

Abstract
The orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII; Nr2f2) is expressed in adipose tissue in vivo and declines during differentiation. Overexpression of COUP-TFII prevents adipogenesis, whereas shRNA-mediated reduction of COUP-TFII promotes differentiation, as shown by increased lipid accumulation and elevated expression of fat cell marker proteins. Furthermore, reduction of COUP-TFII allows uncommitted fibroblasts to be differentiated into fat cells. COUP-TFII represses the expression of a number of proadipogenic factors in adipocytes, with direct action noted at the CAAT enhancer-binding protein α promoter. We show that COUP-TFII acts downstream of hedgehog signaling and is required for the full antiadipogenic effect of this pathway. This effect is mediated in part by interaction with GATA factors. COUP-TFII and GATA2 are physically associated and repress target gene expression in an additive manner. Taken together, our data demonstrate that COUP-TFII represents an endogenous suppressor of adipogenesis, linking antiadipogenic extracellular signals to the core transcriptional cascade.
Keywords: Repressor, differentiation, Nr2f2, GATA
Adipocyte differentiation is a highly regulated process controlled by a complex transcriptional cascade (1–4). A wide array of transcription factors participate in adipogenesis, although most attention has focused on several members of the CAAT enhancer-binding protein (C/EBP) family and the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ). C/EBPβ and C/EBPδ are induced very early during differentiation (5). These early regulators in turn activate two critical proadipogenic transcription factors, PPARγ and C/EBPα, which mutually stimulate each other and drive the transition of preadipocytes to mature adipocytes by activating a variety of genes required for maintaining the adipocyte phenotype (6). Recently, a number of transcription factors have been identified as regulators of adipogenesis, including GATA2 and GATA3 (7, 8), certain members of the Krüppel-like factor (KLF) family (9–11), and early B cell factors (EBF) 1 and 2 (12, 13).
We have sought to identify transcriptional pathways in adipogenesis by using a systematic approach based on DNase-hypersensitivity analysis (51). Briefly, we used an integrated experimental and computational strategy to identify overrepresented motifs in differentiation-dependent DNase-hypersensitive sites flanking adipocyte-selective genes. Among these motifs were sequences with a high degree of similarity to binding sites for the orphan nuclear receptor chicken ovalbumin upstream promoter transcription factor (COUP-TF). First identified as an activator of the chicken ovalbumin gene, COUP-TF was shown to bind to an imperfect direct repeat of the AGGTCA motif (14, 15). Shortly thereafter, three mammalian orthologs were identified, COUP-TFI (also known as Nr2f1 or EAR3), COUP-TFII (ARP-1, Nr2f2), and the more distantly related COUP-TFIII (also known as Nr2f6 or EAR2) (16–20).
We focused our attention on COUP-TFII because expression data suggested a role for this isoform in adipocyte biology (see below). COUP-TFII can act as either a positive or negative regulator of transcription, although the latter appears to be more typical (21). COUP-TFII is expressed widely during development (22), and loss-of-function studies in mice have confirmed a critical role in organogenesis. COUP-TFII-deficient mice die in utero with defects in heart development and angiogenesis (23). Tissue-specific knockout studies show that COUP-TFII is required for the development of limb, skeletal muscles, and stomach (24, 25) and plays a critical role in determining vein identity (26).
Here, we show that COUP-TFII is expressed in adipose tissues and in cultured adipocyte models. Gain-of-function and loss-of-function studies demonstrate that COUP-TFII is a dominant repressor of differentiation in adipocytes. We also show that COUP-TFII is required for other antiadipogenic factors, including sonic hedgehog (Shh) and GATA, to exert their full effect. COUP-TFII acts, in part, through physical and functional interactions with GATA factors. Taken together, these studies identify a repressor of adipogenesis with links to upstream pathways and downstream effectors of this critical developmental process.
Results
COUP-TFII Is Expressed in Adipose Tissue and Is Developmentally Regulated.
To determine the tissue specificity of COUP-TFII, we isolated mRNA and protein from tissues of wild-type adult male C57BL/6 mice and performed Northern and Western blotting. COUP-TFII mRNA [supporting information (SI) Fig. 5A] and protein (Fig. 1A) are expressed at high levels in a number of tissues, including lung, kidney, and spleen. Moreover, COUP-TFII is expressed abundantly in white adipose tissue and, to a lesser degree, in brown adipose tissue. Fractionation of white adipose tissue by low-speed centrifugation allowed us to identify the stromal–vascular fraction (SVF) as the dominant site of COUP-TFII protein expression within the fat pad (Fig. 1B).
Fig. 1.

