

Abstract
The widely accepted model of G protein-coupled receptor (GPCR) regulation describes a system where the agonist-activated receptors couple to G proteins to induce a cellular response, and are subsequently phosphorylated by a family of kinases called the G protein-coupled receptor kinases (GRKs). The GRK-phosphorylated receptor then acts as a substrate for the binding of a family of proteins called arrestins, which uncouple the receptor and G protein so desensitizing the agonist-induced response. Other kinases, principally the second messenger-dependent protein kinases, are also known to play a role in the desensitization of many GPCR responses. It is now clear that there are subtle and complex interactions between GRKs and second messenger-dependent protein kinases in the regulation of GPCR function. Functional selectivity describes the ability of agonists to stabilize different active conformations of the same GPCR. With regard to desensitization, distinct agonist-activated conformations of a GPCR could undergo different molecular mechanisms of desensitization. An example of this is the μ opioid receptor (MOPr), where the agonists morphine and [D-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO) induce desensitization of the MOPr by different mechanisms, largely protein kinase C (PKC)- or GRK-dependent, respectively. This can be best explained by supposing that these two agonists stabilize distinct conformations of the MOPr, which are nevertheless able to couple to the relevant G-proteins and produce similar responses, yet are sufficiently different to trigger different regulatory processes. There is evidence that other GPCRs also undergo agonist-selective desensitization, but the full therapeutic consequences of this phenomenon await further detailed study.
Keywords: GPCR, GRK, second messenger-dependent protein kinase, PKC, PKA, desensitization, functional selectivity, MOPr
GPCR desensitization: the classical model
The desensitization of a G protein-coupled receptor (GPCR) response can be described as the loss of response subsequent to prolonged or repeated administration of an agonist (Hausdorff et al., 1990). Actually the term ‘prolonged' can be somewhat misleading as experimentally this can represent time periods of as little as a few seconds or as long as several hours or even days. Desensitization can be homologous or heterologous in nature; homologous desensitization refers to the loss of response solely to agonists that act at a particular GPCR subtype, whereas heterologous desensitization refers to a more generalized effect involving the simultaneous loss of agonist responsiveness at multiple GPCR subtypes even in the absence of agonist occupation of the other receptors. Homologous desensitization is usually thought to involve adaptive changes at the level of the GPCR itself, whereas heterologous desensitization may also involve changes in signalling components downstream of the GPCR. Following desensitization, and provided that agonist stimulation is curtailed by removal of agonist or addition of an antagonist, GPCR responsiveness can in most cases be regained by a process called resensitization, although as with desensitization, the rapidity of this process varies between GPCR subtypes and can also depend upon the length of agonist pretreatment in the desensitization phase. Furthermore, in pharmacology, desensitization has a different meaning from downregulation, the latter referring to the proteolytic degradation of GPCRs, often in lysosomes. Thus, although downregulation of a GPCR invariably adds to the overall desensitization of a GPCR response, most GPCRs can undergo extensive desensitization (particularly, following acute agonist addition) without any downregulation being detectable. This review resulted from our presentation entitled ‘At the end of the day, receptors get tired too' at the ‘A Day in the Life of a GPCR' symposium at Life Sciences 2007. Therefore, we can with tongue in cheek suggest that desensitization represents the activated receptor becoming tired and taking a rest.
A major mechanism underlying desensitization is phosphorylation of the GPCR (Stadel et al., 1983; Krupnick and Benovic, 1998; Pitcher et al., 1998; Ferguson, 2001; Willets et al., 2003). Until the mid-1980s, GPCR phosphorylation by second messenger-dependent protein kinases such as protein kinase A (PKA) and protein kinase C (PKC) was regarded as the principal mechanism of GPCR desensitization (Benovic et al., 1985). However, the seminal observation that the β2-adrenoceptor (β2-AR) could be phosphorylated and desensitized in cells lacking functional PKA (Strasser et al., 1986), pointed to the existence of other kinases that could phosphorylate GPCRs. The identification of a novel protein kinase, not a second messenger-dependent protein kinase, with the ability to phosphorylate the agonist-occupied β2-AR, was a landmark in GPCR biology (Benovic et al., 1986). The kinase, originally called β-adrenoceptor kinase (β-ARK), was soon found to be just one of a family of kinases, later termed G protein-coupled receptor kinases (GRKs), with the original β-adrenoceptor kinase assuming the name GRK2. The GRKs have since been shown to play a central role in the agonist-induced phosphorylation and desensitization of many GPCR responses (Premont and Gainetdinov, 2007). However, it was found that GRK phosphorylation of GPCRs was by itself insufficient to produce extensive desensitization of the receptor response (Pitcher et al., 1992). Accordingly, another family of regulatory proteins was identified called arrestins (Lohse et al., 1990b), with the ability to bind with high affinity to the agonist-occupied, GRK-phosphorylated GPCR, uncoupling it from G-protein activation and thus inducing desensitization of the receptor-generated response.
These findings and others led to the development of a ‘classical' model for the agonist-induced desensitization of GPCRs (Krupnick and Benovic, 1998; Pitcher et al., 1998), considered to be generally applicable to most GPCRs (Figure 1a). In this model, the agonist-occupied receptor becomes a substrate for phosphorylation by members of the GRK family, with the GRKs invariably phosphorylating serine or threonine residues on the 3rd intracellular loop or COOH terminus of the receptor. The GRK-phosphorylated receptor exhibits a high affinity for arrestins, which bind to the GPCR and inhibit further coupling to the G proteins, hence desensitizing the response. The ability of GRKs to only phosphorylate agonist-occupied GPCRs neatly explains the phenomenon of homologous desensitization of the GPCR response. A recent study has also underlined the importance of agonist occupation of the GPCR, in addition to initiating GRK-mediated phosphorylation, for arrestin binding. Using Fluorescence Resonance Energy Transfer (FRET) to detect interactions between the β2-AR and arrestins, it was shown that upon agonist removal, arrestin dissociated rapidly from the β2-AR, even though the GPCR was still GRK phosphorylated (Krasel et al., 2005). There are even some GPCRs, such as the leukotriene B4 receptor (Jala et al., 2005), with which arrestins can associate in an agonist-dependent manner without the necessity for receptor phosphorylation.
Figure 1.
