

Abstract
Context
The common P12A polymorphism in PPARG (a target for thiazolidinedione medications) has been consistently associated with type 2 diabetes.
Objective
We examined whether PPARG P12A affects progression from impaired glucose tolerance (IGT) to diabetes, or responses to preventive interventions (lifestyle, metformin or troglitazone versus placebo).
Patients
3,548 Diabetes Prevention Program participants.
Design
We performed Cox regression analysis using genotype at PPARG P12A, intervention, and their interactions as predictors of diabetes incidence. We also genotyped five other PPARG variants implicated in the response to troglitazone and assessed their effect on insulin sensitivity at one year.
Results
Consistent with prior cross-sectional studies, P/P homozygotes at PPARG P12A appeared more likely to develop diabetes than alanine carriers (hazard ratio 1.24, 95% CI 0.99–1.57, P=0.07), with no interaction of genotype with intervention. There was a significant interaction of genotype with body mass index (BMI) and waist circumference (P=0.03 and 0.002 respectively), with the alanine allele conferring less protection in more obese individuals. Neither PPARG P12A nor five other variants significantly affected the impact of troglitazone on insulin sensitivity in 340 participants at one year.
Conclusions
The proline allele at PPARG P12A increases risk for diabetes in persons with IGT, an effect modified by BMI. In addition, PPARG P12A has little or no effect on the beneficial response to troglitazone.
Keywords: Diabetes, PPARG, polymorphism
INTRODUCTION
The peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear hormone receptor preferentially expressed in adipose tissue (1). Activation by its ligand causes it to heterodimerize with the retinoid X receptor, bind specific DNA elements and induce a transcriptional cascade that leads to adipocyte differentiation and increased sensitivity to insulin. The PPARγ molecule is now recognized as the cognate receptor for thiazolidinediones (2). A proline → alanine change in codon 12 of its gene PPARG (P12A) has been reproducibly associated with a decreased risk for type 2 diabetes (3–12); the proline allele confers a ∼20% increased risk under a recessive model. Because of its high frequency in the population, the population attributable risk of this variant nears 25% (4). Although some studies have not achieved statistical significance in their attempt to replicate this finding (13–21), most of them report consistent odds ratios (OR) with overlapping 95% confidence intervals, such that a meta-analysis of all published evidence yields a combined P value that achieves genome-wide significance (22). How this molecular change impairs protein function and leads to an increased risk of type 2 diabetes has not been fully elucidated (23).
The risk of type 2 diabetes conferred by PPARG P12A has also been evaluated prospectively. The Finnish Diabetes Prevention Study (24), which randomized 522 subjects with impaired glucose tolerance (IGT) to either placebo or a lifestyle intervention, reported a two-fold increase in risk of developing type 2 diabetes among alanine carriers in the placebo arm when compared to P/P homozygotes, a result which seemed to contradict the sizeable body of cross-sectional literature described above. On the other hand, the much larger Botnia Prospective Study (N=2,293) documented a hazard ratio (HR) for developing diabetes of 1.7 among P/P homozygotes, a result which was statistically significant (25). Different ascertainment schemes (IGT vs a population sample) and analytical methods (logistic regression vs Cox proportional hazards analysis) may explain some but not all of these discrepancies.
In addition to its role in increasing risk of type 2 diabetes, the P12A variant may also affect therapeutic response; if so, its putative impact on preventive interventions might have clinical utility. In support of this concept, two studies have examined the effect of PPARG P12A on response to thiazolidinediones (26, 27). Blüher and colleagues treated 131 subjects with pioglitazone for 26 weeks; the proportion of responders (defined as >15% decrease in HbA1C levels and/or >20% decrease in fasting blood glucose when compared to baseline after 12 or 26 weeks of pioglitazone) did not differ between P/P homozygotes and alanine carriers (26). Snitker and colleagues examined 93 Hispanic women with a previous history of gestational diabetes enrolled in the Troglitazone in Prevention of Diabetes (TRIPOD) study, and obtained intravenous glucose tolerance tests before and three months after treatment with troglitazone; genotype at PPARG P12A did not explain the variability in insulin sensitivity observed among these women (27).
It is possible that these studies were underpowered or that other variants in PPARG may account for the differential therapeutic response. To examine the second possibility, the TRIPOD investigators genotyped a set of 131 common PPARG variants in the same group of 93 Hispanic women, and reported that eight PPARG polymorphisms were associated with response to troglitazone, defined as an overrepresentation of the minor allele in the upper two tertiles of insulin sensitivity (SI during IVGTT) after three months of troglitazone treatment (28). Two of these SNPs (rs4135263 and rs1152003) also showed nominal associations with changes in SI as a quantitative trait under recessive genetic models (28).
As a next step in clarifying the conflicting literature and evaluating the effect of PPARG P12A on thiazolidinedione response in a large multiethnic sample, we set out to confirm the predictive power of this variant and assess its impact on the lifestyle and pharmacological interventions employed in the Diabetes Prevention Program (DPP) (29). We further examined the five non-redundant SNPs which had shown positive nominal associations with response to troglitazone in the TRIPOD study (28) for a similar effect on troglitazone response in the DPP cohort.