COUP-TFII is expressed in adipose tissue in vivo and in vitro. (A) COUP-TFII protein levels in different tissues of adult C57BL/6 mice were determined by Western blotting. B, brain; H, heart; Lu, lung; K, kidney; Li, liver; Sk, skeletal muscle; Sp, spleen; WAT, white adipose tissue; BAT, brown adipose tissue; T, testis. (B) Ovarian white adipose tissue was further separated into SVF and adipocytes. Protein lysates from both fractions were subjected to Western blotting. (C) The 3T3-L1 preadipocytes were differentiated with DMI mixture. Protein lysates were prepared at the indicated time points and were subjected to Western blotting. Ruby staining (B) and Ponceau S staining (C) were used to demonstrate equal loading.
We next used murine 3T3-L1 cells to assess COUP-TFII expression over the course of adipogenesis. As differentiation proceeds, COUP-TFII mRNA is decreased by ≈50% (SI Fig. 5B). COUP-TFII protein is decreased during adipogenesis as well (Fig. 1C); interestingly, the magnitude of the effect appears to be greater at the protein level than at the mRNA level, implying posttranscriptional regulation of COUP-TFII levels during differentiation. This notion is supported by what appears to be an isoform shift favoring more mobile species of COUP-TFII later in differentiation. It is not yet clear what these different isoforms represent, and this question remains a subject of ongoing investigation. The reduction in COUP-TFII levels seen during adipose conversion is consistent with the results in Fig. 1B showing that the SVF, which includes preadipocytes, is the major site of COUP-TFII expression within the fat pad.
COUP-TFII Is a Potent Suppressor of Adipogenesis.
The developmental pattern of COUP-TFII expression suggests that this factor might act as a repressor of adipogenesis. To test this hypothesis, we first performed gain-of-function experiments by using retroviral delivery of COUP-TFII into 3T3-L1 preadipocytes. As shown in SI Fig. 6A, COUP-TFII protein levels were increased by ≈10-fold in COUP-TFII transgenic cells relative to cells transduced with control virus. Strikingly, overexpression of COUP-TFII in these cells completely blocked adipogenesis (Fig. 2A), as shown by oil red O staining of neutral lipids. Adipocyte markers, such as Glut4, LPL, PPARγ, adiponectin, and aP2 (Fig. 2B), were significantly decreased in COUP-TFII-overexpressing cells, further confirming an antiadipogenic effect.
Fig. 2.

Overexpression of COUP-TFII in 3T3-L1 cells suppresses adipogenesis, whereas RNAi-mediated knockdown of COUP-TFII promotes adipogenesis. (A and B) The 3T3-L1 preadipocytes were transduced with a retrovirus expressing COUP-TFII or empty pMSCV vector. (A) The cells were induced with DMI mixture. Oil red O staining was performed on day 7 after induction. (B) mRNA levels of adipocyte genes Glut4, adiponectin, LPL, aP2, and PPARγ were analyzed with Q-PCR on days 0, 2, 4, and 7 after induction. (C and D) The 3T3-L1 preadipocytes were transduced with a retrovirus expressing a shRNA specific for COUP-TFII (shCOUP) or luciferase (shLuc). (C) Oil red O staining was performed on days 4 and 7 after DMI induction. (D) mRNA levels of Glut4, adiponectin, LPL, aP2, and PPARγ were analyzed with Q-PCR on days 0, 2, 4, and 7 after induction. Data are shown as mean ± SD of three biological replicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Experimental manipulations that inhibit adipogenesis need to be interpreted with caution, for the simple reason that differentiation is a complex process requiring a plethora of factors to turn on and off with precise timing; it is easy to imagine that manipulation might disrupt the process through nonspecific mechanisms. We therefore sought confirmation of COUP-TFII antiadipogenic activity through loss-of-function studies, in which we would predict enhanced adipose conversion. We used retroviral delivery of short-hairpin RNA (shRNA) to knock down COUP-TFII in 3T3-L1 preadipocytes. As shown in SI Fig. 6B, doing so resulted in reduction of COUP-TFII protein by >80%. Cells with reduced COUP-TFII demonstrated enhanced adipogenic potential, including greater lipid accumulation (Fig. 2C) and increased expression of adipocyte marker genes (Fig. 2D).