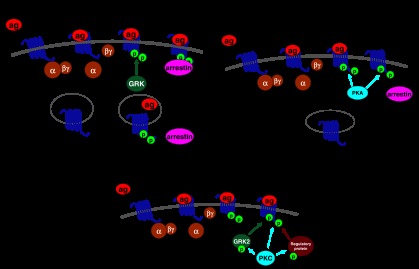
G protein-coupled receptor (GPCR) regulation by G protein-coupled receptor kinases (GRKs) and second messenger-dependent protein kinases. (a) The classical model of GPCR regulation by GRKs and arrestins. The GPCR is activated by agonist (1) leading to G protein coupling (2) and effector modulation. The agonist-occupied GPCR is subsequently phosphorylated by GRK (3), and arrestin binds to the phosphorylated GPCR, leading to receptor desensitization (4), internalization (5), dephosphorylation (6) and recycling (7) of the GPCR. Particularly with longer agonist treatments, internalized GPCR may also be targeted for downregulation. (b) GPCR regulation by second messenger-dependent protein kinases, in this example protein kinase A (PKA). Here, the agonist binds to the GPCR (1) leading to Gs activation (2) and increased levels of cyclic AMP, which activates PKA. The kinase is then able to phosphorylate both agonist occupied (3) and unoccupied (4) GPCRs. This phosphorylation causes desensitization by uncoupling the GPCR from G protein or in the case of the agonist unoccupied receptor by preventing GPCR coupling to G protein. Whether or not GPCR phosphorylation by second messenger-dependent protein kinases leads to arrestin binding or internalization depends upon the particular GPCR subtype in question. (c) Direct and indirect mechanisms of GPCR regulation by second messenger-dependent protein kinases. In this example, activated protein kinase C (PKC) can directly phosphorylate and desensitize the GPCR (1), or PKC can phosphorylate and activate GRK2, which consequently has an enhanced ability to phosphorylate the GPCR (2), or PKC can phosphorylate other, as yet unidentified, regulatory proteins, which then effect GPCR desensitization (3).
In addition to their role in desensitizing the GPCR response, arrestins are also central to GPCR trafficking (Figure 1a). The phosphorylated GPCR/arrestin complex is targeted to clathrin-coated pits, whereupon the GPCRs are internalized and can be either dephosphorylated and recycled to the plasma membrane, or targeted to lysosomes for downregulation (Krupnick and Benovic, 1998; Pitcher et al., 1998). Whether a desensitized and internalized GPCR recycles or is downregulated often depends upon the length of agonist treatment, with many GPCRs undergoing significant downregulation only following hours of agonist treatment. However, some GPCRs such as the δ-opioid receptor downregulate relatively rapidly following agonist addition (Tsao and von Zastrow, 2000), as these GPCRs interact with sorting proteins such as GPCR-associated sorting protein that specifically target the receptors to lysosomes for downregulation (Whistler et al., 2002).
Recent observations have added complexity to the classical model of GPCR desensitization. First, it is clear that, apart from mediating desensitization, GRKs and arrestins are also able to act as signal initiators by acting as multiprotein scaffolds, leading for example to arrestin-dependent activation of mitogen-activated protein kinases (DeWire et al., 2007; Ribas et al., 2007). Interestingly, the phenomenon of signal initiation by an otherwise desensitizing modification can also be observed with second messenger-dependent protein kinases; PKA-mediated phosphorylation of the β2-AR can, apart from uncoupling the GPCR from Gs protein and adenylyl cyclase activation, switch coupling of the β2-AR to Gi proteins (Daaka et al., 1997). Second, in some cases, GRKs are able to mediate phosphorylation-independent as well as arrestin-independent desensitization. This is seen for GRK2 and GRK3 particularly with Gq-coupled receptors, where the kinase binds to the GPCR and also to Gq via the N-terminal regulator of G protein signalling-like region of the GRK, so preventing coupling between GPCR and G protein (Ferguson, 2007). For some GPCRs, such as the group I mGluRs (Dhami et al., 2004) and parathyroid hormone receptor (Dicker et al., 1999), this type of phosphorylation-independent desensitization by GRK2/3 probably represents an important regulatory mechanism in vivo (Ferguson, 2007). Third, even the idea of GPCR internalization being necessary for dephosphorylation and resensitization of the receptor needs reassessment in light of recent findings that the β2-AR (Tran et al., 2007), and the TRH receptor (Jones and Hinkle, 2005) can undergo dephosphorylation at the cell surface, that is, in the absence of internalization. Finally, the phenomenon of dimerization adds complexity to the field of desensitization. Thus, although agonist occupancy is normally required for GRK phosphorylation of a GPCR, a recent study shows that a novel Ca2+ signal generated by a D1–D2 dopamine heterodimer can be desensitized in a GRK-dependent fashion by pretreatment with agonists able to activate only one or the other receptor in the heterodimer (So et al., 2007). The classical view of GPCR regulation is thus being regularly updated, and it is evident that there is much GPCR subtype-dependent variation in the mechanisms involved. Another important variable is the cell type in which the GPCR is expressed. A recent study of the agonist-induced phosphorylation of the M3 muscarinic receptor, probably mediated by a combination of GRKs and casein kinase 2, shows that when the receptor is heterologously expressed in Chinese Hamster Ovary cells, the receptor residues phosphorylated following methacholine treatment are not the same as those phosphorylated in the receptor endogenously expressed in cerebellar granule neurons (Torrecilla et al., 2007).
