

Abstract
Spatiotemporal aspects of ERK activation are stimulus-specific and dictate cellular consequences. They are dependent upon dual specificity phosphatases (DUSPs) that bind ERK via docking domains and can both inactivate and anchor ERK in cellular compartments. Using high throughput fluorescence microscopy in combination with a system where endogenous ERKs are removed and replaced with wild-type or mutated ERK2-green fluorescent protein (GFP), we show that ERK2 activation responses to epidermal growth factor (EGF) and protein kinase C (PKC) are transient and sustained, respectively. PKC-mediated ERK2 activation is associated with prolonged nuclear localization in the dephosphorylated form, whereas EGF-stimulated ERK2 activation mediates only transient nuclear accumulation. By using short inhibitory RNAs to nuclear inducible DUSP1, -2, or -4 (alone or in combination), we demonstrate that all three of these enzymes contribute to the dephosphorylation of PKC (but not EGF)-activated ERK2 in the nucleus but that they have opposing effects on localization. DUSP2 and -4 inactivate and anchor ERK2, whereas DUSP1 dephosphorylates ERK in the nucleus but allows its traffic back to the cytoplasm. Overexpression of DUSP1, -2, or -4 prevented ERK2 activation, but only DUSP2 and -4 caused ERK2-GFP nuclear accumulation or could be immunoprecipitated with ERK2. Furthermore, protein synthesis inhibition or replacement of wild-type ERK2-GFP with docking domain mutants selectively increased PKC effects on ERK activity and altered ERK2-GFP localization. These mutations also impaired the ability of ERK2-GFP to bind DUSP2 and -4. Together, our data reveal a novel, stimulus-specific, and phosphatase-specific mechanism of ERK2 regulation in the nucleus by DUSP1, -2, and -4.
Extracellular signal-regulated kinases 1 and 2 (ERK1/2, referred to as ERK2 herein) are the prototypic members of the mitogen-activated protein kinase (MAPK) family, and are activated by a wide variety of stimuli. The biological outcome of ERK signaling is dependent upon the magnitude, duration, and localization of its activation (1-3). ERK is typically associated with its upstream activator MAPK/ERK kinase (MEK) in the cytosol of quiescent cells. Upon phosphorylation by MEK, ERKs dissociate and typically translocate to the nucleus where they can phosphorylate transcription factors and immediate early gene products, leading to altered gene expression (1-3). Transient versus sustained activation of ERK governs the difference between quiescence and proliferation in fibroblasts and epithelial cells (2-5). Cells in which ERK nuclear translocation is prevented remain quiescent, even if ERK phosphorylation is sustained (6). Similarly, the interruption of sustained ERK phosphorylation with inhibitors abolishes G1/S transition (5).
The activity and localization of ERK are regulated by dual specificity phosphatases (DUSPs), which act by directly binding to and catalyzing the removal of phosphorylated Thr and Tyr within the activation loop (7, 8). A highly related subfamily of DUSPs, termed the nucleus-inducible MAPK phosphatases (MKPs), are rapidly induced by stimuli and localize exclusively to the nucleus. This group consists of DUSP1/MKP-1, DUSP2/PAC1, DUSP4/MKP-2, and DUSP5 (referred to using DUSP nomenclature herein). Each of these enzymes is able to dephosphorylate ERK and, with the exception of DUSP5, is also active with respect to c-Jun N-terminal kinase (JNK) and/or p38 (7-11). Accordingly, they function in negative feedback loops of ERK regulation.
The specificity of ERK binding to phosphatases and other substrates is regulated through a system of docking domains. Two such motifs that govern these interactions are termed D-(docking) and DEF (docking site for ERK, FXFP)-domains. The D-domains are characterized by a string of basic amino acid residues (consensus, (K/R)2–3X1–6(L/I)X(L/I)) that facilitate docking to ERK via the common docking motif, located opposite the catalytic cleft (12, 13). In contrast, DEF-domains have a consensus site of (F/Y)X(F/Y)P, which binds to ERK through a motif adjacent to the catalytic site (14). All of the nuclear inducible MKPs have a D-domain motif, which determines substrate specificity (7, 12, 15), whereas DEF-domains have only been identified in DUSP1 and DUSP4 (7, 16, 17). Where studied, both D- and DEF-motifs have a large influence on phosphatase association and catalysis (10, 11, 15, 17-21). However, little is known about how these motifs influence the spatiotemporal aspects of ERK regulation.
Sustained, mitogenic ERK activation in fibroblasts has been shown to induce expression of nuclear proteins that mediate the dephosphorylation and scaffolding of ERK, resulting in its accumulation in the nucleus (22, 23). This nuclear ERK accumulation is also dependent upon both protein synthesis and D-domain interactions (23) and was therefore postulated to involve the nuclear inducible MKPs. A recent study provided the first direct evidence for this, showing that DUSP5 is rapidly induced by ERK stimuli and when overexpressed can co-immunoprecipitate with ERK and cause its nuclear accumulation in a D-domain-dependent manner (10). In contrast, although DUSP1 contains both D- and DEF-domain binding motifs and can efficiently dephosphorylate ERK (11, 12, 15, 16, 20), it cannot be co-immunoprecipitated with ERK nor cause its nuclear retention on overexpression (10, 15, 24). DUSP2 and DUSP4 have not been studied in this context.
Here we have used siRNA to knock down endogenous ERKs in HeLa cells, and recombinant adenovirus (Ad) to add back either wild-type ERK2-GFP or ERK2-GFP that has been mutated to impair D- or DEF-domain-dependent binding. Using immunofluorescent staining and a semi-automated system for image acquisition and analysis, we now use this model to explore how DUSP1, DUSP2, and DUSP4 shape spatiotemporal aspects of ERK signaling in response to EGF and a PKC-activating phorbol ester, and the dependence of their effects on D- or DEF-domain interactions.
EXPERIMENTAL PROCEDURES
Engineering of Plasmids and Viruses
The pCR3.1 vector was purchased from Invitrogen. pSRα Myc-tagged DUSP1, DUSP2, and DUSP4 vectors were a gift from Prof. Stephen Keyse (Cancer Research UK, University of Dundee). Viral shuttle vectors were constructed initially by subcloning a KpnI-NotI digest of ERK2-GFP in pEGFP-N1 (a gift from Prof. Louis Luttrell, Medical University of South Carolina, Charleston) into a corresponding digest of pacAd5CMV K-N pA (donated by Prof. Beverly Davidson, University of Iowa, Iowa City). Y261A and D319N mutations were introduced using a QuikChange PCR-based mutagenesis kit (Stratagene, Amsterdam, Netherlands) and the following primers: 5′-AAT TTA AAA GCT AGA AAC GCT TTG CTT TCT CTC CCG CAC-3′ and 5′-GCA GTA TTA TGA CCC AAG TAA TGA GCC CAT TGC TGA AGC-3′ along with antisense primers according to manufacturer's recommendations and using the pacAd5CMV ERK2-GFP vector as the template. Viruses were generated from shuttle vectors as described (25). Briefly, 4.5 μg of shuttle vectors were digested alongside 1.5 μg of pacAd5 9.2–100 sub360 backbone vector (donated by Prof. Beverly Davidson, University of Iowa, Iowa City) with PacI. Cut shuttle and backbone vectors were then mixed and transfected into low passage HEK293 cells using Superfect (Qiagen, Crawley, UK). Cells were left to allow recombination between shuttle and backbone vectors. Verification of recombination was performed by restriction digest and sequence analysis, and Ad vectors were grown to high titer and purified according to standard protocols (26).
Cell Culture and Transfection
HeLa cells were cultured in 10% FCS-supplemented Dulbecco's modified Eagle's medium (DMEM). For 96-well plate experiments, cells were harvested by trypsinization and seeded at 5 × 103 cells/well, using RNAiMAX reagent (Invitrogen) and the manufacturer's reverse transfection protocol. Cells were transfected with two siRNA duplexes (Qiagen, Crawley, UK) as follows: for ERK1, 5′-CGU CUA AUA UAU AAA UAU AdTdT-3′,5′-UAU AUU UAU AUA UUA GAC GdGdG-3′, 5′-CCC UGA CCC GUC UAA UAU AdTdT-3′, and 5′-UAU AUU AGA CGG GUC AGG GdAdG-3′; and for ERK2, 5′-CAC UUG UCA AGA AGC GUU AdTdT-3′,5′-UAA CGC UUC UUG ACA AGU GdTdT-3′, 5′-CAU GGU AGU CAC UAA CAU AdTdT-3′, and 5′-UAU GUU AGU GAC UAC CAU GdAdT-3′, which were identified and validated in a recent publication (13). A mixture of all four ERK1/2 duplexes or control siRNA against GFP (Ambion, Warrington, UK) was used in experiments at 1 nm total concentration. For DUSP siRNA transfection, 10 nm SMARTpool or nontargeting control siRNA mixtures (Dharmacon, Cramlington, UK) were included in the transfections. For combination knockdown experiments, each SMARTpool siRNA was added at 10 nm to the ERK1/2 siRNAs and compared with control groups transfected with identical concentrations of control siRNA. Sixteen hours after siRNA transfection, cells were transduced with 2 × 106 pfu/ml Ad wild-type, Y261A, or D319N ERK2-GFP vector in DMEM with 10% FCS. The Ad-containing medium was removed after 4–6 h and replaced with fresh DMEM supplemented with 0.1% FCS. The cells were then maintained for 16–24 h in culture prior to stimulation with EGF (Calbiochem) or phorbol 12,13-dibutyrate (PDBu; Sigma). In some experiments, cells were treated with 30 μm cycloheximide (Sigma) for 30 min prior to stimulation. Expression levels of GFP-tagged fusions were compared using Western blotting techniques (see Fig. 1A and Fig. 3A) as well as comparison of mean cell fluorescence in microscopy assays (not shown).