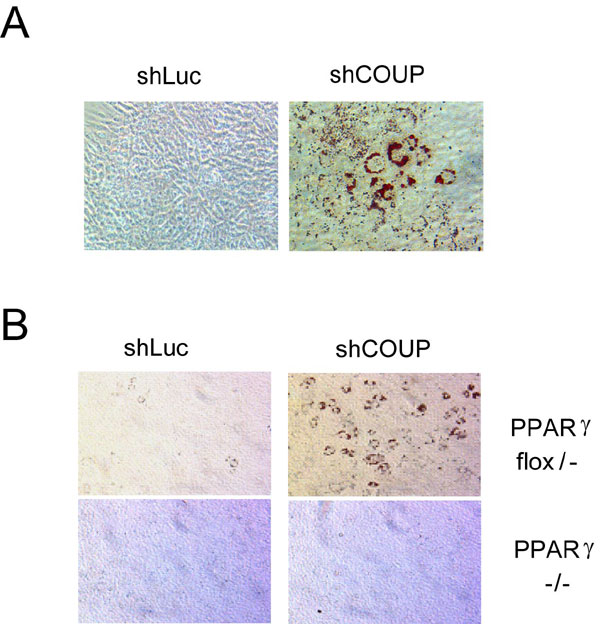
NIH 3T3 and other noncommitted fibroblast lines express high endogenous levels of COUP-TFII (data not shown), leading us to speculate that this expression might explain part of the inability of these cells to form adipocytes upon induction with differentiation mixture. Similar to what we observed in 3T3-L1 preadipocytes, knockdown of COUP-TFII enhanced adipogenesis in NIH 3T3 fibroblasts. Approximately 10% of cells expressing a retrovirus carrying a COUP-TFII-specific shRNA could be induced to differentiate into adipocytes, whereas none of the cells expressing a control shRNA displayed evidence of differentiation (SI Fig. 7A). In addition to the enhanced lipid accumulation, the adipocyte markers adiponectin and Glut4 were dramatically induced in COUP-TFII knockdown cells induced to differentiate (data not shown).
To date, PPARγ is the only protein known to be both necessary and sufficient for adipogenesis. To assess whether reduction of COUP-TFII is sufficient to promote adipogenesis independent of PPARγ, we knocked down COUP-TFII in PPARγflox/− or PPARγ−/− mouse embryonic fibroblasts (MEFs) (27). Knockdown of COUP-TFII in PPARγflox/− cells enhanced adipogenesis to a degree similar to that seen in NIH 3T3 cells but had no effect in PPARγ−/−cells (SI Fig. 7B). This result is consistent with multiple lines of prior data suggesting a preeminent position in the adipogenic cascade for PPARγ.
COUP-TFII Represses a Number of Proadipogenic Factors.
Our data indicate that COUP-TFII is an endogenous suppressor of adipogenesis in multiple cell lines. We next sought to investigate the mechanism by which COUP-TFII negatively regulates adipogenesis. The developmental pattern of COUP-TFII expression in adipogenesis suggests a role in the first few days after the initiation of differentiation, around the time that proadipogenic factors such as C/EBPα, PPARγ, EBF1, EBF2, and others are first expressed. We used transient transfection to overexpress COUP-TFII in mature 3T3-L1 adipocytes and examined mRNA levels of some of these factors. EBF1, which was recently implicated as an activator of adipogenic program (12, 13), was significantly down-regulated upon overexpression of COUP-TFII (SI Fig. 8A), although the equally proadipogenic EBF2 was not (data not shown). Other proadipogenic factors, such as KLF15, SREBP1c, PPARγ1, PPARγ2, and C/EBPα, were also repressed by COUP-TFII.
To address whether COUP-TFII directly represses the expression of some of these key transcription factors, we first performed reporter assays in NIH 3T3 cells by using luciferase constructs driven by the −1.8-kb region of the PPARγ1 promoter, the −900-bp region of the PPARγ2 promoter, and the −300-bp region of the C/EBPα promoter. We found that the promoter activity of C/EBPα was consistently and significantly repressed by COUP-TFII (SI Fig. 8B), but we saw no significant effects on the promoters for PPARγ1 and PPARγ2 (data not shown). COUP-TFII also repressed the positive actions of C/EBPβ on this promoter (SI Fig. 8B).
Computer-assisted searching of the −300-bp region of the C/EBPα promoter does not reveal an obvious COUP-TFII-binding motif. To provide further evidence that this region represents a bona fide COUP-TFII target gene, we performed a chromatin immunoprecipitation (ChIP) assay in 3T3-L1 preadipocytes to test for the presence of endogenous COUP-TFII on the C/EBPα promoter. As shown in SI Fig. 8C, COUP-TFII was specifically immunoprecipitated from this region. COUP-TFII was not precipitated by a nonspecific antibody, nor was it found associated with a distal, irrelevant region of the same gene. It remains unclear whether COUP-TFII directly binds a cryptic binding site in the C/EBPα promoter or whether it associates with the promoter through interactions with other transcription factors.
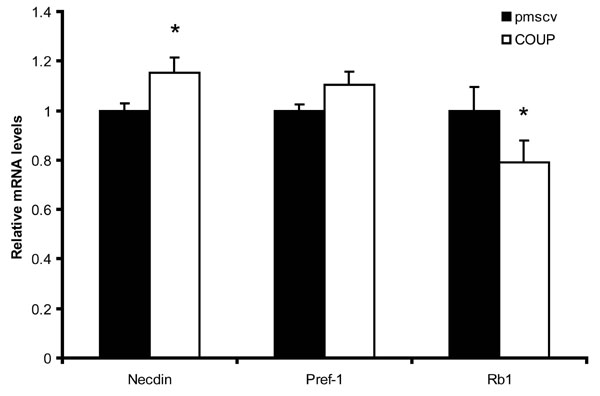
COUP-TFII also represses the proadipogenic pRB protein and increases the level of necdin; these actions may antagonize differentiation (SI Fig. 9).