METHODS
The Diabetes Prevention Program
The details of study design and preventive interventions have been described elsewhere (29–31). The DPP was a 27-center randomized clinical trial that examined whether a lifestyle intervention directed at modifying risk factors for type 2 diabetes (overweight, and sedentary lifestyle), or metformin would prevent or delay the development of diabetes in persons at high risk. The DPP enrolled 3,234 nondiabetic persons with IGT and elevated fasting glucose and randomized them to placebo, metformin 850 mg twice daily, or a lifestyle intervention program; a fourth arm of 585 subjects assigned to treatment with troglitazone 400 mg daily was stopped two years after the trial commenced because of hepatotoxicity (30). The principal endpoint was the development of diabetes, confirmed on a second test using ADA criteria. The lifestyle and metformin interventions reduced the incidence of diabetes in high-risk individuals by 58% (95% CI 48–66) and 31% (95% CI 17–43) respectively versus placebo (29). Diabetes incidence rates were 11.0, 7.8, and 4.8 per 100 person-years in the placebo, metformin, and lifestyle groups respectively; treatment effects were consistent across sex and self-reported ethnicity, and diabetes incidence did not differ across ethnic groups (29). When analyses were restricted to the mean 0.9-year period of active troglitazone treatment, diabetes incidence rates were 12.0, 6.7, 5.1 and 3.0 per 100 person-years in the placebo, metformin, lifestyle and troglitazone groups respectively (P<0.001, troglitazone versus placebo) (32).
Participants
The 3,548 participants included in this study (92.9% of all DPP participants: 2,994 who completed the trial in their originally assigned treatment groups, plus 554 originally randomized to troglitazone) provided informed consent specific to genetic investigation. The study was approved by the relevant Institutional Review Boards at the participating sites. Of the participants in this study, 56.4% were Caucasian, 20.2% were African American, 16.8% were Hispanic, 4.3% were Asian American and 2.4% were American Indian by self report. The participants’ mean age was 51 years and mean body mass index (BMI) was 34.0 kg/m². Subjects had semiannual measurements of fasting glucose and glycated hemoglobin, and an annual 75-g oral glucose tolerance test (OGTT); given the early termination of the troglitazone arm, one-year data were available in only 340 of the participants randomly assigned to troglitazone.
PPARG SNP selection
In addition to P12A (rs1801282), we also genotyped five of the eight SNPs reported by Wolford et al. to have positive nominal associations with response to troglitazone (28). The TRIPOD investigators found that, of those eight SNPs (rs13073869, rs880663, rs4135263, rs1152003, rs6806708, rs13065455, rs13088205 and rs13088214), the first two and the last three were in perfect linkage disequilibrium with each other, respectively (r2=1.0); we therefore selected a non-redundant set of five SNPs for our analyses (rs880663, rs4135263 rs1152003, rs6806708 and rs13065455). We confirmed that these SNPs were indeed non-redundant in our five ethnic groups: with the exception of rs6806708 and rs13065455, which were in near-perfect linkage disequilibrium both in the original publication and in our samples (r2=0.9–1.0), the other SNPs had pairwise r2 ranging from 0.0–0.2 in Caucasians to 0.1–0.4 in American Indians.
Genotyping
DNA was extracted from peripheral blood leukocytes through conventional procedures and quantitated by picogreen analysis (Molecular Probes). Genotyping of PPARG P12A was performed in the forward and reverse directions by allele-specific primer extension of single-plex amplified products, with detection by matrix-assisted laser desorption ionization-time of flight mass spectroscopy on a Sequenom platform as previously described (33); the five other PPARG SNPs were genotyped in the same manner but with single-direction primers only. Our genotyping success rate was 99.8% and there were no discordant genotypes on forward and reverse primer extension. The allele frequencies of all six SNPs in each of the five ethnic groups were in Hardy-Weinberg equilibrium (P>0.01).
Quantitative traits
Data from the baseline and one-year OGTTs were used to calculate measures of insulin secretion and insulin sensitivity, which were expressed using glucose and insulin measured in conventional units (milligrams per deciliter and microunits per milliliter, respectively) as previously described (34). The insulinogenic index (35) was calculated as [(insulin at 30 min) − (insulin at 0 min)]/[(glucose at 30 min) − (glucose at 0 min)]). The insulin sensitivity index (ISI, reciprocal of insulin resistance by the homeostasis model assessment (36)) was calculated as 22.5/[fasting insulin × (fasting glucose/18.01)]. In addition, we examined fasting glucose and 2-hour OGTT glucose at baseline and one year.
Statistical analysis
Time to onset of diabetes was the primary endpoint. Because the previous literature consistently reports a recessive model of risk transmission for proline carriers at PPARG P12A, P/A and A/A individuals were grouped into one genotypic category (A/X). We examined Cox regression models with genotype, intervention and their interactions as the independent variables predicting time to diabetes. These models were also examined with baseline BMI, waist circumference, age, gender and self-reported ethnicity as covariates. Analyses were repeated in the subset of ethnic groups that had comparable allele frequencies (Caucasians, Hispanics and Asian Americans), and in Caucasians only; whether we restricted our analysis to these subgroups or tested for a genotype × ethnicity interaction, we detected no significant effect of self-reported ethnicity in any of our analyses.
For the quantitative trait comparisons, we first obtained baseline measures in the entire cohort according to genotype at PPARG P12A. Differences between means in the two genotypic groups (P/P and A/X) were tested using t tests. For the one-year measurements, a general linear model was examined with and without 3-way interactions (treatment group, genotype, and baseline value of each trait). Least square means were adjusted for baseline values; two-sided nominal P values are reported. The SAS analysis system version 8.2 was used for all analyses (SAS Institute, Inc., Cary, NC).