Role of second messenger-dependent protein kinases
As noted above, second messenger-dependent protein kinases such as PKA and PKC were originally considered to be the principal mediators of GPCR phosphorylation and desensitization (Benovic et al., 1985). Following the discovery of the role of GRKs and arrestins in GPCR desensitization (Benovic et al., 1986; Lohse et al., 1990b), second messenger-dependent protein kinases seemed at times to be relegated to a secondary role. However, their importance in GPCR regulation (Figures 1b and c) has been reassessed in the light of more recent studies. For example, it is now clear that the desensitization of some GPCRs, such as the P2Y1 receptor (Hardy et al., 2005) and the D3 dopamine receptor (Cho et al., 2007), are mediated almost exclusively by feedback phosphorylation of the GPCR by second messenger-dependent protein kinases, with GRKs not playing a significant role. Indeed, the Gi/o coupled GPCR, mGluR4 is neither desensitized nor internalized following agonist activation of the receptor, but does desensitize and internalize when PKC is activated (Mathiesen and Ramirez, 2006); we have observed the same phenomenon with regard to desensitization of another metabotropic glutamate receptor, mGluR2 (S Lennon, PJ Roberts and E Kelly, unpublished observations). How then does second messenger-dependent phosphorylation of the GPCR lead to desensitization? There is little firm evidence to indicate that GPCRs phosphorylated by PKC or PKA are good substrates for the binding of arrestins (Pitcher et al., 1992). However, receptor phosphorylation by PKC may underlie the association of arrestins with some GPCRs such as the D2 dopamine receptor (Namkung and Sibley, 2004), the extracellular Ca2+-sensing receptor (Lorenz et al., 2007) and the δ-opioid receptor (Xiang et al., 2001). In contrast, for the D3 dopamine receptor, PKC phosphorylation of the receptor leads to GRK- and arrestin-independent desensitization and internalization, in a process requiring the interaction of the D3 receptor with the actin-binding protein filamin A (Cho et al., 2007). Otherwise, it is unclear exactly how GPCR phosphorylation by second messenger-dependent protein kinases uncouples the receptor from its G protein, but phosphorylation of the GPCR in a G protein-coupling region may sterically inhibit interaction with the G protein (Benovic et al., 1985). These potential mechanisms of phosphorylation-dependent desensitization warrant further study. One further important difference between second messenger-dependent protein kinase- and GRK-mediated phosphorylation of GPCRs is that activated PKA and PKC are in some instances able to phosphorylate agonist-unoccupied GPCRs, indicating that this type of phosphorylation could underlie some forms of heterologous desensitization (Clark et al., 1988). However, the ability of some Gs- or Gq-coupled GPCRs to be regulated by feedback phosphorylation following PKA or PKC activation in the cellular vicinity of the activated GPCR, suggests that second messenger-dependent phosphorylation is also a major mechanism of homologous desensitization.
In some cases, both second messenger-dependent protein kinases and GRKs are able to phosphorylate and desensitize the same GPCR (Figure 1c) (for example, Hausdorff et al., 1989; Castro et al., 2002; Ally et al., 2003). Such a combination of phosphorylation events by different kinases can lead to additive effects on desensitization (Lohse et al., 1990a; Pitcher et al., 1992), although other interesting interactions are possible, such as the ability of the arrestin interaction with GRK-phosphorylated Complement C5a receptors to inhibit PKC phosphorylation of the GPCR (Pollok-Kopp et al., 2007). The identity and relative roles of the protein kinases involved in the regulation of a particular GPCR has often been difficult to confirm using 32P cell-labelling approaches, but the advent of phosphoreceptor-specific antibodies has allowed significant advances to be made in this area (Pollok-Kopp et al., 2003; Schulz et al., 2004; Tran et al., 2004). The β2-AR for example is known to be phosphorylated by PKA on serine residues in the 3rd intracellular loop, and by GRKs on serine residues in the proximal COOH terminus of the GPCR (Yuan et al., 1994; Seibold et al., 2000). For the β2-AR, these phosphorylation events appear to occur independently of each other (Vaughan et al., 2006), the main difference being the concentration of agonist required to promote phosphorylation; in the case of PKA-mediated phosphorylation and desensitization, low nanomolar concentrations of agonist are sufficient to phosphorylate and desensitize the β2-AR, due to the amplification of the response through cyclic AMP generation and PKA activation. In the case of GRK-mediated phosphorylation, much higher concentrations of agonist are required (Tran et al., 2004), as GRK phosphorylation depends upon agonist occupancy of the GPCR. A similar difference in agonist dependence has been noted for PKC- versus GRK-mediated phosphorylation of the CCR5 chemokine receptor (Pollok-Kopp et al., 2003). As a further consequence, it has been suggested that GRK phosphorylation may represent an important form of β2-AR desensitization at, for example, noradrenergic nerve endings, where the concentration of noradrenaline and hence postsynaptic adrenoceptor occupancy is likely to be high especially during periods of increased neuronal activity. In contrast, PKA phosphorylation of β2-AR may predominate in other tissues such as peripheral blood vessels, where occupancy of β2-AR by circulating adrenaline may be lower in comparison (Arriza et al., 1992).
A further action of second messenger-dependent protein kinases concerns the modulation of GRK function at GPCRs (Figure 1c). For example, it was shown a number of years ago that PKC could phosphorylate and activate GRK2 (Chuang et al., 1995; Krasel et al., 2001), with the PKC phosphorylation enhancing the ability of GRK2 to target the plasma membrane and hence phosphorylate agonist-activated GPCRs. In addition, PKA phosphorylation of GRK2 is known to enhance the ability of the kinase to phosphorylate and desensitize the β2-AR (Cong et al., 2001; Li et al., 2006). In our own studies, we have found that both PKC and PKA regulate GRK2 interaction with group I mGluRs, with PKC activation increasing agonist-induced GRK2 and arrestin association with mGluR1a (Mundell et al., 2004a) and PKA activation inhibiting GRK2 and arrestin association with mGluR1a (Mundell et al., 2004b). Whether or not these effects of PKC and PKA are due to phosphorylation of mGluR1a, which then differentially regulates GRK2 association with the receptor, remains to be determined. A final consideration is that second messenger-dependent protein kinases could phosphorylate other proteins, which subsequently influence GPCR desensitization, such as regulator of G protein signalling proteins (Cunningham et al., 2001) or the Raf kinase inhibitor protein, which upon PKC phosphorylation becomes able to inhibit GRK2 function and hence β-AR desensitization (Lorenz et al., 2003).
Agonist functional selectivity and GPCR desensitization
Agonist activation of a GPCR-mediated response has generally been viewed in ‘linear' terms, that is, the various agonists that activate a particular GPCR do so by stabilizing the same activated conformation of the receptor, which then couples to the same G protein or set of G proteins (or even non-G protein effectors) and hence activate or inhibit the same set of intracellular responses (Figure 2a). However, data obtained more recently show that different agonists that activate the same GPCR are in some cases capable of producing different response profiles through activation of different types of G protein (Figure 2b). This phenomenon has been described by various terms, including ‘collateral efficacy', ‘biased agonism' and ‘stimulus trafficking'; however, recently, the name ‘functional selectivity' was adopted in an authoritative review (Urban et al., 2007) and has therefore been used in the present review. Functional selectivity is thought to be due to the stabilization of different conformations of the GPCR by the agonists, leading to the activation of different sets of responses (Perez and Karnik, 2005; Urban et al., 2007).