COUP-TFII Is Downstream of Hedgehog and Is Required for Hedgehog to Repress Adipogenesis Fully.
Adipogenesis is strongly affected by a myriad of extracellular signals, some of which promote differentiation and some of which antagonize the process. Shh, one of three vertebrate orthologs of the Drosophila hedgehog gene, is particularly interesting in this regard because it represses adipogenesis in multiple mammalian models (28–31), and it strongly induces COUP-TFII expression in P19 embryonal carcinoma cells (32). To investigate whether Shh could also enhance COUP-TFII expression in adipogenic cells, we treated 3T3-L1 preadipocytes with several doses of recombinant Shh. As shown in Fig. 3A, COUP-TFII protein levels responded to Shh administration in a dose-responsive manner. Interestingly, although Tsai et al. (33) suggested that the effect of Shh on COUP-TFII expression in P19 cells is mediated through a Shh response element in the COUP-TFII promoter region, we observed an effect only at the protein level, with no elevation in COUP-TFII mRNA in response to Shh in 3T3-L1 cells (SI Fig. 10).
Fig. 3.

COUP-TFII is required for hedgehog-mediated suppression of adipogenesis. (A) The 3T3-L1 preadipocytes were treated with indicated amount of Shh for 48 h. Cell lysates were subjected to Western blotting for COUP-TFII and β-actin. (B) The 3T3-L1 preadipocytes retrovirally transduced with shRNA specific for COUP-TFII (shCOUP) or luciferase (shLuc) were induced with a mixture of 1.7 μM insulin, 10 μM dexamethasone, and 5 nM IBMX in the absence (−) or presence (+) of 300 ng/ml Shh. (C) The 3T3-L1 preadipocytes transduced with pMSCV empty vector or COUP-TFII were induced with a DMI mixture in the absence (−) or presence (+) of 3.6 μM KAAD-cyclopamine, an antagonist of hedgehog signaling. (B and C) Oil red O staining was performed 7 days after induction.
If COUP-TFII is a downstream mediator of hedgehog signaling on adipogenesis, reduction of COUP-TFII should prevent Shh from fully inhibiting differentiation. We tested this prediction by using our retroviral shRNA delivery system as before. In control cells, 300 ng/ml Shh completely inhibited adipogenesis as measured by oil red O staining (Fig. 3B). In cells with reduced COUP-TFII, however, Shh exhibited a diminished ability to repress adipose conversion, indicating that COUP-TFII contributes to hedgehog-induced suppression of adipogenesis. The effect of Shh was not abolished, however, which suggests that other pathways are operating to decrease adipose conversion downstream of Shh as well.
Cyclopamine is a small molecule that binds to the hedgehog receptor Smoothened and antagonizes Shh signaling (34), leading to enhanced adipogenesis in 3T3-L1 cells (Fig. 3C and ref. 31). In cells that overexpress COUP-TFII, cyclopamine is no longer able to promote adipogenesis (Fig. 3C), further suggesting that COUP-TFII is downstream of hedgehog signaling.
COUP-TFII Mediates the Antiadipogenic Effect of GATA Factors by Physical and Functional Interactions.
The transcription factors GATA2 and GATA3 play a negative regulatory role in adipogenesis. Overexpression of GATA2 and GATA3 traps fat cell precursors at the preadipocyte stage, whereas GATA3-deficient embryonic stem cells display enhanced adipogenic ability (7). GATA factors have been suggested to operate downstream of hedgehog signaling in adipogenesis (31) in a manner analogous to what we have shown here for COUP-TFII, which raised the possibility that COUP-TFII and GATA factors might act cooperatively in this system.
We explored a possible functional relationship between COUP-TFII and GATA by expressing human GATA2 in 3T3-L1 preadipocytes. As expected, GATA2 robustly suppressed adipogenesis in these cells (Fig. 4A). In cells expressing a shRNA directed against COUP-TFII, however, the ability of GATA2 to inhibit differentiation was virtually abolished. This result indicates that GATA actions in adipogenesis require COUP-TFII. We initially hypothesized that COUP-TFII might be downstream of (i.e., induced by) GATA in preadipocytes, but we have been unable to demonstrate any effect of GATA overexpression on COUP-TFII mRNA or protein abundance (data not shown). We therefore postulated that GATA and COUP-TFII might physically interact to create a repressor complex on key target genes. We addressed this possibility with a coimmunoprecipitation (Co-IP) study. FLAG-tagged GATA2, GATA3, and FLAG tag alone were transfected into HEK-293 cells, and the protein lysates were precipitated by using anti-FLAG beads. As shown in Fig. 4B, endogenous COUP-TFII was observed in a complex with either GATA2 or GATA3 but not with FLAG tag alone, suggesting that COUP-TFII interacts with multiple GATA factors. We went on to map the domain required for the COUP-TFII association, using a series of GATA2 deletion mutants (Fig. 4C). These mutant alleles were expressed at comparable levels in 293 cells and were immunoprecipitated with anti-FLAG beads. The only mutant that showed significantly reduced association with COUP-TFII lacked both zinc finger domains and the subsequent 39 aa (construct 5; Fig. 4C). There was also a partial reduction in the COUP-TFII/GATA2 interaction with deletion of the N-terminal 289 aa (construct 2) or the 39 aa immediately after the zinc fingers (construct 6). Taken together, these data demonstrate a physical interaction between these two antiadipogenic factors.
