine effects of desipramine in response to the FST are compromised in CB1 mutant mice
In order to investigate whether the corticosterone elevating effects of CB1 impairment interfered with the behavioral and neuroendocrine actions of antidepressants, we subchronically treated male CB1 mutant mice with desipramine before exposure to the FST.
Two-way ANOVA (Genotype, Treatment) of floating behavior during FST exposure on day 1 (FST-1) failed to show general Genotype (F1,34 = 0.75, p = 0.39) differences, but revealed an almost significant interaction between Genotype and Treatment (F1,34 = 3.6, p = 0.068) reflecting the fact that (i) vehicle-treated CB1+/+ and CB1−/− mice showed similar floating behavior, and that (ii) subchronic desipramine treatment led to a significant decrease of floating in CB1+/+ mice (t20 = 2.2, p < 0.05; planned pair-wise comparison using Student’s t-test; Fig. 5a), but not in CB1−/− mice. No significant effect of desipramine treatment or geneotype was detected regarding floating behavior during FST on day 2 (FST-2), although treatment tended to decrease floating in both CB1+/+ and CB1−/− mice to a similar extent (Fig. 5b; statistics not shown).
Two-way ANOVA (Genotype, Treatment) of struggling behavior during FST exposure on day 1 (FST-1; Fig. 5c) and day 2 (FST-2; Fig. 5d) revealed significant effects of Treatment on both days (F1,34 > 9, p < 0.01), independent of genotype (Genotype x Treatment: F1,34 < 2.8, p > 0.1), with no significant Genotype differences per se (F1,34 < 3.4, p > 0.08). Post hoc analyses revealed that desipramine treatment led to a significant increase of struggling behavior in CB1+/+ mice on both days (p < 0.01, day 1; p < 0.05, day 2), whereas the increase of struggling in CB1−/− mice was less pronounced and did not reach statistical significance on either day.
Two-way ANOVA (Genotype, Treatment) of corticosterone secretion 20 min after FST-2 onset (Fig. 5e) revealed a significant effect of Treatment (F1,34 = 5.6, p < 0.05), but no significant Treatment x Genotype interaction (F1,34 = 0.003, p = 0.96). This reflects the fact that desipramine reduced corticosterone secretion 20 min after FST in both CB1+/+ and CB1−/− mice as compared to the respective vehicle-treated controls. A significant Genotype effect (F1,34 = 4.7, p < 0.05) confirmed our previous observation that CB1−/− mice showed generally higher corticosterone secretion than their CB1+/+ littermates in response to FST stress. Post hoc analyses revealed that only vehicle-treated CB1−/− mice had significantly higher corticosterone levels than their wild-type littermates, whereas this difference failed to reach statistical significance between desipramine-treated CB1−/− and CB1+/+ mice.
Experiment 6: Behavioral and neuroendocrine effects of desipramine in response to the FST remain unaffected by the pharmacological blockade of CB1
In order to assess, whether the slight impairment of antidepressant-like behavioral effects of desipramine seen in the previous experiment in CB1−/− mice represents a possible artifact of developmental changes in these mutant mice due to the life-long absence of the receptor, we repeated the experiment in C57BL/6N mice, but this time under the subchronic pharmacological blockade of CB1.
Two-way ANOVA (Pre-treatment, Treatment) of floating behavior during FST exposure on day 1 (FST-1) revealed a significant Treatment effect of desipramine (F1,43 = 30.1, p < 0.001; Fig. 6a). Contrarily to what we observed in the previous experiment with the genetic deletion of CB1, a non-significant Pre-treatment x Treatment interaction (F1,43 = 0.1, p = 0.75) reflected the fact that this time desipramine exerted its antidepressant-like effects (a reduction in floating) both in vehicle and in SR141716 pre-treated mice to a similar extent. The non-significant Pre-treatment effect (F1,43 = 1.86, p = 0.18) indicated that acute SR141716 treatment per se had no influence on floating behavior on day 1 (FST-1).