FIGURE 1. A knockdown and add-back model for studying ERK regulation.
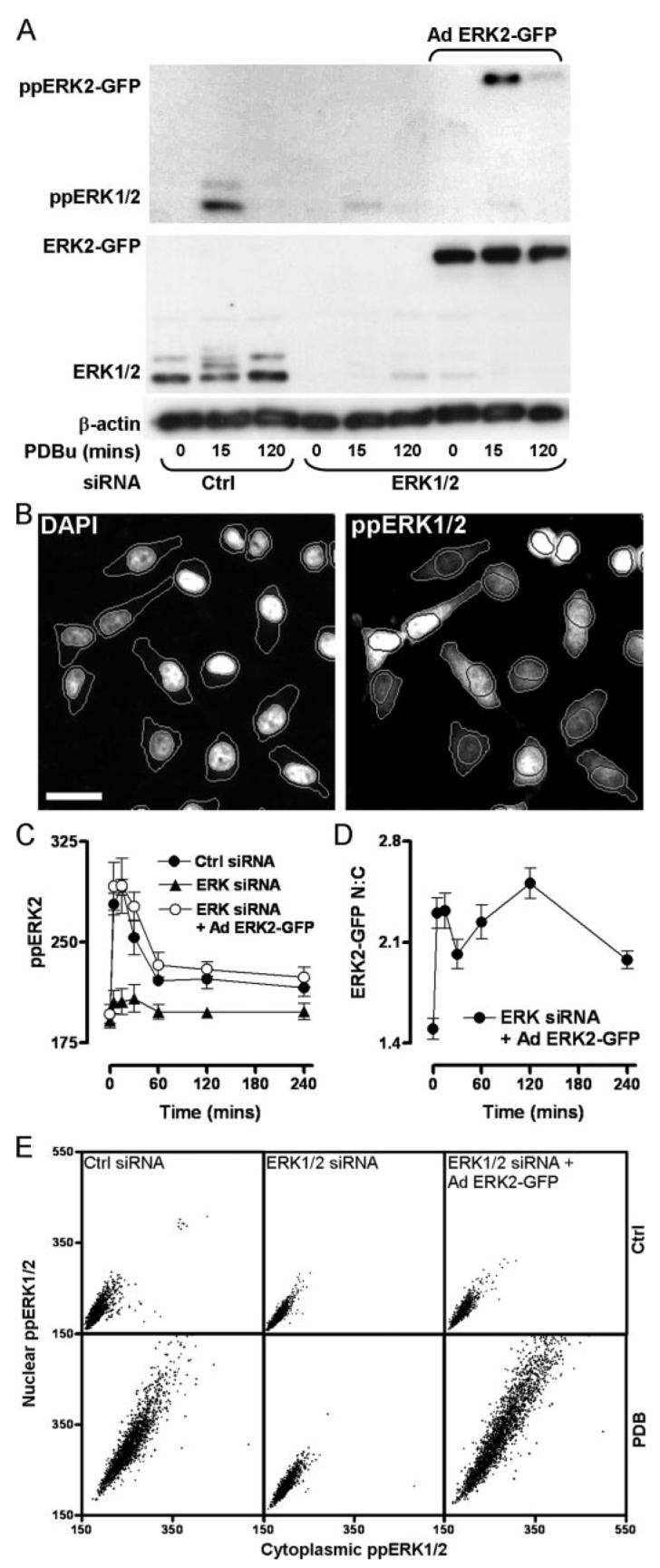
A, cells were transfected with control siRNAs (ctrl), ERK1/2 siRNAs, or ERK1/2 siRNAs and transduced with Ad ERK2-GFP, prior to stimulation with 1 μm PDBu as indicated. Samples were immunoblotted for total ERK1/2 (middle panel) and ppERK1/2 (top panel). Immunoblotting for β-actin (bottom panel) was used as a loading control. ERK1/2 and ppERK1/2 labels denote total or phosphorylated ERK1/2 (44/42 kDa, respectively). ERK2-GFP and ppERK2-GFP labels denote total and phosphorylated bands of ERK2-GFP (69 kDa), respectively. Data shown are representative of three independent experiments showing similar data. B, images of cells transfected in 96-well plates as described for control samples in A prior to 1 μm PDBu stimulation (15 min), staining, and automated image acquisition. Bar, 20 μm. Outlines denote the segmentation of cells according to DAPI and ppERK1/2 staining using IN Cell Analyzer software. C, cells were transfected with control siRNAs (filled circles), ERK1/2 siRNAs (filled triangles), or ERK1/2 siRNAs as well as Ad ERK2-GFP (open circles) and stimulated with 1 μm PDBu for the times indicated. Cells were fixed and stained with DAPI and for ppERK1/2 prior to image acquisition in duplicate wells. The graph shows average ppERK1/2 signal intensity (whole cells) from four independent experiments (mean ± S.E., n = 4). D, cells imaged for ppERK intensity in C were simultaneously imaged and analyzed for ERK2-GFP intensity in nuclear and cytoplasmic compartments. These values were used to calculate the N:C ERK2-GFP ratio for each cell, and the data were pooled from four separate experiments (mean ± S.E., n = 4). E, scatterplots showing nuclear versus cytoplasmic intensity values from cells transfected as indicated and incubated with or without 1 μm PDBu for 15 min. Each dot represents a single cell, and data were acquired from four images in two separate experiments.
FIGURE 3. Stimulus-specific and ERK-dependent regulation of nuclear inducible DUSP mRNA.
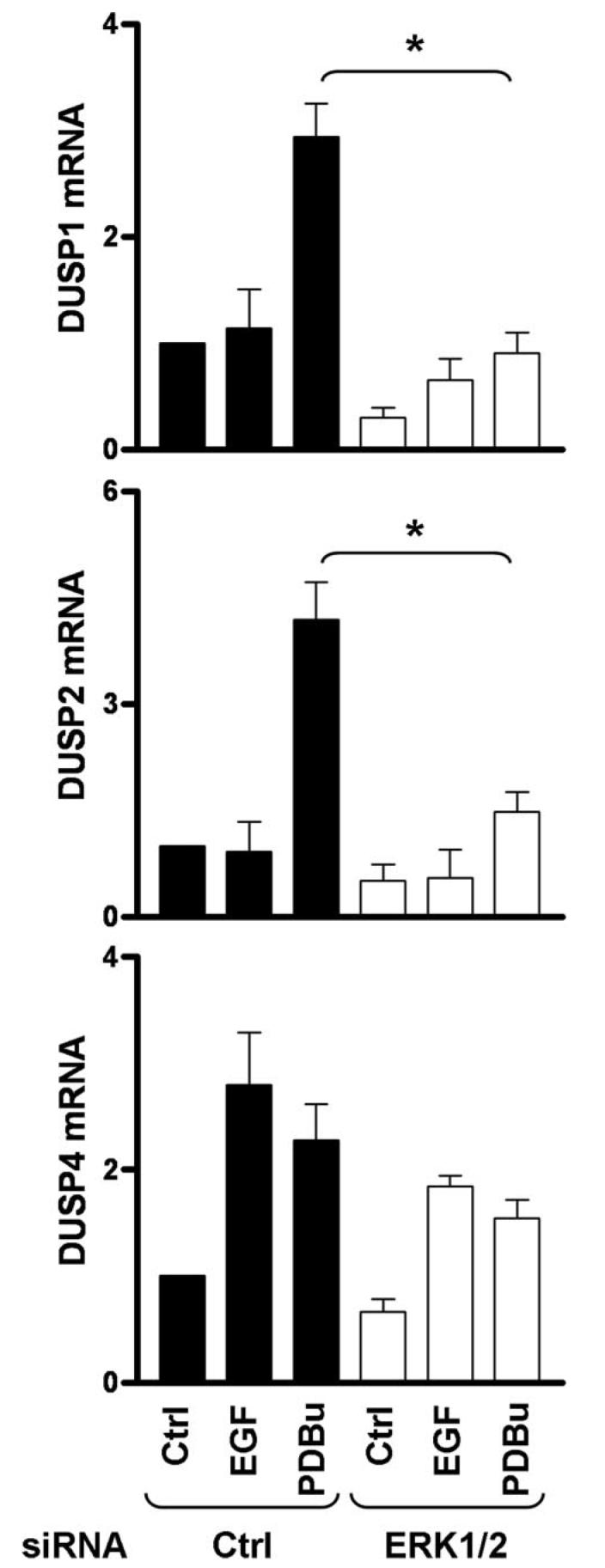
Cells were transfected in 6-well plates with control siRNAs (Ctrl) or ERK1/2 siRNAs prior to stimulation for 120 min with 10 nm EGF or 1 μm PDBu, as indicated. Total RNA isolates were analyzed for relative levels of DUSP1, -2, or -4 mRNA using qPCR as described under “Experimental Procedures.” Data are expressed as average normalized values obtained from three separate experiments, each with duplicate readings (mean ± S.E., n = 3). * = p < 0.05, comparing control siRNA-transfected cells to ERK1/2 siRNA-transfected cells, using Student's t test.
