

Abstract
The common polymorphisms KCNJ11 E23K and ABCC8 A1369S have been consistently associated with type 2 diabetes. We examined whether these variants are also associated with progression from impaired glucose tolerance (IGT) to diabetes and responses to preventive interventions in the Diabetes Prevention Program. We genotyped both variants in 3,534 participants and performed Cox regression analysis using genotype, intervention, and their interactions as predictors of diabetes incidence over ~3 years. We also assessed the effect of genotype on insulin secretion and insulin sensitivity at 1 year. As previously shown in other studies, lysine carriers at KCNJ11 E23K had reduced insulin secretion at baseline; however, they were less likely to develop diabetes than E/E homozygotes. Lysine carriers were less protected by 1-year metformin treatment than E/E homozygotes (P < 0.02). Results for ABCC8 A1369S were essentially identical to those for KCNJ11 E23K. We conclude that the lysine variant in KCNJ11 E23K leads to diminished insulin secretion in individuals with IGT. Given our contrasting results compared with case-control analyses, we hypothesize that its effect on diabetes risk may occur before the IGT-to-diabetes transition. We further hypothesize that the diabetes-preventive effect of metformin may interact with the impact of these variants on insulin regulation. Diabetes 56: 531–536, 2007
The KCNJ11 gene encodes the islet ATP-sensitive potassium channel Kir6.2. Severe activating mutations in KCNJ11 cause a novel form of monogenic neonatal diabetes (1). A common glutamate (E) → lysine (K) change at position 23 (E23K) has been consistently associated with type 2 diabetes, with an overall allelic odds ratio (OR) near 1.15 (2–9) for the comparison of diabetic individuals with nondiabetic control subjects. Moreover, we and others (5,8,10) have shown that normoglycemic lysine carriers consistently display a defect in insulin secretion. In vitro, the lysine risk allele seems to affect potassium channel properties (11,12). Interestingly, this variant is in strong linkage disequilibrium with the upstream missense single nucleotide polymorphism (SNP) A1369S in the adjacent gene ABCC8, which encodes the functionally related sulfonylurea receptor SUR1 (7,8,13); thus, in all examined populations, lysine carriers at KCNJ11 E23K almost invariably carry the alanine allele at ABCC8 A1369S, and it remains possible that either or both variants are actually required to mediate these effects.
The risk of type 2 diabetes conferred by KCNJ11 E23K has been evaluated prospectively. The Finnish Diabetes Prevention Study (14) randomized 522 subjects with impaired glucose tolerance (IGT) to either placebo or a lifestyle intervention and found that lysine carriers at KCNJ11 E23K appeared more likely to develop diabetes over time than E/E homozygotes, although the difference was not statistically significant. In contrast, the larger Botnia Prospective Study (10) suggested that the lysine allele was protective, although, again, this effect was not statistically significant. Whether statistical fluctuations or differences in ascertainment schemes (IGT vs. a population sample) and analytical methods (logistic regression vs. Cox proportional hazards analysis) explain these discrepancies is not yet clear.
Two studies have examined the effect of the KCNJ11 E23K variant on response to sulfonylurea therapy. Gloyn et al. (3) studied 364 subjects randomized to sulfonylurea therapy in the UK Prospective Diabetes Study (UKPDS) and determined that the presence of the lysine allele did not predict failure to treatment with sulfonylureas at 1 year. Recently, Sesti et al. (15) reported a higher proportion of lysine carriers among 208 subjects who failed sulfonylurea-metformin combination therapy (defined as a rise in fasting plasma glucose >300 mg/dl); interestingly, islets isolated from lysine carriers showed a diminished insulin response to glyburide.
Given this divergent (and contradictory) literature, we set out to investigate the effect of this variant on glycemic parameters in obese individuals at higher risk for type 2 diabetes (i.e., IGT or elevated fasting glucose), to prospectively examine its impact on the development of diabetes, and to assess whether it influences the efficacy of the lifestyle or pharmacological interventions used in the Diabetes Prevention Program (DPP) (16).
