

Two of the most enigmatic and challenging problems in neuroscience are the search for the function of sleep and understanding the mechanism by which volatile chemicals can induce general anesthesia. Despite the apparent similarity of sleep and anesthesia to the neophyte, it is widely argued that these brain states are actually apples and oranges, sleep being readily reversible (thankfully), whereas anesthesia is irreversible. In a recent issue of PNAS, Kelz et al. (1) demonstrate that disruption of a specific neural locus involved in normal sleep–wake regulation selectively affects emergence from, but not induction of, general anesthesia. That article demonstrates that waking up from anesthesia uses neural circuits distinct from those necessary to become anesthetized. It also further solidifies the connection between anesthesia and sleep, implicating wake-promoting neural circuitry in selectively contributing to emergence from anesthesia.
Neural loci have been identified that are important for initiation of and emergence from sleep (Fig. 1 and refs. 2 and 3). For example, GABAergic neurons in the hypothalamic ventrolateral preoptic (VLPO) nucleus promote sleep in part by inhibiting arousal-promoting circuits, such as the hypothalamic tuberomammillary nucleus (TMN). Another critical arousal-promoting or -stabilizing locus is the nucleus of orexin-producing neurons in the lateral hypothalamus (4). Progressive loss of orexin-producing neurons results in the human sleep disorder narcolepsy (5, 6). Individuals affected with narcolepsy transition from wakefulness directly to rapid eye movement (REM) sleep, often with accompanying cataplexy (abrupt loss of muscle tone).
Fig. 1.
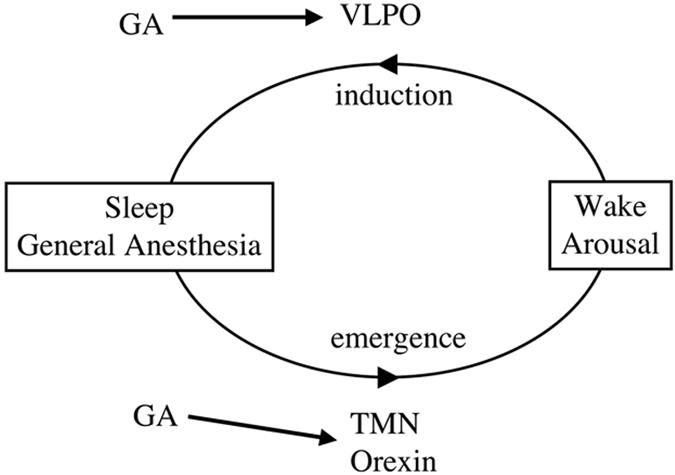
A model linking sleep and general anesthesia. The VLPO area of the hypothalamus promotes sleep, whereas the TMN and the orexin neurons of the lateral hypothalamus promote or stabilize wakefulness. GAs may also activate the VLPO, which in turn inhibits arousal-promoting circuits (13). General anesthetics (GA) affect, or act through, the TMN and orexin neurons. Kelz et al. (1) demonstrate an important role for the orexin neurons, particularly in waking up from anesthesia.
Accumulating evidence indicates sleep, especially non-REM sleep, and general anesthesia use common neuronal and genetic substrates. Both seem to reduce or abolish spontaneous movement and sensory responsiveness. Sleep loss increases the drive to sleep, reflecting homeostatic regulation. Similarly, sleep loss also enhances the action of the volatile anesthetic isoflurane (7). Sleep deprivation/prolonged wakefulness increases concentration of adenosine, an ATP breakdown product, which in turn is thought act as a homeostatic mediator to promote sleep (8). Sleep deprivation effects on isoflurane anesthesia can be partially reversed by adenosine receptor antagonists (9). Anesthetics also appear to act through sleep neural circuits. Anesthetics induce a similar EEG state to that of non-REM sleep (10, 11). Brain imaging studies indicate general anesthetics act on sleep–wake neural circuitry (12). Importantly, the sedative effects of the i.v. anesthetic propofol require GABAA receptor function in the wake promoting TMN (13).
The orexin system that regulates sleep is also linked to general anesthesia. Orexin agonists decrease the duration or depth of anesthesia, whereas administration of the orexin receptor-1 antagonist (SB-334867-A) increases anesthetic duration (14, 15). Changes in duration could be secondary to induction or emergence kinetics. As these studies assessed anesthetic efficacy after bolus administration, steady-state levels were never achieved, and independent effects on emergence and induction could not be assessed.
To address this issue, Kelz et al. (1) used continuous delivery of the volatile anesthetics, isoflurane and sevoflurane and then monitored the effects of perturbing orexin function on induction and emergence times by using loss of righting reflex (LORR). They perturbed orexin function by using mice bearing a preproorexin promoter-ataxin3 fusion transgene (16). Their orexin neurons undergo selective degeneration at 4–6 weeks of age, exhibiting features of narcolepsy, including cataplexy (16). In addition, they administered the orexin 1 receptor antagonist SB-334867-A, linking observed effects to orexin rather than another product of orexin neurons. Surprisingly, they observed that anesthesia induction was unaffected by reducing orexin signaling. On the other hand, potent delays in emergence were noted in these animals.