Western Blotting
HeLa cells were simultaneously plated and transfected in 6-well plates (2.5 × 105 cells/well) with 1 nm ERK1/2 siRNAs and 10 nm control or SMARTpool siRNAs prior to Ad transduction as above. Following treatment noted in figure legends, cells were lysed as described (27), prior to Western blotting. Total and ppERK1/2 were detected using polyclonal rabbit anti-total ERK1/2 and rabbit anti-ppERK1/2 (1:1000; Cell Signaling Technology, Hitchin, UK), respectively. Loading controls were assayed by staining parallel blots with mouse anti-β-actin (AC-15, 1:5000; Sigma). Antibodies were visualized by using horseradish peroxidase-linked secondary antibody and enhanced chemiluminescence kit (GE Healthcare).
Co-immunoprecipitation
HeLa cells were simultaneously plated and transfected in 9-cm plates (2 × 106 cells/plate) with 1 nm control or ERK1/2 siRNAs as described above. Sixteen hours after siRNA transfection, cells were transfected with 10 μg of pCR3.1, pMyc-DUSP1, pMyc-DUSP2, or pMyc-DUSP4 using Superfect (Qiagen) according to manufacturer's instructions, and in add-back experiments simultaneously transduced with 2 × 106 pfu/ml Ad wild-type, Y261A, or D319N ERK2-GFP vector in DMEM with 10% FCS. The Ad-containing medium was removed after 3 h and replaced with fresh DMEM supplemented with 0.1% FCS. The cells were then maintained for 16–24 h in culture prior to lysis and immunoprecipitation using a mammalian Myc tag Co-IP kit (Pierce) according to manufacturer's instructions. Whole cell lysate controls and Myc immunoprecipitates were assayed for ERK1/2, ERK2-GFP, and Myc-DUSP content by Western blotting with polyclonal rabbit anti-total ERK1/2 (Cell Signaling Technology) and mouse monoclonal anti-Myc (9E10, Santa Cruz Biotechnology, Santa Cruz, CA).
Quantitative PCR
HeLa cells were simultaneously plated and transfected in 6-well plates (2.5 × 105 cells/well) with 1 nm ERK1/2 siRNAs and 10 nm control or SMARTpool siRNAs prior to Ad transduction and serum starvation as described above. Cells were stimulated with 10 nm EGF or 1 μm PDBu before extraction of total RNA using the RNeasy kit according to the manufacturer's instructions (Qiagen). Contaminating genomic DNA was removed from columns using an additional DNase (Qiagen) digestion step. Complementary DNA was then prepared for 1 μg of each total RNA sample using a cloned avian myeloblastosis virus first-strand synthesis kit according to the manufacturer's instructions (Invitrogen). cDNAs were then quantified relative to expression of human GTPase-activating protein using the following primers: 5′-CAA CGA GGC CAT TGA CTT CAT AG-3′ and 5′-CAA ACA CCC TTC CTC CAG CA-3′ for DUSP1; 5′-AAA ACC AGC CGC TCC GAC-3′ and 5′-CCA GGA ACA GGT AGG GCA AG-3′ for DUSP2; 5′-CTG GTT CAT GGA AGC CAT AGA GT-3′ and 5′-CGC CCA CGG CAG TCC-3′ for DUSP4; and 5′-GGG AAG GTG AAG GTC GGA GT-3′ and 5′-GAG TTA AAA GCA GCC CTG GTG A-3′ for the human GTPase-activating protein internal control. PCR primers were mixed with 50 ng of reverse transcription-PCR template and SYBR Green PCR master mix (Applied Biosystems, Warrington, UK), and the comparative CT method was used to detect relative expression curves on an ABI PRISM 7500 detection system (Applied Biosystems).
Semi-automated Image Acquisition and Analysis
Cells were transfected with siRNA, transduced with Ad vectors, and plated as described above on Costar plain black-wall 96-well plates (Corning, Arlington, UK). For anti-Myc immunostaining experiments, cells were transfected with pCR3.1, pMyc-DUSP1, pMyc-DUSP2, or pMyc-DUSP4 using Superfect (Qiagen), according to manufacturer's instructions, simultaneously with viral transduction. Following treatment as noted in the figure legends, cells were washed in ice-cold phosphate-buffered saline (PBS) before fixation in 4% paraformaldehyde/PBS and permeabilization in −20 °C methanol. After blocking in 5% normal goat serum/PBS, cells were probed with rabbit anti-ppERK1/2 polyclonal antibody (1:300, Cell Signaling Technology) or mouse anti-Myc (Santa Cruz Biotechnology) in PBS. Alexa 546-conjugated goat anti-rabbit or Alexa 546-conjugated goat anti-mouse secondary antibody (1:300, Invitrogen) was used to visualize ppERK1/2 and Myc antibody binding, respectively. 4′,6-Diamidino-2-phenylindole (DAPI; 600 nm) in PBS was used to stain nuclei. Image acquisition in each well was performed on an IN Cell Analyzer 1000 (GE Healthcare) microscope, using a ×10 objective and 360-nm (DAPI), 475-nm (GFP), and 535-nm (Alexa 546) excitation filters, and monitored through 460-, 535-, and 620-nm emission filters, respectively, with a 61002 trichroic mirror (GE Healthcare). Analysis of ppERK1/2 activity and localization was performed using the Object Intensity algorithm in the IN Cell Analyzer Workstation (GE Healthcare) with images collected from 460- and 620-nm emission filters to define nuclear and cytoplasmic regions, respectively. ERK2-GFP localization and ppERK2 activity were simultaneously analyzed using the Multitarget Analysis algorithm (IN Cell Analyzer Investigator; GE Healthcare) with images collected from 460- and 535-nm emission filters to define nuclear and cytoplasmic regions, respectively. Single cells with levels of ERK2-GFP intensity yielding superphysiological ppERK2 responses in comparison with the endogenous ERK1/2 were deemed to be overexpressed and excluded from analysis (∼20% of cells) to prevent misleading localization data. Similarly, subpopulations of Myc-DUSP expressing cells were selected using filters for Myc-DUSP staining intensity within nuclei to ensure comparison of equal DUSP expression levels in single cells. 300–500 cells per field were typically analyzed, and up to four fields per well were captured in experiments performed in duplicate or quadruplicate, meaning that in each experiment data were normally derived from at least 1000 individual cells per time point. Imaging data are reported as ppERK intensity (mean fluorescence intensity per cell) or as a ratio of nuclear to cytoplasmic intensity (N:C ratio).
RESULTS AND DISCUSSION
Endogenous ERK1/2 Knockdown and Reconstitution with ERK2-GFP Fusion Protein
We have previously shown that EGF causes a transient activation of ERK (ppERK1/2 maximal at 5 min, returned to basal by 60 min), whereas stimulation of PKC with PDBu causes a more sustained ERK activation (maximal at 5–15 min, above basal for >4 h) in HeLa cells. These stimuli also caused ERK translocation to the nucleus as revealed by the increase in nuclear translocation of endogenous ERK1/2 or ERK2-GFP in cells transduced with Ad ERK2-GFP (28). However, the techniques used (Western blotting and confocal microscopy) are labor-intensive, and the assay necessitated co-transduction with MEK to recapitulate the normal cytoplasmic localization of ERK2 (in serum-starved cells) despite its overexpression. Accordingly, the initial aim of these experiments was to develop a more efficient model for exploring spatial and temporal aspects of ERK2 regulation without the overexpression of ERK2 binding partners. To do so, we have used siRNAs to knock down endogenous ERK1/2, recombinant Ad to add back fusion proteins of wild-type or mutated ERK2 and GFP, fluorescent labeling of ppERK1/2, and a wide field fluorescence microscope based platform for semi-automated image acquisition and analysis. Transfection with siRNA sequences targeted to noncoding regions of ERK1 and -2 (ERK1/2) transcripts reduced ERK1/2 expression and PDBu-induced phosphorylation (ppERK1/2) responses by >95%, as judged by Western blotting (Fig. 1A, bottom panel). Subsequent transduction with Ad ERK2-GFP (2 × 106 pfu/ml) restored ERK expression levels and the phosphorylation response (Fig. 1A, note the higher molecular weight of the ERK2-GFP fusion protein), which had comparable magnitude and kinetics to the response in control cells (Fig. 1A, top panel and data not shown).
We next assessed ERK activation and localization by quantitative fluorescence microscopy, defining nuclear regions with a DAPI stain so that ERK2-GFP or dual phosphorylated ERK2-GFP (ppERK2) could be measured in the nucleus and cytoplasm (as well as in whole cells). Outlines of nuclei and cells defined by automated analysis algorithms using DAPI and ppERK1/2 stains are shown in Fig. 1B. As expected, PDBu caused a rapid and sustained increase in whole cell ppERK1/2 levels in control siRNA-transfected cells. No such effect was seen in ERK1/2 siRNA-transfected cells, and the response seen in cells transfected with siRNA and transduced with Ad ERK2-GFP had comparable magnitude and kinetics to that seen in control cells (Fig. 1C). This activation of ERK was paralleled by a rapid and sustained increase in the nuclear to cytoplasmic (N:C) ratio of ERK2-GFP (Fig. 1D). Scatter plots of ppERK1/2 location within single cells revealed that PDBu increased ppERK1/2 labeling in the cytoplasm and nucleus of control cells (Fig. 1E). It had no such effect in cells transfected with ERK1/2 siRNAs alone, but it increased ppERK1/2 within both compartments of cells transfected with ERK1/2 siRNAs and transduced with Ad ERK2-GFP (Fig. 1E). Thus, semi-automated image acquisition and analysis provide an efficient means of monitoring spatial and temporal aspects of ERK activation, and the knockdown and add-back model recapitulates the key features of endogenous ERK activation (ERK2 expression level, ppERK2 level, and N:C distribution) that we considered necessary for further exploration of these response characteristics.