RESEARCH DESIGN AND METHODS
The details of the DPP study design have been described elsewhere (16–18). The DPP was a multicenter randomized clinical trial that hypothesized that modifying risk factors for type 2 diabetes (elevated fasting and postload plasma glucose concentrations, overweight, and sedentary lifestyle) with lifestyle intervention or treatment with metformin would prevent or delay the development of diabetes. The clinical trial was conducted at 27 centers, each of which obtained institutional review board approval. The DPP enrolled 3,234 nondiabetic individuals with IGT and elevated fasting glucose and randomized them to placebo, 850 mg metformin twice daily, or a lifestyle intervention program aimed at ≥7% weight loss and ≥150 min of physical activity per week; a fourth arm of 585 subjects assigned to 400 mg troglitazone daily was stopped 2 years after the trial commenced because of hepatotoxicity (17). The principal end point was the development of diabetes by confirmed oral glucose tolerance tests (OGTTs). Over an average of 3 years, the lifestyle and metformin interventions reduced the incidence of diabetes in high-risk individuals by 58% (95% CI 48–66) and 31% (17–43), respectively, versus placebo; diabetes incidence rates were 11.0, 7.8, and 4.8 per 100 person-years in the placebo, metformin, and lifestyle groups, respectively (16).
The 3,548 participants included in this study (92.9% of all DPP participants: 2,994 who completed the trial in their originally assigned treatment groups, plus 554 originally randomized to troglitazone) provided informed consent specific to genetic investigation. Of the participants in this genetic study, 56.4% were Caucasian, 20.2% were African American, 16.8% were Hispanic, 4.3% were Asian American, and 2.4% were American Indian by self-report. Similar to the entire DPP cohort, the participants’ mean age was 51 years and mean BMI was 34.0 kg/m2. Treatment effects were consistent across sex and self-reported ethnicity.
Genotyping
DNA was extracted from peripheral blood leukocytes through conventional procedures and quantitated by picogreen analysis. Genotyping was performed by allele-specific primer extension of single-plex amplified products, with detection by matrix-assisted laser desorption ionization–time-of-flight mass spectroscopy on a Sequenom platform (19,20). Genotyping success rate was 99.3%. The allele frequencies of both SNPs in each of the five ethnic groups were in Hardy-Weinberg equilibrium (P > 0.05).
Quantitative glycemic measures
Data from the baseline and 1-year OGTTs were used to calculate one measure of insulin secretion and two measures of insulin sensitivity. The insulinogenic index (21) was calculated as [(insulin at 30 min) − (insulin at 0 min)]/[(glucose at 30 min) − (glucose at 0 min)]. The insulin sensitivity index (reciprocal of insulin resistance by the homeostasis model assessment [22]) was calculated as 22.5/[fasting insulin × (fasting glucose/18.01)]. We have previously shown, in the same cohort, that these measures correlate strongly with the corrected insulin response (insulin secretion) and the inverse of fasting insulin (insulin sensitivity), respectively (23).
We again elected to analyze follow-up quantitative traits at 1 rather than 3 years (23). While a very high proportion of DPP participants were diabetes free and had an OGTT at 1 year, this percentage was much lower at 3 years (many participants had developed diabetes by this time, and those who enrolled in the latter half of the recruitment period did not have to undergo the 3-year exam). Since these quantitative traits were calculated in nondiabetic subjects, so as to avoid confounding by treatment, power is greater at 1 year. In addition, weight loss was maximal at 6–12 months (16); therefore, one might expect to see the greatest effect of the lifestyle intervention in the 1-year data.
Statistical analysis
Time to onset of diabetes was the primary end point. We examined Cox regression models with genotype, intervention, and genotype-by-intervention interactions as the independent variables predicting time to diabetes. These models were also examined with BMI at randomization, age at randomization, sex, and self-reported ethnicity as covariates. When allele frequencies differed significantly across self-reported ethnic groups, the analyses were repeated only in those populations that had comparable allele frequencies; no significant effect of self-reported ethnicity was detected in any of our analyses.
In this study, we addressed five distinct hypotheses, limiting subsequent analyses to their further refinement: 1) genotype at KCNJ11 E23K (or at ABCC8 A1369S, as both are highly correlated) influences diabetes incidence, 2) this genetic effect is modified by a lifestyle intervention or 3) by metformin treatment, 4) genotype at KCNJ11 E23K affects insulin secretion, and 5) genotype at KCNJ11 E23K does not affect insulin resistance. Thus, in order to account for the multiple hypotheses tested and to obtain a conservative estimate of true statistical significance, we applied a Bonferroni correction factor of 5 to the nominal two-sided P values.
