

Abstract
DNA interstrand crosslinks are processed by multiple mechanisms whose relationships to each other are unclear. Xeroderma pigmentosum-variant (XP-V) cells lacking DNA polymerase η are sensitive to psoralen photoadducts created under conditions favoring crosslink formation, suggesting a role for translesion synthesis in crosslink repair. Because crosslinks can lead to double strand breaks, we monitored phosphorylated H2AX (γ-H2AX), which is typically generated near double-strand breaks but also in response to single-stranded DNA, following psoralen photoadduct formation in XP-V fibroblasts to assess whether polymerase η is involved in processing crosslinks. In contrast to conditions favoring monoadducts, conditions favoring psoralen crosslinks induced γ-H2AX levels in both XP-V and nucleotide excision repair-deficient XP-A cells relative to control repair-proficient cells; ectopic expression of polymerase η in XP-V cells normalized the γ-H2AX response. In response to psoralen crosslinking, γ-H2AX as well as 53BP1 formed co-incident foci that were more numerous and intense in XP-V and XP-A cells than in controls. Psoralen photoadducts induced γ-H2AX throughout the cell cycle in XP-V cells. These results indicate that polymerase η is important in responding to psoralen crosslinks, and are consistent with a model in which nucleotide excision repair and polymerase η are involved in processing crosslinks and avoiding γ-H2AX associated with double strand breaks and single-stranded DNA in human cells.
Keywords: interstrand crosslink, psoralen, polymerase η, double-strand break, γH2AX
Introduction
DNA interstrand crosslinks (ICL) are an important class of DNA damage that is created by multiple drugs used in cancer chemotherapy. For example, psoralens in conjunction with ultraviolet A (UVA) radiation are often studied as representative ICL-forming agents and are commonly used to treat cutaneous T-cell lymphomas as well as various inflammatory dermatoses, [1, 2]. Psoralens are linear furocoumarins that intercalate between DNA base pairs and upon absorption of single ultraviolet A (UVA) photons are capable of forming monoadducts with pyrimidine bases. Following absorption of a second UVA photon, a furan-sided psoralen monoadduct can further react to form an ICL, primarily at 5’TpA3’ sequences. Unfortunately, as with other forms of chemotherapy involving ICL-forming agents, the use of psoralen and UVA for psoriasis has also been linked to an increased risk for secondary malignancies [3, 4].
Despite the importance of ICL-forming agents in chemotherapy and in iatrogenic carcinogenesis, the mechanisms by which ICL are repaired are still unclear in mammalian systems, particularly in humans. In yeast and mammalian cells, nucleotide excision repair (NER), recombination, and translesion synthesis, as well as certain mismatch repair proteins, have been reported to participate in ICL repair [2, 5-10]. Recombinational repair is in part thought to be a response to formation of double-strand breaks (DSBs) at ICL that appear to be unique to eukaryotes and that arise either as a result of collapsed replication forks blocked at ICL, or as true repair intermediates [8, 9, 11-13].
Another major pathway for ICL processing appears to be mediated by DNA polymerases that perform translesion synthesis (TLS). Several reports have indicated that TLS by polymerase ζ plays a key role in repairing ICL [11, 14-16]. However, another polymerase involved in TLS, polymerase η (pol η), has also been variably implicated in ICL repair. This polymerase is mutated in cells from individuals with the variant form of xeroderma pigmentosum (XP-V) who are prone to skin cancers yet are NER-proficient. While rad30-deficient yeast lacking pol η are not dramatically more sensitive to some ICL agents [5], studies in XP-V cells have suggested that pol η is important for ICL repair in humans. XP-V cells are hypersensitive to psoralen photoadducts, including ICL [17], exhibit increased mutagenesis in response to psoralen ICL associated with triplex-forming oligonucleotides [18], are moderately impaired in reactivation of psoralen crosslinked reporter plasmids [19], and are defective in recombination-independent ICL repair of mitomycin C-induced ICL in a plasmid reporter reactivation assay [20]. However, almost all of these studies have been limited to studying ICL in non-replicating plasmids rather than in native genomic DNA [18-20]. Further, it is unclear how TLS mediated by pol η might be related to pathways that involve DSB products or intermediates resulting from ICL.
γ-H2AX is a phosphorylated uncommon histone variant that forms rapidly after DSB formation in eukaryotic cells and that has commonly been employed as a marker for DSB, including studies of ICL [21-23], though it has also been reported to form in response to single stranded DNA (ssDNA) resulting from incomplete NER [24]. Recently, we observed that, in response to psoralen ICL, repair-proficient GM637 fibroblasts formed significantly less γ-H2AX than NER-deficient cells that lack the XPA protein [23]. This result suggested that cells possess an NER-dependent mechanism for processing ICLs that does not require generating a DSB intermediate as reflected by γ-H2AX. In this study, we demonstrate that, similar to XP-A cells, XP-V cells exhibit enhanced γ-H2AX levels following ICL formation, and that this response exists throughout the cell cycle. Our results are consistent with a model in which NER and TLS involving pol η are key components of an ICL repair pathway in native DNA that prevents formation or accumulation of γ-H2AX associated with DSB and ssDNA intermediates.
Materials and Methods
Chemicals
The psoralen derivative, 4’-hydroxymethyl-3,4,5’-trimethylpsoralen (HMT), as well as angelicin were used as obtained from the manufacturer (Sigma, St Louis, MO), and were dissolved in ethanol to form stock solutions that were stored at −20°C.