Figure 2.

Functional selectivity of agonist action at G protein-coupled receptors (GPCRs). (a) The traditional ‘linear' model of agonist action at a GPCR. In this case, the red and yellow agonists, as well as any other agonist at this GPCR, produce the same set of cellular responses as they each stabilize the same active conformation of the receptor, which couples to the same set of G proteins. (b) Functional selectivity: the red and yellow agonists each stabilizes a different active conformation of the GPCR, which couples to a different array of G proteins. These two agonists therefore produce different cellular response profiles in this tissue. It is interesting to note, however, that functional selectivity would be missed if only the response to the G protein illustrated in brown was measured in a screen for agonist activity.
An extension of this concept is that different agonists could also lead to different mechanisms of desensitization, as each agonist-induced GPCR conformation could possess different affinities for regulatory proteins such as the GRKs or arrestins. It has long been known, for example, that whereas many MOPr agonists such as [D-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO) and etorphine are able to induce arrestin interaction with the MOPr and lead to extensive MOPr internalization, other agonists such as morphine are not (Keith et al., 1996; Zhang et al., 1998). It was thought that morphine behaved in this fashion because in many tissues and cell systems, it is a partial agonist at the MOPr (Zhang et al., 1996; Borgland et al., 2003; Bailey et al., 2004; Connor et al., 2004), and so it was assumed that its low efficacy at MOPr rendered it inefficient in promoting GRK phosphorylation of the receptor and hence produced little arrestin association. This view was also supported by studies showing that in the presence of overexpressed GRK2, morphine is able to induce both translocation of arrestins to the plasma membrane and MOPr internalization (Bohn et al., 2004). However, recent evidence suggests a more complex picture; the agonist herkinorin, a non-nitrogenous compound related to the psychoactive plant extract salvinorin A, is reported to be a full agonist at MOPr, yet it is unable to promote the translocation of arrestins or induce MOPr internalization, even in the presence of overexpressed GRK2 (Groer et al., 2007; Xu et al., 2007). Our recent studies (Johnson et al., 2006) have provided evidence that morphine and DAMGO induce desensitization of MOPr by different mechanisms. When expressed heterologously in Human Embryonic Kidney 293 cells along with a G protein-coupled inwardly rectifying K+ channel, both morphine and DAMGO activate a K+ current, which in each case rapidly desensitizes (Johnson et al., 2006). However, the mechanisms underlying this desensitization are not the same for the two agonists; whereas morphine-induced desensitization can be reduced by inclusion of inhibitors of PKC, DAMGO-induced desensitization is not but is rather inhibited by co-transfection with a dominant negative mutant form of GRK2, which is able to reduce the functional activity of endogenous GRK2 in the cells. In addition, we found that MOPr is phosphorylated under basal conditions in the cells, almost certainly by PKC. This would explain how PKC might be involved in morphine-induced MOPr regulation, because there is little evidence to indicate that the Gi/Go-coupled MOPr can couple directly to PKC activation.
We have also demonstrated that morphine and DAMGO induce different mechanisms of MOPr desensitization in mature neurons of the rat locus coeruleus (LC; Bailey et al., 2004). In these experiments, where the functional response measured was again MOPr activation of a G protein-coupled inwardly rectifying K+ current (Bailey et al., 2003), DAMGO induced rapid PKC-independent desensitization of the MOPr response, whereas morphine could only do so when PKC activity in the neurons was elevated, either by activating Gq-coupled muscarinic acetylcholine receptors on the neurons or by addition of a phorbol ester to directly activate PKC (Bailey et al., 2004, 2006). How to rationalize these findings? If we assume that morphine and DAMGO stabilize different conformations of the MOPr, which can both couple to G proteins and activate the G protein-coupled inwardly rectifying K+ channel, yet have different affinities for PKC versus GRK2, then this could explain these data. This is shown diagrammatically in Figure 3, where the action of these agonists is depicted in two ways; a sequential model similar to that devised for agonist binding to the β2-AR (Kobilka, 2007) or a non-sequential model. Of course, there may be any number of agonist-induced MOPr conformations, but the simplest assumption based on the available data is that there are two, one stabilized by morphine that is susceptible mainly to PKC regulation, and one stabilized by DAMGO and similar agonists such as Met-enkephalin and etorphine that is susceptible mainly to GRK/arrestin regulation. It is possible that herkinorin, the full MOPr agonist, which does not recruit arrestin to the receptor, stabilizes the same conformational state as morphine, or that it stabilizes yet another distinct active conformation of the MOPr. Other opioid agonists may have varying abilities to stabilize active conformations of MOPr (Pineyro and Archer-Lahlou, 2007); for example, we have found evidence that under certain conditions, Met-Enkephalin-induced desensitization in LC neurons can display a significant PKC component, whereas DAMGO-induced desensitization seems to be independent of PKC (Bailey et al., 2004). However, it is unlikely that morphine-induced MOPr desensitization is entirely PKC dependent, and Met-Enkephalin-induced MOPr desensitization is entirely GRK dependent. This is depicted schematically in Figure 4. Thus, in neurons with high levels of GRK activity, or in experimental cell systems where GRKs have been overexpressed, GRK-mediated desensitization may now play a significant role in morphine-mediated MOPr desensitization; conversely in cells with very low GRK levels, PKC may play a significant role in Met-Enkephalin- and DAMGO-induced MOPr desensitization. Indeed, the mechanism of morphine-induced desensitization could be influenced not only by the relative levels of PKC/GRK activity in a particular cell, but also by the subcellular localizations of these enzymes. Evidence suggesting that tissue-dependent variation in MOPr regulatory behaviour does exist, as morphine induces arrestin-dependent MOPr internalization in the dendrites but not the somata of some neuronal types but not others (Haberstock-Debic et al., 2005).
Figure 3.

Models of functional selectivity in the agonist-induced desensitization of μ-opioid receptor (MOPr). (a) Depicts the sequential activation of the receptor, with morphine being able to induce only the MOPr* state of the receptor, whereas DAMGO is capable of inducing MOPr* and then MOPr**. The MOPr* conformation undergoes protein kinase C (PKC)-mediated desensitization, whereas the MOPr** conformation undergoes G protein-coupled receptor kinase (GRK)/arrestin-mediated desensitization. In (b), morphine and DAMGO directly stabilize different active conformations of MOPr, MOPr* and MOPr**, respectively. MOPr* and MOPr** both couple to G protein-coupled inwardly rectifying K+ current, but undergo different mechanisms of desensitization. It is also possible that MOPr* and MOPr** regulate other signalling pathways differentially, such as arrestin-dependent MAP kinase activity.