For the quantitative trait analyses, we first compared baseline measures in the entire cohort according to genotype at each of the two SNPs. General linear models were used to test for mean differences between the quantitative traits. Means were compared, and the P values were further adjusted for additional comparisons across three genotypic groups (within each trait) using the Holm procedure (24). This modification of the Bonferroni adjustment ranks Pi values, and each Pi is compared with α/(n − i + 1) for rejection. Starting with the smallest P value, one continues applying these comparisons (from i = 1 and proceeding in order) until the first nonrejection; thus, the rejected hypotheses Hi (at α = 0.05) are those for which Pj ≤ α/(n − j + 1) for all j ≤ i. The SAS analysis system version 9.1 was used for all analyses (SAS Institute, Cary, NC).
RESULTS
Allele frequency distribution
The frequency of the minor lysine allele at KCNJ11 E23K in DPP U.S. Caucasians (0.37) was comparable with that previously reported in other Caucasian populations. We found significant differences in minor allele frequencies only in African Americans (0.08) when compared with Caucasians; therefore, analyses for incidence of diabetes were performed with and without this ethnic group. We found strong linkage disequilibrium between ABCC8 A1369S and the downstream variant KCNJ11 E23K in the five ethnic groups: Lewontin’s D′ (25) and r2 were 0.97/0.93, 0.98/0.93, 0.99/0.95, 0.99/0.88, and 0.97/0.95 in U.S. Caucasians, African Americans, Hispanic Americans, Asian Americans, and American Indians, respectively. Thus, not surprisingly, our findings in carriers of the alanine allele at ABCC8 A1369S were essentially the same as those for lysine carriers at KCNJ11 E23K in all of the analyses reported below. For simplicity of presentation, given the relative focus on KCNJ11 E23K in the literature and the functional data on this allele, we will highlight results on that SNP in this report.
At KCNJ11 E23K, genotypic frequencies were equally distributed among the four treatment arms and two sex groups. We found no significant differences in baseline age, BMI, or waist circumference by genotype (Table 1).
TABLE 1.
Demographic characteristics of the DPP cohort, according to genotype at KCNJ11 E23K
| E/E | E/K | K/K | P* | |
|---|---|---|---|---|
| n | 1,690 | 1,476 | 374 | |
| Treatment | ||||
| Placebo | 487 (48.9) | 400 (40.2) | 108 (10.9) | 0.82 |
| Metformin | 474 (47.9) | 419 (42.4) | 96 (9.7) | |
| Lifestyle | 465 (46.4) | 423 (42.2) | 114 (11.4) | |
| Troglitazone | 264 (47.7) | 234 (42.2) | 56 (10.1) | |
| Male | 540 (46.2) | 495 (42.3) | 135 (11.5) | 0.27 |
| Age (years) | 51.0 ± 10.3 | 50.6 ± 10.8 | 50.4 ± 10.9 | 0.49 |
| BMI (kg/m2) | 34.2 ± 6.8 | 33.8 ± 6.5 | 33.9 ± 6.9 | 0.22 |
| Waist circumference (cm) | 105 ± 14.7 | 105 ± 14.3 | 105 ± 14.3 | 0.84 |
Data are means ± SD or n (%).
Based on ANOVA for continuous variables and χ2 for categorical variables.
Baseline quantitative glycemic traits
We examined whether individuals with IGT also have a detectable defect in insulin release. As expected, lysine carriers at KCNJ11 E23K had reduced insulin release at baseline, compared with E/E homozgyotes, in proportion to the number of lysine alleles (Bonferroni-corrected P = 0.015 for the pairwise comparison between both homozygous genotypes). Insulin sensitivity at baseline did not differ among genotypic groups (Table 2).
TABLE 2.