Cells
GM637 is an SV40-transformed fibroblast cell line from a normal individual that is proficient in the removal of psoralen monoadducts and ICL [25, 26]. XP12RO is an SV40-transformed fibroblast cell line derived from a xeroderma pigmentosum complementation group A patient [27]. XP30RO and XP-var1 are SV40-transformed fibroblast cell lines derived from unrelated XP-V patients with distinct mutations in pol η [28, 29]. XP30RO/pol η cl5 and XP30RO/cDNA3 are XP30RO cells that respectively contain either an expression vector containing the pol η cDNA or an empty vector [30-32]. All cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum at 37°C, 5% CO2 and were actively growing during subsequent experiments.
Treatment of cells
Cells were incubated in Hanks Balanced Salt Solution (HBSS) without phenol red and supplemented with HMT or angelicin for 20 minutes, then washed in HBSS and irradiated as previously reported [23]. For most experiments with HMT, a split-dose UVA irradiation method was used in which cells were initially irradiated with 5 kJ/m2, immediately rinsed twice with HBSS to remove free HMT, and then re-irradiated respectively with 15 kJ/m2. In some cases, 20 kJ/m2 was delivered continuously in a single dose. Following all treatments, cells were washed with HBSS and allowed to incubate in fresh medium for up to 9 hours before harvesting for analysis. Gamma irradiation was performed as previously described [23].
Cytotoxicity assay
Cells were plated in triplicate in 24-well plates (2 × 104 cells/well) and incubated at 37° C. After 18 hr, the medium was replaced with serum-free medium containing various concentrations of HMT, and cells were irradiated with either a single dose or split dose of UVA. Cytotoxicity was evaluated 72 hr after irradiation, as previously described, by the ability of cells to metabolize thiazolyl blue [33]. Results presented are the averages and standard deviations of three independent experiments assayed in triplicate.
Western immunoblotting
Cells were harvested and processed as previously described [23]. Blots were probed with monoclonal antibodies to γ-H2AX (clone JBW301,1:1000 dilution, Upstate, Lake Placid, NY), pol η (clone ab17725, 1:1000 dilution, Abcam Inc., Cambridge, MA), or β-actin (clone AC-74,1:3000 dilution, Sigma, St Louis, MO). In some cases, blots were stripped (Restore buffer, Pierce Biotechnology, Rockford, IL), and then reprobed. Intensities of bands were quantified using an Image Analyzer LAS-3000 (Fuji, Tokyo, Japan) and quantified using MultiGauge software (Fuji). γ-H2AX levels were normalized to those of β-actin. Plotted results represent the averages and standard deviations of three independent experiments.
Immunofluorescence
Cells were plated on chambered coverslips (Titertek) and incubated with 0.1 mM HMT in PBS supplemented with 5% fetal bovine serum for 10 minutes. Cells were irradiated with 0.1 J/m2 UVA, washed twice with PBS, and then re-irradiated with an additional 0.4 J/cm2 UVA. Cells were then allowed to incubate in DMEM supplemented with 10% FBS for 3 or 6 hours. For γ-H2AX and 53BP1 detection, cells were then fixed and stained exactly as previously described by Marti et al. [34] using antibodies to γ-H2AX and 53BP1 (Cell Signalling Technology, Beverly, MA). For bromodeoxyuridine (BrdU) experiments, cells were incubated with 10 μM BrdU (BDBiosciences, San Jose, CA) immediately following UVA treatment for 3 hours, then fixed and permeabilized using reagents from a BrdU Flow Kit (BDBioisciences) according to manufacturer instructions. The fixed and permeabilized cells were then incubated at room temperature with a rabbit polyclonal antibody to BrdU (Abcam, 1:1000 dilution) and/or the mouse monoclonal antibody to γ-H2AX for 20 min., washed 3 times in PBS containing 10% fetal bovine serum and 1% bovine serum albumin, and incubated for 1 hour at room temperature with anti-rabbit antibody conjugated to AlexaFluor 488 (Invitrogen, 1:1000 dilution) and/or anti-mouse antibody conjugated to AlexaFluor 594 (Invitrogen, 1:1000 dilution), followed by four washes in PBS. All slides were mounted with Prolong Gold Antifade Reagent with DAPI (Invitrogen) and visualized with an Axiovert 200 (Carl Zeiss).
Flow cytometry
As previously described [23], cells were trypsinized, washed with PBS, and fixed in TBS containing 2.0% (v/v) formaldehyde and 0.5−0.8% (v/v) methanol for 20 minutes on ice, then washed with 0.1% (w/v) saponin. Cells were then gently resuspended in permeabilization solution (0.5% saponin, 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2) and incubated with a fluorescein-conjugated monoclonal antibody to γ-H2AX (Upstate, 1:1000 dilution) for 20 minutes. After washing to remove excess antibody, cells were incubated with 10 μg/mL propidium iodide and 100 μg/mL ribonuclease A for 4 h at 4 C° [34]. Cells were analyzed with a FACScan (Becton Dickinson, San Jose, CA).