Two-way ANOVA (Pre-treatment, Treatment) of floating behavior during FST exposure on day 2 (FST-2) also revealed a significant Treatment effect of desipramine (F1,42 = 13.3, p < 0.001; Fig. 6b). A non-significant Pre-treatment x Treatment interaction (F1,42 = 1.54, p = 0.22) suggested that desipramine exerted its antidepressant-like effects (a reduction in floating) both in vehicle and in SR141716 pre-treated mice. However, a significant Pre-treatment effect (F1,42 = 4.98, p = 0.031) indicated that SR141716 pre-treatment per se, in contrast to day 1, reduced floating on day 2 (FST-2).
Two-way ANOVA (Pre-treatment, Treatment) of struggling behavior during FST exposure on day 1 (FST-1; Fig. 6c) and day 2 (FST-2; Fig. 6d), respectively, revealed significant effects of Treatment on both days (F1,42 > 13.3, p < 0.001) reflecting the fact that desipramine significantly increased struggling behavior. A non-significant Pre-treatment x Treatment interaction on day 1 (F1,43 = 0.61, p = 0.44) and a significant Pre-treatment x Treatment interaction on day 2 (F1,42 = 4.07, p < 0.05) suggested that desipramine exerted its effect on struggling in both vehicle and SR141716 pre-treated mice, and even induced a slightly stronger increase in struggling in the SR141716 pre-treated group on day 2 as compared to vehicle pre-treated mice (Fig. 6d). Significant Pre-treatment effects on both days (F1,42 > 16.1, p < 0.001) revealed that SR141716 pre-treatment per se induced an increase of struggling behavior, which reached statistical significance in the post hoc test only on day 1.
Two-way ANOVA (Pre-treatment, Treatment) of corticosterone secretion 20 min after FST-2 (Fig. 6e) revealed no significant effect of Treatment (F1,42 = 0.43, p = 0.52) reflecting the fact that under the current experimental settings (two consecutive injections before FST), in contrast to previous results obtained in CB1+/+ and CB1−/− mice (Experiment 5; Fig. 5e), desipramine did not influence FST stress-induced corticosterone secretion. A significant effect of Pre-treatment (F1,42 = 31.2, p < 0.001) confirmed that SR141716 pre-treatment increased FST stress-induced corticosterone secretion both in vehicle- and in desipramine-treated mice, independent of treatment (Pre-treatment x Treatment: F1,42 = 1.7, p = 0.2).
Experiment 7: Effects of chronic SR141716 pre-treatment on corticosterone secretion in response to an acute SR141716 challenge
Having consistently demonstrated corticosterone elevating effects of SR141716 in the aforementioned experiments, we conducted another experiment in order to assess the extent to which chronic SR141716 administration affects HPA axis activity.
Two-way ANOVA (Pre-treatment, Challenge) revealed significant effects of Pre-treatment (F1,22 = 46.7, p < 0.001; Fig. 7) with SR141716 for 9 days, of acute Challenge (F1,22 = 49.7, p < 0.001) with SR141716 on the tenth day, and a significant Pre-treatment x Challenge interaction (F1,22 = 32.6, p < 0.001). These results reflect that (i) chronic SR141716 pre-treatment for 9 days did not significantly influence vehicle injection stress-induced corticosterone secretion on day 10 (SR-Veh = Veh-Veh), suggesting that chronic SR141716 pre-treatment per se did not alter HPA axis reactivity in absence of an acute pharmacological challenge; (ii) acute SR141716 administration on day 10 in chronically vehicle pre-treated mice led to increased corticosterone levels (Veh-SR > Veh-Veh), similarly to previous findings after acute (Fig. 1, left panel) or subchronic administration of the antagonist (Fig. 4, injection stress levels); (iii) chronic SR141716 pre-treatment led to “tolerance” to its acute stimulatory effects on corticosterone secretion (SR-SR = SR-Veh < Veh-SR), assessed 2 h after injection.
Experiment 8: Effects of chronic pharmacological blockade of CB1 on behavioral and neuroendocrine responses to the FST
Having demonstrated that chronic SR141716 treatment leads to reduced responsiveness to its acute corticosterone stimulatory effects 2 h after intraperitoneal injection, we wondered whether chronic administration would also alter the behavioral and neuroendocrine response to the FST following an acute challenge with SR141716.