Baseline measures of insulin secretion and sensitivity according to KCNJ11 E23K and ABCC8 A1369S genotypes in the DPP
| KCNJ11 E23K | E/E | E/K | K/K | P value (E/E vs. E/K)* | P value (E/E vs. K/K)* |
|---|---|---|---|---|---|
| n | 1,658 | 1,445 | 364 | ||
| Ins index [(μU/ml)/(mg/dl)] | 1.07 (0.94) | 1.04 (0.87) | 0.97 (0.79) | 0.046 | 0.003 |
| ISI [(μU/ml) × (mmol/l)]−1 | 0.161 (0.121) | 0.162 (0.124) | 0.169 (0.144) | 0.98 | 0.76 |
| Fasting insulin (μU/ml) | 24 (16) | 24 (17) | 23 (18) | 0.85 | 0.73 |
| ABCC8 A1369S | S/S | A/S | A/A | P value (S/S vs. A/S)* | P value (S/S vs. A/A)* |
|
| |||||
| n | 1,635 | 1,424 | 378 | ||
| Ins index [(μU/ml)/(mg/dl)] | 1.07 (0.95) | 1.03 (0.86) | 0.97 (0.81) | 0.02 | <0.01 |
| ISI [(μU/ml) × (mmol/l)]−1 | 0.162 (0.121) | 0.162 (0.123) | 0.172 (0.145) | 0.87 | 0.67 |
| Fasting insulin (μU/ml) | 24 (17) | 24 (17) | 22 (17) | 0.73 | 0.88 |
Data are median (interquartile range). The number of subjects for whom full OGTT data were available to calculate the insulinogenic index and insulin sensitivity index are displayed; a few additional subjects had fasting values only, which were used to calculate insulin sensitivity index.
Nominal two-sided P values are displayed; to correct for the number of hypotheses tested, a Bonferroni correction factor of ×5 can be applied (see text for details). Ins, insulinogenic; ISI, insulin sensitivity index.
Incidence of diabetes
Because we detected a nominally significant interaction between genotype and metformin treatment (see below), we proceeded to analyses stratified by treatment arm. In previous cross-sectional studies, the lysine allele has been associated with type 2 diabetes; however, in the placebo arm of the DPP, E/K heterozygotes with IGT appeared 29% less likely to develop diabetes than E/E homozygotes (hazard ratio [HR] 0.71 [95% CI 0.55–0.92], nominal P = 0.01; Bonferroni-corrected P = 0.053). K/K homozygotes had a similar but nonsignificant extent of protection from diabetes (0.81 [0.54–1.22], P = 0.31). HRs in the lifestyle arm were not statistically significant (E/K vs. E/E: 1.09 [0.78–1.54], P = 61; K/K vs. E/E: 0.56 [0.28–1.09], P = 0.08). Adjustment for BMI at randomization, age at randomization, sex, and ethnicity did not alter the results.
Metformin effect
As stated above, we observed a nominally significant interaction of metformin and genotype at KCNJ11 E23K (nominal P = 0.017; Bonferroni-corrected P = 0.085), such that lysine carriers did not seem to benefit from the preventive effect of metformin (HR 0.89 [95% CI 0.66–1.19] for E/K heterozygotes; 0.95 [0.54–1.67] for K/K homozygotes, both vs. placebo); E/E homozygotes had a greater preventive effect (0.55 [0.42–0.71], nominal P < 0.0001 vs. placebo) (Fig. 1). In contrast, there was no significant interaction between the lifestyle intervention and genotype. We observed no significant longitudinal changes in weight or in fasting glucose levels across genotypic groups in the metformin arm, which could explain these findings. Examination of quantitative glycemic measures suggested that the lack of protection by metformin in K/K homozygotes may have been due to a suppression of the beneficial effect of metformin on insulin sensitivity at 1 year (Table 3).
FIG. 1.