Figure 4.

Diagrammatic representation of the possible effects of changes in the cellular concentrations of regulatory proteins on the mechanism of μ-opioid receptor (MOPr) desensitization. In (a), morphine desensitization of MOPr is mediated principally by protein kinase C (PKC), whereas desensitization of MOPr by agonists such as Met-Enkephalin and DAMGO is mediated predominantly by G protein-coupled receptor kinases (GRKs). (b) In a neuron with high levels of GRKs or in an experimental cell system where GRKs have been overexpressed, GRK-mediated desensitization may now play a significant role in morphine-mediated MOPr desensitization; however, Met-Enkephalin- and DAMGO-mediated desensitization will still involve predominantly GRKs.
Behavioural studies in the intact organism also suggest differences in the mechanism of opioid agonist-induced tolerance. In mice, deletion of the GRK3 gene suppresses tolerance to analgesia induced by fentanyl, but has much less effect on morphine tolerance (Terman et al., 2004). In addition, morphine tolerance in mice can be reduced by deletion of the PKCγ gene (Zeitz et al., 2001), or by intrathecal injection of antisense oligonucleotides for PKCα (Hua et al., 2002), and also by i.c.v. injection of highly selective inhibitors of PKCα, PKCγ and PKCɛ (Smith et al., 2007).
Do these findings have any therapeutic significance? It is interesting that morphine, although it has lower efficacy at the MOPr than a number of other opioid agonists, is known to induce significant tolerance in both cellular models of tolerance (Koch et al., 2005) and behavioural studies in rodents (Smith et al., 1999). However, given the fact that in most systems morphine is a partial agonist (Borgland et al., 2003; Connor et al., 2004), these findings may seem surprising on the basis of classical receptor theory because for many GPCRs, there is a good correlation between agonist efficacy and the ability to induce receptor desensitization (Clark et al., 1999). However, a lack of correlation between agonist efficacy and the ability to induce desensitization has been observed for the CB1 cannabinoid receptor (Luk et al., 2004). If the molecular mechanism of desensitization of MOPr is agonist dependent, then, the kinetics and extent of desensitization and indeed, resensitization and downregulation, may well also be agonist dependent and therefore different. This scenario is supported by a study where the morphine-induced phosphorylation and subsequent dephosphorylation and resensitization of MOPr were shown to proceed much more slowly than those due to agonists such as DAMGO (Schulz et al., 2004). This could account for the profound tolerance observed with morphine, as in experimental cell systems the morphine-desensitized MOPr dephosphorylates very slowly in comparison to MOPr desensitized by other agonists such as DAMGO (Schulz et al., 2004). However, at present, we should exercise caution in assuming that MOPr desensitization accounts completely for tolerance in vivo, as other mechanisms including altered activity in neuronal pathways or changes in gene expression could also be involved (Williams et al., 2001). To pursue these important questions, the molecular analysis of MOPr desensitization in neurons and intact brain in response to a range of agonists of different structure and efficacy is clearly required.
Other evidence regarding functional selectivity of desensitization of GPCRs comes from studies of the β2-AR. In this case, distinct conformational changes in the receptor have been detected by fluorescence spectroscopy studies and by intramolecular Fluorescence Resonance Energy Transfer techniques (reviewed in Kobilka, 2007). Most interestingly with regard to desensitization, agonists have been shown to induce different patterns of phosphorylation in the COOH terminus of the β2-AR (Trester-Zedlitz et al., 2005), while most recently, again using intramolecular Fluorescence Resonance Energy Transfer, different agonist-dependent changes in the conformation of the COOH terminus of the β2-AR have been detected (Granier et al., 2007). These findings may have therapeutic significance because in experimental cell systems, the β2-AR agonist salmeterol, which is used in the treatment of asthma, does not trigger arrestin translocation nor induce significant β2-AR internalization. However, salmeterol can induce GRK phosphorylation of the β2-AR, although more slowly than full agonists such as adrenaline (Moore et al., 2007). Thus, salmeterol induces an active conformational state of the β2-AR that can be GRK phosphorylated yet is unable to interact with arrestins to a significant extent. Perhaps this is because, as discussed above, in most cases the high-affinity binding of arrestins to a GPCR requires a combination of GRK phosphorylation of the GPCR plus an agonist-induced change in GPCR conformation (Krasel et al., 2005). The likely clinical consequence of salmeterol's interaction with the β2-AR is that it is able to provide effective bronchodilation for prolonged periods in asthma because the β2-AR is not significantly desensitized or downregulated by this agonist.
Other GPCRs are also subject to functional selectivity in relation to desensitization. The 5-HT2C receptor couples to both phospholipase C and phospholipase A2-mediated arachidonic acid release. Using a range of different agonists, it was shown that not only is there functional selectivity in the ability of the agonists to activate these cellular responses (Berg et al., 2001), but also in the ability of the agonists to induce desensitization of the two responses (Stout et al., 2002). In another example, two agonists at the CC chemokine receptor 7, CCL19 and CCL21, are each able to activate the cellular response in human T cell lymphoma cells, Ca2+ mobilization (Kohout et al., 2004). However, whereas CCL19 induces robust phosphorylation and arrestin-dependent desensitization of the receptor response, CCL21 is unable to do so (Kohout et al., 2004). This example is particularly important because both the agonists are endogenous ligands, indicating that the phenomenon of functional selectivity in desensitization is not simply due to non-physiological receptor conformations induced by synthetic ligands. Finally, in yet another example of functional selectivity in desensitization, the platelet-activating factor receptor undergoes phosphorylation, desensitization and internalisation in response to both the endogenous agonist platelet-activating factor and the inverse agonist WEB2086 (Dupre et al., 2007). This is unexpected as the regulatory consequences of inverse agonist binding to GPCRs are often the opposite of that seen with conventional agonists (Pula et al., 2004). Most interestingly, although both GRK and PKC contribute to platelet-activating factor-mediated desensitization, only PKC mediates WEB2086 desensitization, yet it is different PKC isoforms that mediate the platelet-activating factor versus WEB2086 desensitization (Dupre et al., 2007). This highlights the subtle differences in mechanism that functional selectivity is capable of creating, and which may be missed in many widely used assays of receptor function. An interesting corollary is that GPCRs might also undergo phosphorylation by different GRK isoforms in a ligand-dependent manner.