Comparison of EGF- and PKC-induced ERK2-GFP Activation and Trafficking
We next used the imaging system to define effects of EGF and PDBu on ERK2 signaling. In time course experiments, EGF caused a rapid and transient increase in whole cell ppERK2 levels with a maximum response at 5 min and reduction to basal levels within 60–120 min. PDBu caused a comparable rapid response (maximum at 5–15 min) with a subsequent reduction to ∼30% of peak values by 60 min (Fig. 2A), which was maintained for the duration of the experiment. Thus, the key distinction between these stimuli was the relatively sustained response to PDBu, which is similar in profile to other studies comparing transient and sustained kinetics of ERK activity (4, 5). Both stimuli also increased the proportion of ppERK2 within the nucleus, but responses were transient (having returned to basal values within 60 min), and similar to previous reports (29), the effect of PKC activation was much greater than that of EGF. This distinction is evident in time course experiments using data pooled from repeated experiments (Fig. 2C) and also in single cell data (Fig. 2, B, and ppERK2 panels from E), which illustrate the more pronounced effect of PDBu on nuclear ppERK2. When the distribution of ERK2-GFP was examined, all stimuli caused pronounced initial increases in N:C ERK2-GFP ratio, but the responses were very different after the first 5 min (Fig. 2D). PDBu induced a sustained but biphasic increase in N:C ERK2-GFP, with peaks at 5–15 and 120 min. However, the PDBu response did not drop below 70% of maximal at any time, whereas EGF caused only a transient increase with a peak at 5 min, reducing to <50% of the maximal response within 15 min (Fig. 2D). Thus, although EGF and PDBu cause comparable maximal activation of ERK2, the PDBu-induced ppERK2 response is relatively sustained (Fig. 2A), and it is associated with very high and sustained nuclear localization of ERK2-GFP in the dephosphorylated form (compare localization of active and total ERK2-GFP in Fig. 2, C and D). These stimulus-specific response profiles (Fig. 2, A, C and D) are indicative of stimulus-specific mechanisms controlling termination and compartmentalization of ERK signaling.
FIGURE 2. Spatiotemporal characteristics of EGF- and PDBu-stimulated ERK regulation.
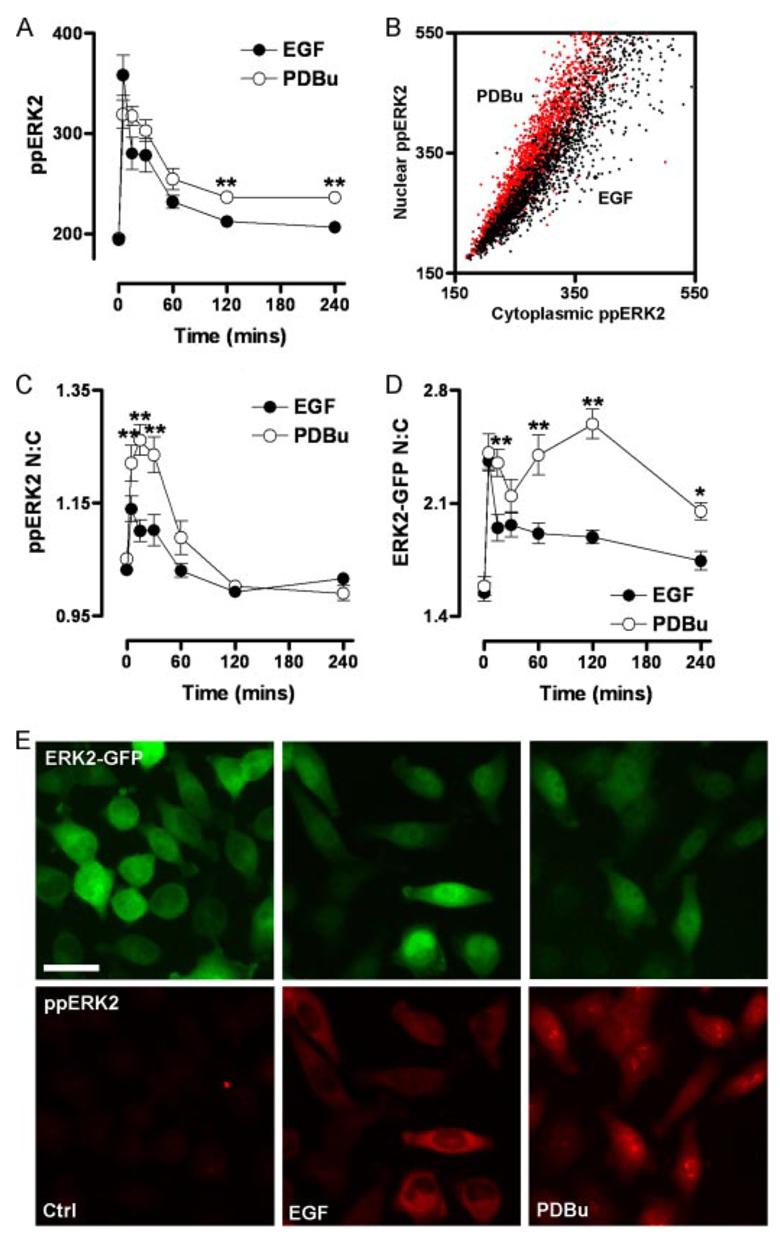
Cells were transfected in 96-well plates with ERK1/2 siRNAs and transduced with Ad ERK2-GFP prior to stimulation with 10 nm EGF or 1 μm PDBu for the times indicated. Cells were fixed and stained before image acquisition and analysis (as described under Fig. 1) for the calculation of whole-cell ppERK2 intensity (A), the N:C ppERK2 ratio (C), and the N:C ERK2-GFP ratio (D). The scatterplot (B) shows the relationship between nuclear and cytoplasmic ppERK2 staining for individual cells stimulated with EGF or PDBu, as indicated, for 5 min. E, representative images of cells in the ERK2-GFP expression range used in analysis are shown of unstimulated cells (Ctrl) or cells treated with 10 nm EGF (5 min) or 1 μm PDBu (15 min) as indicated for fields acquired from ERK2-GFP and ppERK2 signals. Bar, 20 μm. Data shown in A, C, and D were acquired from seven separate experiments, each with duplicate wells (mean ± S.E., n = 7). Each individual experiment included internal control cells transfected with control GFP siRNAs or ERK1/2 siRNAs without addition of Ad ERK2-GFP. In all cases, the ppERK1/2 signal was inhibited >95% by ERK1/2 siRNAs and was recovered by Ad ERK2-GFP transduction at all time points of stimulation by all stimuli (not shown). For figures A, C, and D, * = p < 0.05 and ** = p < 0.01, comparing EGF with PDBu-treated cells using two-way ANOVA and Bonferroni post hoc tests.
Stimulus- and ERK-dependent Regulation of Nuclear Inducible DUSP Transcripts
Because differences in transient and sustained ERK regulation in the nucleus are thought to involve the nuclear inducible MKPs (7, 8, 23), we tested for effects of EGF and PDBu on expression of DUSP1, -2, and -4 by quantitative PCR and also tested for mediation by ERK using ERK1/2 siRNAs. As shown, EGF had little or no effect on transcription of DUSP1 or -2 but increased DUSP4 mRNA almost 3-fold in cells treated with control siRNA (Fig. 3). In contrast, PDBu caused robust increases in levels of all three mRNAs in control cells (Fig. 3). The effect of PDBu on DUSP1 and -2 mRNA was decreased by transfection with ERK1/2 siRNAs (Fig. 3), whereas the induction of DUSP4 was not significantly reduced, indicating ERK-independent activation of DUSP4 transcription.
DUSP1, -2, and -4 Coordinately Regulate the PKC-mediated Nuclear Shuttling and Dephosphorylation of ERK
The data in Fig. 3 suggests that DUSP1, -2, and -4 transcription may play a role in stimulus-specific regulation of ERK2, so we tested the roles of these phosphatases using siRNAs to reduce their expression (see Fig. 6 for validation of mRNA knockdown). siRNAs against DUSP1, -2, or -4 had no measurable effect on ppERK2 responses to PDBu or EGF in cells expressing ERK2-GFP (Fig. 4, upper panels, and data not shown). However, the effect of PDBu on the N:C ERK2-GFP ratio was inhibited by DUSP2 siRNA at 60–120 min and by DUSP4 siRNA at 5–120 min but was actually increased by DUSP1 siRNA at 5–240 min. With such profound stimulus-specific effects on ERK2-GFP distribution, we suspected that the lack of effect on ppERK2 levels could be due to compensation or redundancy and therefore determined the effect of the combined knockdown. As shown (Fig. 4, lower panels), treatment with siRNAs targeting DUSP1, -2, and -4 in combination significantly increased the effect of PDBu on ppERK2 levels (at 5–60 min) and reduced its effect on N:C ERK2-GFP ratio, revealing that the three DUSPs mediate both dephosphorylation and nuclear retention of ERK2 in PDBu-stimulated cells. The combined DUSP1, -2, and -4 knockdown did not significantly alter ERK2-GFP N:C ratios in response to EGF (not shown), revealing that this regulation is stimulus-specific.
FIGURE 6. Regulation of DUSP transcription by DUSP siRNAs.