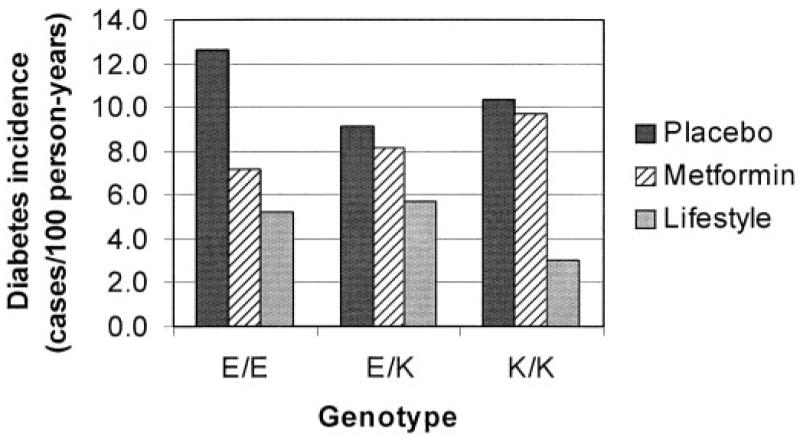
Incidence rates of diabetes per genotypic group and treatment arm in the DPP (cases/100 person-years). Metformin does not seem to protect lysine carriers at KCNJ11 E23K from developing diabetes.
TABLE 3.
One-year measures of insulin secretion and sensitivity according to KCNJ11 E23K genotypes by treatment arm in the DPP
| E/E | E/K | K/K | P value* | |
|---|---|---|---|---|
| Ins index [(μU/ml)/(mg/dl)] | ||||
| n | 1,658 | 1,445 | 364 | |
| Placebo | 1.26 (1.18–1.35) | 1.15 (1.06–1.25) | 1.20 (1.02–1.39) | 0.24 |
| Metformin | 1.30 (1.21–1.39) | 1.09 (1.00–1.18) | 1.16 (0.95–1.36) | 0.005 |
| Lifestyle | 1.17 (1.08–1.26) | 1.26 (1.17–1.35) | 1.09 (0.91–1.27) | 0.18 |
| Troglitazone | 1.18 (1.02–1.34) | 1.26 (1.10–1.41) | 1.20 (0.87–1.53) | 0.78 |
| ISI [(μU/ml) × (mmol/l)]−1 | ||||
| n | 1,688 | 1,474 | 374 | |
| Placebo | 0.184 (0.175–0.194) | 0.192 (0.181–0.202) | 0.213 (0.193–0.232) | 0.04 |
| Metformin | 0.235 (0.223–0.247) | 0.241 (0.229–0.254) | 0.194 (0.167–0.221) | 0.007 |
| Lifestyle | 0.265 (0.250–0.281) | 0.279 (0.263–0.295) | 0.271 (0.240–0.302) | 0.47 |
| Troglitazone | 0.274 (0.246–0.302) | 0.275 (0.248–0.302) | 0.256 (0.199–0.313) | 0.83 |
| Fasting insulin (μU/ml) | ||||
| n | 1,688 | 1,474 | 374 | |
| Placebo | 28 (26–29) | 28 (26–29) | 26 (23–29) | 0.53 |
| Metformin | 24 (23–25) | 24 (22–25) | 27 (24–29) | 0.04 |
| Lifestyle | 22 (21–23) | 21 (20–23) | 20 (18–23) | 0.61 |
| Troglitazone | 21 (19–22) | 21 (20–23) | 21 (17–24) | 0.90 |
Data are least-squares means (95% CI), adjusted for baseline values.
Nominal two-sided P values for the effect of E23K genotype are displayed; to correct for the number of hypotheses tested, a Bonferroni correction factor of ×5 can be applied (see text for details). Ins, insulinogenic; ISI, insulin sensitivity index.
DISCUSSION
The documented and reproducible association of selected polymorphisms in genes that encode drug targets with type 2 diabetes highlights the possible application of human genomics to medicine (26). This avenue can be immediately tested in relevant clinical trials such as the DPP. In contrast to other diabetes prevention trials (27,28), the DPP includes both lifestyle and pharmacological interventions. In addition, its multiethnic design reflects the diversity of the U.S. population, and its large sample size makes it adequate for genetic studies where variants are thought to confer modest risk. Two important distinctions between the DPP and other large observational trials (10) are its interventional design and the exclusive enrollment of individuals with IGT, which suggests the presence of some degree of genetic risk at baseline and imposes constraints in ascertainment. On the other hand, because the DPP did not include a sulfonylurea arm, we could not directly evaluate the effect of these variants on sulfonylurea therapy.