Conclusion
The desensitization of GPCRs involves a number of different mechanisms, the exact pattern of which is GPCR subtype dependent and may also be cell context dependent. A major mechanism of desensitization involves GPCR phosphorylation by GRKs, second messenger-dependent protein kinases, or some combination of these. However, with the advent of functional selectivity, it is likely that a single subtype of GPCR can undergo agonist-selective desensitization, involving different active states of the GPCR. It also seems that in certain cases, GPCR antagonists and inverse agonists are capable of inducing conformations of the receptor that are inactive or which reduce activity in terms of coupling, yet are still capable of undergoing phosphorylation and desensitization. We predict that functional selectivity in desensitization will not be confined to the relatively small number of GPCR examples studied to date, but that many GPCRs will be susceptible to this phenomenon, and that this may have important therapeutic consequences. With the advent of advanced molecular techniques such as high-affinity phosphoreceptor antibodies, mass spectrometry and Fluorescence Resonance Energy Transfer, it should not be too long before we can start to glimpse the full extent and significance of ligand-dependent desensitization.
Acknowledgments
The work on MOPr desensitization in the authors' laboratory was funded by the Wellcome Trust, the Medical Research Council and the National Institute on Drug Abuse.
Glossary
- β2-AR
β2-adrenoceptor
- DAMGO
[D-Ala2,N-MePhe4,Gly-ol5]enkephalin
- FRET
Fluorescence Resonance Energy Transfer
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- MOPr
μ-opioid receptor
- PKA
protein kinase A
- PKC
protein kinase C
Footnotes
Conflict of interest
The authors state no conflict of interest.
References
- Ally RA, Ives KL, Traube E, Eltounsi I, Chen PW, Cahill PJ, et al. Agonist- and protein kinase C-induced phosphorylation have similar functional consequences for gastrin-releasing peptide receptor signaling via Gq. Mol Pharmacol. 2003;64:890–904. doi: 10.1124/mol.64.4.890. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Dawson TM, Simerly RB, Martin LJ, Caron MG, Snyder SH, et al. The G-protein-coupled receptor kinases beta ARK1 and beta ARK2 are widely distributed at synapses in rat brain. J Neurosci. 1992;12:4045–4055. doi: 10.1523/JNEUROSCI.12-10-04045.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G. Mu-opioid receptor desensitisation in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci. 2003;23:10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitisation of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. doi: 10.1124/mol.104.004747. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G. How important is protein kinase C in mu-opioid receptor desensitisation and morphine tolerance. Trends Pharmacol Sci. 2006;27:558–565. doi: 10.1016/j.tips.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Pike LJ, Cerione RJ, Staniszewski C, Yoshimasa T, Codina J, et al. Phosphorylation of the mammalian beta-adrenergic receptor by cyclic AMP-dependent protein kinase. Regulation of the rate of receptor phosphorylation and dephosphorylation by agonist occupancy and effects on coupling of the receptor to the stimulatory guanine nucleotide regulatory protein. J Biol Chem. 1985;260:7094–7101. [PubMed] [Google Scholar]
- Benovic JL, Strasser RH, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc Natl Acad Sci USA. 1986;83:2797–2801. doi: 10.1073/pnas.83.9.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Cropper JD, Niswender CM, Sanders-Bush E, Emeson RB, Clarke WP. RNA-editing of the 5-HT(2C) receptor alters agonist-receptor-effector coupling specificity. Br J Pharmacol. 2001;134:386–392. doi: 10.1038/sj.bjp.0704255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitisation, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- Castro M, Dicker F, Vilardara JP, Krasel C, Bernhardt M, Lohse MJ. Dual regulation of the parathyroid hormone (PTH)/PTH-related peptide receptor signaling by protein kinase C and beta-arrestins. Endocrinology. 2002;143:3854–3865. doi: 10.1210/en.2002-220232. [DOI] [PubMed] [Google Scholar]
- Cho EY, Cho DI, Park JH, Kurose H, Caron MG, Kim KM. Roles of protein kinase C and actin binding protein 280 in the regulation of intracellular trafficking of dopamine D3 receptor. Mol Endocrinol. 2007;21:2242–2254. doi: 10.1210/me.2007-0202. [DOI] [PubMed] [Google Scholar]
- Chuang TT, LeVine H, De Blasi A. Phosphorylation and activation of beta-adrenergic receptor kinase by protein kinase C. J Biol Chem. 1995;270:18660–18665. doi: 10.1074/jbc.270.31.18660. [DOI] [PubMed] [Google Scholar]
- Clark RB, Knoll BJ, Barber R. Partial agonists and G protein-coupled receptor desensitisation. Trends Pharmacol Sci. 1999;20:279–286. doi: 10.1016/s0165-6147(99)01351-6. [DOI] [PubMed] [Google Scholar]
- Clark RB, Kunkel MW, Friedman J, Goka TJ, Johnson JA. Activation of cAMP-dependent protein kinase is required for heterologous desensitization of adenylyl cyclase in S49 wild-type lymphoma cells. Proc Natl Acad Sci USA. 1988;85:1442–1446. doi: 10.1073/pnas.85.5.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong M, Perry SJ, Lin FT, Fraser ID, Hu LA, Chen W, et al. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J Biol Chem. 2001;276:15192–15199. doi: 10.1074/jbc.M009130200. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. Mu-opioid receptor desensitisation: is morphine different. Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham ML, Waldo GL, Hollinger S, Hepler JR, Harden TK. Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Galpha 11 signaling. J Biol Chem. 2001;276:5438–5444. doi: 10.1074/jbc.M007699200. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta 2-adrenergic receptor to different G-proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signalling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Dhami GK, Dale LB, Anborgh PH, O'Connor-Halligan KE, Sterne-Marr R, Ferguson SS. G Protein-coupled receptor kinase 2 regulator of G protein signaling homology domain binds to both metabotropic glutamate receptor 1a and Galphaq to attenuate signaling. J Biol Chem. 2004;279:16614–16620. doi: 10.1074/jbc.M314090200. [DOI] [PubMed] [Google Scholar]
