

Abstract
Accelerating the drug discovery process requires predictive computational protocols capable of reducing or simplifying the synthetic and/or combinatorial challenge. Docking-based virtual screening methods have been developed and successfully applied to a number of pharmaceutical targets. In this review, we first present the current status of docking and scoring methods, with exhaustive lists of these. We next discuss reported comparative studies, outlining criteria for their interpretation. In the final section, we describe some of the remaining developments that would potentially lead to a universally applicable docking/scoring method.
Keywords: docking, scoring, computer-aided drug design, virtual screening, comparative studies
Introduction
Molecular discovery: high-throughput screening vs virtual screening
Since the advent of the human genome project and functional genomics, there has been a continual increase in the number of new therapeutic targets available for drug design. During this time, advances in crystallography and nuclear magnetic resonance spectroscopy have contributed many structural details of protein and protein–ligand complexes. In response to these newly discovered targets and their structural elucidation, cost- and time-efficient design and syntheses of bioactive compounds targeting these became a priority for the field of drug design. In recent years, this field has partially turned away from the traditional approaches, such as rational and semi-rational design, to the high-throughput screening of large combinatorial libraries. However, the identification of novel lead compounds via these traditional approaches has been more fruitful compared with the low hit rates observed with combinatorial methods (Shoichet et al., 2002). In practice, identifying leads or hits in silico rather than via library generation and screening is both faster and more economical, while being easier to setup. Indeed, screening of large libraries has been used in combination with (or in parallel to) or sometimes substituted by computational approaches. Among the most commonly used virtual screening (VS) tools are docking methods, which have been successfully used to predict the binding modes and affinities of many potent enzyme inhibitors as well as receptor antagonists. As a result, many drugs developed in part by computer-aided structure-based drug design methods are in late-stage clinical trials or have now reached the market (Borman, 2005). Following these success stories, the pharmaceutical companies are increasingly relying on computational methods as one of the primary platforms for designing new potential leads.
Current and future challenges
With the ever-growing interest in using computational structure-based drug design tools, there is a need to scrutinize the field and examine its current state and its future. An overview of the latest progress in the structure-based drug design field shows that significant improvements must still be achieved in order to develop highly accurate molecular docking and VS methods (Taylor et al., 2002; Krumrine et al., 2003; Brooijmans and Kuntz, 2003; Mohan et al., 2005; Ferrara et al., 2006; Klebe, 2006; Rester, 2006; Sousa et al., 2006). As most of the discussed programmes have been reported, we will refer the reader to the many reviews cited throughout this review and original articles for more information about each piece of software.
Instead, we review herein the state-of-the-art of docking/scoring methods and discuss the achievements covering the literature until June 2007, the challenges tackled over the recent years and the remaining bottlenecks. This review does not cover low-throughput approaches including molecular dynamics simulation-based approaches (Aqvist et al., 1994; Jones-Hertzog and Jorgensen, 1997; Gervasio et al., 2005) and de novo design programmes such as LEGEND (Nishibata and Itai, 1993), DREAM++ (Makino et al., 1999) and LUDI (Bohm, 1992).
The available docking and scoring methods
Docking and scoring methods
To date, over 60 docking programmes (Table 1) and more than 30 scoring functions (SFs) (Table 2) have been disclosed (see Supplementary Information for more detailed tables). However, only some of these programmes were made available and a limited number of them are widely used (AutoDock, DOCK, FlexX, FRED, Glide, GOLD, ICM, QXP/Flo+, Surflex). In the following sections, the references will appear only in tables and will not be repeated when a programme or a SF is given as an example.
Table 1. Reported docking programmes and, when available, the latest version number, the key references and web sites.
Abbreviation: N/A, not available.
DE, differential evolution; EA, evolutionary algorithm; GA, genetic algorithm; IC, incremental construction; MA, matching algorithm; MD, molecular dynamics; RBD, rigid-body docking; SA, simulated annealing; TS, Tabu search.
Table 2. Reported SFs.
| Scoring function | Software implementations | Class | Reference(s) |
|---|---|---|---|
| ChemScore | GOLD, FRED, CScore, PRO_LEADS | Empirical | (Eldridge et al., 1997) |
| eHiTS SF | eHiTS | Empirical | (Zsoldos et al., 2003) |
| FlexX SF | FlexX | Empirical | (Rarey et al., 1996) |
| Fresno | Standalone | Empirical | (Rognan et al., 1999) |
| GlideScore | Glide | Empirical | (Friesner et al., 2004, 2006) |
| Hammerhead SF | Hammerhead, Surflex, LigandFit | Empirical | (Jain, 1996) |
| HINT | Standalone | Empirical | (Cozzini et al., 2002) |
| LigScore | LigandFit | Empirical | (Krammer et al., 2005) |
| PLP | LigandFit, FRED, DockIt | Empirical | (Gehlhaar et al., 1995) |
| RankScore | FITTED | Empirical/FF | (Moitessier et al., 2006a) |
| SAFE_p | None | Empirical/FF | (Sussman et al., 2002) |
| SCORE | Standalone | Empirical | (Wang et al., 1998) |
| SCORE 3.0 | PSI-DOCK | Empirical | (Pei et al., 2006) |
| SCORE1 | LUDI | Empirical | (Böhm, 1994) |
| SCORE2 | LUDI | Empirical | (Böhm, 1998) |
| ScreenScore | FRED | Empirical/consensus | (Stahl and Rarey, 2001) |
| SIE | Standalone | Empirical/FF | (Naim et al., 2007) |
| SLIDE SCORE | SLIDE | Empirical | (Schnecke and Kuhn, 2000) |
| VALIDATE | Standalone | Empirical/FF | (Head et al., 1996) |
| X-Score | Standalone | Empirical/consensus | (Wang et al., 2002) |
| AutoDock SF | AutoDock, SODOCK | FF/empirical | (Morris et al., 1998) |
| DockScore | DOCK, CScore | FF | (Meng et al., 1992) |
| GoldScore | GOLD, CScore | FF | (Jones et al., 1997) |
| HADDOCK Score | HADDOCK | FF | (van Dijk et al., 2006b) |
| ICM SF | ICM | FF | (Abagyan et al., 1994b) |
| QXP SF | QXP/MCDOCK | FF | (McMartin and Bohacek, 1997) |
| BLEEP | Standalone | Knowledge based | (Mitchell et al., 1999) |
| DrugScoreCSD | Standalone | Knowledge based | (Velec et al., 2005) |
| DrugScorePDB | Standalone | Knowledge based | (Gohlke et al., 2000) |
| M-Score | Standalone | Knowledge based | (Yang et al., 2006) |
| PMF | CScore, LigandFit, BioMedCAChe, DockIt | Knowledge based | (Muegge and Martin, 1999; Muegge, 2006) |
| SMoG | SMoG | Knowledge based | (DeWitte and Shakhnovich, 1996; Ishchenko and Shakhnovich, 2002) |
Abbreviations: FF, force field; SF, scoring function.
Each docking programme relies on two complementary components: (1) a method to explore the conformational space of the ligand and/or the protein target and (2) a SF to evaluate the proposed binding modes referred to as poses. An SF should first assign the best score to the ‘correct pose' (that is, the native pose observed in crystal structures), thus ‘guiding' the conformational sampling algorithm. This first aspect is critical to accurately predict the binding mode. Second, the docked poses of highly active compounds should be attributed better scores than those of non-binders or poor binders. This second aspect is critical in lead optimization and in VS, where potential hits are to be extracted from large libraries. In fact, some programmes use multiple SFs, such as a crude SF to direct the docking and a more refined SF for scoring the final poses.
Whenever a docking programme is selected, two inversely correlated factors are to be considered. While speed is essential for effective virtual high-throughput screening of large libraries, accuracy is critical for lead optimization.
Conformational sampling methods
Multiconformer docking algorithms
Rigid-body docking methods use either a single conformation or multiconformer libraries to consider ligand flexibility. These approaches often dock the small molecules using shape complementarity or interaction site matching algorithms (for example, ADAM, DOCK, FRED, FTDock, LIGIN, SANDOCK and YUCCA). In matching algorithms (Figure 1a), a pharmacophore representing the protein is initially developed and used to guide the docking. An initial ligand conformation is generated and a ligand pharmacophore is derived from this conformation. The distance matrices (listings of all the distances between each of the pharmacophoric points) of the ligand and protein pharmacophore are examined for a match. If there is a match, rotational and translational vectors are calculated and applied to the ligand. These vectors position the ligand in the same frame of reference as the protein. While speed is the major strength of these approaches, their predictive power is reduced as ligand poses are not fully refined, making it difficult for the correct poses to be found or for an accurate score to be assigned. Post-processing of selected poses using an additional local search algorithm addresses the former issue. Owing to their great speed, this first class of programmes is very useful in selecting medium sized libraries from extremely large libraries with acceptable enrichment in active compounds.
Figure 1.

Schematic representation of a matching algorithm (a) and an incremental construction algorithm (b) in the context of docking of a flexible ligand.
Incremental construction
Docking programmes implementing incremental construction (Figure 1b) methods build up the ligand on-the-fly in the active site, often relying on libraries of preferred conformations (for example, MIMUMBA) to connect the fragments and to consider the ligand flexibility. To do so, the ligand is split into a set of fragments, one of which is selected as an anchor and rigidly docked to the binding site using methods such as matching algorithms (for example, DOCK, FlexX, FLOG, Hammerhead, Surflex). Another variation is the docking of fragments followed by reconnection, exemplified by the algorithm implemented in eHiTS.
Stochastic methods
The second class of conformational sampling methods explores the ligand conformational space on the fly. There are many different methods to search stochastically with the most common methods being genetic algorithms (GAs) and Monte Carlo (MC) search (Figure 2). Many programmes make use of GAs (for example, AutoDock, DARWIN, DIVALI, GOLD, EADock, FITTED and PSI-DOCK.), first implemented in docking programmes in the mid-90s to dock flexible ligands (Clark and Ajay, 1995; Jones et al., 1995; Oshiro et al., 1995). GAs (Figure 2a) are based on Darwin's theory of evolution. The pose of the ligand is represented by a chromosome, which is made up of genes. These genes code for each torsional angle, as well as the ligand's rotation and translation in space. These poses then evolve through transmission of genetic information (reproduction), altered over time by genetic operators such as crossover and mutation. There are many different methods for the selection of the next generation, but the most popular is the survival of the fittest, where the two lowest scoring conformations are passed to the next generation. Modifications of GAs (Lamarckian GA in AutoDock or tabu search-enhanced GA in PSI-DOCK) have been found to efficiently explore the conformational space of the ligands.
Figure 2.

Schematic representation of a genetic algorithm (a) and a Monte Carlo search (b) in the context of docking a flexible ligand.