In order to determine the potential effects of genotype on responsiveness to troglitazone, we calculated the ISI in the 340 DPP participants who completed one year of troglitazone treatment at baseline and one year. In accordance with the previous classification (28), we divided this group into tertiles of change in ISI (one-year ISI minus baseline ISI) and assigned the top two tertiles as “responders” and the bottom tertile as “non-responders”; we then examined allelic frequency differences between the two groups by chi square analysis. In addition, we compared change in ISI as a quantitative trait according to genotype at all five loci by means of the non-parametric Kruskal-Wallis test; if nominally significant differences were found, pairwise comparisons between genotypic groups were performed with a Wilcoxon test, with P values adjusted by Holm’s procedure as previously described (37). We also compared one-year ISI as a quantitative trait according to genotypic group at all five loci, adjusted for baseline ISI, under the additive and recessive models. Finally, in order to control for allele frequency differences among populations, we repeated these analyses in the largest group (Caucasians) only.
RESULTS
Allele frequency distribution
For PPARG P12A, the frequency of the minor alanine allele in DPP US Caucasians (0.10) was comparable to that previously reported in other Caucasian populations (4, 6, 8, 16). We found significant differences in minor allele frequencies in African Americans (0.02) and American Indians (0.19) when compared to Caucasians; therefore, analyses for incidence of diabetes were performed both with and without these two ethnic groups.
At PPARG P12A, genotypic frequencies were equally distributed among the four treatment arms and two gender groups. We found no significant differences in baseline age or BMI, but P/P homozygotes appeared to have a smaller waist circumference (Table 1).
Table 1.
Demographic characteristics and baseline quantitative traits according to genotype at PPARG P12A in the Diabetes Prevention Program
| Genotype
|
|||
|---|---|---|---|
| Baseline trait | P/P (n=2,942) | A/X (n=605) | P* |
| Treatment | |||
| Placebo | 831 (83.1) | 169 (16.9) | 0.14 |
| Metformin | 831 (84.0) | 158 (16.0) | |
| Lifestyle | 839 (83.6) | 165 (16.4) | |
| Troglitazone | 441 (79.6) | 113 (20.4) | |
|
| |||
| Male | 957 (81.7) | 214 (18.3) | 0.18 |
| Age in years | 50.8 ± 10.5 | 50.9 ± 10.9 | 0.83 |
| Body mass index (kg/m2) | 34.0 ± 6.7 | 34.2 ± 6.4 | 0.41 |
| Waist (cm) | 105 ± 14.5 | 107 ± 14.5 | 0.002 |
|
| |||
| Glycemic trait | |||
| Ins Index [(μU/ml)/(mg/dl)] | 1.24 ± 0.93 | 1.23 ± 0.90 | 0.73 |
| ISI [(μU/ml)x(mmol/L)]−1 | 0.194 ± 0.130 | 0.194 ± 0.126 | 0.93 |
| Fasting glucose (mg/dl) | 106.9 ± 8.2 | 106.7 ± 8.1 | 0.60 |
| 2-hour glucose (mg/dl) | 164.8 ± 17.0 | 164.5 ± 17.3 | 0.66 |
Plus-minus values are means ± SD, other values are n (%).
Based on t-tests for continuous variables and chi-square for categorical variables; traits were log transformed for statistical analyses where appropriate. Ins Index, insulinogenic index; ISI, insulin sensitivity index. One sample failed genotyping.
Incidence of diabetes
Consistent with previous cross-sectional case/control results, the DPP showed that individuals who were homozygous for the proline allele appeared to progress more rapidly from IGT to diabetes than alanine carriers (HR 1.24, 95% CI 0.99–1.57, P=0.07). We found no interaction between genotype and intervention (P value for the genotype × metformin interaction, 0.89; P value for the genotype × lifestyle interaction, 0.61). Hazard ratios were similar across all treatment arms (Fig. 1). In the placebo group, the hazard ratio was slightly higher but had wider 95% confidence intervals (HR 1.28, 95% CI 0.90–1.82, P=0.17). When the sample was restricted to the Caucasian group only, the overall hazard ratio for all treatment groups combined was statistically indistinguishable, but again with wide 95% confidence intervals possibly due to the smaller sample size (HR 1.18, 95% CI 0.89–1.57, P=0.24).
Figure 1.

Incidence of diabetes per treatment arm and genotype at PPARG P12A in the Diabetes Prevention Program. A, all arms; B, placebo; C, metformin; D, lifestyle intervention.
When baseline BMI was added to the model, we noted a nominally significant genotype × BMI interaction (P=0.03), such that alanine carriers were more susceptible to the deleterious effect of BMI on diabetes incidence than proline homozygotes (Fig. 2). Addition of the BMI interaction term to the model did not significantly alter the overall effect of genotype. Similar effects were noted for waist circumference (which is highly correlated with BMI in this cohort): there was a nominally significant interaction between genotype and waist circumference (P=0.002), and in a model adjusting for baseline waist circumference P/P homozygotes were more likely to progress to diabetes than alanine carriers (HR 1.27, 95% CI 1.01–1.60, P=0.04).
Figure 2.
