

Abstract
Almost all existing models for G-protein-coupled receptors (GPCRs) are based on the occurrence of monomers. Recent studies show that many GPCRs are dimers. Therefore for some receptors dimers and not monomers are the main species interacting with hormones/neurotransmitters/drugs. There are reasons for equivocal interpretations of the data fitting to receptor dimers assuming they are monomers. Fitting data using a dimer-based model gives not only the equilibrium dissociation constants for high and low affinity binding to receptor dimers but also a ‘cooperativity index' that reflects the molecular communication between monomers within the dimer. The dimer cooperativity index (DC) is a valuable tool that enables to interpret and quantify, for instance, the effect of allosteric regulators. For different receptors heteromerization confers a specific functional property for the receptor heteromer that can be considered as a ‘dimer fingerprint'. The occurrence of heteromers with different pharmacological and signalling properties opens a complete new field to search for novel drug targets useful to combat a variety of diseases and potentially with fewer side effects. Antagonists, which are quite common marketed drugs targeting GPCRs, display variable affinities when a given receptor is expressed with different heteromeric partners. This fact should be taken into account in the development of new drugs.
Keywords: adenosine, dopamine, opioid, fitting data, receptor dimers, receptor oligomers, allosteric regulation, cooperativity, drug targets
Introduction: basic concepts in receptor homo- and heteromerization
An increasing number of G-protein-coupled heptaspanning-membrane receptors (GPCRs) are reported to be expressed on the plasma membrane as dimers (Bouvier, 2001; Devi, 2001; Rios et al., 2001; Agnati et al., 2003; Franco et al., 2003; Terrillon and Bouvier, 2004; Bulenger et al., 2005; Prinster et al., 2005; Herrick-Davis et al., 2006; Milligan, 2006, 2007; Milligan and Kostenis, 2006; Pin et al., 2007). Quite often, dimers are constitutively expressed in a given cell, and a given cell can express both homodimers and a variety of heterodimers for different receptors. From our work and that of others on homo and heteromerization of receptors present in striatal neurons, it has been feasible to demonstrate some consequences of homo and heteromerization in terms of both ligand pharmacology and receptor function. Cells sense neurotransmitters (or hormones) in a different way expressing one or another set of receptors that assemble into heterodimers. In fact, signalling and/or desensitization can vary depending on whether one or both the receptors of the heteromer are activated, and quite often the resulting signal is not simply the addition of the signals given by individual activation of the receptors; even there are instances in which heteromer-mediated signalling becomes qualitatively different (Ferré et al., 2007a).
Receptor homo- or heteromerization leads to the so-called intramembrane (or horizontal) interactions, which means that the pharmacology for agonists and/or antagonists of a given receptor usually changes (i) when it forms heteromers with another receptor and/or (ii) when the partner receptor in the heteromer is activated (Figure 1; see Ferré et al., 2007a for examples). This is due to conformational changes in the receptors transmitted within the receptor–receptor interface at the plane of the membrane bilayer, not excluding a G-protein-mediated cooperativity in the plane of the membrane. In this regard, new models considering receptor dimers have been recently developed (Durroux, 2005; Franco et al., 2005, 2006; Albizu et al., 2006). Until now, the approaches for fitting ligand-binding data have been based on the existence of receptor monomers. From a recently devised model for receptor homodimers, there exists a new approach for fitting data that gives more accurate and physiologically relevant parameters (Casadó et al., 2007). Fitting data using the new procedure (see below) give not only the equilibrium dissociation constants (KDs) for high- and low-affinity binding to receptor dimer, but also a new parameter reflecting the molecular communication within the dimer (Casadó et al., 2007, and references therein). For the pharmaceutical industry, the message is that dimers are usually the real targets in therapy. For scientists at academic institutions or at pharmaceutical companies, the dimer cooperativity index (DC) is a very interesting tool that enables to interpret and quantify the effect of allosteric regulators on the binding of ligands to orthosteric centres. According to the two-state dimer model, an allosteric modulator can be any molecule that binds to a non-orthosteric centre, or another protein that interacts with the receptor and affects its binding characteristics. A comprehensive way to fit binding data from saturation isotherms and competition assays to a dimer receptor model is now possible (see below), as it is also possible to give actual values for the concentration giving 50% reduction in radioligand binding when performing competition experiments. These values are much more reliable to establish binding potency orders than the IC50 values reported from competition experiments using monomer-based approaches.
Figure 1.

Pharmacological changes upon heteromerization. The affinity of the green ligand for the green receptor may vary in positive (b, d) or negative fashion (a, c) by either cotransfecting the partner—red receptor (a, b), or by transfecting the red receptor and occupying its orthosteric centre with the red ligand (c, d).