With Monte Carlo (for example, DockVision, ICM, MCDOCK, ProDOCK, SLIDE) (Figure 2b), the pose of the ligand is sequentially modified through bond rotation, translation and/or rigid body rotation, one or more parameters at a time, and the new conformation is then evaluated. If the new conformation has a lower score, it is kept. If a conformation is higher in energy, one can reject it or use a selection criterion such as the Metropolis criterion. Metropolis allows for higher energy conformation to exist by allowing a temperature dependence (the higher the temperature, the more likely that a higher energy conformation is kept), resulting in the crossing of energy barriers on the potential energy surface. If the new pose does not pass the Metropolis criterion, then it is discarded and another set of modifications is tried. Other stochastic optimization algorithms, inspired by analogy to biology, are the ant colony optimization (PLANTS) and the particle swarm optimization (SODOCK/AutoDock) algorithms, both of which are combined with a local search algorithm. Tabu searches (Pro_Leads) have also been employed to consider ligand flexibility.
Multistep procedures
Using one or more of the algorithms mentioned above in conjunction with refinement of the poses has also been performed. For instance, EUDOC and Glide combine the random generation of poses with their refinement to obtain increased accuracy.
Consensus docking
All the methods described so far have shown great success in predicting the binding modes of co-crystallized ligands. However, none of them has been identified as a universally applicable method. Furthermore, CONS_DOCK, a consensus docking method using FlexX, GOLD and DOCK (Paul and Rognan, 2002); AutoxX, combining AutoDock and FlexX interaction models (Wolf et al., 2007) and FITTED, combining a GA and interaction site matching, have been found to be more accurate than the individual search methods. Another way to improve a programme's performance is to provide additional information such as pharmacophore(s) to orient the docking (Hindle et al., 2002; Moitessier et al., 2004; Verdonk et al., 2004), as well as any other knowledge about the target that may reduce the number of false positives and false negatives in VS studies (Jansen and Martin, 2004).
Scoring functions
As previously discussed, SFs have a twofold function: to direct the docking and to predict the binding affinity of the final pose. In this section, only functions that are developed to score the final poses will be discussed. Customized SFs (developed for a specific target only) and functions used to improve the docking accuracy will not be considered. In the past 15 years, a number of SFs have been reported and implemented in docking programmes. Table 2 summarizes the literature in the field (see also Supplementary Information for more detailed data). This table contains SFs used to score the final pose while scoring functions used to orient the docking algorithm towards the correct pose (e.g., knowledge-based interaction fingerprint scoring – (Mpamhanga et al., 2006)) are not included. In general, SFs attempt to predict the binding free energy or to rank-order compounds by their bioactivity. These SFs are classified as force field (FF)-based, empirical and knowledge-based (Tame, 1999; Schulz-Gasch and Stahl, 2004; Mohan et al., 2005; Tame, 2005; Jain, 2006). As SF accuracy is critical for a successful VS campaign (Klebe, 2006), considerable progress over the years has been made. However, commonly used SFs are known to have limitations as the protein–ligand complex formation often includes subtleties not captured by SFs (Gohlke and Klebe, 2002). This in turn causes poor ranking of compounds by predicted activities. Many oversimplifications, discussed in the following sections (for example, solvation and entropy contributions to the binding free energy), are believed to be the main cause of this pitfall. In addition, the SFs tend to be more accurate with the proteins and the docking programmes used to calibrate them. Indeed, customized SFs calibrated for specific targets have also been devised (Catana and Stouten, 2007). To overcome this hurdle, more than one SF can be alternatively employed with docking programmes. For instance, GoldScore and ChemScore have been implemented in GOLD, while ChemScore, ScreenScore and PLP can be used with FRED. As for consensus docking, consensus scoring, which combines several scoring methods, has been found to be superior to the use of a single function in some cases (X-Score, formerly known as X-Cscore; Wang et al., 2002).
Empirical SFs
Since the pioneering work of Böhm (1994) in the development of LUDI, a significant amount of work has been devoted to the development and improvement of empirical SFs. With empirical SFs, the evaluation of the energetics of the ligand binding (essentially from protein/ligand crystal structures) is decomposed into simpler, scalable contributions arising from, for example, hydrogen bonds, metal ligation, hydrophobic effects and freezing of rotatable bonds (Equation (1)). The various scaling factors (ΔGi in Equation (1)) are then defined by regression to fit experimentally determined protein–ligand affinities.
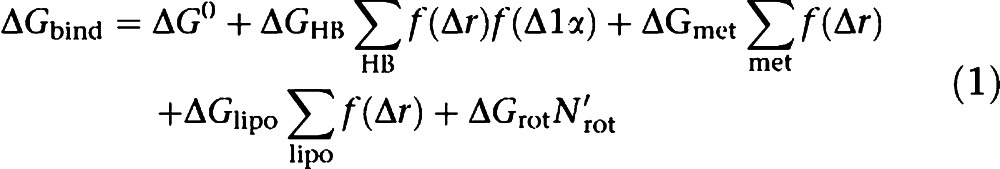 |
Among the most commonly used SFs is ChemScore, which has been implemented in various docking programmes (for example, GOLD, FRED). Standalone SFs have also been devised and include X-Score, DrugScore, VALIDATE and HINT. Each empirical SF differs by the number and nature of the terms used to make up its equation. For instance, several include an explicit directional hydrogen bond energy term (for example, ChemScore, X-Score and the SFs implemented in eHiTS, FlexX, Surflex), while only a few include an explicit directional metal–ligand interaction term (for example, eHiTS, Surflex and X-Score). Functions such as the eHiTS and PLP SFs evaluate the internal energy of the ligand in its bound conformation, while solvation and/or predicted captured water molecules (within GlideScore) are computed in a different manner. Many empirical SFs take into account the hydrophobic effect in the binding, mostly either by computing the hydrophobic surface buried in the complex (for example, SCORE1/2, LigScore), or by evaluating the match of the hydrophobicity of an atom with its environment (for example, FlexX, SCORE, SLIDE), while several combine both approaches (for example, eHiTS, GlideScore, HammerHead, X-Score). On the other hand, HINT computes the logP of the ligand as a measure of its water solvation. The entropic contribution to the binding energy due to the freezing of torsional degrees of freedom upon binding is often estimated by a term proportional to the number of sp3–sp3 and sp2–sp3 rotatable bonds. In some cases (for example, ChemScore, GlideScore, VALIDATE, X-Score), the environment of a bond is taken into consideration to assess the extent of its effect, while RankScore attempts to include the freezing of protein side chains by scaling the interaction with flexible side chains.
FF-based SFs
Force fields were originally developed to reproduce conformational behaviour and thermodynamic and kinetic properties of small molecules and macromolecules. When applied to protein–ligand complexes, FFs are often found to significantly overestimate the binding affinity (Equation (2)) even when applied in conjunction with highly accurate, time-consuming techniques (for example, Linear Interaction Energy method), which consider the bulk water either explicitly or implicitly (for example, PB/SA, GB/SA; Michel et al., 2006). Scaling factors applied to the non-bonded terms (van der Waals and electrostatics) were found to restore the predictiveness of FFs in this area.
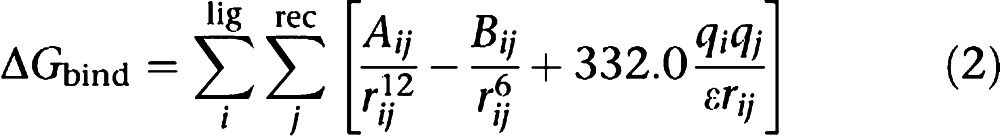 |
When compared to empirical SFs, a smaller number of SFs were developed exclusively from FFs. More commonly, FF terms (illustrated in Equation (2)) are combined with terms from empirical SFs, such as the solvation and ligand entropy terms in the AutoDock SF. The choice of FF parameters is varied; AutoDock, DOCK and RankScore SFs combine the van der Waals, electrostatic and hydrogen bond interaction energy computed using the AMBER FF, while GoldScore makes use of the Tripos parameters and ICM implements a hybrid AMBER-ECEPP/2 approach.
Knowledge-based SFs
Other popular SFs, such as DrugScore and PMF, have been developed from statistical analysis of crystal structures of ligand–protein complexes. These analyses report the distribution of ligand–protein atom-type pairs (histogram in Figure 3) and convert these data into pairwise potentials (blue and green curves in Figure 3). In the interaction between charged species (blue line), there is a sharp minimum at a relatively close distance and a secondary minimum at a larger separation, accounting for the interaction via a bridging water molecule. In contrast, the potential for a pair of aliphatic carbons (green line) shows little preference over a wide range of interatomic distances. The score is calculated by the sum of all interaction pairs between each ligand and protein atom lying within a sphere of a given cutoff (usually 6–12 Å). Although these functions are expected to capture all the data needed for predicting the free energy of binding, some of the interactions are underrepresented in the available crystal structures (for example interactions with metals and/or halogens) and are not well parameterized. As for FF-based SFs, correcting/additional terms were implemented as exemplified by the solvation term included in DrugScore.
Figure 3.

Potentials of mean force for a pair of aliphatic carbons (green) and a pair composed of a positively charged nitrogen and a negatively charged oxygen (blue).
Comparative studies
Comparative studies
Due to the rapid evolution of docking and scoring algorithms, comparing their efficiency was needed. In the past decade, many comparative studies have been reported that evaluate the relative performance of the most popular programmes (Westhead et al., 1997; Bissantz et al., 2000; Perez and Ortiz, 2001; Stahl and Rarey, 2001; Terp et al., 2001; Doman et al., 2002; Diller and Li, 2003; Jenkins et al., 2003; Schulz-Gasch and Stahl, 2003; Wang et al., 2003, 2004; Ferrara et al., 2004; Kellenberger et al., 2004; Kontoyianni et al., 2004; Kroemer et al., 2004; Perola et al., 2004, 2007; Chen et al., 2006; Cummings et al., 2005; Kontoyianni et al., 2005; Warren et al., 2006). These studies can be classified into target-oriented studies and broad comparative studies. This first class is of great interest for the medicinal chemistry community, and aims to help identify the best programme for a specific target. For instance, programmes were evaluated for their ability to dock to matrix metalloproteases (Ha et al., 2000; Hanessian et al., 2001; Hu et al., 2004) and to score BACE-1 inhibitors (Schafferhans and Klebe, 2001; Moitessier et al., 2006a; Holloway et al., 2007) or mannosidase inhibitors (Englebienne et al., 2007). The goal of the broader studies is to evaluate the programmes' accuracy on a set of proteins and is threefold. They aim to evaluate and compare the ability of different docking programs and/or SFs (either separately or in combination) to (1) properly dock compounds to proteins, (2) predict the ligand binding affinities and (3) extract active compounds from libraries of decoys. In Figures 4, 5 and 6, selected comparative studies are shown. Table 3 lists reported comparative studies. While this manuscript was under revision, other comparative studies were published (McGaughey et al., 2007; Onodera et al., 2007; Zhou et al., 2007) and are included in Table 3 but are not discussed.