Chronic treatment with SR141716 for 8 days, followed by an acute challenge 2 h before FST-1 on day 9 and 2 h before FST-2 on day 10 resulted in a significant decrease of floating behavior [Treatment: F1,28 = 6.8, p < 0.05; two-way ANOVA (Day, Treatment); Fig. 8a] and, in parallel, in a significant increase of struggling behavior [Treatment: F1,28 = 7.6, p < 0.05; two-way ANOVA (Day, Treatment); Fig. 8b]. These behavioral effects of SR141716 were maintained throughout the first (FST-1) and second (FST-2) FST exposure (on day 9 and day 10 of chronic SR141716 treatment, respectively), although struggling behavior was almost absent during the second exposure (FST-2). Corticosterone secretion 30 min after the second FST exposure on day 2 (FST-2; day 10 of chronic treatment) was significantly elevated in SR1411716-treated animals as compared to vehicle-treated controls (t26 = 5.6, p < 0.001; Student’s t-test; Fig. 8c) and was not different from FST-induced corticosterone levels of subchronically SR141716-treated mice (compare 30 min time point in Fig. 4a; statistics not shown).
DISCUSSION
Preclinical and clinical research strongly suggests that disturbances of the HPA axis can play a major causal role in depression disorders (De Kloet et al., 2005), and recent findings in rodents imply that the endocannabinoid system is an important HPA axis modulator (Pagotto et al., 2006). Blockade of endocannabinoid signaling has been suggested as a novel antidepressant treatment by some authors (Witkin et al., 2005; Griebel et al., 2005), whereas others regard CB1-deficient mice as an animal model of depression (Hill and Gorzalka, 2005a). The present study was designed to compare neuroendocrine and behavioral consequences of CB1 impairment in response to FST exposure. We showed that (i) the genetic inactivation as well as the acute, subchronic and chronic pharmacological blockade of CB1 lead to increased basal and stress-induced corticosterone secretion; (ii) chronic SR141716 administration causes reduced responsiveness to its corticosterone stimulatory effects following an acute SR141716 challenge; (iii) the subchronic and chronic administration of SR141716 produces antidepressant-like behavioral effects in the FST similar to desipramine; (iv) desipramine-induced antidepressant-like behavioral effects are absent in CB1-deficient mice, but unaffected by pharmacological blockade of CB1; (v) desipramine-induced neuroendocrine effects are not mediated via CB1. Together these results demonstrate that antidepressant-like behavioral effects of SR141716 coincide with exaggerated stress-induced corticosterone secretion.
Acute (Figs. 1,2), subchronic (Figs. 4,6) and chronic SR141716 treatment (Fig. 8), as well as genetic inactivation of CB1 (Figs. 2,4,5), increased levels of corticosterone secretion 10, 20 and 30 min after FST stress, which is in accordance to previous findings from the literature (Patel et al., 2004; Barna et al., 2004). Moreover, our results confirm that HPA axis dysregulations in CB1-deficient mice are not due to developmental deficits, as they could be reliably mimicked by the pharmacological blockade of CB1. They also demonstrate that CB1 inhibition confers similar stress-induced corticosterone elevating effects both in male and female mice. Therefore, our results help to establish CB1 signaling as a universal mechanism for constraining stress-induced corticosterone secretion, largely independent of sex, genetic background, type of stressor or type of CB1 impairment (genetic or pharmacological).
Some reports have raised doubts about the specificity of SR141716 for CB1 as, for instance, the antagonist still exerted behavioral effects in CB1-deficient mutants on a CD1 background (Haller et al., 2002). However, in our mutant line acute SR141716 administration had no effect on immediate FST stress-induced corticosterone secretion (Fig. 2), whereas it increased FST stress-induced corticosterone secretion in CB1+/+ mice to the same levels that were seen in vehicle-treated CB1−/− mice. These results illustrate the similarities between the pharmacological and genetic impairment of CB1 and suggest that SR141716-mediated effects on corticosterone are CB1-specific.