The protection from development of diabetes that we found in carriers of the lysine allele at KCNJ11 E23K does not confirm previous reports and was unexpected: We cannot exclude that it may be a spurious result, given the marginal P value and multiple hypotheses examined. We note that the DPP has 73% power to detect the published HR of ~1.35 when comparing K/K with E/E homozygotes and only 51% power to detect the published HR of ~1.15 when comparing E/K heterozygotes with E/E homozygotes (29). As expected, our power drops further if the analysis is restricted to the placebo arm.
The smaller Finnish Diabetes Prevention Study (14), which also exclusively enrolled individuals with IGT, reported a nonsignificant unadjusted OR of 1.61 (95% CI 0.86–3.00), suggesting increased risk for lysine allele carriers. The Botnia Prospective Study (10), whose enrollment included 31% of subjects with either impaired fasting glucose or IGT, noted a protective but nonsignificant HR of 0.7 (95% CI 0.5–1.1) for lysine carriers versus E/E homozygotes. In a larger prospective Swedish cohort recently examined by the same investigators, the lysine allele conferred a modest but statistically significant risk of diabetes (V. Lyssenko, L. Groop, personal communication). The 95% CIs for this and the two published prospective studies overlap (albeit considering different genetic models and populations), suggesting that they may not be mutually exclusive. Since the totality of the published case/control data establishes the lysine allele as the risk variant influencing the general transition from normoglycemia to type 2 diabetes, although in this study lysine carriers in the placebo arm appeared to be protected against the development of diabetes (at a nominal P = 0.01) when starting from a baseline of IGT, our result raises the possibility that E23K may play its pathogenic role earlier in the course of the disease (i.e., from normoglycemia to IGT, rather than in progressing from IGT to overt diabetes), whereas other genetic and/or environmental factors may be necessary for diabetes to declare itself. This hypothesis could be tested in adequately powered prospective population cohorts. Moreover, these results illustrate the need for caution when comparing studies of different designs (cross-sectional vs. prospective), heterogeneous populations, and divergent subgroups (normoglycemia vs. IGT).
The apparent failure of metformin to protect those individuals carrying the lysine allele at KCNJ11 E23K from developing diabetes was also unexpected and not predictable from prior biological knowledge. The effect appears to be proportional to gene dosage, arguing against a mere statistical fluctuation. A possible mechanistic explanation for this phenomenon is illustrated by the failure of K/K homozygotes to improve their insulin sensitivity after 1 year of metformin treatment, in contrast to their robust response after a lifestyle intervention or troglitazone treatment (Table 3). Why the lysine variant in a β-cell channel would lead to a differential response to metformin in insulin sensitivity requires physiologic studies and validation in an independent cohort. Interestingly, Marchetti et al. (30) reported a beneficial effect of metformin on diabetic islets when exposed to high glucose, although the possible contribution of KCNJ11 E23K genotype was not explored.
In conclusion, we have extended the finding of impaired insulin secretion among individuals carrying the lysine variant to a multiethnic population with IGT. Our results suggest that the lysine allele may manifest its deleterious effects at earlier stages in the evolution of type 2 diabetes (i.e., during the progression from normoglycemia to IGT). Moreover, lysine carriers seem to respond less well to the protective effect of metformin than E/E homozygotes. This result, as well as the impact of this polymorphism on sulfonylurea therapy, requires validation in specifically designed pharmacogenetic studies.
Acknowledgments
J.C.F. is supported by National Institutes of Health (NIH) Research Career Award 1 K23 DK65978-03. The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the NIH provided funding to the clinical centers and the Coordinating Center for the design and conduct of the study, as well as collection, management, analysis, and interpretation of the data. The Southwestern American Indian Centers were supported directly by the NIDDK and the Indian Health Service. The General Clinical Research Center Program, National Center for Research Resources supported data collection at many of the clinical centers. Funding for data collection and participant support was also provided by the Office of Research on Minority Health, the National Institute of Child Health and Human Development, the National Institute on Aging, the Centers for Disease Control and Prevention, Office of Research on Women’s Health, and the American Diabetes Association. This research was also supported, in part, by the intramural research program of the NIDDK.