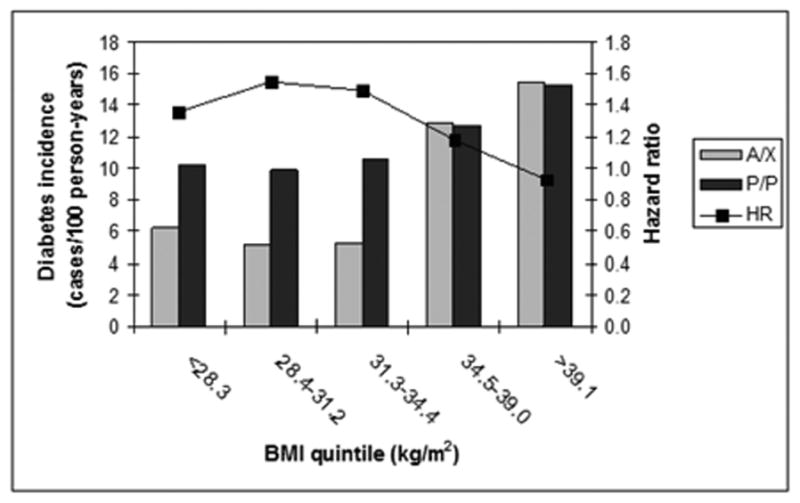
Interaction of BMI with genotype at PPARG P12A on diabetes risk. The bar plot (left axis) shows incidence of diabetes (cases/100 person-years) in the placebo arm by quintile of baseline BMI for either alanine carriers or proline homozygotes at PPARG P12A. The line plot (right axis) shows the hazard ratio (HR, P/P versus A/X) in the full DPP cohort by quintile of baseline BMI. The protective effect of alanine seems to disappear at BMI >34.5 kg/m2.
Quantitative traits
At baseline, proline homozygotes and alanine carriers had indistinguishable indices of insulin sensitivity and insulin secretion (Table 1). At one year, the lifestyle intervention, metformin and troglitazone all led to significant improvements in insulin sensitivity, as previously reported (32, 34); however, there were no significant differences in the magnitude of these improvements by genotype at PPARG P12A (Table 2). Examination of fasting and 2-hour glucose levels after OGTT both at baseline and at one year did not reveal significant differences between proline homozygotes and alanine carriers across all treatment groups (Table 1 and data not shown).
Table 2.
One-year quantitative glycemic traits according to PPARG P12A genotypes by treatment arm in the DPP
| Genotype
|
|||
|---|---|---|---|
| Ins Index [(μU/ml)/(mg/dl)] | P/P | A/X | P value |
| Placebo | 1.21 (1.14–1.27) | 1.24 (1.09–1.38) | 0.72 |
| Metformin | 1.20 (1.14–1.27) | 1.16 (1.01–1.31) | 0.62 |
| Lifestyle | 1.18 (1.12–1.25) | 1.29 (1.14–1.43) | 0.20 |
| Troglitazone | 1.25 (1.13–1.37) | 1.10 (0.86–1.33) | 0.25 |
| ISI [(μU/ml)x(mmol/L)]−1 | P/P | A/X | P value |
|
| |||
| Placebo | 0.190 (0.183–0.198) | 0.192 (0.176–0.208) | 0.87 |
| Metformin | 0.234 (0.225–0.243) | 0.234 (0.214–0.254) | 0.99 |
| Lifestyle | 0.268 (0.257–0.279) | 0.292 (0.267–0.318) | 0.09 |
| Troglitazone | 0.268 (0.248–0.289) | 0.289 (0.249–0.328) | 0.37 |
Values are expressed as least-squares means (adjusted for baseline values) with 95% confidence intervals. Ins Index, insulinogenic index; ISI, insulin sensitivity index.
Response to troglitazone
In addition to PPARG P12A, we examined the five PPARG SNPs rs880663, rs4135263 rs1152003, rs6806708 and rs13065455 for association with response to troglitazone. The median (25th –75th percentile) ISI (expressed in [(μU/ml) × (mmol/L)]−1) for participants randomly assigned to troglitazone treatment at baseline was 0.163 (0.119–0.232). After one year of troglitazone treatment, participants in the bottom tertile of change in ISI (“non-responders”) did not show any improvement in ISI (one-year ISI minus baseline ISI, −0.070 ± 0.088, mean ± SD), whereas “responders” in the middle and upper tertiles did (one-year ISI minus baseline ISI, +0.047 ± 0.028 and +0.252 ± 0.180, respectively). There were no significant allele frequency differences at any of the five loci between troglitazone “responders” and troglitazone ”non-responders” after one year (Table 3).
Table 3.