Results
XP-V cells are hypersensitive to interstrand crosslinks but not monoadducts
The XP-V cell line, XP30RO, possesses mutations in pol η that result in a severe truncation of the protein [35, 36]. XP30RO/pol η is an XP-V cell line transfected with a vector expressing wild-type pol η cDNA, while XP30RO/cDNA3 is an empty vector-treated control cell line [31, 32]. As shown in Fig. 1A, XP30RO/pol η cells expressed detectable levels of pol η while XP30RO/cDNA3 cells did not. To determine whether pol η is important for survival following psoralen ICL formation, we measured the sensitivity of these cell lines to UVA photoadducts of the psoralen derivative, HMT. Cells were exposed to a range of HMT concentrations and treated either with a single 5 kJ/m2 UVA dose in which most lesions are monoadducts or with a split-dose UVA protocol designed to enrich for ICL to the point where they constitute a majority of the photoadducts [37]. Importantly, the single and split-dose UVA protocols generate an identical number of HMT photoadducts and only differ in the ratio of monoadducts to ICL. As shown in Fig. 1B, relative to XP30RO/pol η cells, XP30RO/cDNA cells were much more sensitive to HMT photoadducts generated with either the single or split-dose protocols. However, at HMT doses at which XP30RO/pol η cells showed no significant difference in sensitivity to single or split-dose UVA (i.e., 0.1 and 0.3 M), uncorrected XP30RO/cDNA cells were 2−3-fold more sensitive to the split-dose than the single dose protocol. These results suggest that pol η contributes to cellular survival following psoralen ICL relative to monoadducts, and may be preferentially involved in responding to psoralen ICLs.
Figure 1.

Hypersensitivity of XP-V cells to psoralen photoadducts. A) Whole cell lysates of XP30RO/c15 (XP-V/pol η) and XP30RO/cDNA3 (XP-V/cDNA) cells were analyzed by Western immunoblotting for pol η expression, using actin as a loading control. B) Cytotoxicity following HMT and UVA treatment. XP-V/pol η(•, ■) and XP-V/cDNA (○, □) were exposed to increasing concentrations of either HMT, and then irradiated with either a single dose of 5 kJ/m2 UVA (•,○), or a split-dose UVA protocol consisting of 5 kJ/m2, followed by rinsing with HBSS to remove unbound HMT and then re-irradiation with 15 kJ/m2 (■,□)). Cells were then replaced in media and incubated for 3 days before survival was assessed. Error bars denote standard deviation.
Psoralen photoadducts induce γ-H2AX in XP-A and XP-V cells
To determine the relative dependence of the γ-H2AX response to ICL on NER and pol η, we assessed the response of XP-A and XP-V cells following psoralen photoadduct formation. Both wild-type GM637 and NER-deficient XP-A cells possess detectable pol η protein, while two separate XP-V cell lines, XP30RO and XPvar-1 do not (Fig. 2A, D). Cells were treated with HMT and split-dose UVA irradiation, and γ-H2AX levels were measured. When assayed by Western immunoblotting, γ-H2AX was undetectable in untreated cells, in cells treated with UVA alone, as well as immediately following treatment with both HMT and UVA ([23] and data not shown). Three hours following treatment with HMT and split-dose UVA, γ-H2AX was detectable in all cells. Relative to GM637, γ-H2AX was almost two-fold greater in XP-A cells, as previously observed [23], and a similar induction of γ-H2AX occurred in the XP-V cells (Fig. 2B and 2E). An identical dose of HMT and of total UVA delivered in a single continuous irradiation, creating more overall