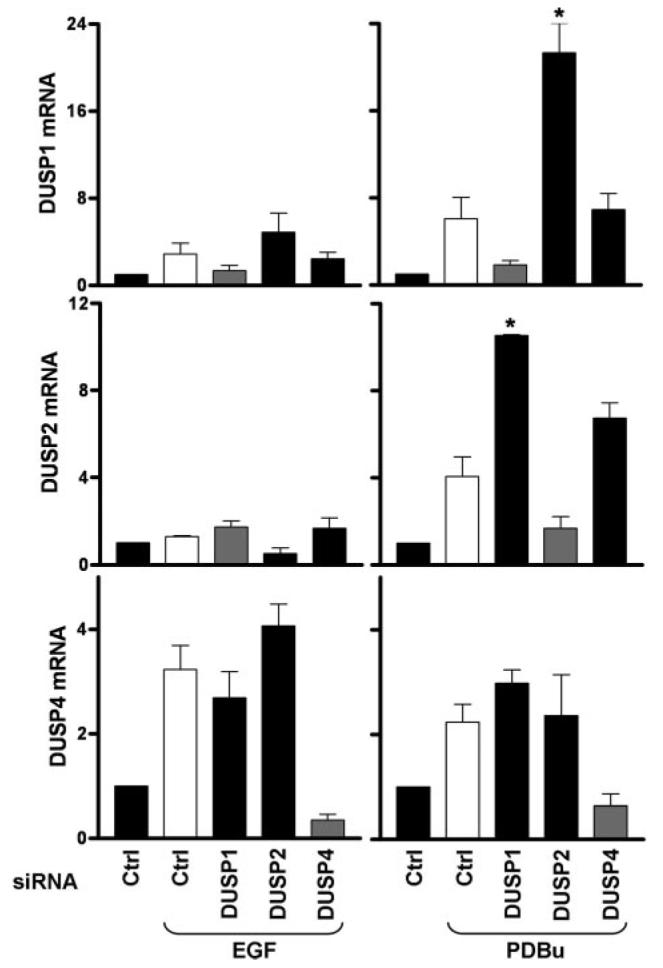
Cells were transfected in 6-well plates with ERK1/2 siRNAs and 10 nm control (Ctrl) or individual DUSP1, -2, or -4 siRNAs (as indicated) before addition of Ad ERK2-GFP and stimulation with 10 nm EGF or 1 μm PDBu as indicated for 120 min. Total RNA isolates were analyzed for relative levels of DUSP1, -2, or -4 mRNA using qPCR protocols as described under “Experimental Procedures.” Data are expressed as average normalized values obtained from three separate experiments, each with duplicate readings (mean ± S.E., n = 3). * = p < 0.05 comparing control siRNA-transfected cells to DUSP siRNA-transfected cells stimulated with the same ligand, using Student's t test.
FIGURE 4. Stimulus-specific effects of nuclear inducible DUSP siRNAs alone and in combination.
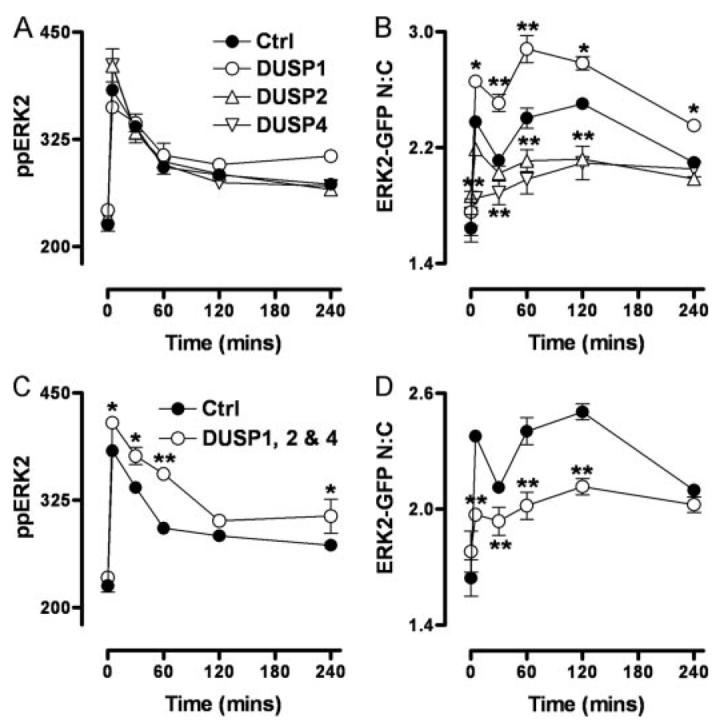
A and B, cells were transfected in 96-well plates with ERK1/2 siRNAs alongside control (Ctrl) or individual DUSP1, -2, or -4 siRNA SMARTpools as indicated, prior to transduction with ERK2-GFP. For triple knockdowns (C and D), cells were transfected with ERK1/2 siRNAs and DUSP1, -2, and -4 siRNA SMARTpools in combination (as indicated) before addition of Ad ERK2-GFP. Cells were stimulated with 1 μm PDBu (circles) for the times indicated, prior to staining and imaging (as described under Fig. 1). Data obtained from control siRNA-transfected cells are included in all plots for clarity. Data shown are ppERK2 intensity values (A and C) and N:C ERK2-GFP ratios (B and D) from four separate experiments (mean ± S.E., n = 4). * = p < 0.05 and ** = p < 0.01, comparing control siRNA-transfected cells to DUSP siRNA-transfected cells using two-way ANOVA and Bonferroni post hoc tests.
Effects of DUSP Overexpression on ERK2-GFP Activation and Trafficking
To determine the effects of DUSP overexpression on ERK responses, HeLa cells were transiently transfected with Myc-tagged DUSP1, -2, or -4. Automated imaging revealed a transfection efficiency of ∼50%, as well as comparable expression levels and the expected nuclear localization of the tagged phosphatases (not shown). PDBu caused the expected robust increase in ppERK1/2 staining, and this was abolished in cells expressing Myc-tagged DUSP1, -2, or -4 (not shown), which echoes published work (11). In cells transfected with siRNAs against ERK1/2 and transduced with ERK2-GFP, expression of DUSP2 or -4 significantly increased the N:C ERK2-GFP ratio, whereas ratios were unaltered by expression of DUSP1 (Fig. 5B). We also used immunoprecipitation (anti-Myc) and Western blotting (anti-ERK and anti-Myc) to explore binding of ERK to these DUSPs. This again revealed comparable expression levels for the tagged phosphatases and robust co-immunoprecipitation of ERK2-GFP with DUSP2 and DUSP4 but not with DUSP1 (Fig. 5A). This pattern was also seen with endogenous ERK1/2 (not shown). Thus, although all three of these DUSPs cause comparable inhibition of PDBu-stimulated ERK phosphorylation, this selective immunoprecipitation suggests that, like DUSP5 (10), DUSP2 and -4 have higher affinity for dephosphorylated ERK than DUSP1. This distinction presumably explains why overexpression of DUSP2 and -4 (but not DUSP1) increases the nuclear localization of ERK2-GFP in intact cells (Fig. 5B), just as siRNA knockdown of DUSP2 and -4 (but not DUSP1) reduces the PDBu-stimulated nuclear translocation of ERK2-GFP (Fig. 4).
FIGURE 5. Differential binding and nuclear accumulation of ERK2-GFP by Myc-tagged DUSPs.
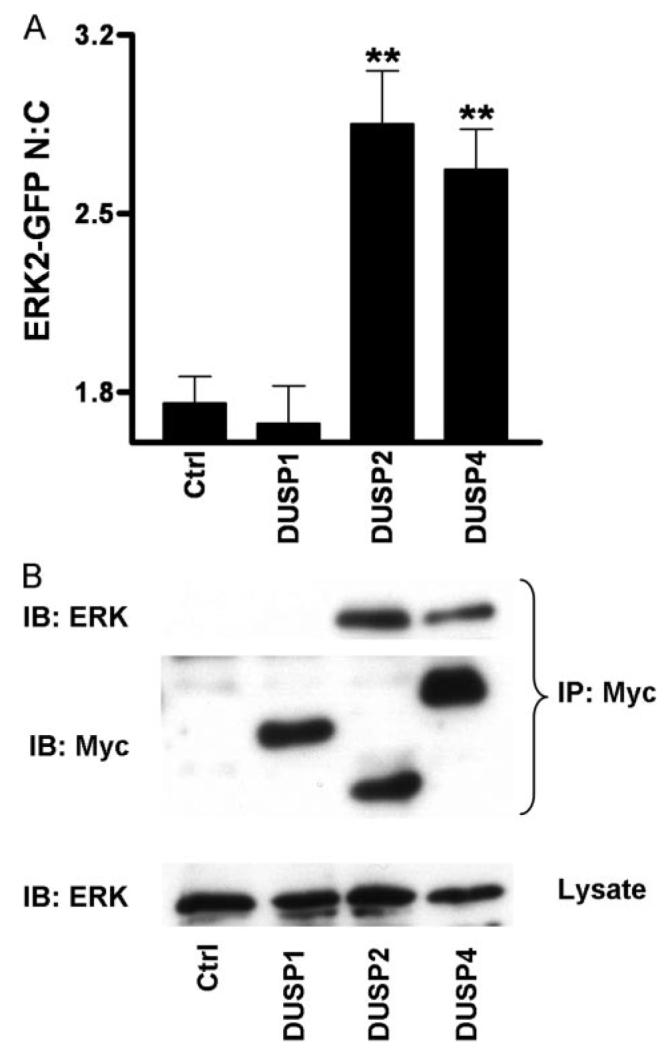
A, cells were transfected in 96-well plates with ERK1/2 siRNAs prior to being simultaneously transduced with Ad ERK2-GFP and transfected with pCR3.1 (Ctrl), or Myc-tagged DUSP1, DUSP2, or DUSP4 constructs as indicated, prior to fixation and staining with anti-Myc antibodies as described under “Experimental Procedures.” Cells expressing comparable levels of Myc-tagged DUSP1, -2, and -4 were compared with control cells in four independent experiments (mean ± S.E., n = 4). ** = p < 0.01, comparing control (ctrl) cells to Myc-DUSP-transfected cells using one-way ANOVA and Dunnet's post hoc test. B, cells were transfected as above in 9-cm plates with ERK1/2 siRNAs prior to being simultaneously transduced with Ad ERK2-GFP and transfected with pCR3.1 (Ctrl), or Myc-tagged DUSP1, DUSP, or DUSP4 constructs as indicated. Cells were lysed and immunoprecipitated with immobilized anti-Myc as described under “Experimental Procedures.” Representative immunoblots for Myc (IB: Myc) and for ERK (IB: ERK) from anti-Myc immunoprecipitates (IP: Myc) and whole cell lysates (lysate) are shown from four independent experiments showing similar data.