Figure 4.

Comparative studies of docking programmes. A criterion of r.m.s.d. <2 Å has been used in all the studies except in Kontoyianni and co-workers, where a more suggestive criterion has been used (see text). R.m.s.d., root mean-square deviation.
Figure 5.

Comparative studies of docking-based VS programmes. Top panel: average enrichment factors computed for a variety of enzymes and programmes; bottom left panel: application of MVP to various enzymes; bottom right panel: evaluation of SFs for identifying actives in large libraries. SF, scoring function; VS, virtual screening.
Figure 6.

Comparative studies of docking/scoring programmes. Top panel: average Spearman coefficient factors computed for a variety of enzymes and programmes; bottom panel: Spearman coefficient factors computed for a set of scoring functions and consensus SFs. SF, scoring function.
Table 3. Reported comparative studies.
| Programs | Scoring function (SF) | Reference |
|---|---|---|
| Glide, GOLD, DOCK | Native scoring functions | (Zhou et al., 2007) |
| FLOG, FRED, Glide | Native scoring functions | (McGaughey et al., 2007) |
| DOCK, AutoDock, GOLD | Native scoring functions | (Onodera et al., 2007) |
| N/A | MMFFs, LigScore1, LigScore2, PLP1, PLP2, PMF, LUDI, X-Score | (Holloway et al., 2007) |
| GOLD, Glide, FlexX, AutoDock, eHiTS, LigandFit, FITTED | Native scoring functions | (Englebienne et al., 2007) |
| FlexX, GOLD, Glide, ICM | Native scoring functions | (Chen et al., 2006) |
| DOCK, DockIt, FlexX, Flo, Fred, Glide, Gold, LigFit, MOE, MVP | Native scoring functions, FlexX, DrugScore, Mcdock, Mcdock+, Fulldock, Sdock, Zdock, ChemScore, ScreenScore, CVFF,DOCKPLP, PMF. Dockit-Score, Cscore, DockScore, GoldScore, Sdock+, Fulldock+, LigScore1, LigScore2, PLP1, Dreiding, DOCK-Energy, DOCK-Chemical, DOCK-Contact | (Warren et al., 2006) |
| N/A | PLP1, PLP2, LUDI, PMF, DockScore, GoldScore, ChemScore, LigScore1, LigScore2, CFF91, AMBER94, RankScore | (Moitessier et al., 2006a) |
| Glide, LigandFit, FlexX, DOCK | LigScore1, LigScore2, PMF, PMF, ChemScore, GoldScore, GoldScore, FlexX, PLP1, PLP2 | (Kontoyianni et al., 2005) |
| DOCK, DOCKVISION, Glide, GOLD | Native scoring functions, consensus scoring | (Cummings et al., 2005) |
| FlexX, DOCK, GOLD, LigandFit, Glide | Native scoring functions | (Kontoyianni et al., 2004) |
| DOCK, FlexX, FRED, Glide, GOLD, SLIDE, SurFlex, QXP | Native scoring functions | (Kellenberger et al., 2004) |
| Glide, GOLD, ICM | ChemScore, GlideScore, OPLS-AA | (Perola et al., 2004) |
| N/A | BLEEP, PMF, GoldScore, DockScore, ChemScore | (Marsden et al., 2004) |
| DOCK, FlexX, GOLD, AutoDock | Native scoring functions, DrugScore, X-Cscore | (Hu et al., 2004) |
| N/A | X-Score, DrugScore, D-Score, PMF-Score, GoldScore, ChemScore, F-Score, LigScore, PLP, PMF, LUDI, HINT | (Wang et al., 2004) |
| N/A | CHARMm, DOCK-Energy, DOCK-Chemical, DOCK-contact, DrugScore, ChemScore, AutoDock, PMF, GoldScore | (Ferrara et al., 2004) |
| FlexX, GOLD, ICM, LigandFit, the Northwestern University version of DOCK, QXP | Native scoring functions | (Kroemer et al., 2004) |
| Glide, FRED | FlexX, ScreenScore, GlideScore, GlideComp, ChemScore, PLP | (Schulz-Gasch and Stahl, 2003) |
| DockVision, GOLD | LUDI, GoldScore | (Jenkins et al., 2003) |
| AutoDock, DOCK, FlexX, GOLD, ICM | Native scoring functions | (Bursulaya et al., 2003) |
| FlexX | FlexX, PLP, DrugScore, PMF, ScreenScore | (Stahl and Rarey, 2001) |
| GOLD | SCORE, LUDI, GRID, PMF, DockScore, GoldScore, ChemScore, F-Score | (Terp et al., 2001) |
| DOCK, AutoDock | Native scoring functions | (Hanessian et al., 2001) |
| DOCK, FlexX | PMF, FF | (Ha et al., 2000) |
| DOCK, FlexX, GOLD | Chemscore, DockScore, FlexX, Fresno, GoldScore, Pmf, Score | (Bissantz et al., 2000) |
Abbreviation: N/A, not available.
Limitations of comparative studies
While comparative studies provide insight into the various programmes' accuracy, speed, applicability to a range of targets and other factors, they should be considered with great care for many reasons.
First, the versions of the programmes in question are not always specified and may vary from one study to another.
Second, the accuracy is greatly dependent on the settings employed and fine-tuning parameters may lead to significant changes in accuracy. As a result, greater expertise with a specific programme may bias the study. Comparative studies published as part of the development of new programmes will not be discussed herein, as the authors are more proficient with their own programme, thus lowering the objectivity of the study. We believe that the most useful programmes for medicinal chemists are those that can provide accurate and reliable results without extensive training and/or labour-intensive optimizations.
Third, the preparation of receptors and ligands for docking requires knowledge of the active site and careful consideration of all possible isomers (for example, tautomers) and protonation states. To address this issue, Warren and co-workers have divided the work between chemists with expertise on each of the proteins studied, and others with expertise on each of the programmes considered. This strategy significantly reduces bias, making this comparative study very fair and objective.
Fourth, as pointed out by Cole et al. (2005), using the root mean-square deviation (r.m.s.d.) as a criterion of docking accuracy is questionable. For instance, an r.m.s.d. of less than 2.0 Å used as a criterion of success yields misleading results when specific interactions such as directional hydrogen bonds or floppy solvent exposed groups are to be considered. In fact, Kontoyianni used a criterion that was more subjective yet more representative of the true accuracy of the programmes. In this study, in addition to examining the r.m.s.ds, all of the poses were also visually inspected and success of the docking was then described as ‘close' (correct pose), ‘active site' (fair pose) or ‘inaccurate' (wrong pose). Unfortunately, this criterion would be difficult to implement in programmes and therefore cannot be applied to large testing sets. During the same year, interaction-based accuracy classification was also proposed and found to be a more meaningful criterion of success than the r.m.s.d. (Kroemer et al., 2004). Recently, Marcou and Rognan (2007) came to the same conclusion when they used interaction fingerprints combined with a Tanimoto coefficient. Additionally, the metrics for success in virtual screening (usually enrichment factors and ROC curves) are sometimes difficult to interpret (Truchon and Bayly, 2007).
Fifth, self-docking is a good indication of a programme's ability to identify native poses amongst several others, but provides little information about the accuracy in a real drug discovery scenario. In practice, cross-docking is a more valuable experiment. In fact, although Warren and co-workers pointed out that a careful selection of the protein structure can reduce the impact of the induced fit upon binding, many other studies have shown the significant impact of protein flexibility on the docking accuracy (Claußen et al., 2001; Carlson, 2002; Osterberg et al., 2002; Zentgraf et al., 2006; Corbeil et al., 2007).
Sixth, in certain cases, no mention is made (or insufficient details are provided) regarding the CPU time required to run the study. As many programmes (for example, GOLD and Glide) provide various levels of speed and accuracy, a comparative study should reveal the accuracy to expect within a specified period of time. For instance, the early version of our own programme, FITTED, performs as well as other programmes, but was found to be much slower and therefore not competitive.
For medicinal chemists, the best indicator of a programme's accuracy is its ability to identify novel compounds in VS studies that are then experimentally confirmed. However, it would be difficult to compare studies as they often deal with different proteins and compound libraries. A number of successful applications can be found in (Kitchen et al., 2004; Ghosh et al., 2006; Muegge and Oloff, 2006). To date, the available programmes (that is, AutoDock, DOCK, GOLD, FlexX, Glide) have been very successful.
Docking accuracy
Several conclusions can be drawn from the fair and exhaustive comparative study by Warren and co-workers. First, as can be seen in Figure 4 (top panel), the accuracy is highly dependent on the protein considered. Second, none of the 10 programmes assessed clearly outperforms all the others. However, in this study, MVP, Glide, GOLD and FlexX may be considered the best four, while DOCK, DockIt and MOE reproduced the native pose in only a few cases. Other comparative studies showed that Glide, GOLD and FlexX are among the best programmes. Chen and co-workers reported that ICM outperformed the other three programmes investigated. Interestingly, Perola et al. (2007) commented on this same article that they were not able to reproduce the high accuracy of ICM, although they found that ICM is still at least as accurate as the other programmes assessed. A series of comparative studies looked at metalloenzymes and revealed that the metal coordination is still not well reproduced by many docking programmes. Englebienne et al. recently looked at the α-mannosidase and reported Glide as the best of the seven assessed programmes (Englebienne et al., 2007).
Virtual screening accuracy
Comparing programmes for their ability to identify actives within libraries is an even more difficult task. In fact, each of these types of comparative studies must be considered separately. Figure 5 summarizes some of the data from selected comparative studies. From a more global point of view, one can draw three major conclusions. First, none of the programmes is universally accurate as the data indicate that the accuracy is highly protein dependent. Second, until recently, none of the assessed programmes were consistently better than others, as most of them exhibit similar overall accuracies. However, the recent study by Warren and co-workers, which appeared to be the largest study of this kind, clearly showed that MVP stood out. Further investigation of this result on other proteins is needed to confirm this important discovery. Third, all the programmes, except for MOEDock, provide enrichments of the top of the hit list in active compounds. This shows that in virtually every case, it is worth running a VS to guide the development of a focused library as enrichment is likely to be obtained. Similarly, studies were reported that identified the best SFs in the context of VS as illustrated in Figure 5 (bottom right panel).