photoadducts but relatively fewer ICL and more monoadducts, resulted in γ-H2AX levels in XP-V cells that were intermediate between those of GM637 and XP-A (Fig. 2C). Since XP-A cells are completely NER-deficient for both monoadducts and ICL [26], while XP-V cells are NER-proficient, these results indicate that both NER and pol η are important in avoiding γ-H2AX in response to psoralen ICL. Moreover, the results suggest that the elevated γ-H2AX levels observed in XP-V cells are largely due to unprocessed ICL and not monoadducts which, as long as NER is intact, should be fully repairable without forming DSB.
Figure 2.
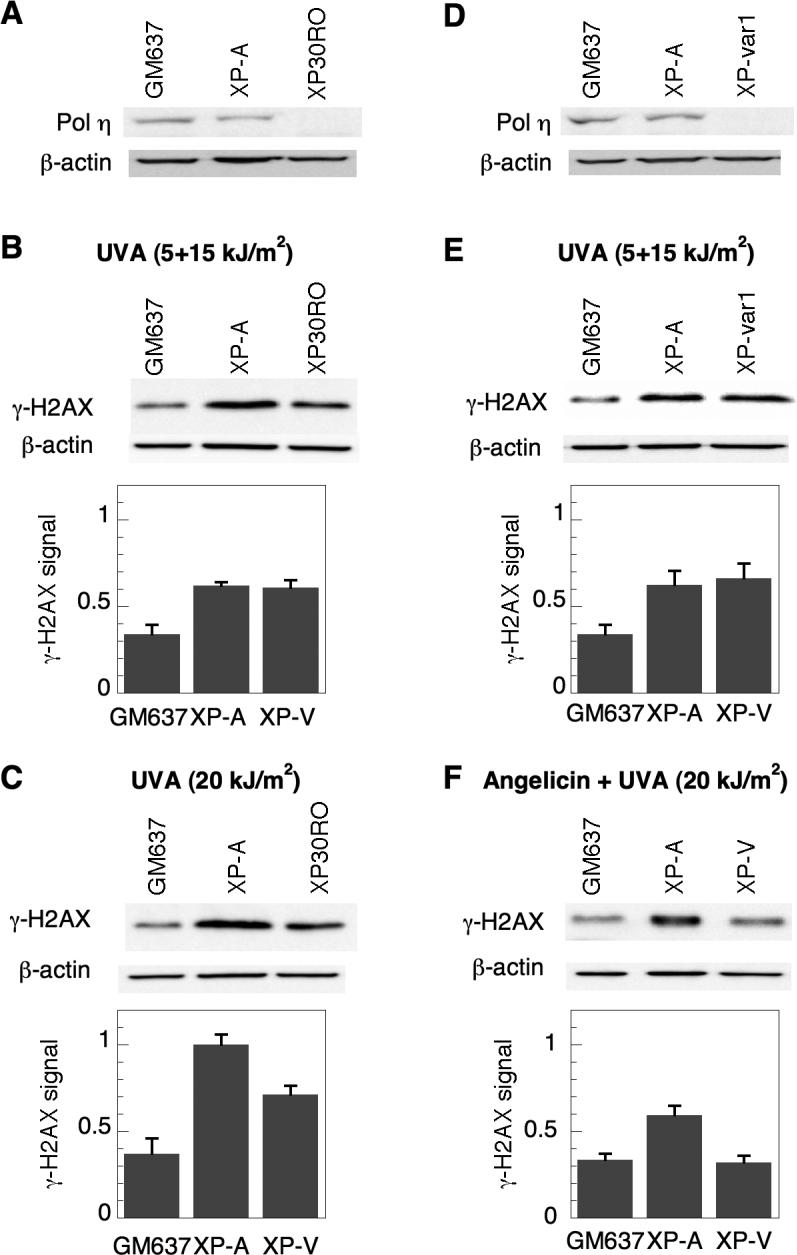
Induction of γ-H2AX in XP-A and XP-V cells following psoralen photoadducts. A) pol η expression was analyzed by Western immunoblotting in GM637, XP-A (XP12RO) and XP-V (XP30RO) cells. B) Cells were treated with 10 μM HMT and split-dose UVA, incubated in media for 3 hours, and analyzed by Western immunoblotting for γ-H2AX expression. C) Cells were also treated with 10 μM HMT and continuously irradiated with 20 kJ/m2 UVA. Identical experiments were performed with a separate XP-V cell line (XPvar-1) and compared to GM637 and XP-A cells to assess D) pol η expression, and E) γ-H2AX expression following treatment with HMT and split-dose UVA. F) γ-H2AX expression following angelicin and UVA. GM637, XP-A and XP-V (XP30RO) cells were treated with 10 μM angelicin and irradiated with 20 kJ/m2 UVA, allowed to incubate for 6 hours, and then harvested for Western immunoblotting to detect and quantify γ-H2AX and β-actin levels. Actin served as a loading control in all experiments. Error bars in graphs denote standard deviation.
As a further test of the selective role of ICL and not monoadducts in inducing γ-H2AX in XP-V cells, γ-H2AX formation following angelicin and UVA was measured. Angelicin is a furocoumarin that, unlike psoralens, forms only monoadducts and is incapable of forming ICL. As shown in Fig. 2F, following treatment with angelicin and UVA, γ-H2AX levels in XP-V cells were not elevated relative to GM637, while XP-A cells exhibited γ-H2AX levels that remained almost twice those of GM637. These results further suggest that a lack of pol η leads to excess γ-H2AX formation principally after ICL, and not monoadducts.
Restoration of pol η in XP-V cells reduces γ-H2AX induction following psoralen photoadducts
To further confirm that pol η deficiency and not an idiosyncratic characteristic of the XP-V cell lines was responsible for the observed increases in γ-H2AX following psoralen ICL formation, we compared XP30RO/cDNA3 cells which contain an empty vector and XP30RO/pol η cells in which XP30RO has been corrected with a vector expressing pol η (Fig. 1A). Following treatment with HMT and split-dose UVA, XP30RO/cDNA3 cells formed approximately twice as much γ-H2AX as control GM637 cells, as observed for the parental XP30RO cells (Fig. 3). As expected, restoration of pol η in XP30RO/pol η cells reduced γ-H2AX levels to normal, suggesting that loss of pol η is specifically responsible for elevating γ-H2AX levels in response to ICL in these cells.
Figure 3.
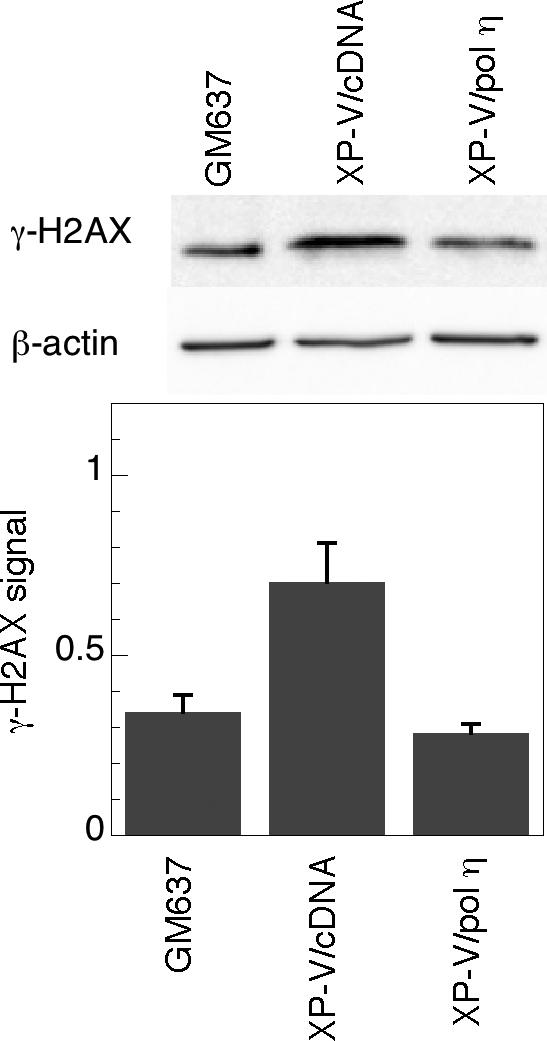
Restoration of pol η expression suppresses γ-H2AX induction following psoralen-induced ICL. GM637, XP30RO/c15 (XP-V/pol η), and XP30RO/cDNA3 (XP-V/cDNA) were treated with 10 μM HMT and split-dose UVA, incubated in media for 3 hours, and then analyzed by Western immunoblotting for γ-H2AX expression. Actin served as a loading control.