Association testing of five PPARG SNPs for response to troglitazone at one year in the Diabetes Prevention Program
| Responders vs. Non-responders | Change in ISI at one year [median (range)] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Alleles (M/m) | MAF | OR (95% CI) | P * | M/M | M/m | m/m | P † | P ‡ |
| rs880663 | T/C | 0.29 | 1.18 (0.83–1.69) | 0.36 | 0.034 (−0.573/+0.778) | 0.062 (−0.286/+1.069) | 0.016 (−0.149/+0.938) | 0.005 | TT v CT: 0.02 CT v CC: 0.04 TT v CC: 0.35 |
| rs4135263 | T/C | 0.17 | 0.88 (0.58–1.34) | 0.55 | 0.050 (−0.573/+0.997) | 0.039 (−0.333/+1.069) | 0.021 (−0.052/+0.186) | 0.94 | |
| rs1152003 | C/G | 0.45 | 1.02 (0.74–1.40) | 0.91 | 0.046 (−0.286/+0.778) | 0.051 (−0.337/+1.069) | 0.028 (−0.573/+0.938) | 0.94 | |
| rs6806708 | G/T | 0.35 | 0.96 (0.68–1.33) | 0.79 | 0.043 (−0.337/+0.659) | 0.048 (−0.573/+0.997) | 0.056 (−0.142/+1.069) | 0.86 | |
| rs13065455 | C/A | 0.34 | 0.93 (0.67–1.31) | 0.69 | 0.047 (−0.337/+0.659) | 0.047 (−0.573/+0.997) | 0.056 (−0.142/+1.069) | 0.86 | |
Five non-redundant PPARG single nucleotide polymorphisms (SNPs) were tested for association with response to troglitazone in the DPP after one year of treatment. M/m, major allele/minor allele; MAF, minor allele frequency; OR, odds ratio; CI, confidence interval; ISI, insulin sensitivity index; P *, P value for the allelic association of the minor allele with response to troglitazone, defined as a larger proportion of minor alleles in the upper two tertiles of change in ISI by chi square analysis; P †, P value (non-parametric Kruskal-Wallis test) of the comparison of change in ISI after one year across all three genotypic groups; P ‡, P value (Wilcoxon test) of the pairwise comparison between genotypic groups, performed if the initial test was nominally significant and corrected for multiple comparisons by the Holm procedure.
When we analyzed change in ISI at one year as a continuous trait, we noted a nominally significant higher change in ISI in rs880663 heterozygotes when compared to homozygotes for either allele (P=0.02–0.04); no other nominally significant differences were found at any of the four remaining SNPs (Table 3). Similar results were obtained when we compared one-year ISI (adjusted for baseline ISI) across genotypic groups. Furthermore, in contrast with the results of Wolford et al. (28), homozygotes for the minor allele at all five SNPs had one-year ISI levels (adjusted for baseline ISI) indistinguishable from major allele carriers (P=0.10–0.84). Adjustment for gender, baseline age, baseline BMI or self-reported ethnicity did not change the results. Analyses restricted to the largest ethnic group (Caucasians only, N=201) did not reveal any statistically significant differences in the response to troglitazone.
DISCUSSION
A limited number of common genetic variants have been consistently associated with type 2 diabetes (22): these include PPARG P12A, the E23K polymorphism in the gene encoding the islet ATP-sensitive potassium channel Kir6.2 (KCNJ11) and SNP44 in the gene that encodes calpain 10 (CAPN10). More recently, a common allele in the TCF7L2 gene has been convincingly associated with type 2 diabetes, with an estimated allelic relative risk of 1.56 and high statistical significance (38). While these validated associations have been usually tested in case/control samples, few studies have examined them prospectively and/or in regard to their effect on therapeutic interventions.
The DPP is a unique study in which to carry out such analyses. It differs from other diabetes prevention trials (39, 40) in that it included both lifestyle and pharmacological interventions; in addition, its multiethnic design reflects the diversity of the US population. Moreover, its large sample size makes it adequate for genetic studies where variants are thought to confer modest risk. An important distinction with other large observational trials (25) is the DPP’s interventional design and the exclusive enrollment of individuals with IGT, which suggests the presence of some degree of genetic risk at baseline and may introduce selection bias by imposing constraints at ascertainment. We have recently validated the association of common variants in TCF7L2 with development of diabetes in this cohort (33).
Genetic studies in multiethnic cohorts raise the issue of population stratification (41). We have addressed this possible confounder by repeating the analyses in the ethnic groups which have comparable allele frequencies, further restricting the analyses to the largest ethnic group alone, and explicitly testing for a genotype × ethnicity interaction. In addition, we note that in the short interval and high-risk population studied in the DPP there were no significant differences in diabetes incidence across ethnic groups (29); thus, it is unlikely that differences in allele frequencies across populations have confounded our phenotypic results.
In agreement with both the Botnia Prospective Study (25) and the preponderance of the cross-sectional literature (and in contrast with the Finnish Diabetes Prevention Study (24)), we also observed a modest genetic risk conferred by the homozygous P/P genotype at PPARG P12A. Although the P values we obtained do not quite reach conventional statistical significance, the very high prior probability that PPARG P12A increases risk of type 2 diabetes makes us believe that the hazard ratios we have noted here represent a real effect. Possible reasons for its lower magnitude in the DPP include the initial high-risk characteristics of the DPP cohort and the limited 3-year window of the IGT-to-diabetes transition examined during this study. It is also possible that this variant may exert a stronger effect on the transition from normoglycemia to IGT, rather than in the progression from IGT to diabetes.
By detecting a strong genotype × obesity interaction we have been able to clarify some of the heterogeneity found in the literature, where studies conducted in leaner populations tend to report higher odds ratios for risk associated with the P/P genotype (3, 5). Our data show that the protective effect of the alanine allele disappears at BMIs above ∼35 kg/m2. Indeed, this might partially explain the apparent lack of a protective effect of the alanine allele in the Finnish Diabetes Prevention Study (24), where A/A homozygotes were more obese than P/P homozygotes at baseline (BMI 33.0 ± 6.3 versus 31.1 ± 4.4 kg/m2 [mean ± SD], respectively). This interaction of PPARG P12A with BMI is also consistent with the increased skeletal muscle glucose uptake seen in lean but not obese (BMI >27 kg/m2) alanine carriers when compared to P/P homozygotes (42).