SF accuracy
Other comparative studies aim to identify the most accurate SF in rank-ordering compounds by affinity. The Spearman coefficient describing the ability to reproduce a rank-ordered list has often been used. Figure 6 summarizes the accuracy of various SFs as determined by a number of different comparative studies. Comparing SFs for their ability to identify actives within libraries is an even more difficult task. In contrast to the docking programmes, which often demonstrate similar accuracies, these published studies revealed a wide range of accuracies. For instance, Bissantz and co-workers determined that the poses produced by GOLD were the most suitable for their VS targets (thymidine kinase and oestrogen receptor), while identifying which SF to use proved to be a very difficult task. Two of the SFs (PMF and FlexX SF) that performed well for the thymidine kinase screening were among the worst for the oestrogen receptor screening, highlighting the target specificity of SFs. Bissantz et al. (2000) have also shown that regardless of the docking programme, GoldScore was often the most accurate SF of their set. It has also been shown that consensus scoring is often better (not always significantly) than the individual SFs (Wang et al., 2004; Figure 6).
The challenges of docking/scoring programmes
Although considerable efforts have been devoted to the development of accurate and fast docking/scoring methods, a more universal method is yet to be developed. Although developing a universal method might not be the major goal, increasing the transferability from one protein to another of existing methods is certainly to be improved. Among the issues that remain to be addressed and those that are currently being addressed are the treatment of protein flexibility, and in particular the scoring of modelled protein conformations, and the presence of water molecules including the evaluation of their binding free energy. Ligands forming covalent complexes and macromolecules such as nucleic acids and metal-containing enzymes are subjects in need of further study. Each challenge requires examination of both docking and scoring. Considering that the free energy of binding is a combination of subtle enthalpic and entropic effects but that high-throughput scoring is desired, careful approximations have to be made when developing SFs. Unfortunately these approximations inevitably add noise to the SFs, resulting in reduced accuracy. In the following section we will describe what we believe are the remaining issues facing the developers. To illustrate the state-of-the-art, examples are given in each of these sections. However, the following descriptions are limited to the reported features of each programme, although we are aware that some features such as the docking of covalent inhibitors with AutoDock are described only in user manuals and/or on web pages.
Exhaustive sampling vs speed
Despite the considerable efforts to address the problem of conformational sampling using a large panel of algorithms, one is always left with the task of balancing speed and accuracy. On one end of the spectrum, one can apply crude but fast docking (for example, rigid-body docking) and scoring (for example, shape complementarity) approaches. In practice, many fast programmes that only consider discrete values for the many degrees of freedom were enhanced by local search algorithms (simplex, Solis and Wets in AutoDock, conjugate gradients), although significantly extending the time required for the computations. On the other end of the spectrum, one can apply molecular dynamics-based approaches that would consider the Boltzmann's distribution of conformations (for example, Linear Energy Interaction). While being accurate in predicting relative binding affinity of congeneric compounds, these latter methods cannot be applied to large libraries of ligands (for example 100 000 compounds) due to their speed.
Protein flexibility
Another major factor that significantly limits the accuracy of today's docking methods is protein flexibility upon ligand binding (Bursavich and Rich, 2002). Najmanovich et al. (2000) pointed out in a statistical analysis of Brookhaven protein databank (PDB) structures that 85% of the proteins contain one to three flexible residues. Although this number of flexible side chains can be considered small, a slight adjustment of the protein can have a significant effect on the molecular recognition process. As a result, the success rate drops significantly when going from self- to cross-docking (Osterberg et al., 2002). Due to the large fraction of flexible proteins reported, flexibility cannot be ignored. Many approaches were proposed and recently reviewed (Cavasotto et al., 2005), although very few of these have been made available to the greater medicinal chemistry community.
Among these approaches, the docking of ligands to soft structures (reduced van der Waals penalties at short distances; for example, LJ 8-4 in GOLD) simulates slight adjustments of the protein. A similar practice reduces the van der Waals optimal distances thereby uniformly enlarging the binding site (smooth potential in AutoDock 3.0 and vdW offset in ADAM). Local optimization of hydrogens is also possible (polar hydrogens in GOLD, all hydrogens in PLANTS). Overall, these approaches were found to improve the docking accuracy but not when considerable side chain rotations are observed. The first attempt to account for larger moves has been reported by Leach (1994) and relied on libraries of side chain rotamers. Similar approaches have since been devised (Frimurer et al., 2003) and even implemented in available programmes. ICM offers the use of biased probability moves of the side chains coupled with Monte Carlo search of the ligand pose (Totrov and Abagyan, 1997). Docking to multiple structures can also account for side chain movement (EUDOC) while significantly increasing the required CPU time.
Ideally, one would like to dock ligands to virtual proteins where the flexibility, including backbone adjustments and side chain rotamers, is considered on-the-fly. To this effect, two other major approaches have been proposed. For programmes relying on grids for scoring, combination of grids derived from more than one protein conformations can be either combined leading to one (for example, DOCK; Knegtel et al., 1997) or to a set of individual grids (for example, AutoDock; Osterberg et al., 2002) or aligned into a larger grid modelling the conformational ensemble (Sotriffer and Dramburg, 2005). The great advantage of this approach is that the time required for a single run is roughly similar to the time necessary to dock to a rigid protein. For programmes using atomic representations, docking to composite structure ensembles has been shown to significantly enhance the docking accuracy (for example, FlexE, FITTED). These composite structure ensembles can be implemented in different ways. Within FlexE, multiple crystal structures are merged where similar, while the dissimilar areas are marked as different alternatives. Upon docking, the ligand is compared with each alternative and the best scoring protein structure is selected. Within FITTED, the evolution of the individual allows for the crossover of the conformation of the backbone and active site residues independently of each other. An induced-fit docking method has also been developed (SLIDE), removing clashes between the ligand and the protein by directed, single bond rotation of either the ligand or the side chains of the protein.
Although conformational flexibility of proteins is now implemented in some programmes, one major challenge remains to be addressed. As soon as the correct protein structure is found, it must be attributed a score. Considering the subtle protein entropy and enthalpy changes upon binding is very challenging. To our knowledge, ICM is the only programme that approximates these two contributions (Abagyan and Totrov, 1994a), while FITTED implicitly considers the entropy cost by reducing the interaction energy between the docked ligand and flexible residue side chains but disregards the protein enthalpy change. ROSETTALIGAND computes all the intramolecular interactions of the proteins when reconstructing and evaluating side-chain conformations.
Water molecules
The presence of water molecules critical to the binding of ligands adds another dimension to the docking problem (Ladbury, 1996; van Dijk and Bonvin, 2006a; Barillari et al., 2007; Li and Lazaridis, 2007). Although these water molecules are often removed, other options exist. First, one can keep them explicitly. This approach has the drawback of preventing the docking of ligands requiring displacement of a water molecule. Another approach is to implement a protocol that will select the best option (keep or displace each of the water molecules). Such methods have only been implemented in GOLD, FlexX and SLIDE in which waters are toggled on or off, and in FITTED, which makes use of a specific potential energy term. The FlexX method is unique in the way it ‘predicts' potential locations of water molecules, thereby not relying on their crystallographic positions.
Unfortunately, these approaches have not been extensively applied and validated by independent studies. In addition, water locations should be optimized upon docking of ligands, a property that has only partially been addressed by SLIDE. The latter translates the water molecule to remove collisions with protein or ligand atoms but, to our knowledge, does not consider the energetics associated with these moves. Once more, scoring the presence/absence of water molecules is a challenge that remains to be dealt with. The key issue in this context is the consideration of the binding free energy of these specific water molecules. For instance, FITTED, GOLD and SLIDE add a penalty for each displaced water molecule to the final score. However, each water molecule has a different free energy of binding that cannot be accurately represented by a single value (Gohlke and Klebe, 2002).
Covalently bound inhibitors
Although many enzymes are targeted by covalent inhibitors (reversible or irreversible), little effort has been dedicated to this specific area. For instance, GOLD (Jones et al., 1997), FlexX (Kramer et al., 1999) and AutoDock allow the user to manually select which atoms of the ligand and protein are to be joined by a covalent bond and add the protein atom to the ligand input file. However, these methods are hardly amenable to VS of large libraries of compounds given that the user must manually locate the covalent bond and insert it in each individual input file. MacDOCK combines ligand–protein docking (DOCK 4.0) with ligand–ligand superposition to mimic the pose of a covalently bound inhibitor (using the module MIMIC). Within MacDOCK, anchor-guided AG-DOCK includes the location and directionality of the atoms involved in the covalent bond in the docking (Fradera et al., 2004). The functionalities within the ligand that can form covalent bonds and the site of covalent binding of the protein are automatically identified. The geometrical arrangement is then modified to match that of the product structure. If several functional groups can be used, poses are generated for each one and docked to generate a set of product structures, which are then compared. The SF is modified to ignore the van der Waals clashes at the site of formation of the covalent bond. To our knowledge, no other programmes consider the formation of a covalent bond upon ligand binding, and scoring the formation of this bond remains to be explored.
Nucleic acids
Proteins have been the major targets of docking methods. However, nucleic acids are also targets for medicinal chemistry and should be further investigated. One of the major challenges lies in the fact that binding pockets on nucleic acids are highly charged and more solvent exposed than the binding sites of most proteins, which can be deeply buried. Docking DNA intercalators is even more challenging, as a major opening of the bases must occur to bind the ligand. The first attempts for docking to nucleic acids relied on the use of existing programmes originally developed for docking small molecules to proteins. For instance, Chen et al. (1997) used DOCK 3.5 to identify possible binders to the RNA major groove, while Leclerc and Karplus (1999) used the MCSS method. The authors noted that the effect of solvent on the ligand–RNA interaction was not evaluated and represented a necessary improvement. In just a few cases, existing programmes were modified to account for the high polarity of nucleic acids (Pan et al., 2003; Kang et al., 2004).
In addition to the high charge density, flexibility of the nucleic acid strands and presence of key water molecules have been some of the hurdles to the successful applications of docking programmes in this area. In fact, flexibility and water molecules were found to be necessary to improve docking accuracy (Moitessier et al., 2006b). DNA flexibility has been addressed using the HADDOCK programme and semi-flexible refinement using simulated annealing methods following rigid docking (van Dijk et al., 2006b). To our knowledge, few SFs have been specifically designed to score RNA binders. HADDOCK has its own SF, HADDOCK Score, which includes FF terms, solvation energies and a buried surface area term. The DNA conformations are scored using deformation energies calculated from statistical preferences. RiboDOCK evaluates specific nucleic acid–ligand interactions such as aromatic stacking onto the nucleic acid bases and interaction between guanidinium and carbonyl functions. In these two programmes, the water molecules are not directly addressed (Morley and Afshar, 2004). Chen et al. (2004) developed an SF that can identify native protein–RNA structures from the incorrectly docked decoys based on hydrogen-bonding geometries and scores, which they believe to be nucleic acid specific. Finally, Ge and co-workers developed a knowledge-based SF that considers the binding patterns of ligands around individual bases. This approach does not directly address the presence of water, instead considers the frequency of the observed water at a hydration site (Ge et al., 2005).