Bristol-Myers Squibb and Parke-Davis provided medication. LifeScan, Health O Meter, Hoechst Marion Roussel, Merck-Medco Managed Care, Merck, Nike Sports Marketing, Slim Fast Foods, and Quaker Oats donated materials, equipment, or medicines for concomitant conditions. McKesson BioServices, Matthews Media Group, and the Henry M. Jackson Foundation provided support services under subcontract with the Coordinating Center.
The investigators gratefully acknowledge the commitment and dedication of the participants of the DPP. We thank Santica Marcovina and Greg Strylewicz for careful processing of the DNA samples and Mark Daly, Noël Burtt, and our colleagues at the Broad Institute Genetic Analysis Platform for excellent assistance.
The opinions expressed are those of the investigators and do not necessarily reflect the views of the Indian Health Service or other funding agencies.
Abbreviations
- DPP
Diabetes Prevention Program
- IGT
impaired glucose tolerance
- OGTT
oral glucose tolerance test
- SNP
single nucleotide polymorphism
References
- 1.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JMCL, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JPH, Sumnik Z, van Rhijn A, Wales JKH, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 2.Hani EH, Boutin P, Durand E, Inoue H, Permutt MA, Velho G, Froguel P. Missense mutations in the pancreatic islet β cell inwardly rectifying K+ channel gene (KIR6.2/BIR): a meta-analysis suggests a role in the polygenic basis of type II diabetes mellitus in Caucasians. Diabetologia. 1998;41:1511–1515. doi: 10.1007/s001250051098. [DOI] [PubMed] [Google Scholar]
- 3.Gloyn AL, Hashim Y, Ashcroft SJ, Ashfield R, Wiltshire S, Turner RC. Association studies of variants in promoter and coding regions of β-cell ATP-sensitive K-channel genes SUR1 and Kir6.2 with type 2 diabetes mellitus (UKPDS 53) Diabet Med. 2001;18:206–212. doi: 10.1046/j.1464-5491.2001.00449.x. [DOI] [PubMed] [Google Scholar]
- 4.Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen E-MD, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glumer C, Thorsteinsson B, Borch-Johnsen K, Hansen T, Pedersen O. The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003;52:573–577. doi: 10.2337/diabetes.52.2.573. [DOI] [PubMed] [Google Scholar]
- 6.Love-Gregory L, Wasson J, Lin J, Skolnick G, Suarez B, Permutt MA. E23K single nucleotide polymorphism in the islet ATP-sensitive potassium channel gene (Kir6.2) contributes as much to the risk of type II diabetes in Caucasians as the PPARγ Pro12Alα variant. Diabetologia. 2003;46:136–137. doi: 10.1007/s00125-002-0947-x. [DOI] [PubMed] [Google Scholar]
- 7.Barroso I, Luan J, Middelberg RPS, Harding A-H, Franks PW, Jakes RW, Clayton D, Schafer AJ, O’Rahilly S, Wareham NJ. Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-cell function as well as insulin action. PLoS Biol. 2003;1:e20. doi: 10.1371/journal.pbio.0000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Florez JC, Burtt N, de Bakker PIW, Almgren P, Tuomi T, Holmkvist J, Gaudet D, Hudson TJ, Schaffner SF, Daly MJ, Hirschhorn JN, Groop L, Altshuler D. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004;53:1360–1368. doi: 10.2337/diabetes.53.5.1360. [DOI] [PubMed] [Google Scholar]
- 9.van Dam RM, Hoebee B, Seidell JC, Schaap MM, de Bruin TWA, Feskens EJM. Common variants in the ATP-sensitive K+ channel genes KCNJ11 (Kir6.2) and ABCC8 (SUR1) in relation to glucose intolerance: population-based studies and meta-analyses. Diabet Med. 2005;22:590–598. doi: 10.1111/j.1464-5491.2005.01465.x. [DOI] [PubMed] [Google Scholar]
- 10.Lyssenko V, Almgren P, Anevski D, Orho-Melander M, Sjögren M, Saloranta C, Tuomi T, Groop L the Botnia Study Group. Genetic prediction of future type 2 diabetes. PLoS Med. 2005;2:e345. doi: 10.1371/journal.pmed.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwanstecher C, Meyer U, Schwanstecher M. KIR6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic β-cell ATP-sensitive K+ channels. Diabetes. 2002;51:875–879. doi: 10.2337/diabetes.51.3.875. [DOI] [PubMed] [Google Scholar]