Untreated male CB1−/− mice had also elevated corticosterone levels as compared to their wild-type littermates under basal conditions (Fig. 3a). This underscores the notion of an HPA axis-modulating endocannabinoid tone, which is present under basal non-stress conditions (Cota et al., 2007; Patel et al., 2004). The lack of genotype differences in basal corticosterone levels in female CB1 mutant mice (Fig. 3b) is in contrast to our previous observations made at another time of the day (Cota et al., 2007), which might be explained by sex-specific differences in the circadian rhythm of endocannabinoid action. In fact, Valenti and co-workers recently demonstrated that the levels of the two major endocannabinoids 2-arachidonoyl-glycerol and anandamide undergo strong diurnal variations (Valenti et al., 2004), which might be differentially regulated between male and female mice. It has to be noted that with the pharmacological application of SR141716 it was impossible to investigate true basal corticosterone levels, as drug administration is invariably conferring mild stress to the animals. Nevertheless, apart from Experiments 1 and 2, where we first tested the drug, in the following experiments we injected SR141716 two hours prior to any additional stress exposure or blood sampling in order to minimize residual stress effects of the injection procedure.
Unexpectedly, chronic SR141716 administration led to a reduction of the corticosterone stimulating effects induced by an acute SR141716 challenge (Fig. 7), which is in contrast to results from another group (Wade et al., 2006). Noteworthy, corticosterone elevating effects of an acute SR141716 challenge after subchronic treatment (compare the “Injection” bars of Fig. 4) were already lower than those after acute administration (compare the Veh-SR bar of Fig. 7,), but still significantly higher than after chronic treatment (compare the SR-SR bar of Fig. 7). This suggests that tolerance to the acute corticosterone stimulatory effects of SR141716 develops gradually over the time period of 10 days, which could explain why Wade and co-workers did not yet observe any “tolerance” effect after 5 days of treatment (Wade et al., 2006).
Despite these “tolerance” effects following intraperitoneal injection, chronic SR141716 administration did not lead to “tolerance” to its stimulatory effects on FST stress-induced corticosterone secretion (Fig. 8c), which were comparable to those observed after subchronic (Fig. 4) or acute SR141716 treatment (Fig. 1). One possible explanation is that “tolerance” might only develop towards a mild stressor (IP injection), which has been repeatedly applied over the 10 days period, and that this “tolerance” can be overridden by exposure to a novel, severe stressor such as FST. Another possibility is that “tolerance” only develops in those pathways and processes, which are involved in re-setting of the HPA axis (i.e., two hours after injection stress), but not in those responsible for the early corticosterone response (i.e., 30 min after FST stress). Such a scenario would provide another explanation for the different findings from Wade and co-workers, who measured plasma corticosterone one hour, but not two hours, after administration of SR141716. Nevertheless, certainly more detailed studies will be required in the future in order to elucidate the precise mechanisms underlying “tolerance” to corticosterone-stimulating effects of chronic SR141716 administration.
We have chosen the FST as stressor in the present study, because this test is frequently applied to investigate antidepressant-like potentials of novel drugs (Cryan and Holmes, 2005). However, in this specific function, not the endocrine responses, but the behavioral performance during the test procedure is usually considered. Hence, we also investigated the behavioral stress coping of SR141716-treated mice. Acute, subchronic and chronic SR141716 administration (Figs. 6c,d and 8b) increased struggling behavior, whereas only subchronic and chronic (Figs. 6b, 8a), but not acute (Fig. 6a), SR141716 administration decreased floating in the FST. These results are in agreement with our previous findings, where we found no effect of acute SR141716 administration on total floating time at the first FST exposure on day 1, but a slight decrease following subchronic administration during repeated FST exposure on day 2 (Steiner et al., in press). The lack of acute drug effects on floating behavior are at odds with other reports that demonstrated antidepressant-like effects of acute treatment with the CB1 antagonists AM251 and SR141716 in the FST (Shearman et al., 2003; Tzavara et al., 2003; Griebel et al., 2005). Irrespective of the reasons for these discrepancies, our data imply that under our current experimental conditions (injection two hours prior to FST) the antidepressant-like effects of SR141716 with regard to floating behavior develop gradually, from no effect after acute SR141716 administration (Fig. 6a) via slight effects after subchronic (Fig. 6b) towards solid effects after chronic administration (Fig. 8a). Thus, in combination with our consistent findings on struggling, these results favor an antidepressant-like potential of chronic SR141716 administration at the behavioral level, in particular, as subchronic SR141716 (10 mg/kg) treatment was as effective as the classical antidepressant desipramine (20 mg/kg) in increasing struggling and decreasing floating behavior (Figs. 6b,d). In view of the loss of SR141716-induced corticosterone elevation after chronic injection (compare SR-SR bar in Fig. 7), one may argue that high pre-FST corticosterone levels caused by acute SR141716 injection could have prevented the development of antidepressant-like behavioral effects of the drug, which became more pronounced after subchronic and chronic SR141716 administration, when SR141716 injection-induced pre-FST corticosterone levels were reduced or absent. However, present data from our laboratory (unpublished results) do not support such a hypothesis, as we found clear acute antidepressant-like behavioral effects of SR141716 in the FST, when administered 1 h before the test, when pre-FST corticosterone levels were very high (compare Fig. 1).