Entropy
Computational evaluation of the entropy is often based on normal mode analysis or other CPU-expensive approaches. Using such approaches would significantly reduce the throughput of docking methods. To date, the translational and rotational entropy loss estimated in the range of 15 to 20 kJ mol−1 is ignored (Murray and Verdonk, 2002) and most SFs consider the number of rotatable bonds as an estimate of the entropic contribution to the binding free energy. However, not all the bonds are equal and penalties should be assigned accordingly. Indeed, ChemScore considers rotatable bonds in the context of their hydrophobicity; apolar portions tend be more flexible in a binding site than polar fragments and are less penalized. A more advanced approach has recently been proposed that evaluates the restriction of the ligand conformational space by considering a large number of poses (Ruvinsky, 2007) at the expense of CPU time. As mentioned above, the changes in protein entropy are seldom implemented in available programmes (for example, ICM and FITTED).
Solvation
Apart from the bridging water molecules, the effect of solvation by bulk water is a key player in ligand protein binding. Although fairly accurate methods have been reported such as GB/SA and PB/SA (Taylor et al., 2003; Morreale et al., 2007), their implementation in a given docking programme affects the time required to perform a single run unless pre-computed on a grid (DOCK 5) or on the unbound protein (FITTED 2.0). In order to reduce the CPU cost, one can look at ligand lipophilicity as a measure of its desolvation without considering the protein desolvation, which can be assumed to be constant regardless of the pose of the docked ligand. However, larger ligands imply larger desolvation of the binding site and various poses for a single ligand can require the desolvation of different residues. Approaches with higher throughput have been devised, such as the use of atom-based solvation parameters evaluating the buried surface, as exemplified by the AutoDock and Hammerhead SFs, which also penalize the hydrogen bond-capable sites that are not fulfilled. Solvation is known to be partly proportional to the change in solvent-exposed surface area. Based on this concept, DrugScore and ICM evaluate the solvation contribution to the free energy of binding using a term proportional to this area.
Metals
Many groups have looked closely at the scoring of metalloprotease inhibitors (Hu et al., 2004; Irwin et al., 2005; Khandelwal et al., 2005; Englebienne et al., 2007; Jain and Jayaram, 2007) and identified the scoring of metal coordination as a major issue in SFs. In fact, metals can adopt various coordination geometries and docking to metals remains a challenge. Coordination templates have been exploited by GOLD to orient the docking towards potential coordination geometries. However, this has been found to increase only marginally the docking accuracy (Englebienne et al., 2007). As soon as the pose is generated, scoring the binding to the metal is the next issue, and very few SFs such as the eHiTS SF include a specific metal binding term. Only eHiTS, SLIDE and Surflex SF take the geometry restraints into account. Again, evaluation of the impact of these additional terms has not been often investigated, and one cannot state that they significantly improve the docking/scoring accuracy.
Specific interactions
Most of the developed programmes account for the most common interactions such as hydrogen bonds, hydrophobic and ionic interactions. However, the seldom studied and observed interactions, such as cation–π interactions (Scharer et al., 2005) or CH–π interactions (Gil et al., 2007), are not captured by the available SFs and specific functions remain to be developed. This lack is due in part to the low number of complexes featuring these interactions in the PDB. Other interactions such as weak hydrogen bonds required considerable efforts to be considered by SFs or docking programmes (Verdonk et al., 2004).
Training set quality
An accurate SF has to be trained and tested against a high quality set of protein ligand complexes and care must be taken to select the protein–ligand complexes (Hartshorn et al., 2007). Efforts are being made to prepare large databases of high-quality protein–ligand complexes (for example, PDBbind, BindingDB, Relibase) to calibrate SFs. However, automated preparation of some of these publicly available large data sets does not allow for their refined preparation. For instance, a careful identification and orientation of bridging water molecules or the optimal protonation state of each residue and ligand has rarely been carried out, even though it has a great impact on the accuracy of the derived SF. In addition, some of the biological data collected is difficult to interpret and use, such as the very different affinity values reported for an adenosine deaminase inhibitor (PDB codes 1fkx and 2ada; Ki=6 mM and 0.1 pM, respectively).
The PDBbind-refined set of 800 complexes has been used to assess the accuracy of available SFs (Wang et al., 2004). In contrast to our unpublished study, Wang and co-workers have found that all the SFs were poorly predictive for HIV-1 protease inhibitors. In fact, this particular enzyme binds ligands with the need of a water molecule and ligand-dependent protonation state of the two catalytic aspartic acids (Yamazaki et al., 1994; Kulkarni and Kulkarni, 1999; Czodrowski et al., 2007). However, in their protocol, water molecules were all removed and protonation was automatically assigned to ionized aspartic acids.
A disadvantage of using crystal structures of protein–ligand complexes to derive predictive methods is that for the former to exist, the ligand needs to bind to the protein to a certain extent. As a result, SFs are not trained with non-binders. As obvious as it sounds, SF should be trained against completely inactive compounds as well in order to be able to discriminate the latter. In a recent development, Pham and co-workers recently showed that docked compounds can help train an SF against inactive compounds (Pham and Jain, 2006).
Although various sets have been reported and made available, the ultimate benchmark set should be a set for cross-docking studies. As mentioned above, self-docking is not representative of a true medicinal chemistry scenario, while cross-docking is a more reliable evaluation of a programme's performance.
Docking to homology models
It has been observed that moving from crystal structures to homology models often leads to a decrease in accuracy. Solutions to reduce this discrepancy by inclusion of additional information (QSAR data in DRAGHOME; Schafferhans and Klebe, 2001, and SAR information in DoMCoSAR; Vieth and Cummins, 2000) have been proposed although not extensively validated. Rognan and co-workers successfully used homology models of G-protein-coupled receptors to dock antagonists to three human G-protein-coupled receptors; the docking of agonists remains elusive (Bissantz et al., 2003). More recently, Ferrara and Jacoby (2007) evaluated the docking accuracy on homology models and showed that the accuracy can in some cases be as high as with crystal structures.
Conclusion
We have reviewed herein the state-of-the-art in the field of small-molecule-protein docking and scoring methods. Comparative studies, the only available tools to evaluate relative performance of docking programmes, are often carried out by chemists with varying expertise and using different metrics to define accuracy/success. Conclusions and results are difficult to extrapolate to other proteins. An interesting proposal would be to set up a large blind competition analogous to CAPRI in the field of protein–protein docking (http://capri.ebi.ac.uk/). Overall, these comparative studies indicate that none of the docking programmes truly outperforms the others, and the high accuracy observed for ICM and MVP in the two largest and most recent comparative studies remains to be cross-validated.
Ultimately, docking/scoring programmes should be able to identify novel potential ‘binders' very accurately. However, to reach this goal, many issues, including those discussed throughout this review, have yet to be addressed. Currently other strategies, such as post-docking strategies or smart selection of docked compounds, are used to reduce the number of false positive and negatives.
Acknowledgments
We thank Virochem Pharma as well as the Canadian Foundation for Innovation (New Opportunities Fund), NSERC and CIHR for financial support. CC, PE and JL were supported by scholarships from the Canadian Institutes of Health Research (Strategic Training Initiative in Chemical Biology) and a GW McConnell Memorial Fellowship (PE).
Glossary
- FF
force field
- PDB
Brookhaven protein databank
- r.m.s.d.
root mean-square deviation
- SF
scoring function
- VS
virtual screening
Footnotes
Supplementary Information accompanies the paper on British Journal of Pharmacology website (http://www.nature.com/bjp)
Conflict of interest
The authors state no conflict of interest.
Supplementary Material
References
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994a;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Abagyan R, Totrov M, Kuznetsov D. ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994b;15:488–506. [Google Scholar]
- Alberts IL, Todorov NP, Dean PM. Receptor flexibility in de novo ligand design and docking. J Med Chem. 2005;48:6585–6596. doi: 10.1021/jm050196j. [DOI] [PubMed] [Google Scholar]
- Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Barillari C, Taylor J, Viner R, Essex JW. Classification of water molecules in protein binding sites. J Am Chem Soc. 2007;129:2577–2587. doi: 10.1021/ja066980q. [DOI] [PubMed] [Google Scholar]
- Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-protein coupled receptors suitable targets. Proteins Struct Funct Bioinf. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- Böhm H-J. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des. 1998;12:309–309. doi: 10.1023/a:1007999920146. [DOI] [PubMed] [Google Scholar]
- Bohm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- Böhm HJ. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J Comput Aided Mol Des. 1994;8:243. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- Borman S. Drugs by design. Chem Eng News. 2005;83:28–30. [Google Scholar]
- Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- Budin N, Majeux N, Caflisch A. Fragment-based flexible ligand docking by evolutionary optimization. Biol Chem. 2001;382:1365–1372. doi: 10.1515/BC.2001.168. [DOI] [PubMed] [Google Scholar]
- Burkhard P, Taylor P, Walkinshaw MD. An example of a protein ligand found by database mining: description of the docking method and its verification by a 2.3 Å X-ray structure of a thrombin–ligand complex. J Mol Biol. 1998;277:449–466. doi: 10.1006/jmbi.1997.1608. [DOI] [PubMed] [Google Scholar]
- Bursavich MG, Rich H. Designing non-peptide peptidomimetics in the 21st century: inhibitors targeting conformational ensembles. J Med Chem. 2002;45:541–558. doi: 10.1021/jm010425b. [DOI] [PubMed] [Google Scholar]
- Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- Catana C, Stouten PFW. Novel, customizable scoring functions, parameterized using N-PLS, for structure-based drug discovery. J Chem Inf Model. 2007;47:85–91. doi: 10.1021/ci600357t. [DOI] [PubMed] [Google Scholar]
- Cavasotto CN, Orry AJW, Abagyan RA. The challenge of considering receptor flexibility in ligand docking and virtual screening. Curr Comput Aided Drug Des. 2005;1:423–440. [Google Scholar]
- Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- Chen HM, Liu BF, Huang HL, Hwang SF, Ho SY. SODOCK: Swarm optimization for highly flexible protein–ligand docking. J Comput Chem. 2007;28:612–623. doi: 10.1002/jcc.20542. [DOI] [PubMed] [Google Scholar]