Psoralen photoadducts induce formation of γ-H2AX foci in XP-V and XP-A cells
To determine the spatial pattern and confirm the overall increases in γ-H2AX levels in XP-V and XP-A cells observed by Western immunoblotting, γ-H2AX immunofluorescence was performed on wild-type GM637, XP30RO and XP12RO cells treated with HMT and split dose UVA (Fig. 4). As a positive control, GM637 cells treated with γ-rays, which directly generate DSB, exhibited nuclear foci of γ-H2AX as well as of 53BP1, another protein that localizes to DSB, whereas no foci of γ-H2AX and only diffuse 53BP1 staining were observed in unirradiated cells (Fig. 4A, B) [38]. Because immunofluorescence proved to be much more sensitive than Western immunoblotting, it was necessary to reduce the HMT concentration to 0.1 μM. Following treatment with split-dose UVA in the absence of HMT, all cells showed weak γ-H2AX immunofluorescence and few foci (Fig. 4E, C, E, G). Following treatment with both HMT and UVA, GM637 cells remained comparable to untreated controls (Fig. 4D) whereas γ-H2AX intensity increased dramatically in both XP-A and XP-V cells (Fig. 4F, H). In addition, the γ-H2AX immunofluorescence pattern in XP-A and XP-V cells consisted of numerous discrete, bright foci rather than diffuse nuclear staining. Immunofluorescent foci of 53BP1 also increased in number in GM637 cells treated with γ-rays, and in HMT-UVA-treated XP-A and XP-V cells, and largely co-localized with γ-H2AX foci. For GM637, XP-A, and XP-V cells treated with split-dose UVA alone or with HMT and split-dose UVA, γ-H2AX foci were quantified in 100 cells (Fig. 4I), confirming the increase in γ-H2AX in XP-A and XP-V cells relative to wild-type GM637 following HMT and UVA treatment, but not following UVA alone.
Figure 4.
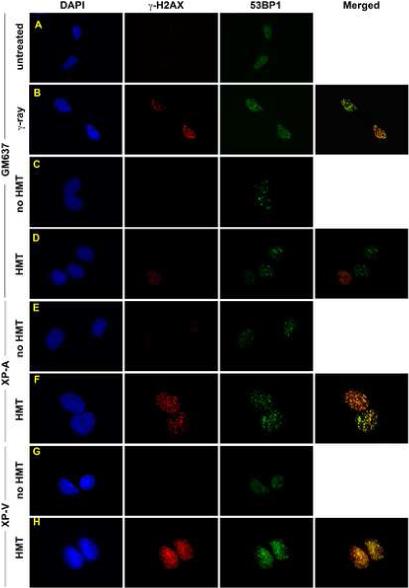
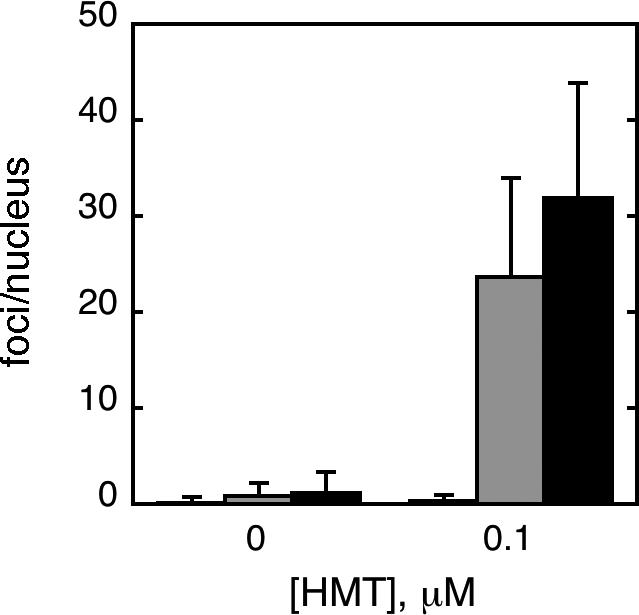
Immunofluorescence of γ-H2AX following HMT and UVA. GM637 cells that were A) untreated, or B) irradiated with 6 Gy of γ-rays and fixed within 15 minutes. GM637 (C, D), XP12RO (E,F), and XP30RO (G,H) cells were incubated either with no HMT (C, E, G) or with 0.1 μM HMT followed by split dose UVA. Cells were allowed to incubate in medium for 6 hours before fixation. All cells were stained with antibodies to γ-H2AX and 53BP1, counterstained with DAPI, and epifluorescence was imaged with a 100× objective. In merged images, yellow color represents sites of coincident γ-H2AX and 53BP1 antibody staining. I) γ-H2AX foci were quantified in nuclei of one hundred randomly selected GM637 (white bars), XP-A (gray bars) and XP-V (black bars) cells treated with or without HMT followed by split-dose UVA to determine the average number of foci per nucleus. Error bars denote standard deviations.