- Dicker F, Quitterer U, Winstel R, Honold K, Lohse MJ. Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc Natl Acad Sci USA. 1999;96:5476–5481. doi: 10.1073/pnas.96.10.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre DJ, Thompson C, Chen Z, Rollin S, Larrivee J-F, Le Gouill C, et al. Inverse agonist-induced signalling and down-regulation of the platelet-activating factor receptor. Cell Signal. 2007;19:2068–2079. doi: 10.1016/j.cellsig.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signalling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Ferguson SS. Phosphorylation-independent attenuation of GPCR signalling. Trends Pharmacol Sci. 2007;28:173–179. doi: 10.1016/j.tips.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Granier S, Kim S, Shafer AM, Ratnala VRP, Fung JJ, Zare RN, et al. Structure and conformational changes in the C-terminus domain of the β2-adrenoceptor. J Biol Chem. 2007;282:13895–13905. doi: 10.1074/jbc.M611904200. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, et al. An opioid agonist that does not induce micro-opioid receptor—arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. Morphine promotes rapid, arrestin-dependent endocytosis of mu-opioid receptors in striatal neurons. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy AR, Conley PB, Luo J, Benovic JL, Poole AW, Mundell SJ. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood. 2005;105:3552–3560. doi: 10.1182/blood-2004-07-2893. [DOI] [PubMed] [Google Scholar]
- Hausdorff WP, Bouvier M, O'Dowd BF, Irons GP, Caron MG, Lefkowitz RJ. Phosphorylation sites on two domains of the beta 2-adrenergic receptor are involved in distinct pathways of receptor desensitisation. J Biol Chem. 1989;264:12657–12665. [PubMed] [Google Scholar]
- Hausdorff WP, Caron MG, Lefkowitz RJ. Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 1990;4:2881–2889. [PubMed] [Google Scholar]
- Hua XY, Moore A, Malkmus S, Murray SF, Dean N, Yaksh TL, et al. Inhibition of spinal protein kinase Calpha expression by an antisense oligonucleotide attenuates morphine infusion-induced tolerance. Neuroscience. 2002;113:99–107. doi: 10.1016/s0306-4522(02)00157-4. [DOI] [PubMed] [Google Scholar]
- Jala VR, Shao WH, Haribabu B. Phosphorylation-independent beta-arrestin translocation and internalization of leukotriene B4 receptors. J Biol Chem. 2005;280:4880–4887. doi: 10.1074/jbc.M409821200. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, et al. Agonist-selective mechanisms of mu-opioid receptor desensitisation in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Jones BW, Hinkle PM. β-Arrestin mediates desensitization and internalization but does not affect dephosphorylation of the thyrotropin-releasing hormone receptor. J Biol Chem. 2005;280:38346–38354. doi: 10.1074/jbc.M502918200. [DOI] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, et al. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitisation, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- Krasel C, Bunemann M, Lorenz K, Lohse MJ. Beta-arrestin binding to the beta2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J Biol Chem. 2005;280:9528–9535. doi: 10.1074/jbc.M413078200. [DOI] [PubMed] [Google Scholar]
- Krasel C, Dammeier S, Winstel R, Brockmann J, Mischack H, Lohse MJ. Phosphorylation of GRK2 by protein kinase C abolishes its inhibition by calmodulin. J Biol Chem. 2001;276:1911–1915. doi: 10.1074/jbc.M008773200. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- Li X, Huston E, Lynch MJ, Houslay MD, Baillie GS. Phosphodiesterase-4 influences the PKA phosphorylation status and membrane translocation of G-protein receptor kinase 2 (GRK2) in HEK-293beta2 cells and cardiac myocytes. Biochem J. 2006;394:427–435. doi: 10.1042/BJ20051560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Caron MG, Lefkowitz RJ. Multiple pathways of rapid beta 2-adrenergic receptor desensitization. Delineation with specific inhibitors. J Biol Chem. 1990a;265:3202–3211. [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990b;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- Lorenz S, Frenzel R, Paschke R, Breitwieser GE, Miedlich SU. Functional desensitization of the extracellular calcium-sensing receptor is regulated via distinct mechanisms: role of G protein-coupled receptor kinases, protein kinase C and β-arrestins. Endocrinology. 2007;148:2398–2404. doi: 10.1210/en.2006-1035. [DOI] [PubMed] [Google Scholar]
- Luk T, Jin W, Zvonok A, Lu D, Lin X-Z, Chavkin C, et al. Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol. 2004;142:495–500. doi: 10.1038/sj.bjp.0705792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen JM, Ramirez MT. The metabotropic glutamate receptor 4 is internalised and desensitised upon protein kinase C activation. Br J Pharmacol. 2006;148:279–290. doi: 10.1038/sj.bjp.0706733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RH, Millman EE, Godines V, Hanania NA, Tran TM, Peng H, et al. Salmeterol stimulation dissociates beta2-adrenergic receptor phosphorylation and internalization. Am J Respir Cell Mol Biol. 2007;36:254–261. doi: 10.1165/rcmb.2006-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundell SJ, Pula G, McIlhinney RA, Roberts PJ, Kelly E. Desensitization and internalization of metabotropic glutamate receptor 1a following activation of heterologous Gq/11-coupled receptors. Biochemistry. 2004a;43:7541–7551. doi: 10.1021/bi0359022. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Pula G, More JC, Jane DE, Roberts PJ, Kelly E. Activation of cyclic AMP-dependent protein kinase inhibits the desensitization and internalization of metabotropic glutamate receptors 1a and 1b. Mol Pharmacol. 2004b;65:1507–1516. doi: 10.1124/mol.65.6.1507. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Sibley DR. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J Biol Chem. 2004;279:49533–49541. doi: 10.1074/jbc.M408319200. [DOI] [PubMed] [Google Scholar]
- Perez DM, Karnik SS. Multiple signalling states of G protein-coupled receptors. Pharmacol Rev. 2005;57:147–161. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- Pineyro G, Archer-Lahlou E. Ligand-specific receptor states: implication for opiate receptor signalling and regulation. Cell Signal. 2007;19:8–19. doi: 10.1016/j.cellsig.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Pitcher J, Lohse MJ, Codina J, Caron MG, Lefkowitz RJ. Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry. 1992;31:3193–3197. doi: 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Pollok-Kopp B, Huttenrauch F, Rethorn S, Oppermann M. Dynamics of protein kinase C-mediated phosphorylation of the complement C5a receptor on serine 334. J Biol Chem. 2007;282:4345–4353. doi: 10.1074/jbc.M601317200. [DOI] [PubMed] [Google Scholar]