- Chen Q, Shafer RH, Kuntz ID. Structure-based discovery of ligands targeted to the RNA double helix. Biochemistry. 1997;36:11402–11407. doi: 10.1021/bi970756j. [DOI] [PubMed] [Google Scholar]
- Chen Y, Kortemme T, Robertson T, Baker D, Varani G. A new hydrogen-bonding potential for the design of protein–RNA interactions predicts specific contacts and discriminates decoys. Nucleic Acids Res. 2004;32:5147–5162. doi: 10.1093/nar/gkh785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi V. YUCCA: an efficient algorithm for small-molecule docking. Chem Biodivers. 2005;2:1517–1524. doi: 10.1002/cbdv.200590123. [DOI] [PubMed] [Google Scholar]
- Clark KP, Ajay Flexible ligand docking without parameter adjustment across four ligand–receptor complexes. J Comput Chem. 1995;16:1210–1226. [Google Scholar]
- Claußen H, Buning C, Rarey M, Lengauer T. FLEXE: efficient molecular docking considering protein structure variations. J Mol Biol. 2001;308:377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- Cole JC, Murray CW, Nissink JWM, Taylor RD, Taylor R. Comparing protein–ligand docking programs is difficult. Proteins Struct Funct Genet. 2005;60:325–332. doi: 10.1002/prot.20497. [DOI] [PubMed] [Google Scholar]
- Corbeil CR, Englebienne P, Moitessier N. Docking ligands into flexible and solvated macromolecules. 1. Development and validation of FITTED 1.0. J Chem Inf Model. 2007;47:435–449. doi: 10.1021/ci6002637. [DOI] [PubMed] [Google Scholar]
- Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, Mozzarelli A. Simple, intuitive calculations of free energy of binding for protein–ligand complexes. 1. Models without explicit constrained water. J Med Chem. 2002;45:2469–2483. doi: 10.1021/jm0200299. [DOI] [PubMed] [Google Scholar]
- Cummings MD, DesJarlais RL, Gibbs AC, Mohan V, Jaeger EP. Comparison of automated docking programs as virtual screening tools. J Med Chem. 2005;48:962–976. doi: 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- Czodrowski P, Sotriffer CA, Klebe G. Atypical protonation states in the active site of HIV-1 protease: a computational study. J Chem Inf Model. 2007;47:1590–1598. doi: 10.1021/ci600522c. [DOI] [PubMed] [Google Scholar]
- DeWitte RS, Shakhnovich EI. SMoG: de novo design method based on simple, fast, and accurate free energy estimates. 1. Methodology and supporting evidence. J Am Chem Soc. 1996;118:11733–11744. [Google Scholar]
- Diller DJ, Li R. Kinases, homology models, and high throughput docking. J Med Chem. 2003;46:4638–4647. doi: 10.1021/jm020503a. [DOI] [PubMed] [Google Scholar]
- Diller DJ, Merz KM., Jr High throughput docking for library design and library prioritization. Proteins Struct Funct Genet. 2001;43:113–124. doi: 10.1002/1097-0134(20010501)43:2<113::aid-prot1023>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, et al. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- Englebienne P, Fiaux H, Kuntz DA, Corbeil CR, Gerber-Lemaire S, Rose DR, et al. Evaluation of docking programs for predicting binding of Golgi alpha-mannosidase ii inhibitors: a comparison with crystallography. Proteins Struct Funct Bioinf. 2007;69:160–176. doi: 10.1002/prot.21479. [DOI] [PubMed] [Google Scholar]
- Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III Assessing scoring functions for protein–ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- Ferrara P, Jacoby E. Evaluation of the utility of homology models in high throughput docking. J Mol Mod. 2007;13:897–905. doi: 10.1007/s00894-007-0207-6. [DOI] [PubMed] [Google Scholar]
- Ferrara P, Priestle JP, Vangrevelinghe E, Jacoby E. New developments and applications of docking and high-throughput docking for drug design and in silico screening. Curr Comput Aided Drug Des. 2006;2:83–91. [Google Scholar]
- Floriano WB, Vaidehi N, Zamanakos G, Goddard WA., III HierVLS hierarchical docking protocol for virtual ligand screening of large-molecule databases. J Med Chem. 2004;47:56–71. doi: 10.1021/jm030271v. [DOI] [PubMed] [Google Scholar]
- Fradera X, Kaur J, Mestres J. Unsupervised guided docking of covalently bound ligands. J Comput Aided Mol Des. 2004;18:635–650. doi: 10.1007/s10822-004-5291-4. [DOI] [PubMed] [Google Scholar]
- Fradera X, Knegtel RMA, Mestres J. Similarity-driven flexible ligand docking. Proteins Struct Funct Genet. 2000;40:623–636. doi: 10.1002/1097-0134(20000901)40:4<623::aid-prot70>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Frimurer TM, Peters GH, Iversen LF, Andersen HS, Møller NPH, Olsen OH. Ligand-induced conformational changes: improved predictions of ligand binding conformations and affinities. Biophys J. 2003;84:2273–2281. doi: 10.1016/S0006-3495(03)75033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabb HA, Jackson RM, Sternberg MJE. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- Ge W, Schneider B, Olson WK. Knowledge-based elastic potentials for docking drugs or proteins with nucleic acids. Biophys J. 2005;88:1166–1190. doi: 10.1529/biophysj.104.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, et al. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol. 1995;2:317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Gervasio FL, Laio A, Parrinello M. Flexible docking in solution using metadynamics. J Am Chem Soc. 2005;127:2600–2607. doi: 10.1021/ja0445950. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10:194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Gil A, Branchadell V, Bertran J, Oliva A. CH/π; interactions in DNA and proteins. A theoretical study. J Phys Chem B. 2007;111:9372–9379. doi: 10.1021/jp0717847. [DOI] [PubMed] [Google Scholar]
- Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein–ligand interactions. J Mol Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed Engl. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Goto J, Kataoka R, Hirayama N. Ph4Dock: pharmacophore-based protein–ligand docking. J Med Chem. 2004;47:6804–6811. doi: 10.1021/jm0493818. [DOI] [PubMed] [Google Scholar]
- Grosdidier A, Zoete V, Michielin O. EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins Struct Funct Genet. 2007;67:1010–1025. doi: 10.1002/prot.21367. [DOI] [PubMed] [Google Scholar]
- Ha S, Andreani R, Robbins A, Muegge I. Evaluation of docking/scoring approaches: a comparative study based on MMP3 inhibitors. J Comput Aided Mol Des. 2000;14:435–448. doi: 10.1023/a:1008137707965. [DOI] [PubMed] [Google Scholar]
- Hanessian S, Moitessier N, Therrien E. A comparative docking study and the design of potentially selective MMP inhibitors. J Comput Aided Mol Des. 2001;15:873–881. doi: 10.1023/a:1014356529909. [DOI] [PubMed] [Google Scholar]
- Hart TN, Read RJ. A multiple-start Monte Carlo docking method. Proteins Struct Funct Genet. 1992;13:206–222. doi: 10.1002/prot.340130304. [DOI] [PubMed] [Google Scholar]
- Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WTM, Mortenson PN, et al. Diverse, high-quality test set for the validation of protein–ligand docking performance. J Med Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- Head RD, Smythe ML, Oprea TI, Waller CL, Green SM, Marshall GR. VALIDATE: a new method for the receptor-based prediction of binding affinities of novel ligands. J Am Chem Soc. 1996;118:3959–3969. [Google Scholar]
- Hindle SA, Rarey M, Buning C, Lengauer T. Flexible docking under pharmacophore type constraints. J Comput Aided Mol Des. 2002;16:129–149. doi: 10.1023/a:1016399411208. [DOI] [PubMed] [Google Scholar]
- Holloway MK, McGaughey GB, Coburn CA, Stachel SJ, Jones KG, Stanton EL, et al. Evaluating scoring functions for docking and designing β-secretase inhibitors. Bioorg Med Chem Lett. 2007;17:823–827. doi: 10.1016/j.bmcl.2006.10.051. [DOI] [PubMed] [Google Scholar]
- Hu X, Balaz S, Shelver WH. A practical approach to docking of zinc metalloproteinase inhibitors. J Mol Graph Model. 2004;22:293–307. doi: 10.1016/j.jmgm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- Ishchenko AV, Shakhnovich EI. SMall Molecule Growth 2001 (SMoG2001): an improved knowledge-based scoring function for protein–ligand interactions. J Med Chem. 2002;45:2770–2780. doi: 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- Jackson RM. Q-fit: a probabilistic method for docking molecular fragments by sampling low energy conformational space. J Comput Aided Mol Des. 2002;16:43–57. doi: 10.1023/a:1016307520660. [DOI] [PubMed] [Google Scholar]
- Jain AN. Scoring noncovalent protein–ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des. 1996;10:427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- Jain AN. Scoring functions for protein–ligand docking. Curr Protein Pept Sci. 2006;7:407–420. doi: 10.2174/138920306778559395. [DOI] [PubMed] [Google Scholar]
- Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- Jain T, Jayaram B. Computational protocol for predicting the binding affinities of zinc containing metalloprotein–ligand complexes. Proteins Struct Funct Genet. 2007;67:1167–1178. doi: 10.1002/prot.21332. [DOI] [PubMed] [Google Scholar]
- Jansen JM, Martin EJ. Target-biased scoring approaches and expert systems in structure-based virtual screening. Curr Opin Chem Biol. 2004;8:359–364. doi: 10.1016/j.cbpa.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Jenkins JL, Kao RYT, Shapiro R. Virtual screening to enrich hit lists from high-throughput screening: a case study on small-molecule inhibitors of angiogenin. Proteins Struct Funct Genet. 2003;50:81–93. doi: 10.1002/prot.10270. [DOI] [PubMed] [Google Scholar]
- Jones-Hertzog DK, Jorgensen WL. Binding affinities for sulfonamide inhibitors with human thrombin using Monte Carlo simulations with a linear response method. J Med Chem. 1997;40:1539–1549. doi: 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Joseph-McCarthy D, Thomas BE, IV, Belmarsh M, Moustakas D, Alvarez JC. Pharmacophore-based molecular docking to account for ligand flexibility. Proteins Struct Funct Genet. 2003;51:172–188. doi: 10.1002/prot.10266. [DOI] [PubMed] [Google Scholar]
- Kang X, Shafer RH, Kuntz ID. Calculation of ligand-nucleic acid binding free energies with the generalized-born model in DOCK. Biopolymers. 2004;73:192–204. doi: 10.1002/bip.10541. [DOI] [PubMed] [Google Scholar]
- Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins Struct Funct Genet. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- Khandelwal A, Lukacova V, Comez D, Kroll DM, Raha S, Balaz S. A combination of docking, QM/MM methods, and MD simulation for binding affinity estimation of metalloprotein ligands. J Med Chem. 2005;48:5437–5447. doi: 10.1021/jm049050v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening of drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knegtel RMA, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- Kolb P, Caflisch A. Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking. J Med Chem. 2006;49:7384–7392. doi: 10.1021/jm060838i. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M, McClellan LM, Sokol GS. Evaluation of docking performance: comparative data on docking algorithms. J Med Chem. 2004;47:558–565. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M, Sokol GS, McClellan LM. Evaluation of library ranking efficacy in virtual screening. J Comput Chem. 2005;26:11–22. doi: 10.1002/jcc.20141. [DOI] [PubMed] [Google Scholar]
- Korb O, Stutzle T, Exner TE. Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) Brussels; 2006. PLANTS: application of ant colony optimization to structure-based drug design; pp. 247–258. [Google Scholar]
- Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins Struct Funct Genet. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein–ligand docking. Proteins Struct Funct Genet. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. LigScore: a novel scoring function for predicting binding affinities. J Mol Graph Model. 2005;23:395–407. doi: 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Kroemer RT, Vulpetti A, McDonald JJ, Rohrer DC, Trosset JY, Giordanetto F, et al. Assessment of docking poses: interactions-based accuracy classification (IBAC) versus crystal structure deviations. J Chem Inf Comput Sci. 2004;44:871–881. doi: 10.1021/ci049970m. [DOI] [PubMed] [Google Scholar]