The fact that we did not observe any behavioral differences in the FST between vehicle-treated CB1+/+ and CB1−/− mice (Figs. 5a-d) is in agreement with another study (Shearman et al., 2003) and suggests compensatory changes in CB1−/− mice due to the lifelong absence of CB1. On the other hand, behavioral genotype differences might have been masked by preceding vehicle injection of the animals, since we have recently observed increased floating behavior of CB1−/− mice in comparison with CB1+/+ mice without any prior treatment (Steiner et al., in press). Apparently, there is a discrepancy between the genetic inactivation and the pharmacological blockade of CB1 with regard to the behavioral performance in the FST, whereas the FST stress-induced corticosterone secretion is almost identically affected by the pharmacological blockade and the genetic inactivation of CB1.
Recently, Gobshtis and co-workers demonstrated that SR141716 and desipramine mediate their effects on floating behavior in the FST independently from each other (Gobshtis et al., 2007). Hill and co-workers, however, proposed that CB1 signaling is necessary for chronic desipramine treatment to exert its dampening effects on corticosterone secretion after FST exposure in rats (Hill et al., 2006). Although we failed to observe acute antidepressant-like behavioral effects of desipramine in CB1−/− mice (Figs. 5a,c), we demonstrated that the pharmacological blockade of CB1 did not interfere with behavioral effects of desipramine in the FST (Figs. 6a,c). This is in line with the first study (Gobshtis et al., 2007) and suggests that the discrepant findings obtained in CB1−/− mice are due to potential developmental changes. In this context, it has to be mentioned that Shearman and co-workers as well as our laboratory previously found intact antidepressant-like behavioral effects of desipramine in the FST in female CB1-deficient mice (Shearman et al., 2003; Steiner et al., in press). This adds another layer of complexity to the role of CB1 in the behavioral response to the FST, because it suggests that potential developmental changes in CB1-deficient mice that may lead to impaired desipramine responsiveness in the FST only occur in male mice and, thus, are sex-dependent.
Other than for the shared antidepressant-like behavioral effects, CB1 impairment and desipramine treatment differed in their effects on FST stress-induced corticosterone secretion. Whereas desipramine either had no effects on corticosterone secretion following FST in vehicle pre-treated mice (Fig. 6e), or slightly decreased corticosterone secretion in CB1+/+ mice (Fig. 5e), both the pharmacological blockade and the genetic knockout of CB1 increased corticosterone secretion (Figs. 5e, 6e). We found no interaction between impaired CB1 signaling and desipramine treatment, suggesting that CB1 is not mediating the neuroendocrine effects of subchronic desipramine treatment in mice. The discrepant findings observed by us and Hill and co-workers (Hill et al., 2006) might be due to species-specific differences (mice vs. rats) or to different desipramine treatment schedules (subchronic vs. chronic).
In conclusion, we demonstrate that CB1 signaling represents a general constraint mechanism for the secretion of corticosterone under basal conditions as well as in response to stress in male and female mice. Furthermore, we show that desipramine-induced neuroendocrine effects are unaffected by the absence of CB1 signaling, whereas desipramine-induced behavioral effects are compromised by the genetic but not the pharmacological impairment of CB1 signaling. Considering that antidepressant-like behavioral effects of subchronic and chronic SR141716 administration coincide with corticosterone elevating effects in the mouse FST, one might suggest that SR141716 could be of potential benefit for the treatment of atypical depression, which is characterized by a general HPA axis hypoactivity (Gold and Chrousos, 2002). However, further evaluation of SR141716 is required, before its general applicability for the treatment of depression or for the subtype of melancholic depression, which is characterized by HPA axis hyperactivity (Gold and Chrousos, 2002), should be envisioned (De Kloet et al., 2005; Witkin et al., 2005).