- 12.Riedel MJ, Light PE. Saturated and cis/trans unsaturated acyl CoA esters differentially regulate wild-type and polymorphic β-cell ATP-sensitive K+ channels. Diabetes. 2005;54:2070–2079. doi: 10.2337/diabetes.54.7.2070. [DOI] [PubMed] [Google Scholar]
- 13.Inoue H, Ferrer J, Warren-Perry M, Zhang Y, Millns H, Turner RC, Elbein SC, Hampe CL, Suarez BK, Inagaki N, Seino S, Permutt MA. Sequence variants in the pancreatic islet β-cell inwardly rectifying K+ channel Kir6.2 (Bir) gene: identification and lack of role in Caucasian patients with NIDDM. Diabetes. 1997;46:502–507. doi: 10.2337/diab.46.3.502. [DOI] [PubMed] [Google Scholar]
- 14.Laukkanen O, Pihlajamaki J, Lindstrom J, Eriksson J, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Tuomilehto J, Uusitupa M, Laakso M the Finnish Diabetes Prevention Study Group. Polymorphisms of the SUR1 (ABCC8) and Kir6.2 (KCNJ11) genes predict the conversion from impaired glucose tolerance to type 2 diabetes: the Finnish Diabetes Prevention Study. J Clin Endocrinol Metab. 2004;89:6286–6290. doi: 10.1210/jc.2004-1204. [DOI] [PubMed] [Google Scholar]
- 15.Sesti G, Laratta E, Cardellini M, Andreozzi F, Del Guerra S, Irace C, Gnasso A, Grupillo M, Lauro R, Hribal ML, Perticone F, Marchetti P. The E23K variant of KCNJ11 encoding the pancreatic β-cell KATP channel subunits Kir6.2 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:2334–2339. doi: 10.1210/jc.2005-2323. [DOI] [PubMed] [Google Scholar]
- 16.Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diabetes Prevention Program Research Group. The Diabetes Prevention Program: design and methods for a clinical trial in the prevention of type 2 diabetes. Diabetes Care. 1999;22:623–634. doi: 10.2337/diacare.22.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diabetes Prevention Program Research Group. The Diabetes Prevention Program: baseline characteristics of the randomized cohort: the Diabetes Prevention Program Research Group. Diabetes Care. 2000;23:1619–1629. doi: 10.2337/diacare.23.11.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 20.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci U S A. 1999;96:10016–10020. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne CD, Wareham NJ, Brown DC, Clark PM, Cox LJ, Day NE, Palmer CR, Wang TW, Williams DR, Hales CN. Hypertriglyceridemia in subjects with normal and abnormal glucose tolerance: relative contributions of insulin secretion, insulin resistance and suppression of plasma non-esterified fatty acids. Diabetologia. 1994;37:889–896. doi: 10.1007/BF00400944. [DOI] [PubMed] [Google Scholar]
- 22.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 23.Diabetes Prevention Program Research Group. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the Diabetes Prevention Program: effects of lifestyle intervention and metformin. Diabetes. 2005;54:2404–2414. doi: 10.2337/diabetes.54.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 25.Lewontin RC. On measures of gametic disequilibrium. Genetics. 1988;120:849–852. doi: 10.1093/genetics/120.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guttmacher AE, Collins FS. Realizing the promise of genomics in biomedical research. JAMA. 2005;294:1399–1402. doi: 10.1001/jama.294.11.1399. [DOI] [PubMed] [Google Scholar]
- 27.Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, Hu ZX, Lin J, Xiao JZ, Cao HB, Liu PA, Jiang XG, Jiang YY, Wang JP, Zheng H, Zhang H, Bennett PH, Howard BV. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance: the Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- 28.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa M. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh FY, Lavori PW. Sample-size calculations for the Cox proportional hazards model with nonbinary covariates. Control Clin Trials. 2000;21:552–560. doi: 10.1016/s0197-2456(00)00104-5. [DOI] [PubMed] [Google Scholar]
- 30.Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, Bugliani M, Boggi U, Vistoli F, Mosca F, Del Prato S. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–5541. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]