Effects of DUSP Knockdown on EGF- and PDBu-stimulated DUSP Expression
We suspected that DUSP knockdown might cause compensatory changes in the effects of EGF or PDBu on expression of other DUSPs and explored this by quantitative PCR. EGF had little or no effect on DUSP1 and -2 mRNA levels but caused a pronounced increase in DUSP4, whereas PDBu caused marked increases in expression of all three mRNAs (Fig. 6, see also Fig. 4). Each siRNA mixture prevented any significant induction of the mRNA to which it was targeted without reducing levels of the other mRNAs, demonstrating the efficacy and specificity of these knockdowns. Interestingly, pronounced compensatory increases in transcription were seen under some conditions. Thus, knockdown of DUSP2 actually increased the effect of PDBu on transcription of DUSP1 (Fig. 6, top right panel). Similarly, knockdown of DUSP1 increased the effect of PDBu on DUSP2 transcription (Fig. 6, middle right panel). Thus, the lack of effect of the single DUSP knockdowns on PDBu-stimulated ppERK levels (Fig. 4) may be, at least in part, because of compensatory increases in expression of the other DUSPs (Fig. 6), and this may explain why it is necessary to knock down all three of these DUSPs to enhance the PDBu effect on ERK phosphorylation (Fig. 4).
Effects of Docking Domain Mutation on ERK2-GFP Activation and Trafficking
ERKs bind to many proteins containing modular D- or DEF-motifs, and because these proteins include scaffolds and anchors, perturbation of such binding can provide insight into the roles of these binding partners in ERK compartmentalization and signaling. D319N mutation interferes with the ability of ERK to form D-domain interactions with phosphatases and other substrates, and it is analogous to the seven-maker gain of function mutation in Drosophila (10-13, 18, 20, 30). Y261A mutation of ERK impairs binding to substrate DEF-domains (13, 14), some of which are predicted on DUSPs (16, 17). These mutations do not affect the intrinsic kinase activity of ppERK2 (13), and so we introduced D319N and Y261A mutations in our ERK2-GFP sequence to examine the molecular mechanisms involved in DUSP regulation of ERK. Cells transfected with ERK1/2 siRNAs were transduced with Ad expressing wild-type (WT), Y261A, or D319N ERK2-GFP. Western blotting revealed similar expression levels for endogenous ERK1/2 and all three exogenous ERK2-GFP constructs, as well as the lack of stimulus-independent activity and clear activation by EGF (Fig. 7A). Cell imaging revealed that stimulation with EGF or PDBu caused a pronounced and dose-dependent increase in ppERK2 staining and ERK2-GFP translocation after knockdown of endogenous ERKs and add-back of WT ERK2-GFP, whereas no measurable increase in ppERK2 staining was seen in cells receiving the ERK1/2 siRNAs alone (Fig. 7B and not shown). Similar dose-dependent increases in ppERK2 were seen with add-back of the WT and mutated fusion proteins (Fig. 7B and not shown). For all stimuli, minimum, maximum, and EC50 values were all unaffected by the ERK mutations (logEC50 values for ppERK2 activation in cells expressing WT ERK2-GFP and stimulated with EGF or PDBu were −10.3 ± 0.5 and −6.6 ± 0.3, respectively), confirming that these mutations do not impede ERK activation (13). These mutations did, however, influence either basal (unstimulated) ERK2-GFP distribution or its response to activation (Fig. 7C). Thus, the Y261A mutation increased the N:C ERK2-GFP ratio from 1.45 to 1.83 in control cells without preventing the response to stimulus (i.e. the increment in N:C ratio caused by EGF was indistinguishable for WT and Y261A ERK2-GFP). This indicates that DEF-domain interactions are important in stimulus-independent compartmentalization of ERK2 and is in accord with recent studies showing the importance of this domain for phosphorylation-independent association with nuclear shuttling machinery (31, 32). In contrast, the D319N mutation had no measurable effect on basal N:C ratio but reduced the translocation response to stimulus, with the increment in N:C ratio caused by EGF activating D319N ERK2-GFP only 50% of that seen with activation of WT ERK2-GFP (Fig. 7C). Similar data were obtained when PDBu was used as the stimulus, and ligand potencies (log EC50 values) were not significantly different for the three ERK2-GFP constructs (data not shown).
FIGURE 7. Influence of docking domains on ERK signaling.
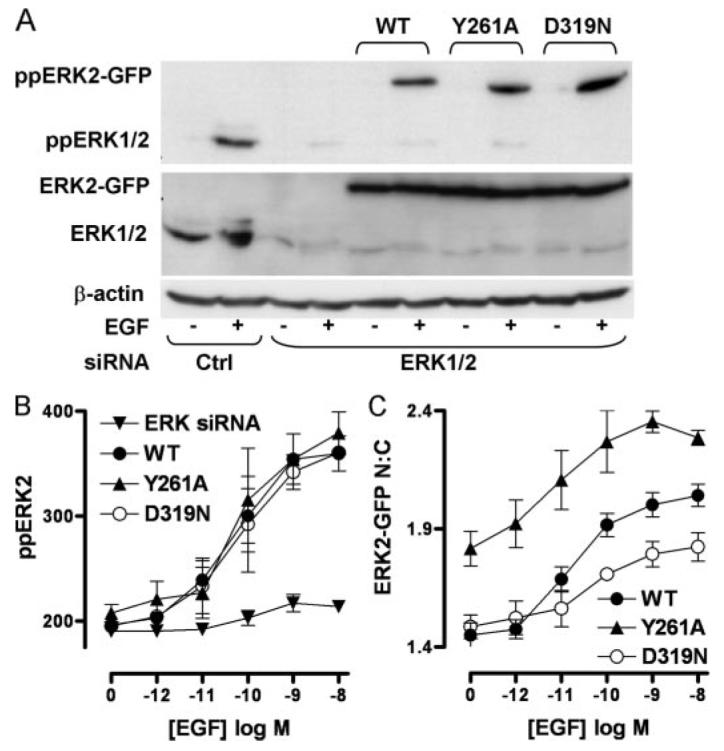
Cells transfected with control siRNAs (Ctrl) or ERK1/2 siRNAs were transduced with Ad WT or Y261A or D319N-mutated ERK2-GFP and analyzed for activation and localization as follows. A, cells were harvested following stimulation with (+) or without (−) 10 nm EGF (5 min). ERK1/2 protein levels and ppERK1/2 activation were assessed by immunoblotting with anti-ERK1/2 (middle panel) and anti-ppERK1/2, respectively. Immunoblotting for β-actin (bottom panel) was used as a loading control. Data shown are representative of three independent experiments showing similar data. B and C, cells were stimulated in 96-well plates with the indicated concentrations of EGF for 5 min and stained before image acquisition and analysis (as described under Fig. 1) for the calculation of whole-cell ppERK2 intensity (B), and the N:C ERK2-GFP ratio (C). Data shown in B and C are pooled from three independent experiments, each performed in triplicate (mean ± S.E., n = 3).
Stimulus-specific Spatiotemporal Regulation of ERK2 Is Mediated through Docking Domains
The D- and DEF-domains of ERK binding partners play key roles in determining cellular responses. For example, interference with D-domain binding not only reduces the ability of DUSP1, -2, and -4 to dephosphorylate ERK (11) but also potentiates serum-induced ERK phosphorylation (23) and prevents DUSP5 from causing its nuclear accumulation (10). DUSP1 and -4 are also predicted to bind ERK via DEF-domains, but their role has not been examined in this context. To further examine the functional relevance of these regions in nuclear inducible MKPs, we used our model to test for effects of protein synthesis inhibition (using cycloheximide, CHX) and blockade of D- or DEF-domains. As shown (Fig. 8A, top panels), CHX had no effect on basal ppERK2 levels in cells expressing WT ERK2-GFP and also failed to influence the amplitude or kinetics of the response to EGF. It also had no influence on the initial response to PDBu but caused a marked prolongation of the effect, completely preventing the reduction in ppERK2 seen between 30 and 60 min and thereby significantly increasing ppERK2 levels at 60–240 min. When DEF-domain binding was impaired (i.e. in cells expressing Y261A ERK2-GFP), the amplitude and kinetics of the ppERK2 response to EGF was unaltered, whereas PDBu-induced ppERK2 levels were significantly increased with Y261A ERK2-GFP (Fig. 8A, middle panels). In contrast to this ligand-dependent effect however, inhibition of D-domain binding (D319N mutation) caused more sustained responses to both stimuli (Fig. 8A, bottom panels), which is consistent with its widespread or universal role in phosphatase interaction with ERK2.
FIGURE 8. Effects of CHX and docking domain mutation on ERK2-GFP distribution and induction of DUSP mRNA.