The occurrence of heteromers with different pharmacological and signalling properties opens a completely new field to search for novel targets. One relevant example is given by targeting adenosine receptors in Parkinson's disease whose main target are dopamine receptors. Further consideration is derived from the fact that drugs (agonists or antagonists) display different affinities for their receptors depending on whether the targeted receptor forms heteromers with one or another receptor. These changes in affinity would help to clarify why different compounds for the same receptor might have different therapeutic profiles.
Fitting radioligand binding data to receptor homo- and heterodimers
Traditionally, biphasic binding isotherms have been interpreted as being due to the existence of two monomeric receptor species with different affinities (high and low) for the radioligand. Therefore, two KD constants for the binding of the radioligand to these two independent sites are usually provided. These constants, KDH and KDL, would then account for the affinity of, respectively, the high and low affinity sites. Although one must recognize that this approach has been really useful, when a radioligand binds to a receptor dimer, another interpretation is possible. On the one hand, cooperativity is naturally explained by assuming that binding of the first ligand to the dimer modifies the equilibrium parameters of binding of the second ligand molecule to the dimer, and concave upward Scatchard plots would imply negative cooperativity, whereas concave downward Scatchard plots would indicate positive cooperativity (see Franco et al., 2005, 2006 for details). On the other hand, when contemplating the two-state dimer model (Figure 2) and for conceptual and comparative purposes, it is also necessary to calculate the ‘macroscopic' constants, for instance, dissociation constants for binding to each of the two protomers, KD1 and KD2, which define the dissociation equilibria involved in the binding of a ligand to the receptor dimer as a whole. In the two-state dimer receptor model, there are two macroscopic KDs describing the binding of an agonist (or antagonist or inverse agonist): KD1 for binding of the first ligand molecule to a dimer and KD2 for binding of the second ligand molecule to the dimer, according to the equation
 |
where A represents the radioligand concentration and RT the total amount of receptor dimers. Obviously, the traditional Bmax value would be 2 RT.
Figure 2.
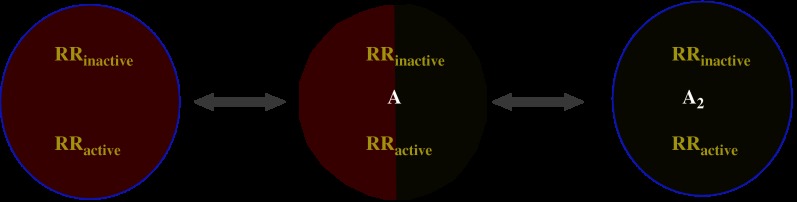
Scheme of the two-state dimer model (modified from Casadó et al., 2007). The dimer can be inactive or active, and can be empty or occupied by one or two molecules of ligand. Macroscopically, two KDs define the equilibrium for the binding of the first A molecule (KD1) and for the binding of the second A molecule (KD2) to the receptor dimer. See Franco et al. (2005), (2006) and Casadó et al. (2007) for details. KD, dissociation constant.
Although saturation isotherms are more adequate to describe the binding to receptors, there are a number of instances where this type of assays cannot be performed. In particular, newly developed agonist or antagonist compounds are not available in radioactive form and, therefore, have to be assayed by means of competition curves of standard radioligand binding. As in saturation experiments, competition curves are often biphasic. Taking into account the two-state dimer model as the simplest one, and the competition equilibrium derived from it (see Franco et al., 2006 and Casadó et al., 2007 for details), data should be fitted using the following equation, which is deduced for the case when the radioligand, for instance an antagonist, displays non-complex, that is, non-cooperative behaviour:
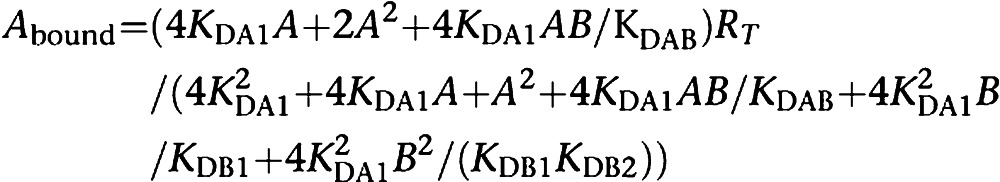 |
where KDA1 is already known (see above), A is the fixed concentration of the radioligand, B is the variable concentration of the assayed competing compound and KDB1 and KDB2 are respectively the KDs of the first and second bindings of B; KDAB can be described as a hybrid KD, that is, the dissociation constant of B binding to a receptor dimer semi-occupied by A. Reciprocally, KDBA value, which is the KD of A binding to a receptor dimer semi-occupied by B, can be deduced. The relationship between these parameters is shown by the following equation:
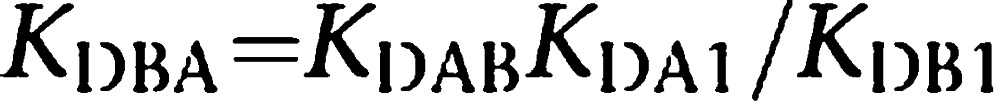 |
Apart from providing actual KD values of binding to receptor dimers (KDB1 and KDB2) from competition experiments, the dimer model allows direct calculation of the concentration providing half saturation (B50) for the tested compounds. B50 is easily calculated as follows:
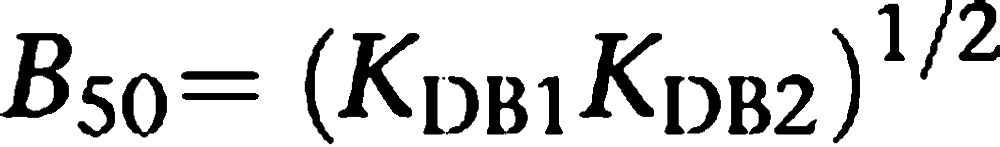 |
These B50 values are, in our opinion, more suitable than the usually employed IC50 values for ordering the compounds according to their binding potency to a receptor dimer. From competition experiments (including those giving biphasic curves), the B50 value, obtained from fitting to the present model, corresponds exactly to the concentration of B that displaces 50% of the binding of the radioligand. In contrast, in a biphasic displacement curve, two IC50 values are obtained with an ambiguous molecular meaning and they represent apparent constants that do not necessarily correspond to the concentration of compound that displaces 50% of the binding of the radioligand.