Fig. 4.

COUP-TFII is required for GATA-mediated suppression of adipogenesis. (A) The 3T3-L1 preadipocytes were transduced with GATA2-pMSCV or empty vector, selected, and then transduced a second time with a virus expressing shRNA specific for luciferase (shLuc) or COUP-TFII (shCOUP). Oil red O staining was performed 7 days after DMI induction. (B) HEK-293 cells were transfected with FLAG empty vector (Flag), FLAG-GATA2, or FLAG-GATA3. Co-IP analysis was performed with anti-FLAG beads. Ten percent input and the SDS eluate were subjected to Western blotting with polyclonal antibodies against FLAG or COUP-TFII. (C) The 293 cells were transfected with FLAG empty vector (EV) or FLAG-tagged deletion mutants of GATA2. Co-IP analysis was performed as described above. (D) The 3T3-L1 adipocytes (day 5 after DMI induction) were transfected with 1 μg of pCDNA3 (EV), 1 μg of COUP-TFII (COUP), 1 μg of GATA2 (GATA), or 0.5 μg of COUP-TFII plus 0.5 μg of GATA2 (COUP + GATA). Relative mRNA levels of C/EBPα and Glut4 were analyzed by using Q-PCR. Data are shown as mean ± SD of three biological replicates. *, P < 0.05; **, P < 0.01.
Because COUP-TFII and GATA2 are tightly associated with each other both functionally (Fig. 4A) and physically (Fig. 4 B and C), we next asked whether they can cooperate in the repression of individual adipocyte target genes. We found that COUP-TFII and GATA2 were individually able to repress the expression of endogenous C/EBPα and Glut4 (Fig. 4D). When cotransfected (taking care to keep total repressor levels equivalent), GATA2 and COUP-TFII were able to reduce expression of these genes to a greater degree than either factor alone.
Discussion
We have sought to identify transcriptional pathways in adipogenesis by using multiple experimental and computational techniques. These studies have led us to consider a role for the orphan nuclear receptor COUP-TFII in adipocyte differentiation. COUP-TFII is well known to play a significant role in the development of multiple organs and tissues, including heart, blood vessels, muscle, limb, and stomach (23–26). In the present work, we show that COUP-TFII is expressed in adipose tissues and in cultured adipocytes. Gain-of-function and loss-of-function experiments were performed in several models of adipogenesis. Overexpression of COUP-TFII in 3T3-L1 preadipocytes suppresses adipogenesis. Conversely, knockdown of COUP-TFII in 3T3-L1 preadipocytes, NIH 3T3 fibroblasts, and MEFs enhances fat differentiation (Fig. 2 and SI Fig. 7). NIH 3T3 cells and MEFs are generally nonadipogenic and have been shown to differentiate into fat cells only when strongly proadipogenic transcription factors are ectopically overexpressed (35). The fact that knockdown of COUP-TFII promotes adipogenesis in these cells indicates that COUP-TFII is a potent suppressor of adipogenesis. Taken together, our data from multiple models prove that COUP-TFII represents an endogenous suppressor of adipogenesis.
We have shown that levels of many proadipogenic transcription factors are reduced by transfection of COUP-TFII, although at present we do not know whether these actions are immediately downstream of COUP-TFII. COUP-TFII exerts a repressive effect on the C/EBPα promoter in a reporter assay and localizes to this region in 3T3-L1 preadipocytes. Nonetheless, our inability to locate an obvious COUP-TF response element in this region makes it unclear whether the effect on C/EBPα transcription is mediated by direct COUP-TFII binding or by an indirect action through other binding partners. Both mechanisms have been shown to be relevant to COUP-TFII action in other cell types (36–41). COUP-TFII has been shown to inhibit the ability of PPARγ to induce phosphoenolpyruvate carboxykinase expression in NIH 3T3 fibroblasts, perhaps by direct competition for the PPARγ response element (42). It is also worth pointing out that an effect on the C/EBPα promoter is unlikely to account fully for the antiadipogenic action of COUP-TFII because shRNA-mediated reduction of COUP-TFII in NIH 3T3 cells, which are functionally C/EBPα-deficient (43), is able to promote adipogenesis. The identification of direct COUP-TFII target genes relevant to adipocyte differentiation is currently a priority for our laboratory.