Psoralen photoadducts induce γ-H2AX throughout the cell cycle
Because another bypass polymerase, pol ζ, in yeast has been reported to be involved in ICL processing but restricted to G1 of the cell cycle [15], we sought to determine the dependence of γ-H2AX formation on pol η during the cell cycle. Cells were labeled with BrdU following UVA irradiation for 3 hours to identify cells that had passed through S-phase during the experiment. While over 80% of XP-V cells exhibited γ-H2AX foci following HMT and UVA treatment, less than 50% labeled with bromodeoxyuridine following the treatment, as illustrated by Fig. 5A in which not all cells with γ-H2AX foci are labeled with BrdU, suggesting that γ-H2AX formed largely outside of S-phase. To analyze the cell cycle distribution further, XP30RO/cDNA3 and XP30RO/pol η cells were treated with HMT and split-dose UVA, and then assayed by flow cytometry for DNA content and for γ-H2AX expression (Fig. 5B). UVA irradiation alone did not induce significant levels of γ-H2AX in either XP30RO/cDNA3 or XP30RO/pol η cells (Fig. 5B, panels a,f,c,h). However, when XP30RO/cDNA3 cells were treated with HMT and UVA, significant levels of γ-H2AX were induced in all phases of the cell cycle in 38% of cells (Fig. 5B, panels b,c,e). In contrast, replacement of pol η in XP30RO/pol η cells reduced γ-H2AX induction throughout the cell cycle such that only 9% of cells exhibited γ-H2AX fluorescence above background (Fig. 5B, panels g,h,j). Notably, a significant sub-G1 population of cells did not occur, suggesting that apoptosis is not occurring during the time scale of these experiments.
Figure 5.

Cell cycle dependence of γ-H2AX expression following HMT and UVA. A) XP30RO cells were treated with 0.1 μM HMT and split dose UVA, and immediately incubated in medium containing BrdU for 3 hours followed by staining with DAPI and antibodies to γ-H2AX and BrdU. B) XP30RO/ cDNA (panels a-e) and XP30RO/cl5 (panels f-j) were treated with either UVA alone (a, d, f, i) or 3 μM HMT and split dose-UVA irradiation (b, e, g, j), incubated for 3 hours, and then harvested and co-stained with fluorescent monoclonal antibodies to γ-H2AX and propidium iodide for analysis by flow cytometry. Panels a, b, f, and g display propidium iodide signal on the x-axis, and γ-H2AX signal on the y-axis. Panels d, e, i, and j represent histograms of the propidium iodide signal, while panels C and H represent histograms of the γ-H2AX signal. In panels a, b, f and g, the horizontal line represents the background threshold for γ-H2AX levels. In panels c and h, thin curves represent UVA alone and bold curves represent HMT+UVA.
Discussion
Although results from yeast suggest that pol η does not confer resistance to ICL by cisplatin [5], XP-V cells have been reported to be hypersensitive to and impaired in the repair of a number of DNA damaging agents, including ICL-forming agents, suggesting that TLS by pol η is important in lesion tolerance and survival following ICL in humans [17, 20, 32, 39]. However, most prior studies of pol η and ICLs have been performed utilizing plasmid substrates, rather than analyzing genomic DNA [18-20]. We confirmed that loss of pol η renders cells moderately hypersensitive to genomic photoadducts of the psoralen derivative, HMT, under split-dose UVA conditions designed to generate significant quantities of ICL. Further, unlike single dose irradiation of HMT or of angelicin, which generate mostly or exclusively monoadducts, split-dose UVA induces elevated γ-H2AX levels in XP-V cells relative to wild-type GM637 cells, and this induction returns to normal levels when pol η is ectopically expressed in XP-V cells, indicating that pol η is indeed involved in the cellular response to psoralen ICL in genomic DNA.
The observed reduction of γ-H2AX throughout the cell cycle in the presence of pol η in response to ICL also suggests that pol η can mediate a response to ICL or its repair intermediates that are not necessarily at blocked replication forks. Because there is considerable evidence that γ-H2AX is a marker of DSB [21-23, 40], one interpretation of our results is that pol η may contribute to suppression of DSB in response to ICL throughout the cell cycle. Although there is evidence from mammalian cell extracts in which replication is likely not occurring that DSB may still be generated at psoralen ICL [8], the preponderance of reports have shown that ICL induce DSB during replication [12, 13, 21, 41]. Thus, an alternative interpretation of the data is that the observed γ-H2AX induction, particularly outside of S-phase, is not entirely in response to DSB formation. Recently, it has been reported that following ultraviolet radiation γ-H2AX does not necessarily reflect DSB during the G1 phase of the cell cycle [42], as well as in growth-arrested cells in G0 [24]. Interestingly, ICL-forming agents have been reported to generate ssDNA foci that depend on NER, and ssDNA resulting from impaired gap-filling during nucleotide excision repair has recently been reported to be associated with γ-H2AX induction [24, 43].
Our results suggest that pol η participates in a psoralen ICL repair pathway that avoids accumulation of DSB or ssDNA intermediates that serve to trigger induction of γ-H2AX (Fig. 6). While both DSB and ssDNA have been identified as potential intermediate products of replication arrested at a DNA lesion and likely account for a γ-H2AX signal during S-phase, ssDNA associated with incomplete NER may account for γ-H2AX generated during other phases of the cell cycle. NER may operate to uncouple one strand of the ICL, followed by pol η-mediated TLS using the remaining intact strand as a template, as has been generally proposed for other polymerases previously [15, 16, 20]. While a role for gap-filling during NER by pol η at lesions outside of a replication fork has not been specifically described, polymerase κ, another bypass polymerase in the Y-family to which pol η also belongs, has been proposed to function in this role in NER [44]. Loss of NER may direct more ICL into another pathway, specifically dependent on XPF/ERCC1 but not NER, that processes the lesions to DSB [21, 23]. Such a model also accounts for the extreme hypersensitivity of cells deficient in XPF or ERCC1 which appear to be involved in both DSB-forming and NER/TLS repair pathways [21, 23].