- Pollok-Kopp B, Schwarze K, Baradari VK, Oppermann M. Analysis of ligand-stimulated CC chemokine receptor 5 (CCR5) phosphorylation in intact cells using phosphosite-specific antibodies. J Biol Chem. 2003;278:2190–2198. doi: 10.1074/jbc.M209844200. [DOI] [PubMed] [Google Scholar]
- Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- Pula G, Mundell SJ, Roberts PJ, Kelly E. Agonist-independent internalization of metabotropic glutamate receptor 1a is arrestin- and clathrin-dependent and is suppressed by receptor inverse agonists. J Neurochem. 2004;89:1009–1020. doi: 10.1111/j.1471-4159.2004.02387.x. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, et al. The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signalling. Biochim Biophys Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal μ-opioid receptor desensitisation by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold A, Williams B, Huang ZF, Friedman J, Moore RH, Knoll BJ, et al. Localization of the sites mediating desensitization of the beta(2)-adrenergic receptor by the GRK pathway. Mol Pharmacol. 2000;58:1162–1173. doi: 10.1124/mol.58.5.1162. [DOI] [PubMed] [Google Scholar]
- Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL. Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain. 2007;127:129–139. doi: 10.1016/j.pain.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Smith FL, Lohmann AB, Dewey WL. Involvement of phospholipid signal transduction pathways in morphine tolerance in mice. Br J Pharmacol. 1999;128:220–226. doi: 10.1038/sj.bjp.0702771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So CH, Verma V, O'Dowd BF, George SR. Desensitization of the dopamine D1 and D2 receptor hetero-oligomer mediated calcium signal by agonist occupancy of either receptor. Mol Pharmacol. 2007;72:450–462. doi: 10.1124/mol.107.034884. [DOI] [PubMed] [Google Scholar]
- Stadel JM, Nambi P, Shorr RGL, Sawyer DF, Caron MG, Lefkowitz RJ. Catecholamine-induced desensitization of turkey erythrocyte adenylate cyclase is associated with phosphorylation of the â-adrenergic receptor. Proc Natl Acad Sci USA. 1983;80:3173–3177. doi: 10.1073/pnas.80.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout BD, Clarke WP, Berg KA. Rapid desensitization of the serotonin(2C) receptor system: effector pathway and agonist dependence. J Pharmacol Exp Ther. 2002;302:957–962. doi: 10.1124/jpet.302.3.957. [DOI] [PubMed] [Google Scholar]
- Strasser RH, Sibley DR, Lefkowitz RJ. A novel catecholamine-activated adenosine cyclic 3′,5′-phosphate independent pathway for beta-adrenergic receptor phosphorylation in wild-type and mutant S49 lymphoma cells: mechanism of homologous desensitization of adenylate cyclase. Biochemistry. 1986;25:1371–1377. doi: 10.1021/bi00354a027. [DOI] [PubMed] [Google Scholar]
- Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, et al. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla I, Spragg EJ, Poulin B, McWilliams PJ, Mistry SC, Blaukat A, et al. Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J Cell Biol. 2007;177:127–137. doi: 10.1083/jcb.200610018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TM, Friedman J, Baameur F, Knoll BJ, Moore RH, Clark RB. Characterization of beta2-adrenergic receptor dephosphorylation: comparison with the rate of resensitization. Mol Pharmacol. 2007;71:47–60. doi: 10.1124/mol.106.028456. [DOI] [PubMed] [Google Scholar]
- Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65:196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- Trester-Zedlitz M, Burlingame A, Kobilka B, von Zastrow M. Mass spectrometric analysis of agonist effects on posttranslational modifications of the β2-adrenoceptor in mammalian cells. Biochemistry. 2005;44:6133–6143. doi: 10.1021/bi0475469. [DOI] [PubMed] [Google Scholar]
- Tsao PI, von Zastrow M. Type-specific sorting of G protein-coupled receptors after endocytosis. J Biol Chem. 2000;275:11130–11140. doi: 10.1074/jbc.275.15.11130. [DOI] [PubMed] [Google Scholar]
- Urban JW, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vaughan DJ, Millman EE, Godines V, Friedman J, Tran TM, Dai W, et al. Role of the G protein-coupled receptor kinase site serine cluster in beta2-adrenergic receptor internalization, desensitization, and beta-arrestin translocation. J Biol Chem. 2006;281:7684–7692. doi: 10.1074/jbc.M500328200. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P, et al. Modulation of postendocytic sorting of G protein-coupled receptors. Science. 2002;297:615–620. doi: 10.1126/science.1073308. [DOI] [PubMed] [Google Scholar]
- Willets JM, Challiss RA, Nahorski SR. Non-visual GRKs: are we seeing the whole picture. Trends Pharmacol Sci. 2003;24:626–633. doi: 10.1016/j.tips.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Xiang B, Yu G-H, Guo J, Chen L, Hu W, Pei G, et al. Heterologous activation of protein kinase C stimulates phosphorylation of δ-opioid receptor at serine 344, resulting in β-arrestin- and clathrin-mediated receptor internalisation. J Biol Chem. 2001;276:4709–4716. doi: 10.1074/jbc.M006187200. [DOI] [PubMed] [Google Scholar]
- Xu H, Partilla JS, Wang X, Rutherford JM, Tidgewell K, Prisinzano TE, et al. A comparison of noninternalizing (herkinorin) and internalizing (DAMGO) mu-opioid agonists on cellular markers related to opioid tolerance and dependence. Synapse. 2007;61:166–175. doi: 10.1002/syn.20356. [DOI] [PubMed] [Google Scholar]
- Yuan N, Friedman J, Whaley BS, Clark RB. cAMP-dependent protein kinase and protein kinase C consensus site mutations of the beta-adrenergic receptor. Effect on desensitization and stimulation of adenylylcyclase. J Biol Chem. 1994;269:23032–23038. [PubMed] [Google Scholar]
- Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI. Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKC gamma mutant mice. Pain. 2001;94:245–253. doi: 10.1016/S0304-3959(01)00353-0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, et al. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu Y, Mackin S, Weight FF, Uhl GR, Wang JB. Differential mu opiate receptor phosphorylation and desensitisation induced by agonists and phorbol esters. J Biol Chem. 1996;271:11449–11454. doi: 10.1074/jbc.271.19.11449. [DOI] [PubMed] [Google Scholar]