One relevant feature of the dimer model, as devised previously (Franco et al., 2005, 2006; Casadó et al., 2007), is the possibility to directly estimate the degree of binding cooperativity. For this reason, DC was introduced. This index is very useful to know the extent to which binding of one neurotransmitter to one orthosteric centre in the dimer is sensed in the second orthosteric centre present in the partner receptor. Therefore, DC is a measure of the intramolecular cross-talk (considering the dimer as a single molecule). It is of interest for pharmacological purposes to know the degree of cooperativity for the binding of different molecules (different agonists and eventually, different inverse agonists or antagonists) to receptor dimers. DC is defined as log (4KD1/KD2). For the binding of ligand A, DCA would be calculated as follows:
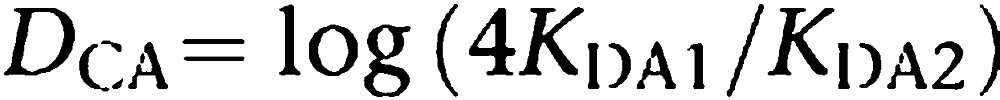 |
The index is ‘0' for non-cooperative binding. In the absence of cooperativity, microscopic/intrinsic constants are identical (k1=k2 and k−1=k−2) (Franco et al., 2005, 2006). Macroscopic dissociation constants (KD1 and KD2; Figure 2) reflect the fact that the ligand binds to a dimer and they are not equal in the absence of cooperativity. The first molecule entering the dimer has two possibilities: to enter into one subunit or into the other. When this molecule is bound to a monomer there is only possible dissociation event. To shift from a semi-occupied receptor to a fully occupied receptor, the second ligand molecule has only one option, which is to enter into the empty subunit. In contrast, a fully occupied state has two possibilities to arrive to a semi-occupied dimer depending on which monomer releases the ligand. Then there is a factor of 2 for each of the two binding processes. Taking into account dissociation constants, there is a dividing factor in the first binding and a multiplying factor in the second binding. Therefore, KD2/KD1 equals 4 even if microscopically the binding occurs with the same affinity for the two subunits in the dimer (see Franco et al., 2005, 2006). This explains why KD1 and KD2, which are the ones easily measurable from binding data (Casadó et al., 2007), are different even if there is no intramolecular cross-talk upon binding. In summary, the way the index is defined is such that positive values of DC indicate positive cooperativity, whereas negative values imply negative cooperativity. The usefulness of DC, compared with Hill coefficient, is that DC just depends on the KD values and therefore is constant for binding of a specific ligand to a specific receptor dimer. Moreover, DC values are highly sensitive to changes in equilibrium binding constants and consequently it is a valuable tool to detect the smallest ligand-induced change in the cooperativity within the subunits of the dimer. As mentioned, DC can be calculated from agonist or antagonist binding isotherms and also from competition curves.
A new way to understand allosteric effects
The assumption of GPCR dimerization opens a new way to understand allosteric modulators, a variety of compounds or macromolecules that bind to the receptor to a non-orthosteric centre and affect their pharmacology, and their physiological function (May et al., 2007). The most paradigmatic example is the heterotrimeric G protein. This protein is known to bind to the intracellular loops of receptors that are therefore called GPCRs. In practice, the heterotrimeric G protein is an allosteric modulator of receptor dimers that can eventually modify the binding characteristics of the orthosteric centres in the dimer (Hepler and Gilman, 1992). Variations in the binding characteristics due to the binding of small or big molecules to allosteric centres can be measured according to a receptor dimer model in two ways. On the one hand, an allosteric effector may change the affinity of the agonist(s), but on the other hand, allosteric effectors may change DC (Casadó et al., 2007). In a typical example, addition of GTP, which activates the heterotrimeric G protein, leads to disappearance of cooperativity. In such a case, GTP will shift the DC to a zero value.