Figure 6.

Model for psoralen ICL repair pathways involving polymerase η and their relationship to γ-H2AX.
An alternate but not mutually exclusive possibility consistent with our results is that pol η functions in a separate pathway downstream of ICL-induced γ-H2AX associated with DSB, and that loss of pol η leads to an accumulation of irreparable DSB, leading to enhanced γ-H2AX levels (Fig. 6). Recently, it has been proposed that, following DSB formation by a blocked replication fork at an ICL, TLS occurs as a necessary prelude for homologous recombination [45], and pol η could be the TLS polymerase involved in this model. In addition, pol η has also been reported to be involved in homologous recombination with a polymerase activity independent of its TLS functions [46, 47] and could function in recombinational repair of DSB resulting from ICL.
The relative importance of pol η and other TLS polymerases in ICL repair is unclear. Recent studies have suggested that polymerase ζ is the predominant determinant of ICL repair [14-16]. However, in yeast, REV3 activity in ICL repair was restricted exclusively to the G1 phase of the cell cycle, leaving open the possibility that other TLS polymerases might fulfill similar roles in other phases of the cell cycle [15]. Our data suggest that pol η may have a more general role in the response to ICL throughout the cell cycle, and may complement the more restricted activity of polymerase ζ. In addition, pol η and pol ζ may function cooperatively in TLS at an ICL. Pol η is unique in its ability to perform translesion synthesis opposite cyclobutane pyrmidine dimers presumably due to its extremely open catalytic site. However, pol η often has difficulty extending past bulkier or more distorting DNA lesions [48]. In contrast, pol ζ is extremely poor in replicating opposite a wide variety of DNA lesions, and appears to function best as an extender of 3’ ends opposite a lesion. Thus, both polymerases may be necessary to optimally bypass the unusual structure resulting from an initial ICL uncoupling.
Acknowledgments
We thank J. Cleaver for providing XP30RO-SV40, P. Hanawalt and A. Ganesan for providing XP-var1, and M. O'Connor and A. Lehmann for providing XP30RO/c15 and XP30RO/cDNA3. We thank J. Cleaver and C. Largman for helpful discussions, and H. Kataoka for assistance with flow cytometry. This work was supported by NIH grant CA105958 and a VA Merit Award (D.H.O.).
Abbreviations
- BrdU
bromodeoxyuridine
- DSB
double-strand break
- HMT
4’-hydroxymethyl-3,4,5’-trimethylpsoralen
- ICL
interstrand crosslink
- NER
nucleotide excision repair
- ssDNA
single-strand DNA
- TLS
translesion synthesis
- UVA
ultraviolet A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gasparro FP, editor. Psoralen DNA Photobiology. CRC Press, Inc.; Boca Raton: 1988. [Google Scholar]
- 2.Dronkert MLG, Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 3.Stern RS. Malignant melanoma in patients treated for psoriasis with PUVA. Photodermatol. Photoimmunol. Photomed. 1999;15:37–38. doi: 10.1111/j.1600-0781.1999.tb00052.x. [DOI] [PubMed] [Google Scholar]
- 4.Stern RS, Laird N, Melski J, Parrish JA, Fitzpatrick TB, Bleigh HL. Cutaneous squamous-cell carcinoma in patients treated with PUVA. N. Engl. J. Med. 1984;310:1156–1161. doi: 10.1056/NEJM198405033101805. [DOI] [PubMed] [Google Scholar]
- 5.Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE. S. cerevisiae has three pathways for DNA interstrand crosslink repair. Mutat. Res. 2001;487:73–83. doi: 10.1016/s0921-8777(01)00106-9. [DOI] [PubMed] [Google Scholar]
- 6.Saffran WA, Ahmed S, Bellevue S, Pereira G, Patrick T, Sanchez W, Thomas S, Alberti M, Hearst JE. DNA repair defects channel interstrand DNA cross-links into alternate recombinational and error-prone repair pathways. J. Biol. Chem. 2004;279:36462–36469. doi: 10.1074/jbc.M402323200. [DOI] [PubMed] [Google Scholar]
- 7.Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol. Cell. Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ. hMutSβ is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol. Cell Biol. 2002;22:2388–2397. doi: 10.1128/MCB.22.7.2388-2397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barber LJ, Ward TA, Hartley JA, McHugh PJ. DNA interstrand cross-link repair in the Saccharomyces cerevisiae cell cycle: Overlapping roles for PSO2(SNM1) with MutS factors and EXO1 during S phase. Mol. Cell. Biol. 2005;25:2297–2309. doi: 10.1128/MCB.25.6.2297-2309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng H, Wang X, Legerski RJ, Glazer PM, Li L. Repair of DNA interstrand cross-links: Interactions between homology-dependent and homology-independent pathways. DNA Repair. 2006 doi: 10.1016/j.dnarep.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 11.McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besso T. Induction of DNA replication-mediated double strand breaks by psoralen DNA interstrand cross-links. J. Biol. Chem. 2003;278:5250–5254. doi: 10.1074/jbc.M212323200. [DOI] [PubMed] [Google Scholar]
- 13.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S, Liu ST, Majumdar A, Liu JL, Nairn RS, Bernier M, Maher V, Sieidman MM. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways. Nucleic Acids Res. 2005;33:5382–5303. doi: 10.1093/nar/gki851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase z. EMBO J. 2006;25:1285–1994. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen X, Jun S, O'Neal LE, Sonoda E, Bemark M, Sale JE, Li L. REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquinated proliferating cell nuclear antigen (PCNA) J. Biol. Chem. 2006;281:13869–13872. doi: 10.1074/jbc.C600071200. [DOI] [PubMed] [Google Scholar]