Despite the well-documented effect that this missense polymorphism (in a gene that encodes the molecular target for thiazolidinedione medications) has on type 2 diabetes, we have been unable to detect a discernible impact of this variant on quantitative glycemic traits such as fasting glucose, 2-hour glucose after an OGTT, or validated measures of insulin secretion and insulin sensitivity. In addition, both a lifestyle intervention and troglitazone treatment for one year improved insulin sensitivity in proline homozygotes and alanine carriers to a similar degree. Our findings support similar results reported in smaller groups of 131 German subjects with type 2 diabetes treated with pioglitazone for 26 weeks (26) or 93 Hispanic women with a history of gestational diabetes treated with troglitazone for 3 months (27), although the length of exposure to thiazolidinediones was modest for all three studies. If the small non-significant differences we observed between genotypic groups are real, we estimate that at least 3,995 subjects would be needed to have 80% power to detect this difference at an alpha of 0.05, which in turn raises the question of its clinical relevance.
It is possible that other genetic variants at PPARG may affect thiazolidinedione response, even though none of them has been convincingly associated with type 2 diabetes. Recently, a comprehensive set of common variants in PPARG was genotyped in the TRIPOD group of 93 Hispanic women with a history of gestational diabetes, and examined for their impact on response to troglitazone. Allele frequencies at eight PPARG SNPs differed between the 63 responders and 30 non-responders, although the sample was small and the P values modest (28). We have been unable to reproduce these findings of association for five of those SNPs in our larger cohort of 340 subjects. This lack of replication may be due to the differences in duration of troglitazone treatment (three months in TRIPOD versus 1 year in the DPP), differing estimates of insulin sensitivity (SI from IVGTT versus ISI from OGTT), phenotypic heterogeneity (gestational diabetes versus IGT), ethnic variation or statistical fluctuations: nevertheless, because the 95% confidence intervals between both studies overlap, we cannot exclude that the results are mutually consistent. The nominally significant higher change in ISI at one year in rs880663 heterozygotes in the DPP does not conform with the published data and does not follow a clear genetic model; given the multiple tests performed, this finding likely represents a false positive result. The next logical step will be to test comprehensively all common variation at PPARG in the various ethnic groups of the DPP.
In summary, we have confirmed the modest protection from type 2 diabetes conferred by the alanine allele at PPARG P12A, we have shown a significant interaction of this variant with BMI and waist circumference, and in examining the largest cohort studied to date we have not detected any significant effect of genotype at PPARG P12A in response to troglitazone.
Acknowledgments
The investigators gratefully acknowledge the commitment and dedication of the participants of the DPP. The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health provided funding to the clinical centers and the Coordinating Center for the design and conduct of the study; collection, management, analysis, and interpretation of the data. The Southwestern American Indian Centers were supported directly by the NIDDK and the Indian Health Service. The General Clinical Research Center Program, National Center for Research Resources supported data collection at many of the clinical centers. Funding for data collection and participant support was also provided by the Office of Research on Minority Health, the National Institute of Child Health and Human Development, the National Institute on Aging, the Centers for Disease Control and Prevention, Office of Research on Women’s Health, and the American Diabetes Association. Bristol-Myers Squibb and Parke-Davis provided medication. This research was also supported, in part, by the intramural research program of the NIDDK. LifeScan Inc., Health O Meter, Hoechst Marion Roussel, Inc., Merck-Medco Managed Care, Inc., Merck and Co., Nike Sports Marketing, Slim Fast Foods Co., and Quaker Oats Co. donated materials, equipment, or medicines for concomitant conditions. McKesson BioServices Corp., Matthews Media Group, Inc., and the Henry M. Jackson Foundation provided support services under subcontract with the Coordinating Center. The opinions expressed are those of the investigators and do not necessarily reflect the views of the Indian Health Service or other funding agencies. A complete list of Centers, investigators, and staff can be found in reference 30. JCF is supported by NIH Research Career Award 1 K23 DK65978-03.
We thank Santica Marcovina and Greg Strylewicz for careful processing of the DNA samples, and Mark Daly, Noël Burtt and our colleagues at the Broad Institute Genetic Analysis Platform for excellent assistance.
Footnotes
“This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.”
Disclosures: The authors have nothing to declare relevant to the contents of this article.