A dimer model for fitting pharmacological data is instrumental to interpret results that, by using the two-independent-site model, would be understood as changes in the proportion of low- and high-affinity sites. These changes have been useful to interpret a large amount of data before demonstration of the occurrence of GPCR dimers. But now there is another approach to analyse the effect of regulators of receptor binding and function. With the development of the two-state dimer receptor model, it is not necessary to assume that a modulator modifies the proportion of high- and low-affinity states, something that is uncertain if the two receptor sites are independent, that is, if no communication can be established between them. On this line of reasoning, we had shown that the reported agonist-dependent conversion of high- to low-affinity sites (Casadó et al., 1991) cannot be explained assuming that the high- and low-affinity sites are independent. In sharp contrast, the two-state dimer model interprets these findings in terms of both, variation of KD(s) and variation of cooperativity, without any a priori assumption about the ‘state' or ‘conformation' of the dimer.
Receptors are susceptible to regulation by allosteric modulators of various types, as recently defined (Costa and Cotecchia, 2005, May et al., 2007). Allosteric regulators may, for instance, affect KD of the binding of an agonist, without modifying cooperativity. In these circumstances, KD1 and KD2 values change in the presence of the modulator, maintaining the DC value. Allosterism in the two-state dimer model has been worked out at the conceptual level in a direct way, that is, by considering that the allosteric modulator modifies KD(s) for the binding of the agonist. Also in cases of complex or biphasic binding, the allosteric modulator would potentially affect the cooperativity of the binding to the orthosteric site. In these cases, the two-state dimer receptor model deals with the binding of allosteric modulators in a qualitatively different way than when considering receptors as monomers. According to the dimer model, an allosteric enhancer would lead to conformational changes in the dimer that would not only be reflected in terms of different KD values for agonists, but also in terms of a different cooperativity on agonist binding. Therefore, allosterically mediated modifications in DC can be of interest to know the magnitude of the changes produced by the allosteric modulator and also to get insight into the mechanism by which the allosteric effect is produced. In summary, the dimer model provides an additional tool, the DC, which allows to quantify and interpret the effects of allosteric regulators upon binding of ligands to the two orthosteric centres in the dimer. It should be noted that with the two-state dimer model, an allosteric modulator could be any molecule (endogenous or synthetic) that is able to bind to a non-orthosteric centre, and is able to directly interact with the receptor and affect its agonist binding characteristics. This strategy is straightforward, since it does not require any assumption concerning modifications in the proportion of high- and low-affinity receptor sites induced by the binding of the modulator.
The ‘dimer fingerprint': pharmacological/biochemical
A dimer fingerprint is a specific property displayed by the receptor heteromer. A biochemical fingerprint of a receptor heterodimer can often be obtained with radioligand-binding techniques. Thus, the affinity for a specific ligand can be modified when the receptor for that ligand forms heterodimers. As an example, we showed the existence of A1–A2A receptor heteromers on the cell surface of cotransfected cells. Radioligand-binding experiments in cotransfected cells and rat striatum showed that a main biochemical characteristic of the A1–A2A receptor heteromer is the ability of A2A receptor activation to reduce the affinity of the A1 receptors for agonists (Figure 3; Ciruela et al., 2006a). Another example is the antagonistic A2A–D2 receptor interaction in which the stimulation of A2A receptor decreases the affinity of D2R for dopamine or its analogues in membrane preparations (Ferré et al., 1991). The intramembrane A2A–D2 receptor interaction implies a close physical interaction between the two receptors. In fact, the pharmacology of D2 receptor is affected by adenosine analogues activating A2A receptor by means of conformational changes at the A2A–D2 receptor heteromer interface (Canals et al., 2003). A common biochemical fingerprint for receptor heterodimers is an intramembrane receptor–receptor interaction (Agnati et al., 2003), which can only be attributed to the existence of a direct or indirect intermolecular interaction between the two receptors (intramolecular, if we consider the heterodimer as one molecule composed of two receptors). In this case, the binding of a ligand (usually an agonist) to one receptor modifies the affinity of the other receptor for the same (in the case of isoreceptors, that is, different receptor subtypes for the same neurotransmitter) or another ligand (see Figure 1). For homodimers, cooperativity found by biphasic kinetics or non-linear Scatchard plots in radioligand-binding experiments may constitute a valid dimer fingerprint.
Figure 3.

Dimer fingerprint of adenosine A1–A2A receptor heteromers (modified from Ciruela et al., 2006a). Binding of the radioligand specific for the A1 receptor is assayed in the presence of variable amounts of the selective A2A receptor agonist (competition assay). The dotted line is the theoretical one assuming that there is no interaction between the receptors. The actual curve reflects intramolecular cross-talk within the A1–A2A receptor heteromer, that is, the dimer fingerprint of this heteromer. The fingerprint is detected in both cotransfected cells and membranes from brain striatum (Ciruela et al., 2006a).