Changes in the kinetics of induction or emergence could be secondary to the distribution or elimination of the drug. For example, delayed emergence could simply be caused by failure to clear the drug. However, brain concentrations of isoflurane were indistinguishable in mice treated with orexin antagonist and those untreated at a time during emergence when LORR is highly affected (1). Moreover, cataplectic episodes were not observed to occur before, during, or after anesthesia, eliminating this alternative explanation for delayed emergence (1). Thus, mice with comparable anesthetic concentrations emerge from anesthesia very differently depending on the orexin system.
In addition to the reported roles of the orexin system in emergence from anesthesia, Kelz et al. (1) demonstrate that orexin neurons themselves are directly or indirectly sensitive to anesthetics. Orexin neuron activation was assessed in response to anesthetic administration by examining c-Fos induction. Anesthetic administration was associated with dramatic reductions of c-Fos, comparable to those observed during non-REM sleep (1, 17). Notably, activation of adjacent melanin-concentrating, hormone-expressing neurons was not affected by anesthetic administration (1). In addition, anesthetics affect orexin receptor function (18), indicating that anesthetics could also affect the function of orexin targets. Given that perturbation of the orexin system has little effect on anesthetic induction, it will be interesting to see what the consequences are, if any, of anesthetic effects on the orexin system. One possibility is that anesthetics act by inhibiting redundant arousal-promoting pathways.
The finding that anesthesia induction and emergence use different neural substrates should lead to a search for other substrates important for these processes. Given the accumulating evidence for ties between sleep and anesthesia, other components of sleep circuitry represent strong candidates. Other wake/arousal-promoting circuits may be important for coming out of anesthesia (Fig. 1). One might also hypothesize that a sleep-promoting locus, such as the VLPO, may play an especially important role in anesthetic induction.
In addition to neural substrates, there has been a substantial search for molecular targets of anesthetics. In light of the data presented here, the genetics of anesthesia needs to be re-examined particularly with respect to genes selectively important for emergence and induction. Again, genes involved in sleep may provide potential candidates to examine as targets or mediators of anesthetics. One such link may be the Shaker potassium channel. The Shaker mutant in Drosophila, isolated because of its leg shaking under ether anesthesia, also exhibits resistance to anesthetics (19) and reduced sleep (20).
Progressive loss of orexin-producing neurons results in the human sleep disorder narcolepsy.
Although it should be emphasized that sleep and anesthesia are distinct, the data presented here demonstrate that the neural circuitry important for keeping us awake may also wake us up from general anesthesia. The issues of emergence and induction are not solely theoretical. The development of agents that promote emergence could be useful clinically for promoting recovery from anesthesia. A better understanding of how we naturally wake and sleep could provide a framework for understanding how general anesthetics put us to sleep.
Footnotes
The author declares no conflict of interest.
See companion paper on page 1309 in issue 4 of volume 105.
References
- 1.Kelz MB, Sun Y, Chen J, Meng QC, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, Yanagisawa M. Proc Natl Acad Sci USA. 2008;105:1309–1314. doi: 10.1073/pnas.0707146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saper CB, Scammell TE, Lu J. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 3.Lydic R, Baghdoyan HA. Anesthesiology. 2005;103:1268–1295. doi: 10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai T. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 5.Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, et al. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 6.Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tung A, Szafran MJ, Bluhm B, Mendelson WB. Anesthesiology. 2002;97:906–911. doi: 10.1097/00000542-200210000-00024. [DOI] [PubMed] [Google Scholar]
- 8.Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tung A, Herrera S, Szafran MJ, Kasza K, Mendelson WB. Anesthesiology. 2005;102:1158–1164. doi: 10.1097/00000542-200506000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Sleigh JW, Andrzejowski J, Steyn-Ross A, Steyn-Ross M. Anesth Analg. 1999;88:659–661. doi: 10.1097/00000539-199903000-00035. [DOI] [PubMed] [Google Scholar]
- 11.Tung A, Lynch JP, Roizen MF. J Clin Monit Comput. 2002;17:37–42. doi: 10.1023/a:1015404803637. [DOI] [PubMed] [Google Scholar]
- 12.Alkire MT, Haier RJ, Fallon JH. Conscious Cogn. 2000;9:370–386. doi: 10.1006/ccog.1999.0423. [DOI] [PubMed] [Google Scholar]
- 13.Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M. Nat Neurosci. 2002;5:979–984. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- 14.Kushikata T, Hirota K, Yoshida H, Kudo M, Lambert DG, Smart D, Jerman JC, Matsuki A. Neuroscience. 2003;121:855–863. doi: 10.1016/s0306-4522(03)00554-2. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda Y, Takeda A, Fukuda S, Suzuki H, Ishimoto M, Mori Y, Eguchi H, Saitoh R, Fujihara H, Honda K, Higuchi T. Anesth Analg. 2003;97:1663–1666. doi: 10.1213/01.ANE.0000089964.85834.EF. [DOI] [PubMed] [Google Scholar]
- 16.Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 17.Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minami K, Uezono Y, Sakurai T, Horishita T, Shiraishi M, Ueta Y. Pharmacology. 2007;79:236–242. doi: 10.1159/000101713. [DOI] [PubMed] [Google Scholar]
- 19.Walcourt A, Scott RL, Nash HA. Anesth Analg. 2001;92:535–541. doi: 10.1097/00000539-200102000-00047. [DOI] [PubMed] [Google Scholar]
- 20.Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]