Transcription factors regulate developmental events such as adipogenesis under the influence of signaling pathways, which include such ancient pathways as Wnt and hedgehog, both of which act to repress adipogenesis in mammalian systems. In most cases, the link between the upstream signaling events and effects on the transcriptional cascades that govern differentiation is poorly understood. We were struck, however, by the reported relationship between COUP-TFII and hedgehog signaling in heterologous cells (32), which suggested that there might be a similar link in developing adipocytes. In fact, this suggestion proved to be true because COUP-TFII acts downstream of hedgehog signaling and is required for the full expression of the antiadipogenic effect driven by Shh. Others have suggested a role for GATA factors as downstream mediators of hedgehog activity in developing adipocytes (31). Interestingly, we were able to show that COUP-TFII and GATA2 demonstrate significant interdependence, with both physical and functional interactions between the two proteins. Thus, GATA2 cannot fully inhibit adipogenesis in the absence of COUP-TFII, and GATA2 and COUP-TFII show additivity in repressing the expression of key adipocyte genes such as C/EBPα and Glut4.
COUP-TFII is exceptionally well conserved among species (22). Mouse and human COUP-TFII are identical, and COUP-TF orthologs ranging from human to Caenorhabditis elegans and Drosophila display the highest degree of interspecies conservation found in the nuclear hormone receptor superfamily (22). Intriguingly, the Drosophila ortholog of COUP-TF, seven-up, has been shown to play an essential role in fat body development in flies (44). Disruption of seven-up results in loss or reduced expression of two terminal marker genes, Adh and Dcg1, specifically in the larval fat body (44). Interestingly, serpent, the Drosophila ortholog of GATA, is also required for fat cell differentiation in flies (45). In particular, serpent was identified as a transcriptional activator of Adh gene (46). Although the molecular connection between seven-up and serpent in Drosophila remains unknown, their mammalian orthologs, COUP-TFII and GATA, appear to function cooperatively in higher organisms, albeit they have the opposite effect on fat development versus that seen in the invertebrate models. The conserved and integrated function of COUP-TFII and GATA during evolution highlights their position as a team of key regulators of fat cell differentiation.
COUP-TFs have been shown to affect gene expression and development by multiple molecular mechanisms, some of which require DNA binding and some of which do not. For example, COUP-TFII can activate or repress gene expression through binding to COUP-TF motifs, such as those found in the ovalbumin gene (47) and the apolipoprotein AI gene (48). Alternatively, COUP-TFII can also bind to other transcription factors and influence gene expression as a cofactor (37–40). Finally, COUP-TFII can also compete for binding to the motifs of other nuclear receptors or can compete for other key cofactors, such as retinoic X receptor. We anticipate that the antiadipogenic effects of COUP-TFII require more than one of these mechanisms.
A major challenge in the field of adipogenesis is integrating transcriptional effectors into the existing signaling and transcriptional cascade. This work establishes COUP-TFII as a critical regulator of adipogenesis in multiple cell autonomous systems. Equally as important, however, is the identification of hedgehog as an upstream inducer of COUP-TFII and the identification of C/EBPα as a downstream target of COUP-TFII. Although it is virtually certain that there will be other targets of COUP-TFII in adipogenesis and other upstream modulators of COUP-TFII expression and activity, this work describes one of the first direct connections between extracellular signals and the classic transcriptional effectors in adipocyte differentiation.
Methods
For additional materials and procedures, see SI Materials and Methods.
Materials and Reagents.
The full-length mouse COUP-TFII cDNA was purchased from American Type Culture Collection. The coding region was excised with SspI and MslI and cloned into pMSCV-puro (Clontech) at the HpaI site by blunt end ligation.
To construct the shRNA plasmids, DNA oligonucleotides were synthesized, annealed, and cloned into pSIREN vector (Clontech) at BamHI and EcoRI sites. Two COUP-TFII shRNA constructs were used in our studies and showed similar effects on adipogenesis (data not shown). The target sequence of the construct used in these experiments is 5′-AGCTCTTGCTTCGTCTCCC. FLAG-tagged GATA2 mutant constructs were the kind gift of Gökhan Hotamisligil (Harvard School of Public Health, Boston).
Rabbit anti-COUP-TFII antibody was a generous gift from Sotirios Karathanasis (Lilly). Rabbit anti-Glut4 antibody was a gift from Barbara Kahn (Beth Israel Deaconess Medical Center, Boston). Antibodies against β-actin, FLAG, and GAPDH were purchased from Santa Cruz Biotechnology.
Cell Culture.