Acknowledgments
We would like to thank Klaus Wanisch, Johanna Stalla, Anja Mederer and Martina Reents (all MPI of Psychiatry) for excellent technical assistance. Part of the study was supported by grants from the National Institute of Mental Health and by a PhD fellowship of the Boehringer Ingelheim Foundation (to M.A. Steiner).
Role of Funding Source
Funding for this study was partly provided by NIMH grants and a PhD stipend of the Boehringer Ingelheim Foundation (to MA Steiner). NIMH and Boehringer Ingelheim Foundation had no further role in study design, in the collection and analysis and interpretation of data, in the writing of the report and in the decision to submit the paper for publication.
Footnotes
Conflict of Interest
All authors declare to have no conflicts of interest.
Contributors
Michel A Steiner contributed to the design of the experiments, carried out the experiments and wrote the first draft of the manuscript. Giovanni Marsicano created the CB1 receptor knock-out mice and supervised the breeding colony. He designed and facilitated all experiments involving CB1 receptor knock-out mice. Eric J Nestler designed and facilitated the social defeat experiment, provided the background information for the social defeat procedure and provided valuable criticism in writing the manuscript. Florian Holsboer contributed to the design, facilitation and interpretation of the desipramine experiment and provided valuable criticism in writing the manuscript. Beat Lutz and Carsten T Wotjak contributed to the design of all experiments, their statistical analysis and interpretation and substantially revised the first draft of the manuscript. All authors contributed to and have approved of the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barna I, Zelena D, Arszovszki AC, Ledent C. The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci. 2004;75:2959–2970. doi: 10.1016/j.lfs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Manqieri RA, Fu J, Kim JH, Arquello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007 doi: 10.1016/J.biopsych.2006.12.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Buckley NE, Hansson S, Harta G, Mezey E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience. 1998;82:1131–1149. doi: 10.1016/s0306-4522(97)00348-5. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Ann Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Conti AC, Cryan JF, Dalvi A, Lucki I, Blendy JA. cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J Neurosci. 2002;22:3262–3268. doi: 10.1523/JNEUROSCI.22-08-03262.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Lutz B, Vicennati V, Stalla GK, Pasquali R, Pagotto U. Endogenous cannabinoid system as a modulator of food intake. Int J Obesity. 2003;27:289–301. doi: 10.1038/sj.ijo.0802250. [DOI] [PubMed] [Google Scholar]
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. doi: 10.1210/en.2005-1649. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Holmes A. Advances in modelling human depression and anxiety. Nat Rev Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Joels M, Holsboer F. Stress and the brain: From adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Fride E, Suris R, Weidenfeld J, Mechoulam R. Differential response to acute and repeated stress in cannabinoid CB1 receptor knockout newborn and adult mice. Behav Pharmacol. 2005;16:431–440. doi: 10.1097/00008877-200509000-00016. [DOI] [PubMed] [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Lefur G, Casellas P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobshtis N, Ben Shabat S, Fride E. Antidepressant-induced undesirable weight gain: Prevention with rimonabant without interference with behavioral effectiveness. Eur J Pharmacol. 2007;554:155–163. doi: 10.1016/j.ejphar.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7:254–275. doi: 10.1038/sj.mp.4001032. [DOI] [PubMed] [Google Scholar]
- Griebel G, Stemmelin J, Scatton B. Effects of the cannabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol Psychiatry. 2005;57:261–267. doi: 10.1016/j.biopsych.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Haller J, Bakos N, Szirmay M, Ledent C, Freund TF. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci. 2002;16:1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- Haller J, Varga B, Ledent C, Barna I, Freund TF. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 2004;19:1906–1912. doi: 10.1111/j.1460-9568.2004.03293.x. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. Is there a role for the endocannabinoid system in the etiology and treatment of melancholic depression. Behav Pharmacol. 2005a;16:333–352. doi: 10.1097/00008877-200509000-00006. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. Pharmacological enhancement of cannabinoid CB1 receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur Neuropsychopharmacology. 2005b;15:593–599. doi: 10.1016/j.euroneuro.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB. Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2006;31:2591–2599. doi: 10.1038/sj.npp.1301092. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, Kunzel HE, Pfennig A, Uhr M, Holsboer F. Combined Dexamethasone/Corticotropin Releasing Hormone Test Predicts Treatment Response in Major Depression-A potential Biomarker? Biol Psychiatry. 2007;62:47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Karanth S, Linthorst ACE, Stalla GK, Barden N, Holsboer F, Reul JMHM. Hypothalamic-pituitary-adrenocortical axis changes in a transgenic mouse with impaired glucocorticoid receptor function. Endocrinology. 1997;138:3476–3485. doi: 10.1210/endo.138.8.5331. [DOI] [PubMed] [Google Scholar]
- Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol. 2005;168:299–325. doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- Manzanares J, Corchero J, Fuentes JA. Opioid and cannabinoid receptor-mediated regulation of the increase in adrenocorticotropin hormone and corticosterone plasma concentrations induced by central administration of Delta(9)-tetrahydrocannabinol in rats. Brain Res. 1999;839:173–179. doi: 10.1016/s0006-8993(99)01756-4. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Neuromodulatory functions of the endocannabinoid system. J Endocrinol Invest. 2006;29:27–46. [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang JR, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Pagotto U, Marsicano G, Fezza F, Theodoropoulou M, Grubler Y, Stalla J, Arzberger T, Milone A, Losa M, Di Marzo V, Lutz B, Stalla GK. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor type 1 and synthesize endogenous cannabinoids: First evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J Clin Endocrinol Metab. 2001;86:2687–2696. doi: 10.1210/jcem.86.6.7565. [DOI] [PubMed] [Google Scholar]
- Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Rosko KM, Fleischer R, Wang J, Xu S, Tong XS, Rocha BA. Antidepressant-like and anorectic effects of the cannabinoid CB1 receptor inverse agonist AM251 in mice. Behav Pharmacol. 2003;14:573–582. doi: 10.1097/00008877-200312000-00001. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Wanisch K, Monory K, Marsicano G, Borroni E, Bächli H, Holsboer F, Lutz B, Wotjak CT. Impaired cannabinoid receptor type 1 signaling interferes with stress-coping behavior in mice. Pharmacogenomics J. 2007 doi: 10.1038/sj.tpj.6500466. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Davis RJ, Perry KW, Li X, Salhoff C, Bymaster FP, Witkin JM, Nomikos GG. The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: implications for therapeutic actions. Br J Pharmacol. 2003;138:544–553. doi: 10.1038/sj.bjp.0705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriguen L, Perez-Rial S, Ledent C, Palomo T, Manzanares J. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology. 2004;46:966–973. doi: 10.1016/j.neuropharm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Valenti M, Vigano D, Casico MG, Rubino T, Steardo L, Parolaro D, Di Marzo V. Differential diurnal variations of anandamide and 2-arachidonoyl-glycerol levels in rat brain. Cell Mol Life Sci. 2004;61:945–950. doi: 10.1007/s00018-003-3453-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade MR, Degroot A, Nomikos GG. Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol. 2006;551:162–167. doi: 10.1016/j.ejphar.2006.08.083. [DOI] [PubMed] [Google Scholar]
- Wei Q, Lu XY, Liu L, Schafer G, Shieh KR, Burke S, Robinson TE, Watson SJ, Seasholtz AF, Akil H. Glucocorticoid receptor overexpression in forebrain: A mouse model of increased emotional lability. Proc Natl Acad Sci USA. 2004;101:11851–11856. doi: 10.1073/pnas.0402208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger T, Fernandez-Ruiz JJ, Ramos JA. Immunocytochemical demonstration of CB1 cannabinoid receptors in the anterior lobe of the pituitary gland. J Neuroendocrinol. 1999;11:873–878. doi: 10.1046/j.1365-2826.1999.00402.x. [DOI] [PubMed] [Google Scholar]
- Wenger T, Ledent C, Tramu G. The endogenous cannabinoid, anandamide, activates the hypothalamo-pituitary-adrenal axis in CB1 cannabinoid receptor knockout mice. Neuroendocrinology. 2003;78:294–300. doi: 10.1159/000074882. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Tzavara ET, Davis RJ, Li X, Nomikos GG. A therapeutic role for cannabinoid CB1 receptor antagonists in major depressive disorders. Trends Pharmacol Sci. 2005;26:609–617. doi: 10.1016/j.tips.2005.10.006. [DOI] [PubMed] [Google Scholar]