References
- 1.Spiegelman BM. PPAR-γ: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 3.Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998;20:284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 4.Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 5.Hara K, Okada T, Tobe K, Yasuda K, Mori Y, Kadowaki H, Hagura R, Akanuma Y, Kimura S, Ito C, Kadowaki T. The Pro12Ala polymorphism in PPARγ2 may confer resistance to type 2 diabetes. Biochem Biophys Res Commun. 2000;271:212–216. doi: 10.1006/bbrc.2000.2605. [DOI] [PubMed] [Google Scholar]
- 6.Ardlie KG, Lunetta KL, Seielstad M. Testing for population subdivision and association in four case-control studies. Am J Hum Genet. 2002;71:304–311. doi: 10.1086/341719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mori H, Ikegami H, Kawaguchi Y, Seino S, Yokoi N, Takeda J, Inoue I, Seino Y, Yasuda K, Hanafusa T, Yamagata K, Awata T, Kadowaki T, Hara K, Yamada N, Gotoda T, Iwasaki N, Iwamoto Y, Sanke T, Nanjo K, Oka Y, Matsutani A, Maeda E, Kasuga M. The Pro12 → Ala substitution in PPAR-γ is associated with resistance to development of diabetes in the general population: possible involvement in impairment of insulin secretion in individuals with type 2 diabetes. Diabetes. 2001;50:891–894. doi: 10.2337/diabetes.50.4.891. [DOI] [PubMed] [Google Scholar]
- 8.Douglas JA, Erdos MR, Watanabe RM, Braun A, Johnston CL, Oeth P, Mohlke KL, Valle TT, Ehnholm C, Buchanan TA, Bergman RN, Collins FS, Boehnke M, Tuomilehto J. The peroxisome proliferator-activated receptor-γ2 Pro12Ala variant: association with type 2 diabetes and trait differences. Diabetes. 2001;50:886–890. doi: 10.2337/diabetes.50.4.886. [DOI] [PubMed] [Google Scholar]
- 9.Memisoglu A, Hu FB, Hankinson SE, Liu S, Meigs JB, Altshuler DM, Hunter DJ, Manson JE. Prospective study of the association between the Proline to Alanine codon 12 polymorphism in the PPARγ gene and type 2 diabetes. Diabetes Care. 2003;26:2915–2917. doi: 10.2337/diacare.26.10.2915. [DOI] [PubMed] [Google Scholar]
- 10.Doney ASF, Fischer B, Cecil JE, Boylan K, McGuigan FE, Ralston SH, Morris AD, Palmer CNA. Association of the Pro12Ala and C1431T variants of PPARG and their haplotypes with susceptibility to Type 2 diabetes. Diabetologia. 2004;47:555–558. doi: 10.1007/s00125-003-1323-1. [DOI] [PubMed] [Google Scholar]
- 11.Ghoussaini M, Meyre D, Lobbens S, Charpentier G, Clement K, Charles M-A, Tauber M, Weill J, Froguel P. Implication of the Pro12Ala polymorphism of the PPAR-γ2 gene in type 2 diabetes and obesity in the French population. BMC Medical Genetics. 2005;6:11. doi: 10.1186/1471-2350-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeggini E, Parkinson JR, Halford S, Owen KR, Walker M, Hitman GA, Levy JC, Sampson MJ, Frayling TM, Hattersley AT, McCarthy MI. Examining the relationships between the Pro12Ala variant in PPARG and Type 2 diabetes-related traits in UK samples. Diabet Med. 2005;22:1696–1700. doi: 10.1111/j.1464-5491.2005.01717.x. [DOI] [PubMed] [Google Scholar]
- 13.Mancini FP, Vaccaro O, Sabatino L, Tufano A, Rivellese AA, Riccardi G, Colantuoni V. Pro12Ala substitution in the peroxisome proliferator-activated receptor-γ2 is not associated with type 2 diabetes. Diabetes. 1999;48:1466–1468. doi: 10.2337/diabetes.48.7.1466. [DOI] [PubMed] [Google Scholar]
- 14.Ringel J, Engeli S, Distler A, Sharma AM. Pro12Ala missense mutation of the peroxisome proliferator activated receptor γ and diabetes mellitus. Biochem Biophys Res Commun. 1999;254:450–453. doi: 10.1006/bbrc.1998.9962. [DOI] [PubMed] [Google Scholar]
- 15.Meirhaeghe A, Fajas L, Helbecque N, Cottel D, Auwerx J, Deeb SS, Amouyel P. Impact of the peroxisome proliferator activated receptor γ2 Pro12Ala polymorphism on adiposity, lipids and non-insulin-dependent diabetes mellitus. Int J Obes Relat Metab Disord. 2000;24:195–199. doi: 10.1038/sj.ijo.0801112. [DOI] [PubMed] [Google Scholar]
- 16.Clement K, Hercberg S, Passinge B, Galan P, Varroud-Vial M, Shuldiner AR, Beamer BA, Charpentier G, Guy-Grand B, Froguel P, Vaisse C. The Pro115Gln and Pro12Ala PPARγ gene mutations in obesity and type 2 diabetes. Int J Obes Relat Metab Disord. 2000;24:391–393. doi: 10.1038/sj.ijo.0801191. [DOI] [PubMed] [Google Scholar]
- 17.Oh EY, Min KM, Chung JH, Min Y-K, Lee M-S, Kim K-W, Lee M-K. Significance of Pro12Ala mutation in peroxisome proliferator-activated receptor-γ 2 in Korean diabetic and obese subjects. J Clin Endocrinol Metab. 2000;85:1801–1804. doi: 10.1210/jcem.85.5.6499. [DOI] [PubMed] [Google Scholar]
- 18.Lei HH, Chen MH, Yang WS, Chiu MC, Chen MC, Tai TY, Chuang LM. Peroxisome proliferator-activated receptor γ2 Pro12Ala gene variant is strongly associated with larger body mass in the Taiwanese. Metabolism. 2000;49:1267–1270. doi: 10.1053/meta.2000.9517. [DOI] [PubMed] [Google Scholar]
- 19.Sramkova D, Kunesova M, Hainer V, Hill M, Vcelak J, Bendlova B. Is a Pro12Ala polymorphism of the PPARγ2 gene related to obesity and type 2 diabetes mellitus in the Czech population? Ann NY Acad Sci. 2002;967:265–273. doi: 10.1111/j.1749-6632.2002.tb04282.x. [DOI] [PubMed] [Google Scholar]