HEK-293 cells and Phoenix packaging cells were maintained in DMEM supplemented with 10% FBS. NIH 3T3 fibroblasts were maintained in DMEM supplemented with 10% calf serum. 3T3-L1 preadipocytes and MEFs were maintained and differentiated as described in refs. 27 and 49. Briefly, 3T3-L1 preadipocytes were grown to confluence in DMEM supplemented with 10% calf serum. Two days after confluence, cells were supplied with differentiation medium [DMEM containing 10% FBS plus 1.7 μM insulin, 10 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine (DMI)]. Forty-eight hours after induction, cells were fed maintenance medium (DMEM containing 10% FBS plus 0.8 μM insulin), and the medium was replaced every 2 days. For differentiation of NIH 3T3 cells and MEFs, differentiation medium was supplemented with 10 μM rosiglitazone, and the induction time was prolonged to 72 h.
Retrovirus Preparation and Infection.
Retrovirus preparation and infection were performed as described in ref. 27. Briefly, pMSCV, pSIREN empty vectors, or their derivatives containing specific cDNA or shRNA, along with group-specific antigens and reverse transcriptase (gag-pol) and VSV-G-expressing plasmids, was transfected into Phoenix packaging cells with the CellPhect transfection kit (Amersham Biosciences). Viral supernatant was collected 48 h after transfection, filtered through 0.45-μm filters, and added to target cells for 12 h along with 8 μg/ml Polybrene. Cells were selected with 4 μg/ml puromycin or 400 μg/ml hygromycin to make stable lines and were maintained in media containing appropriate antibiotics.
Adipocyte Transfection.
3T3-L1 adipocytes were differentiated as described. On day 5 after DMI induction, the cells were trypsinized, and 1 μg of DNA was transferred into 5 × 106 cells with the Amaxa nucleofection device (Amaxa Biosystems) according to manufacturer's instruction. mRNA was extracted 24 h posttransfection.
Adipose Tissue Fractionation.
Ovarian fat pads were dissected from 12-week-old C57BL/6J mice. The isolated white adipose tissue was subjected to a 45-min digestion with 0.12 unit/ml collagenase at 37°C in a shaker at 25 rpm. The samples were then filtered through 300-μm nylon meshes (Spectrum Laboratories) and were subjected to centrifugation at 500 × g for 5 min. The floating fraction (comprised of white adipocytes) and the pellet fraction (containing the SVF) were dissolved in TRIzol reagent (Invitrogen) for RNA preparation or in TNN lysis buffer [50 mM Tris·HCl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P-40] plus a protease inhibitor mixture (Roche) for Western blotting.
RNA Preparation and Quantitative PCR (Q-PCR).
Total RNA was extracted from mouse tissues or cells with TRIzol reagent according to the manufacturer's instructions. cDNA was reverse-transcribed from 2 μg of RNA by using the RETROscript first-strand synthesis kit (Ambion). Q-PCR was performed with Brilliant SYBR Green QPCR Master Mix (Stratagene) and an Mx3000P thermal cycler (Stratagene). The relative amount of mRNA normalized to cyclophilin B was calculated by using the comparative Ct method.
Western Blotting.
Cell lysates were prepared in TNN buffer with protease inhibitor mixture (Roche) unless described otherwise. Twenty micrograms of protein lysate was resolved by 10% SDS/PAGE and transferred onto PVDF membranes. Ponceau S (Boston Bioproducts) staining was performed according to the manufacturer's instructions. In some cases, a separate gel was run in parallel and was subjected to Ruby staining (Invitrogen) according to the manufacturer's instructions. Membranes were blocked in PBS supplemented with 0.5% Tween 20 (PBST) plus 10% nonfat milk for 1 h followed by incubation with primary (1:2,000) and secondary antibodies (1:2,000) for 1 h each with PBST washes in between. Blots were then exposed to enhanced chemiluminescence substrate and exposed to film.
Co-IP Analysis.
Co-IP analysis was performed according to a modified protocol described by Xu et al. (50). Briefly, HEK-293 cells were transfected with FLAG empty vector, FLAG-tagged GATA2, GATA3, or truncated mutants of GATA2 by using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions. Forty-eight hours later, cells were lysed with TD buffer containing 1% Triton X-100, 50 mM Tris (pH 7.5), 250 mM NaCl, 5 mM EDTA, 50 mM NaF, plus protease inhibitor mixture (Roche). Cell lysates were diluted 1:1 with dilution buffer (1% Triton plus 20% glycerol) and were incubated with anti-FLAG beads (Sigma) overnight. The beads were eluted with nonreducing SDS/PAGE loading buffer after extensive washes with Tris-buffered saline and were subjected to SDS/PAGE and Western blotting.
Oil Red O Staining.
Cells were fixed with 4% Formalde-Fresh (Fisher Scientific) for 15 min at room temperature and stained with oil red O solution (0.5% oil red O in isopropyl alcohol/water = 3:2) for 2 h. Cells were washed twice with distilled water before photography.