The ‘dimer fingerprint': functional
Fingerprints for heterodimers may also be detected by changes in signalling triggered by a given neurotransmitter drug. Coactivation of the two receptors in a heterodimer may change the signalling pathway triggered by the neurotransmitter, as well as the traffic of the receptors. Dopamine neurotransmission constitutes a good example of this. Many of the functional consequences of the heteromerization of dopamine (D1 or D2) and adenosine receptors (A1 or A2A) in the direct and indirect striatal pathways are known (Ginés et al., 2000; Hillion et al., 2002; Ferré et al., 2007a, 2007b). These interactions are antagonistic and collectively all the findings have led to propose that adenosine receptor antagonists could be useful in Parkinson's disease. Clinical assays for different adenosine A2A receptor antagonists are underway to check their usefulness in this neurodegenerative condition for which the main treatment is administration of L-DOPA, which is a precursor of dopamine.
Orexin-1-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function (Ellis et al., 2006). The orexin-1 receptor is targeted to the cell surface but becomes internalized following exposure to the peptide agonist orexin A. In contrast, constitutive expression of cannabinoid CB1 receptor results in a spontaneous, agonist-independent internalization. Expression of the orexin-1 receptor in the presence of the CB1 receptor results in both receptors displaying the spontaneous internalization phenotype. Treatment of cells coexpressing the orexin-1 and CB1 receptors with the orexin-1 receptor antagonist or the CB1 receptor antagonist results in re-localization of both receptors to the cell surface. Treatment with CB1 antagonist decreases potency of orexin A to activate the mitogen-activated protein kinases (MAPKs) ERK1/2 only in cells coexpressing the two receptors and, analogously, treatment with orexin-1 receptor antagonist also reduces the potency of a CB1 receptor agonist to phosphorylate ERK1/2 when the two receptors are coexpressed. The authors conclude that the results introduce an entirely novel pharmacological paradigm whereby ligands modulate the function of receptors for which they have no significant inherent affinity by acting as regulators of receptor heterodimers.
In 2004 the group of Susan George showed that dopamine D1 and D2 receptors form heteromers in transfected cells (Lee et al., 2004). This was somewhat unexpected since they are specific receptors for dopamine but one is coupled positively with the cyclase, whereas the other is coupled negatively to the enzyme. Therefore, it is not expected that a given neuron would express the two receptors. But the answer to this conundrum is precisely a different signalling mediated by the D1–D2 receptor heteromer. Instead of coupling to a Gs or Gi protein, the heteromer couples to a different heterotrimeric G protein, Gq/11. In fact, dopamine activating D1 and D2 receptors in the heteromer does not lead to signalling via cAMP and PKA but to calcium mobilization and calmodulin kinase activation. Recently, the same group has demonstrated that these heteromers occur in vivo and that their activation leads to the activation of calmodulin kinase in the nucleus accumbens (Rashid et al., 2007). It is thus likely that the reported coupling between dopamine receptor signalling and calcium occurs only in neurons expressing D1–D2 receptor heterodimers.
Also, recently, our laboratory (Marcellino et al., 2007) has shown the occurrence of D1–D3 heteromers. The demonstration has first been made in heterologous systems and using biophysical techniques of energy transfer, fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET). The functional role of D1–D3 heteromers has been demonstrated using striatal membranes and reserpinized rats. Radioligand-binding assays have allowed detection of the biochemical ‘dimer fingerprint' that confirms the existence of the heteromers in a natural tissue such as striatum. In this case, there is synergism since the affinity of D1 for its agonists increases when the partner D3 receptor is in an agonist-bound state. On the other hand, the experiments with reserpinized animals showed that stimulation of D3 receptor potentiates the behavioural effects that are mediated by D1 receptors; this effect occurs irrespective of D2 receptor activation. All these data show that a relevant role of D3 receptor is to achieve a higher dopaminergic response in striatal neurons coexpressing the two receptors. It is without any doubt interesting the parallelism between heteromerization between striatal dopamine receptor subtypes and that of striatal adenosine A1 and A2A receptor subtypes (see below; Ciruela et al., 2006a, 2006b).
Another recent example of changes in signalling also involves cannabinoid CB1 receptors. It has been for instance described the occurrence of adenosine A2A–cannabinoid CB1 receptor heterodimers. The mechanism of action responsible for the motor depressant effects of cannabinoids, which operate through centrally expressed cannabinoid CB1 receptors, is still a matter of debate. Carriba et al. (2007) have reported that CB1 and adenosine A2A receptors form heteromeric complexes in cotransfected human embryonic kidney-293T (HEK-293T) cells and rat striatum, where they colocalize in fibrillar structures. In a human neuroblastoma cell line, CB1 receptor signalling was found to be completely dependent on A2A receptor activation. Accordingly, blockade of A2A receptors counteracted the motor depressant effects produced by the intrastriatal administration of a cannabinoid CB1 receptor agonist. These biochemical and behavioural findings suggest that the profound motor effects of cannabinoids might be partly dependent on physical and functional interactions between striatal A2A and CB1 receptors (Carriba et al., 2007).