- 20.Malecki MT, Frey J, Klupa T, Skupien J, Walus M, Mlynarski W, Sieradzki J. The Pro12Ala polymorphism of PPARγ2 gene and susceptibility to type 2 diabetes mellitus in a Polish population. Diabetes Research and Clinical Practice. 2003;62:105–111. doi: 10.1016/s0168-8227(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 21.Tai ES, Corella D, Deurenberg-Yap M, Adiconis X, Chew SK, Tan CE, Ordovas JM. Differential effects of the C1431T and Pro12Ala PPARγ gene variants on plasma lipids and diabetes risk in an Asian population. J Lipid Res. 2004;45:674–685. doi: 10.1194/jlr.M300363-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Florez JC, Hirschhorn JN, Altshuler D. The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet. 2003;4:257–291. doi: 10.1146/annurev.genom.4.070802.110436. [DOI] [PubMed] [Google Scholar]
- 23.Knouff C, Auwerx J. Peroxisome proliferator-activated receptor-γ calls for activation in moderation: lessons from genetics and pharmacology. Endocr Rev. 2004;25:899–918. doi: 10.1210/er.2003-0036. [DOI] [PubMed] [Google Scholar]
- 24.Lindi VI, Uusitupa MIJ, Lindstrom J, Louheranta A, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Tuomilehto J. Association of the Pro12Ala polymorphism in the PPAR-γ2 gene with 3-year incidence of type 2 diabetes and body weight change in the Finnish Diabetes Prevention Study. Diabetes. 2002;51:2581–2586. doi: 10.2337/diabetes.51.8.2581. [DOI] [PubMed] [Google Scholar]
- 25.Lyssenko V, Almgren P, Anevski D, Orho-Melander M, Sjögren M, Saloranta C, Tuomi T, Groop L for the Botnia Study Group. Genetic prediction of future type 2 diabetes. PLoS Medicine. 2005;2 doi: 10.1371/journal.pmed.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bluher M, Lubben G, Paschke R. Analysis of the relationship between the Pro12Ala variant in the PPAR-γ2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care. 2003;26:825–831. doi: 10.2337/diacare.26.3.825. [DOI] [PubMed] [Google Scholar]
- 27.Snitker S, Watanabe RM, Ani I, Xiang AH, Marroquin A, Ochoa C, Goico J, Shuldiner AR, Buchanan TA. Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-γ2 gene variant: results from the Troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care. 2004;27:1365–1368. doi: 10.2337/diacare.27.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolford JK, Yeatts KA, Dhanjal SK, Black MH, Xiang AH, Buchanan TA, Watanabe RM. Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes. 2005;54:3319–3325. doi: 10.2337/diabetes.54.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Diabetes Prevention Program Study Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Diabetes Prevention Program Study Group. The Diabetes Prevention Program. Design and methods for a clinical trial in the prevention of type 2 diabetes. Diabetes Care. 1999;22:623–634. doi: 10.2337/diacare.22.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Diabetes Prevention Program Study Group. The Diabetes Prevention Program: baseline characteristics of the randomized cohort. The Diabetes Prevention Program Research Group. Diabetes Care. 2000;23:1619–1629. doi: 10.2337/diacare.23.11.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The Diabetes Prevention Program Study Group. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54:1150–1156. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PIW, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D for the Diabetes Prevention Program Study Group. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med. 2006;355:241–250. doi: 10.1056/NEJMoa062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Diabetes Prevention Program Study Group. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the Diabetes Prevention Program: effects of lifestyle intervention and metformin. Diabetes. 2005;54:2404–2414. doi: 10.2337/diabetes.54.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byrne CD, Wareham NJ, Brown DC, Clark PM, Cox LJ, Day NE, Palmer CR, Wang TW, Williams DR, Hales CN. Hypertriglyceridemia in subjects with normal and abnormal glucose tolerance: relative contributions of insulin secretion, insulin resistance and suppression of plasma non-esterified fatty acids. Diabetologia. 1994;37:889–896. doi: 10.1007/BF00400944. [DOI] [PubMed] [Google Scholar]
- 36.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 37.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:5–70. [Google Scholar]
- 38.Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 39.Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, Hu ZX, Lin J, Xiao JZ, Cao HB, Liu PA, Jiang XG, Jiang YY, Wang JP, Zheng H, Zhang H, Bennett PH, Howard BV. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- 40.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa M. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 41.Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, Pato MT, Petryshen TL, Kolonel LN, Lander ES, Sklar P, Henderson B, Hirschhorn JN, Altshuler D. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 42.Vanttinen M, Nuutila P, Pihlajamaki J, Hallsten K, Virtanen KA, Lautamaki R, Peltoniemi P, Kemppainen J, Takala T, Viljanen APM, Knuuti J, Laakso M. The effect of the Ala12 allele of the peroxisome proliferator-activated receptor-γ2 gene on skeletal muscle glucose uptake depends on obesity: a positron emission tomography study. J Clin Endocrinol Metab. 2005;90:4249–4254. doi: 10.1210/jc.2005-0101. [DOI] [PubMed] [Google Scholar]