- 17.Misra RR, Vos J-MH. Defective replication of psoralen adducts detected at the gene-specific level in xeroderma pigmentosum variant cells. Mol. Cell. Biol. 1993;13:1002–1012. doi: 10.1128/mcb.13.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raha M, Wang G, Seidman MM, Glazer PM. Mutagenesis by third-strand-directed psoralen adducts in repair-deficient human cells: High frequency and altered spectrum in a xeroderma pigmentosum variant. Proc. Natl. Acad. Sci. 1996;93:2941–2946. doi: 10.1073/pnas.93.7.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination independent and error-prone pathway of DNA interstrand cross-link repair. Mol. Cell Biol. 2001;21:713–720. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase h-mediated error-prone removal of mitomycin C interstrand cross-links. Mol. Cell. Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothfuss A, Grompe M. Repair kinetics of genomic interstrand cross-links: Evidence for DNA double-strand break-dependent activation of the Fanconi Anemia/BRCA pathway. Mol. Cell Biol. 2004;24:123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo h.B., Hoeijmakers JHJ, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mogi S, Oh DH. γ-H2AX formation in response to interstrand crosslinks requires XPF in human cells. DNA Repair. 2006;5:731–740. doi: 10.1016/j.dnarep.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto M, Yaginuma K, Igarashi A, Imura M, Hasegawa M, Iwabuchi K, Date T, Mori T, Ishizaki K, Yamashita K, Inobe M, Matsunaga T. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci. 2007;120:1104–1112. doi: 10.1242/jcs.03391. [DOI] [PubMed] [Google Scholar]
- 25.Gruenert DC, Cleaver JE. Repair of psoralen-induced cross-links and monoadducts in normal and repair-deficient human fibroblasts. Cancer Res. 1985;45:5399–5404. [PubMed] [Google Scholar]
- 26.Vuksanovic L, Cleaver JE. Unique cross-link and monoadduct repair characteristics of a xeroderma pigmentosum revertant cell line. Mutat. Res. 1987;184:255–263. doi: 10.1016/0167-8817(87)90024-1. [DOI] [PubMed] [Google Scholar]
- 27.Royer-Pokora B, Peterson J, W.D., Haseltine WA. Biological and biochemical characterization of an SV40-transformed xeroderma pigmentosum cell line. Exp. Cell Res. 1984;151:408–420. doi: 10.1016/0014-4827(84)90391-4. [DOI] [PubMed] [Google Scholar]
- 28.Volpe JP, Cleaver JE. Xeroderma pigmentosum variant cells are resistant to immortalization. Mutat. Res. 1995;337:111–117. doi: 10.1016/0921-8777(95)00015-c. [DOI] [PubMed] [Google Scholar]
- 29.Barbis DP, Schultz RA, Friedberg EC. Isolation and partial characterization of virus-transformed cell lines representing the A, G and variant complementation groups of xeroderma pigmentosum. Mutat. Res. 1986;165:175–184. doi: 10.1016/0167-8817(86)90052-0. [DOI] [PubMed] [Google Scholar]
- 30.Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LHF, Lehmann AR. Domain structure, localization, and function of DNA polymerase η, defective in xeroderma pigmentosum variant cells. Genes and Dev. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stary A, Kannouche P, Lehmann AR, Sarasin A. Role of DNA polymerase η in the UV mutation spectrum in human cells. J Biol. Chem. 2003;278:18767–18775. doi: 10.1074/jbc.M211838200. [DOI] [PubMed] [Google Scholar]
- 32.Albertella MR, Green CM, Lehmann AR, O'Connor MJ. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- 33.Oh DH, Yeh K. Differentiating human keratinocytes are deficient in p53 but retain global nucleotide excision repair following ultraviolet radiation. DNA Repair. 2005;4:1149–1159. doi: 10.1016/j.dnarep.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 36.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yusasa m., Araki M, Iwai S, takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 37.Vos JM, Hanawalt PC. DNA interstrand cross-links promote chromosomal integration of a selected gene in human cells. Mol. Cell. Biol. 1989;9:2897–2905. doi: 10.1128/mcb.9.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y-W, Cleaver JE, Hanaoka F, Chang C-F, Chou K-M. A novel role of DNA polymerase h in modulating cellular sensitivity to chemotherapeutic agents. Mol. Cancer Res. 2006;4:257–265. doi: 10.1158/1541-7786.MCR-05-0118. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 41.Akkari YMN, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol. Cell. Biol. 2000;20:8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cleaver JE, Hefner E, Laposa RR, Karentz D, Marti T. Cockayne syndrome exhibits dysregulation of p21 and other gene products that may be independent of transcription-coupled repair. Neuroscience. 2006 doi: 10.1016/j.neuroscience.2006.08.074. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee Y-J, Park S-J, Ciccone SLM, Kim C-R, Lee SH. An in vivo analysis of MMC-induced DNA damage and repair. Carcinogenesis. 2006;27:446–453. doi: 10.1093/carcin/bgi254. [DOI] [PubMed] [Google Scholar]
- 44.Ogi T, Lehmann A. The Y-family DNA polymerase κ (pol κ) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006;8:640–642. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]
- 45.Cipak L, Watanabe N, Bessho T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nat. Struct. Mol. Biol. 2006;13:729–733. doi: 10.1038/nsmb1120. [DOI] [PubMed] [Google Scholar]
- 46.Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada k., Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, Takeda S. Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Mol. Cell. 2005;20:793–799. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 47.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 48.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]