A and B, cells were transfected in 96-well plates with ERK1/2 siRNAs and transduced with either Ad WT ERK2-GFP, Y261A-mutated ERK2-GFP (Y261A, or D319N-mutated ERK2-GFP (D319N). Cells represented in the top panels were treated with 30 μm CHX for 30 min before all cells were stimulated with 10 nm EGF or 1 μm PDBu as indicated in internally controlled experiments. Data obtained from stimulated WT ERK2-GFP transduced cells are included in all graphs (closed circles) showing comparison with simultaneous CHX treatment (top panels), Y261A mutation (middle panels), and D319N mutation (bottom panels) for clarity (each test condition is represented by open circles). Data shown are ppERK2 intensity (A) and ERK2-GFP N:C ratios (B) obtained from five separate experiments, each with duplicate wells (mean ± S.E., n = 3–5). * = p < 0.05 and ** = p < 0.01, comparing WT to CHX, Y261A, or D319N conditions, according to two-way ANOVA and Bonferroni post hoc tests. C, cells were transfected in 6-well plates with control siRNAs (Ctrl) or ERK1/2 siRNAs and transduced with Ad WT or Y261A or D319N-mutated ERK2-GFP prior to stimulation with 10 nm EGF or 1 μm PDBu as indicated for 120 min. Total RNA isolates were analyzed for relative levels of DUSP2 mRNA using qPCR protocols described under “Experimental Procedures.” Data shown are normalized values obtained from three separate experiments, each with duplicate readings (mean ± S.E., n = 3). ** = p < 0.01, comparing WT ERK2-GFP expressing cells with D319N ERK2-GFP expressing cells, using Student's t test.
Exploring the effects of these manipulations on the distribution of ERK2-GFP, we found that both the transient increase in ERK2-GFP N:C ratio elicited by EGF and the sustained increase caused by PDBu were inhibited by CHX (Fig. 8B, top panels), despite the fact that CHX increased the effect of PDBu on ppERK2 in the same cells (compare top panels in Fig. 8, A and B). In contrast, the Y261A mutation increased both basal and stimulated levels of N:C ERK2-GFP ratio at nearly all time points despite only having effects on ppERK2 levels in response to PDBu (compare middle panels in Fig. 8, A and B), further supporting a role for the DEF-domain in mediating stimulus-independent shuttling of ERK2. Similar to CHX treatment, D319N mutations reduced both EGF- and PDBu-mediated increases in N:C ratio of ERK2-GFP, which is again despite the fact that ppERK2 levels are increased in the same cells (compare bottom panels in Fig. 8, A and B). We also determined the effects of these manipulations in combination and found no additive effects of CHX and the Y261A or D319N mutations on ppERK levels or N:C ERK2-GFP values in control, PDBu-stimulated, or EGF-stimulated cells (not shown).
We found that the influence of these mutations on DUSP mRNA induction by EGF and PDBu were indistinguishable in cells expressing wild-type or Y261A-ERK2-GFP. However, expression of the D319N mutant significantly increased effects of EGF and PDBu on DUSP2 (but not DUSP1 or -4) mRNA levels. This demonstrates that the ability of this mutation to prolong ERK phosphorylation (Fig. 8A, lower panels) is functionally relevant in terms of amplifying this downstream effect.
Influence of D- and DEF-domain Mutations on the Ability of DUSPs to Bind ERK and Influence ERK Compartmentalization
In the final series of experiments we co-expressed the WT and mutant ERK2-GFP constructs with the Myc-tagged DUSPs (1, 2, and 4) and then tested for co-immunoprecipitation and ERK2-GFP localization. As expected, we were able to co-immunoprecipitate the WT ERK2-GFP with DUSP2 and -4 (but not with DUSP1), and expression of these DUSPs increased the proportion of WT ERK2-GFP in the nucleus, whereas expression of DUSP1 failed to do so (Fig. 9, see also Fig. 5). Very similar data were obtained with the DEF-domain mutant (Y261A ERK2-GFP), despite the fact that this mutant was able to enhance sustained ppERK2 responses and that DUSP4 contains a predicted DEF-domain. In contrast, the D-domain mutant (D319N ERK2-GFP) was not co-immunoprecipitated by any of the DUSPs. Moreover, expression of DUSP1 and -4 failed to alter the localization of D319N ERK2-GFP and, although DUSP2 did increase the proportion of this D-domain mutant ERK in the nucleus, the effect was less marked than with the wild-type or DEF-domain mutant (Fig. 9). The simplest interpretation of these data is that, similar to DUSP5 (10), DUSP2 and -4 have higher affinity than DUSP1 for dephosphorylated ERK2 (as evidenced by the selective co-immunoprecipitation and effects on nuclear localization) and that their stable interaction with ERK is dependent upon D-domain s but not DEF-domains (as evidenced by the inhibitory effect of the D319N, but not the Y261A, mutation).
FIGURE 9. Docking domain dependence of binding and nuclear accumulation of ERK2-GFP by Myc-tagged DUSPs.
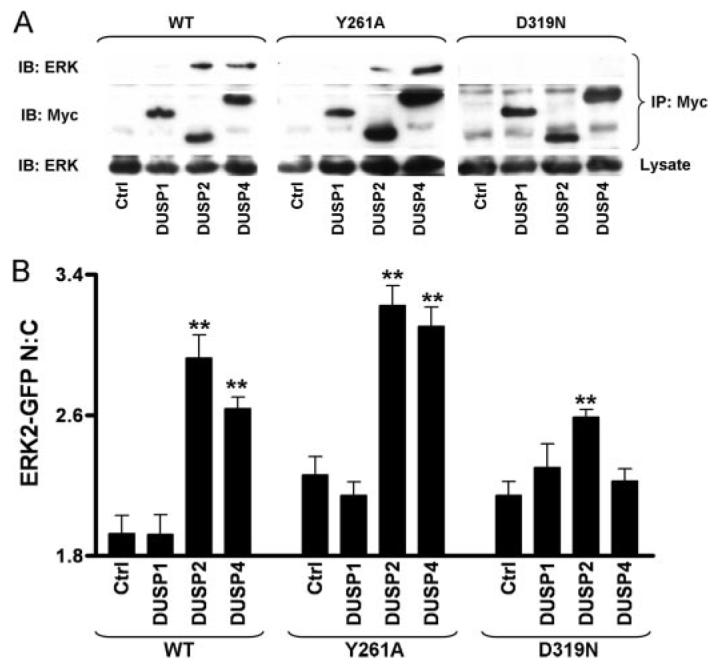
A, cells were transfected in 9-cm plates with ERK1/2 siRNAs prior to being simultaneously transduced with either Ad WT ERK2-GFP, Y261A-mutated ERK2-GFP (Y261A), or D319N-mutated ERK2-GFP (D319N) and transfected with pCR3.1 (Ctrl) or Myc-tagged DUSP1, DUSP2, or DUSP4 constructs as indicated. Cells were lysed and immunoprecipitated with immobilized anti-Myc as described under “Experimental Procedures.” Representative immunoblots for Myc (IB: Myc) and for ERK (IB: ERK) from anti-Myc immunoprecipitates (IP: Myc) and whole cell lysates (lysate) are shown from two independent experiments showing similar data. B, cells were transfected in 96-well plates with ERK1/2 siRNAs prior to being simultaneously transduced with either Ad WT ERK2-GFP, Y261A-mutated ERK2-GFP (Y261A), or D319N-mutated ERK2-GFP (D319N) and transfected with pCR3.1 (Ctrl) or Myc-tagged DUSP1, DUSP2, or DUSP4 constructs as indicated, prior to fixation and staining with anti-Myc antibodies as described under “Experimental Procedures.” Cells expressing comparable levels of Myc-tagged DUSP1, -2, and -4 were compared with control cells in four independent experiments (mean ± S.E., n = 6). ** = p < 0.01, comparing control cells to Myc-DUSP-transfected cells using one-way ANOVA and Dunnet's post hoc test.
Summary and Model
It is now well established that spatial and temporal aspects can dictate the biological outcomes of ERK activation. Transient versus sustained activation of ERK governs the difference between quiescence and proliferation in many cell types, as does the localization of ERK activity within the nucleus or cytosol (2-4, 6, 22, 23). Despite its importance, the mechanisms of ERK nuclear regulation are still poorly understood. Here, we have explored how nuclear inducible MKPs shape ERK responses to EGF and PKC. We show that PKC activation causes a sustained activation and nuclear accumulation of ERK. These characteristics are dictated by the inducible nuclear DUSP1, -2, and -4, as evidenced by the effects of DUSP knockdown and overexpression, as well as by potentiation of ppERK2 signals by CHX, D-, and DEF-domain mutations. In contrast, EGF causes relatively transient activation and nuclear accumulation of ERK that is insensitive to CHX and is presumably therefore terminated by pre-existing (rather than nuclear inducible) phosphatases (Fig. 9) (33).