- Krumrine J, Raubacher F, Brooijmans N, Kuntz I. Principles and methods of docking and ligand design. Methods Biochem Anal. 2003;44:443–476. [PubMed] [Google Scholar]
- Kulkarni SS, Kulkarni VM. Structure based prediction of binding affinity of human immunodeficiency virus-1 protease inhibitors. J Chem Inf Comput Sci. 1999;39:1128–1140. doi: 10.1021/ci990019p. [DOI] [PubMed] [Google Scholar]
- Ladbury JE. Just add water! The effect of water on the specificity of protein–ligand binding sites and its potential application to drug design. Chem Biol. 1996;3:973–980. doi: 10.1016/s1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Davis PC. CLIX: a search algorithm for finding novel ligands capable of binding proteins of known three-dimensional structure. Proteins Struct Funct Genet. 1992;12:31–41. doi: 10.1002/prot.340120105. [DOI] [PubMed] [Google Scholar]
- Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994;235:345–356. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- Leclerc F, Karplus M. MCSS-based predictions of RNA binding sites. Theor Chem Acc. 1999;101:131–137. [Google Scholar]
- Li H, Li C, Gui C, Luo X, Chen K, Shen J, et al. GAsDock: a new approach for rapid flexible docking based on an improved multi-population genetic algorithm. Bioorg Med Chem Lett. 2004;14:4671–4676. doi: 10.1016/j.bmcl.2004.06.091. [DOI] [PubMed] [Google Scholar]
- Li Z, Lazaridis T. Water at biomolecular binding interfaces. Phys Chem Chem Phys. 2007;9:573–581. doi: 10.1039/b612449f. [DOI] [PubMed] [Google Scholar]
- Liu M, Wang S. MCDOCK: a Monte Carlo simulation approach to the molecular docking problem. J Comput Aided Mol Des. 1999;13:435–451. doi: 10.1023/a:1008005918983. [DOI] [PubMed] [Google Scholar]
- Majeux N, Scarsi M, Apostolakis J, Ehrhardt C, Caflisch A. Exhaustive docking of molecular fragments with electrostatic solvation. Proteins Struct Funct Genet. 1999;37:88–105. [PubMed] [Google Scholar]
- Makino S, Ewing TJA, Kuntz ID. DREAM++: flexible docking program for virtual combinatorial libraries. J Comput Aided Mol Des. 1999;13:513–532. doi: 10.1023/a:1008066310669. [DOI] [PubMed] [Google Scholar]
- Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Gaussian docking functions. Biopolymers. 2003;68:76–90. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- McGaughey GB, Sheridan RP, Bayly CI, Culberson JC, Kreatsoulas C, Lindsley S, et al. Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model. 2007;47:1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- McMartin C, Bohacek RS. QXP: powerful, rapid computer algorithms for structure-based drug design. J Comput Aided Mol Des. 1997;11:333–344. doi: 10.1023/a:1007907728892. [DOI] [PubMed] [Google Scholar]
- Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins Struct Funct Genet. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- Michel J, Verdonk ML, Essex JW. Protein–ligand binding affinity predictions by implicit solvent simulations: a tool for lead optimization. J Med Chem. 2006;49:7427–7439. doi: 10.1021/jm061021s. [DOI] [PubMed] [Google Scholar]
- Miller MD, Kearsley SK, Underwood DJ, Sheridan RP. FLOG: a system to select ‘quasi-flexible' ligands complementary to a receptor of known three-dimensional structure. J Comput Aided Mol Des. 1994;8:153–174. doi: 10.1007/BF00119865. [DOI] [PubMed] [Google Scholar]
- Mitchell JBO, Laskowski RA, Alex A, Forster MJ, Thornton JM. BLEEP—potential of mean force describing protein–ligand interactions: II. Calculation of binding energies and comparison with experimental data. J Comput Chem. 1999;20:1177–1185. [Google Scholar]
- Mizutani MY, Takamatsu Y, Ichinose T, Nakamura K, Itai A. Effective handling of induced-fit motion in flexible docking. Proteins Struct Funct Genet. 2006;63:878–891. doi: 10.1002/prot.20931. [DOI] [PubMed] [Google Scholar]
- Mizutani MY, Tomioka N, Itai A. Rational automatic search method for stable docking models of protein and ligand. J Mol Biol. 1994;243:310–326. doi: 10.1006/jmbi.1994.1656. [DOI] [PubMed] [Google Scholar]
- Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11:323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- Moitessier N, Henry C, Maigret B, Chapleur Y. Combining pharmacophore search, automated docking, and molecular dynamics simulations as a novel strategy for flexible docking. Proof of concept: docking of arginine–glycine–aspartic acid-like compounds into the αvβ3 binding site. J Med Chem. 2004;47:4178–4187. doi: 10.1021/jm0311386. [DOI] [PubMed] [Google Scholar]
- Moitessier N, Therrien E, Hanessian S. A method for induced-fit docking, scoring, and ranking of flexible ligands. Application to peptidic and pseudopeptidic β-secretase (BACE 1) inhibitors. J Med Chem. 2006a;49:5885–5894. doi: 10.1021/jm050138y. [DOI] [PubMed] [Google Scholar]
- Moitessier N, Westhof E, Hanessian S. Docking of aminoglycosides to hydrated and flexible RNA. J Med Chem. 2006b;49:1023–1033. doi: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]
- Morley SD, Afshar M. Validation of an empirical RNA–ligand scoring function for fast flexible docking using RiboDock®. J Comput Aided Mol Des. 2004;18:189–208. doi: 10.1023/b:jcam.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- Morreale A, Gil-Redondo R, Ortiz AR. A new implicit solvent model for protein–ligand docking. Proteins Struct Funct Genet. 2007;67:606–616. doi: 10.1002/prot.21269. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, et al. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- Mpamhanga CP, Chen B, McLay IM, Willett P. Knowledge-based interaction fingerprint scoring: A simple method for improving the effectiveness of fast scoring functions. J Chem Inf Model. 2006;46:686–698. doi: 10.1021/ci050420d. [DOI] [PubMed] [Google Scholar]
- Muegge I. PMF scoring revisited. J Med Chem. 2006;49:5895–5902. doi: 10.1021/jm050038s. [DOI] [PubMed] [Google Scholar]
- Muegge I, Martin YC. A general and fast scoring function for protein–ligand interactions: a simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- Muegge I, Oloff S. Advances in virtual screening. Drug Discov Today: Technologies. 2006;3:405–411. doi: 10.1016/j.ddtec.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CW, Baxter CA, Frenkel AD. The sensitivity of the results of molecular docking to induced fit effects: application to thrombin, thermolysin and neuraminidase. J Comput Aided Mol Des. 1999;13:547–562. doi: 10.1023/a:1008015827877. [DOI] [PubMed] [Google Scholar]
- Murray CW, Verdonk ML. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J Comput Aided Mol Des. 2002;16:741–753. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
- Naim M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, et al. Solvated interaction energy (SIE) for scoring protein–ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model. 2007;47:122–133. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]
- Najmanovich R, Kuttner J, Sobolev V, Edelman M. Side-chain flexibility in proteins upon ligand binding. Proteins Struct Funct Genet. 2000;39:261–268. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Nishibata Y, Itai A. Confirmation of usefulness of a structure construction program based on three-dimensional receptor structure for rational lead generation. J Med Chem. 1993;36:2921–2928. doi: 10.1021/jm00072a011. [DOI] [PubMed] [Google Scholar]
- Onodera K, Satou K, Hirota H. Evaluations of molecular docking programs for virtual screening. J Chem Inf Model. 2007;47:1609–1618. doi: 10.1021/ci7000378. [DOI] [PubMed] [Google Scholar]
- Oshiro CM, Kuntz ID, Dixon JS. Flexible ligand docking using a genetic algorithm. J Comput Aided Mol Des. 1995;9:113–130. doi: 10.1007/BF00124402. [DOI] [PubMed] [Google Scholar]
- Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in autodock. Proteins Struct Funct Genet. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci USA. 2003;100:12648–12653. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YP, Perola E, Xu R, Prendergast FG. EUDOC: a computer program for identification of drug interaction sites in macromolecules and drug leads from chemical databases. J Comput Chem. 2001;22:1750–1771. doi: 10.1002/jcc.1129. [DOI] [PubMed] [Google Scholar]
- Paul N, Rognan D. ConsDock: a new program for the consensus analysis of protein–ligand interactions. Proteins Struct Funct Genet. 2002;47:521–533. doi: 10.1002/prot.10119. [DOI] [PubMed] [Google Scholar]
- Pei J, Wang Q, Liu Z, Li Q, Yang K, Lai L. PSI-DOCK: towards highly efficient and accurate flexible ligand docking. Proteins Struct Funct Genet. 2006;62:934–946. doi: 10.1002/prot.20790. [DOI] [PubMed] [Google Scholar]
- Perez C, Ortiz AR. Evaluation of docking functions for protein–ligand docking. J Med Chem. 2001;44:3768–3785. doi: 10.1021/jm010141r. [DOI] [PubMed] [Google Scholar]
- Perola E, Walters WP, Charifson P. Comments on the article ‘on evaluating molecular-docking methods for pose prediction and enrichment factors'. J Chem Inf Model. 2007;47:251–253. doi: 10.1021/ci600460h. [DOI] [PubMed] [Google Scholar]
- Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins Struct Funct Genet. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- Pham TA, Jain AN. Parameter estimation for scoring protein–ligand interactions using negative training data. J Med Chem. 2006;49:5856–5868. doi: 10.1021/jm050040j. [DOI] [PubMed] [Google Scholar]
- Rarey M, Kramer B, Lengauer T. The particle concept: placing discrete water molecules during protein–ligand docking predictions. Proteins Struct Funct Genet. 1999;34:17–28. [PubMed] [Google Scholar]
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Rester U. Dock around the clock—current status of small molecule docking and scoring. QSAR Comb Sci. 2006;25:605–615. [Google Scholar]
- Rognan D, Lauemoller SL, Holm A, Buus S, Tschinke V. Predicting binding affinities of protein ligands from three-dimensional models: application to peptide binding to class I major histocompatibility proteins. J Med Chem. 1999;42:4650–4658. doi: 10.1021/jm9910775. [DOI] [PubMed] [Google Scholar]
- Ruvinsky AM. Role of binding entropy in the refinement of protein–ligand docking predictions: analysis based on the use of 11 scoring functions. J Comput Chem. 2007;28:1364–1372. doi: 10.1002/jcc.20580. [DOI] [PubMed] [Google Scholar]
- Schafferhans A, Klebe G. Docking ligands onto binding site representations derived from proteins built by homology modelling. J Mol Biol. 2001;307:407–427. doi: 10.1006/jmbi.2000.4453. [DOI] [PubMed] [Google Scholar]
- Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, et al. Quantification of cation–π interactions in protein–ligand complexes: crystal-structure analysis of factor Xa bound to a quaternary ammonium ion ligand. Angewandte Chemie—Int Ed. 2005;44:4400–4404. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- Schnecke V, Kuhn LA. Virtual screening with solvation and ligand-induced complementarity. Persp Drug Discov Des. 2000;20:171–190. [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Gasch T, Stahl M. Binding site characteristics in structure-based virtual screening: evaluation of current docking tools. J Mol Mod. 2003;9:47–57. doi: 10.1007/s00894-002-0112-y. [DOI] [PubMed] [Google Scholar]
- Schulz-Gasch T, Stahl M. Scoring functions for protein–ligand interactions: a critical perspective. Drug Discov Today: Technologies. 2004;1:231–239. doi: 10.1016/j.ddtec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Seifert MHJ. ProPose: steered virtual screening by simultaneous protein–ligand docking and ligand–ligand alignment. J Chem Inf Model. 2005;45:449–460. doi: 10.1021/ci0496393. [DOI] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Shoichet BK, McGovern SL, Wei B, Irwin JJ. Hits, leads and artifacts from virtual and high throughput screening. Molecular Informatics: Confronting Complexity May 13th–16th 2002. 2002.