Rios et al. (2006) performed BRET studies using epitope-tagged μ, δ and κ opioid receptors with Renilla luciferase, and CB1 cannabinoid or CCR5 chemokine receptors with yellow fluorescent protein to show that coexpression of opioid receptors with cannabinoid receptors, but not with chemokine receptors, leads to a significant increase in the level of BRET signal, thus suggesting a specific opioid–cannabinoid heteromerization. To examine the implications of these interactions in signalling, GTPγS-binding and MAPK phosphorylation assays were performed revealing that the μ receptor-mediated signalling is attenuated by the CB1 receptor agonist; the effect is reciprocal and is seen in heterologous cells and endogenous tissue expressing both the receptors. Studies on Src and STAT3 phosphorylation and neuritogenesis in Neuro-2A cells showed that the simultaneous activation of μ opioid and CB1 cannabinoid receptors leads to a significant attenuation of the response seen upon activation of individual receptors, probably implicating a role of receptor–receptor interactions in modulating neuritogenesis (Rios et al., 2006).
These results reported by different laboratories provide a common message, namely that the functional role of the heteromer is different from the functional role of the homodimer. Then drugs aimed at treating Parkinson's disease patients, such as L-DOPA and A2A receptor antagonists, target in striatum dopamine receptor homodimers, D1–D2 receptor heteromers, D1–D3 receptor heteromers, D2–A2A receptor heteromers and/or A2A receptor homodimers. Analogously, natural and synthetic opiates not only target opioid receptor homodimers, but also heteromers formed by different opioid receptor subtypes or by opioid and cannabinoid receptors. This adds complexity to drug development tasks, but opens a novel perspective to design drugs acting preferentially in one receptor heteromer but not in another; this would expand the therapeutic profile of ligands acting on a given receptor, but also less side effects would be expected from compounds preferentially targeting cells expressing the heteromer.
GPCR antagonism varies in receptor heteromers
To our knowledge, the first example showing that a natural antagonist displays different affinities depending on which heteromer forms the targeted receptor has been provided for caffeine, which is a nonselective adenosine receptor antagonist. As mentioned above, adenosine A1 and A2A receptors form heteromers and the main biochemical characteristic of the A1–A2A heteromer, the dimer fingerprint, is the ability of A2A receptor activation to reduce the affinity of the A1 receptors for agonists (Figure 3; Ciruela et al., 2006a). Although postsynaptic A1 and A2A receptors are largely present in different striatal neurons, the two receptors are coexpressed presynaptically in more than 60% of striatal glutamatergic terminals. The biochemical dimer fingerprint found in striatal tissue confirmed that the two receptors form heteromers in a significant proportion of striatal neurons. In terms of signalling, it was proven that an increase of adenosine concentration sufficient to activate A2A receptors would in turn shut-off the A1 receptor-mediated signalling. Careful analysis of glutamate release from striatal terminals at different adenosine concentrations clearly indicates that at low adenosine concentrations the A1 receptor-mediated signalling predominates and the read-out is a negative modulation of glutamate release. In contrast, at higher adenosine concentrations, which can happen in low-energy status, for instance due to hypoxia upon high caffeine consumption, the A2A receptor-mediated signalling is prevalent and blocks A1 receptor-mediated signalling in the heteromer, therefore resulting in a positive modulation of glutamate release (Figure 4). The A1–A2A receptor heteromer turns out to be a sensor of adenosine concentration whose effect in the striatum leads to completely opposite read-outs in terms of glutamate release regulation (Ciruela et al., 2006a). Such an exquisite regulation would not be possible if A1 and A2A receptors would not form heterodimers in these glutamatergic striatal neurons. Interestingly, the affinity of the adenosine A2A receptor for caffeine is markedly reduced when it heteromerizes with the adenosine A1 receptor (Ciruela et al., 2006a). No change in affinity is observed for A1 receptor and caffeine in the heterodimer, and also it does not occur when A2A receptors form heterodimers with dopamine D2 receptors. There is solid evidence that this is not a special feature of caffeine and adenosine A2A receptors. We observe changes in affinity in the case of synthetic agonists targeting other heteromers (data in preparation). The relevance of this finding is due to the fact that a significant number of prescribed drugs targeting GPCRs are indeed antagonists. This means that these antagonists target specific receptors but with putative different affinities in different cells expressing different heteromers. It may then happen that two synthetic antagonists having similar potencies according to in vitro assays using single transfected cells may have different therapeutic profiles. This may explain why different antagonists for a given receptor do not necessarily have similar in vivo profiles and similar side effects. A reduction in the concentration of the antagonist assuming that we target a receptor in a given heteromer would reduce the side effects. On the other hand it is also predicted that different antagonists for the same receptor might be useful for different diseases just by preferentially targeting the same receptor but in a different heteromeric context, that is, in different cells/tissues/systems.