To our knowledge, our data show the first definitive evidence that endogenous (rather than overexpressed) nuclear inducible DUSPs shape ERK responses to sustained stimulation. Moreover, they reveal a remarkable degree of adaptation at the level of DUSP expression that is clearly important for data interpretation. Notably, knockdown of DUSP1 increased PDBu-stimulated DUSP2 transcription, just as knockdown of DUSP2 increased PDBu-stimulated DUSP1 transcription. Thus, although knockdown of individual DUSPs did not influence ppERK2 responses to PDBu, compensatory changes in expression of other DUSPs may have obscured the roles of the individual DUSPs, necessitating combined knockdown to reveal the involvement of DUSP1, -2, and -4 in the ppERK2 response to PDBu (Fig. 4). Our data also reveal important functional distinctions between the nuclear inducible MKPs that may underlie their selective effects on ERK activity and compartmentalization. Importantly, we have found that DUSP2 and -4 co-immunoprecipitate ERK2-GFP and cause its accumulation within the nucleus in a manner dependent on D-domain motifs, whereas neither effect is seen with overexpression of DUSP1 (Figs. 5 and 9). This is intriguing given that the D-domain interaction is essential for efficient dephosphorylation of ERK by DUSP1, -2, and -4 (11, 20). This may be explained by a model postulated by Tanoue et al. (15), in which subtle differences in the DUSP D-domain govern the stability of substrate binding and that there is discordance between MAPK binding to DUSPs and their ability to activate the phosphatase catalytically (19, 20). For example, DUSP1 cannot stably interact with ERK but co-immunoprecipitates with its other substrates, JNK and p38 (15), despite having comparable catalytic activity for phosphorylated ERK and p38 (20). Similarly, DUSP4 preferentially dephosphorylates ERK and JNK but stably associates with only ERK and p38 (19). The implication in our data is that the D-domains of DUSP2 and -4 confer the ability to both inactivate and anchor ERK within the nucleus, whereas DUSP1 may dephosphorylate ERK within the nucleus but then release it. The first mechanisms would cause sustained nuclear accumulation of inactive ERK and consequent inhibition of ERK activation, whereas the latter could facilitate reactivation in the cytoplasm in the presence of persistent upstream stimuli (Fig. 10). Consistent with this idea, we found that knockdown of DUSP1 actually increased the nuclear location of ERK2-GFP without altering the whole cell ppERK2 response (Fig. 4). Similarly, knockdown of DUSP2 had no effect on the whole cell ppERK2 response but reduced the nuclear location of ERK2-GFP, presumably because of greater reliance on DUSP1 for dephosphorylation and consequent release (rather than scaffolding) of ERK from the nucleus (Figs. 4 and 6). We suggest that DUSP1, -2, and -4 act together to regulate ERK2 dephosphorylation and compartmentalization (in response to sustained PKC-mediated signaling), and our data argue in favor of distinct but coordinated DUSP effects (rather than functional redundancy). This specificity highlights the need for future work to determine why these termination mechanisms are stimulus-specific.
FIGURE 10. Model of nuclear ERK dephosphorylation and traffic during sustained ERK responses.
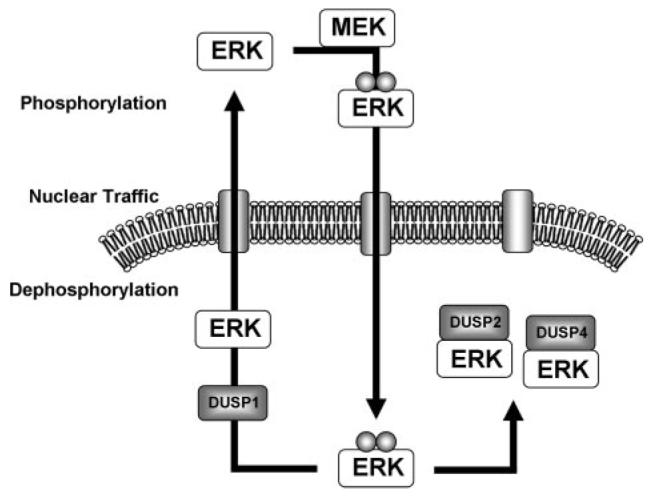
Phosphorylation by MEK in the cytoplasm causes translocation of nontethered phosphorylated ERK to the nucleus. Our data suggest that DUSP1, -2, and -4 transcription is induced, and each protein contributes to the dephosphorylation of ERK whereas DUSP1 releases it. Thus DUSP2 and -4 expression causes nuclear accumulation of ERK as a negative feedback mechanism, whereas DUSP1 allows it to return to the cytosol for reactivation.
Footnotes
This work was supported by Wellcome Trust Project and Equipment Grants 062918, 076557, and 078407.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: ERK, extracellular signal-regulated kinase; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; MEK, MAPK/ERK kinase; EGF, epidermal growth factor; MKP, MAPK phosphatase; DUSP, dual specificity phosphatase; JNK, c-Jun N-terminal kinase; D, docking; DEF, docking site for ERK, FXFP; siRNA, short inhibitory RNA; GFP, (enhanced) green fluorescent protein; PDBu, phorbol 12,13-dibutyrate; CHX, cycloheximide; Ad, adenovirus; PBS, phosphate-buffered saline; qPCR, quantitative PCR; hGAP, human GTPase-activating protein; DAPI, 4′,6-diamidino-2-phenylindole; N:C ratio, nuclear:cytoplasmic ratio; DMEM, Dulbecco's modified Eagle's medium; FCS, fetal calf serum; WT, wild type; ANOVA, analysis of variance; pfu, plaque-forming unit.
REFERENCES
- 1.Caunt CJ, Finch AR, Sedgley KR, McArdle CA. Trends Endocrinol. Metab. 2006;17:276–283. doi: 10.1016/j.tem.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Ebisuya M, Kondoh K, Nishida E. J. Cell Sci. 2005;118:2997–3002. doi: 10.1242/jcs.02505. [DOI] [PubMed] [Google Scholar]
- 3.Murphy LO, Blenis J. Trends Biochem. Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Nat. Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Curr. Biol. 2006;16:1171–1182. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 6.Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Owens DM, Keyse SM. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 8.Jeffrey KL, Camps M, Rommel C, Mackay CR. Nat. Rev. Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- 9.Franklin CC, Kraft AS. J. Biol. Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 10.Mandl M, Slack DN, Keyse SM. Mol. Cell. Biol. 2005;25:1830–1845. doi: 10.1128/MCB.25.5.1830-1845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. J. Biol. Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- 12.Tanoue T, Adachi M, Moriguchi T, Nishida E. Nat. Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 13.Dimitri CA, Dowdle W, MacKeigan JP, Blenis J, Murphy LO. Curr. Biol. 2005;15:1319–1324. doi: 10.1016/j.cub.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 14.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Mol. Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 15.Tanoue T, Yamamoto T, Nishida E. J. Biol. Chem. 2002;277:22942–22949. doi: 10.1074/jbc.M202096200. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Genes Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou B, Wu L, Shen K, Zhang J, Lawrence DS, Zhang ZY. J. Biol. Chem. 2001;276:6506–6515. doi: 10.1074/jbc.M009753200. [DOI] [PubMed] [Google Scholar]
- 18.Bott CM, Thorneycroft SG, Marshall CJ. FEBS Lett. 1994;352:201–205. doi: 10.1016/0014-5793(94)00958-9. [DOI] [PubMed] [Google Scholar]
- 19.Chen P, Hutter D, Yang X, Gorospe M, Davis RJ, Liu Y. J. Biol. Chem. 2001;276:29440–29449. doi: 10.1074/jbc.M103463200. [DOI] [PubMed] [Google Scholar]
- 20.Slack DN, Seternes OM, Gabrielsen M, Keyse SM. J. Biol. Chem. 2001;276:16491–16500. doi: 10.1074/jbc.M010966200. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Zhou B, Zheng CF, Zhang ZY. J. Biol. Chem. 2003;278:29901–29912. doi: 10.1074/jbc.M303909200. [DOI] [PubMed] [Google Scholar]
- 22.Lenormand P, Brondello JM, Brunet A, Pouyssegur J. J. Cell Biol. 1998;142:625–633. doi: 10.1083/jcb.142.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volmat V, Camps M, Arkinstall S, Pouyssegur J, Lenormand P. J. Cell Sci. 2001;114:3433–3443. doi: 10.1242/jcs.114.19.3433. [DOI] [PubMed] [Google Scholar]
- 24.Brondello JM, McKenzie FR, Sun H, Tonks NK, Pouyssegur J. Oncogene. 1995;10:1895–1904. [PubMed] [Google Scholar]
- 25.Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- 26.Hislop JN, Madziva MT, Everest HM, Harding T, Uney JB, Willars GB, Millar RP, Troskie BE, Davidson JS, McArdle CA. Endocrinology. 2000;141:4564–4575. doi: 10.1210/endo.141.12.7813. [DOI] [PubMed] [Google Scholar]
- 27.Hislop JN, Everest HM, Flynn A, Harding T, Uney JB, Troskie BE, Millar RP, McArdle CA. J. Biol. Chem. 2001;276:39685–39694. doi: 10.1074/jbc.M104542200. [DOI] [PubMed] [Google Scholar]
- 28.Caunt CJ, Finch AR, Sedgley KR, Oakely L, Luttrell LM, McArdle CA. J. Biol. Chem. 2006;281:2701–2710. doi: 10.1074/jbc.M507242200. [DOI] [PubMed] [Google Scholar]
- 29.Whitehurst A, Cobb MH, White MA. Mol. Cell. Biol. 2004;24:10145–10150. doi: 10.1128/MCB.24.23.10145-10150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunner D, Oellers N, Szabad J, Biggs WH, III, Zipursky SL, Hafen E. Cell. 1994;76:875–888. doi: 10.1016/0092-8674(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 31.Casar B, Sanz-Moreno V, Yazicioglu MN, Rodriguez J, Berciano MT, Lafarga M, Cobb MH, Crespo P. EMBO J. 2007;26:635–646. doi: 10.1038/sj.emboj.7601523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yazicioglu MN, Goad DL, Ranganathan A, Whitehurst AW, Goldsmith EJ, Cobb MH. J. Biol. Chem. 2007;282:28759–28767. doi: 10.1074/jbc.M703460200. [DOI] [PubMed] [Google Scholar]
- 33.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Curr. Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]