- Sobolev V, Wade RC, Vriend G, Edelman M. Molecular docking using surface complementarity. Proteins Struct Funct Genet. 1996;25:120–129. doi: 10.1002/(SICI)1097-0134(199605)25:1<120::AID-PROT10>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Sotriffer CA, Dramburg I. ‘In situ cross-docking' to simultaneously address multiple targets. J Med Chem. 2005;48:3122–3125. doi: 10.1021/jm050075j. [DOI] [PubMed] [Google Scholar]
- Sousa SF, Fernandes PA, Ramos MJ. Protein–ligand docking: current status and future challenges. Proteins Struct Funct Genet. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- Sussman F, Villaverde MC, Martinez L. Modified solvent accessibility free energy prediction analysis of cyclic urea inhibitors binding to the HIV-1 protease. Protein Eng. 2002;15:707–711. doi: 10.1093/protein/15.9.707. [DOI] [PubMed] [Google Scholar]
- Tame JRH. Scoring functions: a view from the bench. J Comput Aided Mol Des. 1999;13:99–108. doi: 10.1023/a:1008068903544. [DOI] [PubMed] [Google Scholar]
- Tame JRH. Scoring functions—the first 100 years. J Comput Aided Mol Des. 2005;19:445–451. doi: 10.1007/s10822-005-8483-7. [DOI] [PubMed] [Google Scholar]
- Taylor JS, Burnett RM. DARWIN: a program for docking flexible molecules. Proteins Struct Funct Genet. 2000;41:173–191. [PubMed] [Google Scholar]
- Taylor RD, Jewsbury PJ, Essex JW. A review of protein-small molecule docking methods. J Comput Aided Mol Des. 2002;16:151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- Taylor RD, Jewsbury PJ, Essex JW. FDS: flexible ligand and receptor docking with a continuum solvent model and soft-core energy function. J Comput Chem. 2003;24:1637–1656. doi: 10.1002/jcc.10295. [DOI] [PubMed] [Google Scholar]
- Terp GE, Johansen BN, Christensen IT, Jørgensen FS. A new concept for multidimensional selection of ligand conformations (multiselect) and multidimensional scoring (multiscore) of protein–ligand binding affinities. J Med Chem. 2001;44:2333–2343. doi: 10.1021/jm001090l. [DOI] [PubMed] [Google Scholar]
- Thomsen R, Christensen MH. MolDock: A new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Tietze S, Apostolakis J. GlamDock: development and validation of a new docking tool on several thousand protein–ligand complexes. J Chem Inf Model. 2007;47:1657–1672. doi: 10.1021/ci7001236. [DOI] [PubMed] [Google Scholar]
- Tøndel K, Anderssen E, Drabløs F. Protein Alpha Shape (PAS) Dock: A new gaussian-based score function suitable for docking in homology modelled protein structures. J Comput Aided Mol Des. 2006;20:131–144. doi: 10.1007/s10822-006-9041-7. [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Flexible protein–ligand docking by global energy optimization in internal coordinates. Proteins Struct Funct Genet. 1997;29:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- Trabanino RJ, Hall SE, Vaidehi N, Floriano WB, Kam VWT, Goddard WA., III First principles predictions of the structure and function of G-protein-coupled receptors: validation for bovine rhodopsin. Biophys J. 2004;86:1904–1921. doi: 10.1016/S0006-3495(04)74256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosset JY, Scheraga HA. PRODOCK: software package for protein modeling and docking. J Comput Chem. 1999;20:412–427. [Google Scholar]
- Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the ‘early recognition' problem. J Chem Inf Model. 2007;47:488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- van Dijk ADJ, Bonvin AMJJ. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics. 2006a;22:2340–2347. doi: 10.1093/bioinformatics/btl395. [DOI] [PubMed] [Google Scholar]
- van Dijk M, van Dijk AD, Hsu V, Boelens R, Bonvin AM. Information-driven protein–DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006b;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velec HFG, Gohlke H, Klebe G. DrugScoreCSD-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- Verdonk ML, Berdini V, Hartshorn MJ, Mooij WTM, Murray CW, Taylor RD, et al. Virtual screening using protein–ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- Verdonk ML, Chessari G, Cole JC, Hartshorn MJ, Murray CW, Nissink JWM, et al. Modeling water molecules in protein–ligand docking using GOLD. J Med Chem. 2005;48:6504–6515. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins Struct Funct Genet. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Vieth M, Cummins DJ. DoMCoSAR: a novel approach for establishing the docking mode that is consistent with the structure—activity relationship. Application to HIV-1 protease inhibitors and VEGF receptor tyrosine kinase inhibitors. J Med Chem. 2000;43:3020–3032. doi: 10.1021/jm990609e. [DOI] [PubMed] [Google Scholar]
- Vieth M, Hirst JD, Dominy BN, Daigler H, Brooks CL., III Assessing search strategies for flexible docking. J Comput Chem. 1998a;19:1623–1631. [Google Scholar]
- Vieth M, Hirst JD, Kolinski A, Brooks CL., III Assessing energy functions for flexible docking. J Comput Chem. 1998b;19:1612–1622. [Google Scholar]
- Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- Wang R, Liu L, Lai L, Tang Y. SCORE: a new empirical method for estimating the binding affinity of a protein–ligand complex. J Mol Mod. 1998;4:379–394. [Google Scholar]
- Wang R, Lu Y, Fang X, Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein–ligand complexes. J Chem Inf Comput Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Welch W, Ruppert J, Jain AN. Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol. 1996;3:449–462. doi: 10.1016/s1074-5521(96)90093-9. [DOI] [PubMed] [Google Scholar]
- Westhead DR, Clark DE, Murray CW. A comparison of heuristic search algorithms for molecular docking. J Comput Aided Mol Des. 1997;11:209–228. doi: 10.1023/a:1007934310264. [DOI] [PubMed] [Google Scholar]
- Wolf A, Zimmermann M, Hofmann-Apitius M. Alternative to consensus scoring—a new approach toward the qualitative combination of docking algorithms. J Chem Inf Model. 2007;47:1036–1044. doi: 10.1021/ci6004965. [DOI] [PubMed] [Google Scholar]
- Wu G, Robertson DH, Brooks CL, III, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER—a CHARMm-based MD docking algorithm. J Comput Chem. 2003a;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- Wu G, Vieth M. SDOCKER: a method utilizing existing X-ray structures to improve docking accuracy. J Med Chem. 2004;47:3142–3148. doi: 10.1021/jm040015y. [DOI] [PubMed] [Google Scholar]
- Wu SY, McNae I, Kontopidis G, McClue SJ, McInnes C, Stewart KJ, et al. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: structural basis for ligand-induced disordering of the activation loop. Structure. 2003b;11:399–410. doi: 10.1016/s0969-2126(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Yamada M, Itai A. Development of an efficient automated docking method. Chem Pharm Bull. 1993;41:1200–1202. [Google Scholar]
- Yamazaki T, Nicholson LK, Wingfield P, Stahl SJ, Kaufman JD, Eyermann CJ, et al. NMR and X-ray evidence that the HIV protease catalytic aspartyl groups are protonated in the complex formed by the protease and a non-peptide cyclic urea-based inhibitor. J Am Chem Soc. 1994;116:10791–10792. [Google Scholar]
- Yang CY, Wang R, Wang S. M-score: a knowledge-based potential scoring function accounting for protein atom mobility. J Med Chem. 2006;49:5903–5911. doi: 10.1021/jm050043w. [DOI] [PubMed] [Google Scholar]
- Yang JM, Chen CC. GEMDOCK: a generic evolutionary method for molecular docking. Proteins Struct Funct Bioinf. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- Zavodszky MI, Kuhn LA. Side-chain flexibility in protein–ligand binding: the minimal rotation hypothesis. Protein Sci. 2005;14:1104–1114. doi: 10.1110/ps.041153605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentgraf M, Fokkens J, Sotriffer CA. Addressing protein flexibility and ligand selectivity by ‘in situ cross-docking'. ChemMedChem. 2006;1:1355–1359. doi: 10.1002/cmdc.200600073. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sanner MF. FLIPDock: docking flexible ligands into flexible receptors. Proteins Struct Funct Bioinf. 2007;68:726–737. doi: 10.1002/prot.21423. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Felts AK, Friesner RA, Levy RM. Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J Chem Inf Model. 2007;47:1599–1608. doi: 10.1021/ci7000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsoldos Z, Reid D, Simon A, Sadjad BS, Johnson AP. eHiTS: an innovative approach to the docking and scoring function problems. Curr Protein Pept Sci. 2006;7:421–435. doi: 10.2174/138920306778559412. [DOI] [PubMed] [Google Scholar]
- Zsoldos Z, Szabo I, Szabo Z, Johnson AP. Software tools for structure based rational drug design. J Mol Struct Theochem. 2003;666–667:659–665. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.