Figure 4.

Receptor-heteromer-mediated dual regulation of glutamate release by adenosine. At low concentrations, adenosine acts by depressing glutamate release in GABAergic striatal neurons. At high concentrations, adenosine in the same neurons enhances glutamate release. This signalling via A1 receptors at low [adenosine] and via A2A receptors at high [adenosine] is only possible by the occurrence of pre-synaptic A1–A2A receptor heteromers (for details see text and Ciruela et al., 2006a).
Dual and receptor–heteromer-specific drugs
There is interest in targeting heteromers and this can be achieved by different approaches. One is by the development of the so-called dual compounds that would target the two receptors that are partners in the heteromer. In our laboratory, dual compounds have been developed that are ergopeptide derivatives able to interact with both adenosine and dopamine receptors (Vendrell et al., 2007). For the same target, that is, adenosine–dopamine receptors heteromers, which are relevant for the treatment of Parkinson's disease, dual molecules consisting of a xanthine analogue and a dopamine analogue linked by a spacer of variable length are being developed (Ventura et al., 2007, in preparation). Dopamine D1–D2 heteromeric complexes possess a unique pharmacology such that a specific subset of D1 receptor agonists, SKF81297 and SKF83959, can activate the heteromer by acting concurrently on both the D1 receptor and a distinct conformation of the D2 receptor that depends on the presence of the D1 receptor. Whereas SKF83959 activates a Gq protein, it does not activate adenylate cyclase (AC)-coupled D1 or D2 receptors or Gq/11 through D1 receptor homomeric units. Therefore, it seems likely that SKF83959 is in fact a specific agonist for Gq/11-coupled D1–D2 receptor hetero-oligomers (Rashid et al., 2007).
Heteromerization of (Gomes et al., 2004; Waldhoer et al., 2004; Gupta et al., 2006) opioid receptors has been shown to alter opioid ligand properties and affect receptor trafficking in cell culture model systems. Waldhoer et al. (2005) demonstrated that 6′-guanidinonaltrindole has the unique property of selectively activating only opioid receptor heteromers but not homomers. When assayed in vivo, the compound induced analgesia depending on the place of administration. This study constitutes a proof of the concept for tissue-selective drug targeting based on GPCRs.
Conclusions
G-protein-coupled receptors occur as homodimers and/or heterodimers on the cell surface and therefore dimers/oligomers are the real targets for agonists/antagonists and for drugs interacting with these receptors at the orthosteric site. This is a concept that is currently overlooked by pharmaceutical companies, which concentrate on a single receptor whose pharmacological characterization is frequently performed using single-transfected cells in which receptor heteromers cannot occur. Heteromerization affects all aspects of receptor physiology/pharmacology: trafficking, signalling, ligand affinities, etc. On the other hand, models to deal with GPCRs rely on their occurrence as monomers. Recent models consider these receptors as dimers. These models are very useful for obtaining reliable KD values from binding data (from saturation isotherms but also from competition assays) in cases of biphasic kinetics. These models consider intramolecular communication within the dimer that can be quantitated by a newly defined parameter Dc. This index is useful for instance to quantitate but also to give insight about the mechanism of allosteric regulation in GPCRs. Therefore, the occurrence of receptor heterodimer/oligomers opens new perspectives for GPCRs from both the functional and the pharmacological point of view.
An interesting therapeutic approach that is currently being explored in several laboratories is indeed based on the occurrence of receptor heteromers. This consists of designing compounds, which would act in the two receptors in the heteromer. Two possibilities exist for this development of ‘dual' drugs. One involves synthesizing molecules with moderate affinity for the two receptors and the other involves synthesizing ‘dimeric' compounds that would activate simultaneously the two receptors in the heterodimer. The latter would also serve as excellent tools to detect receptor heteromers in natural tissues. Although rational designs for dual compounds, which are able to interact with two receptors simultaneously or not, exist, many receptor–heteromer-specific compounds have been found serendipitously. Other more rational (perhaps modelling based) approaches will likely appear in the future. In the meantime it would be advisable for pharmaceutical companies to screen potential drugs against heteromers of therapeutic interest, such as opioid or cannabinoid receptor heteromers for pain or dopamine/adenosine receptor heteromers for Parkinson's disease.
Acknowledgments
Supported by the Intramural Research funds of the National Institute on Drug Abuse, NIH and by Grants SAF2005–05481 (to EIC) and SAF2006-05481 (to RF) from the Spanish Ministerio de Ciencia y Tecnologia.
Glossary
- B50
concentration of compound B able to displace 50% the binding of a specific radioligand to a given receptor
- BRET
bioluminescence resonance energy transfer
- DC
cooperativity index
- FRET
fluorescence resonance energy transfer
- GPCR
G-protein-coupled receptor
- KD
equilibrium dissociation constant
- MAPK
mitogen-activated protein kinase
- PKA
protein kinase A
- RT
total amount of receptor dimers
Footnotes
Conflict of interest
The authors state no conflict of interest.
References
- Agnati LF, Ferré S, Lluis C, Franco R, Fuxe K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev. 2003;55:509–550. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, et al. Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. doi: 10.1124/mol.106.025684. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, et al. Adenosine A2A–dopamine D2 receptor–receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem. 2003;278:46741–46749. doi: 10.1074/jbc.M306451200. [DOI] [PubMed] [Google Scholar]
- Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32:2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, et al. 2007Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: The receptor-dimer cooperativity index Pharmacol Ther(in press). [DOI] [PubMed]
- Casadó V, Mallol J, Canela EI, Lluis C, Franco R. The binding of [3H]R-PIA to A1 adenosine receptors produces a conversion of the high- to the low-affinity state. FEBS Lett. 1991;286:221–224. doi: 10.1016/0014-5793(91)80978-c. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Lujan R, Burgueño J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J Neurosci. 2006a;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Ferré S, Casadó V, Cortés A, Cunha RA, Lluis C, et al. Heteromeric adenosine receptors: a device to regulate neurotransmitter release. Cell Mol Life Sci. 2006b;73:2427–2431. doi: 10.1007/s00018-006-6216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T, Cotecchia S. Historical review: negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol Sci. 2005;26:618–624. doi: 10.1016/j.tips.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Devi LA. Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol Sci. 2001;22:532–537. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- Durroux T. Principles: a model for the allosteric interactions between ligand binding sites within a dimeric GPCR. Trends Pharmacol Sci. 2005;26:376–384. doi: 10.1016/j.tips.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. Orexin-1 receptor–cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- Ferré S, Agnati LF, Ciruela F, Lluis C, Woods AS, Fuxe K, et al. Neurotransmitter receptor heteromers and their integrative role in ‘local modules': the striatal spine module. Brain Res Rev. 2007b;55:55–67. doi: 10.1016/j.brainresrev.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007a;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Ferré S, von Euler G, Johansson B, Fredholm BB, Fuxe K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci USA. 1991;88:7238–7241. doi: 10.1073/pnas.88.16.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Canals M, Marcellino D, Ferre S, Agnati L, Mallol J, et al. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends Biochem Sci. 2003;28:238–243. doi: 10.1016/S0968-0004(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferre S, Fuxe K, et al. The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol. 2006;69:1905–1912. doi: 10.1124/mol.105.020685. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferre S, Fuxe K, Cortes A, et al. Dimer-based model for heptaspanning membrane receptors. Trends Biochem Sci. 2005;30:360–366. doi: 10.1016/j.tibs.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Gines S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI, et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci USA. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Décaillot FM, Devi LA. Targeting opioid receptor heterodimers: strategies for screening and drug development. AAPS J. 2006;8:E153–E159. doi: 10.1208/aapsj080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992;17:383–387. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Weaver BA, Grinde E, Mazurkiewicz JE. Serotonin 5-HT2C receptor homodimer biogenesis in the endoplasmic reticulum: real-time visualization with confocal fluorescence resonance energy transfer. J Biol Chem. 2006;281:27109–27116. doi: 10.1074/jbc.M604390200. [DOI] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casadó V, Scout R, Terasmaa A, et al. Coaggregation, cointernalization and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277:18091–18097. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lança AJ, et al. Dopamine D1 and D2 receptor co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Ferré S, Casadó V, Cortés A, Le Foll B, Saur O, et al. 2007Identification of dopamine D1–D3 receptor heteromers: indications for a role of synergistic D1–D3 receptor interactions in the striatumrevised version. [DOI] [PMC free article] [PubMed]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Milligan G. G-protein-coupled receptor heterodimers: pharmacology, function and relevance to drug discovery. Drug Discov Today. 2006;11:541–549. doi: 10.1016/j.drudis.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta. 2007;1768:825–835. doi: 10.1016/j.bbamem.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol. 2006;147 Suppl 1:S46–S55. doi: 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, et al. International union of basic and clinical pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- Prinster SC, Hague C, Hall RA. Heterodimerization of G protein-coupled receptors: specificity and functional significance. Pharmacol Rev. 2005;57:289–298. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, et al. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA.2006mu Opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis Br J Pharmacol 148387–395.comment in Br J Pharmacol148, 385–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92:71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Terrillon D, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrell M, Angulo E, Casadó V, Lluis C, Franco R, Albericio F, et al. Novel ergopeptides as dual ligands for adenosine and dopamine receptors. J Med Chem. 2007;50:3062–3069. doi: 10.1021/jm060947x. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Barlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, et al. 2005A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers Proc Natl Acad Sci USA 1029050–9055.comment in Proc Natl Acad Sci USA102, 8793–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]