Supplementary Material
ACKNOWLEDGMENTS.
We are grateful to Dr. Gökhan Hotamisligil for providing FLAG-tagged full-length and deleted GATA constructs and to Drs. Barbara Kahn and Sotirios Karathanasis for their generous gift of reagents. This work was funded by National Institutes of Health Grant DK63906 (to E.D.R.) and an American Heart Association postdoctoral award (to Z.X.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0707082105/DC1.
References
- 1.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 2.Tong Q, Hotamisligil GS. Rev Endocr Metab Disord. 2001;2:349–355. doi: 10.1023/a:1011863414321. [DOI] [PubMed] [Google Scholar]
- 3.Rosen ED, MacDougald OA. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 4.Farmer SR. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Z, Umek RM, McKnight SL. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Mol Cell. 1999;3:151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- 7.Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Science. 2000;290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- 8.Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS. Mol Cell Biol. 2005;25:706–715. doi: 10.1128/MCB.25.2.706-715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J, Srinivasan SV, Neumann JC, Lingrel JB. Biochemistry. 2005;44:11098–11105. doi: 10.1021/bi050166i. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, Kawahara R, Hauner H, Jain MK. J Biol Chem. 2003;278:2581–2584. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- 11.Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, et al. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Akerblad P, Lind U, Liberg D, Bamberg K, Sigvardsson M. Mol Cell Biol. 2002;22:8015–8025. doi: 10.1128/MCB.22.22.8015-8025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jimenez MA, Akerblad P, Sigvardsson M, Rosen ED. Mol Cell Biol. 2007;27:743–757. doi: 10.1128/MCB.01557-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pastorcic M, Wang H, Elbrecht A, Tsai SY, Tsai MJ, O'Malley BW. Mol Cell Biol. 1986;6:2784–2791. doi: 10.1128/mcb.6.8.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang LH, Tsai SY, Sagami I, Tsai MJ, O'Malley BW. J Biol Chem. 1987;262:16080–16086. [PubMed] [Google Scholar]
- 16.Jonk LJ, de Jonge ME, Pals CE, Wissink S, Vervaart JM, Schoorlemmer J, Kruijer W. Mech Dev. 1994;47:81–97. doi: 10.1016/0925-4773(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 17.Qiu Y, Cooney AJ, Kuratani S, DeMayo FJ, Tsai SY, Tsai MJ. Proc Natl Acad Sci USA. 1994;91:4451–4455. doi: 10.1073/pnas.91.10.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang LH, Tsai SY, Cook RG, Beattie WG, Tsai MJ, O'Malley BW. Nature. 1989;340:163–166. doi: 10.1038/340163a0. [DOI] [PubMed] [Google Scholar]
- 19.Wehrenberg U, Ivell R, Walther N. Biochem Biophys Res Commun. 1992;189:496–503. doi: 10.1016/0006-291x(92)91585-e. [DOI] [PubMed] [Google Scholar]
- 20.Miyajima N, Kadowaki Y, Fukushige S, Shimizu S, Semba K, Yamanashi Y, Matsubara K, Toyoshima K, Yamamoto T. Nucleic Acids Res. 1988;16:11057–11074. doi: 10.1093/nar/16.23.11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereira FA, Qiu Y, Tsai MJ, Tsai SY. J Steroid Biochem Mol Biol. 1995;53:503–508. doi: 10.1016/0960-0760(95)00097-j. [DOI] [PubMed] [Google Scholar]
- 22.Qiu Y, Krishnan V, Pereira FA, Tsai SY, Tsai MJ. J Steroid Biochem Mol Biol. 1996;56:81–85. doi: 10.1016/0960-0760(95)00225-1. [DOI] [PubMed] [Google Scholar]
- 23.Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. Genes Dev. 1999;13:1037–1049. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CT, Li L, Takamoto N, Martin JF, Demayo FJ, Tsai MJ, Tsai SY. Mol Cell Biol. 2004;24:10835–10843. doi: 10.1128/MCB.24.24.10835-10843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takamoto N, You LR, Moses K, Chiang C, Zimmer WE, Schwartz RJ, DeMayo FJ, Tsai MJ, Tsai SY. Development. 2005;132:2179–2189. doi: 10.1242/dev.01808. [DOI] [PubMed] [Google Scholar]
- 26.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
- 27.Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zehentner BK, Leser U, Burtscher H. DNA Cell Biol. 2000;19:275–281. doi: 10.1089/10445490050021186. [DOI] [PubMed] [Google Scholar]
- 29.Spinella-Jaegle S, Rawadi G, Kawai S, Gallea S, Faucheu C, Mollat P, Courtois B, Bergaud B, Ramez V, Blanchet AM, et al. J Cell Sci. 2001;114:2085–2094. doi: 10.1242/jcs.114.11.2085. [DOI] [PubMed] [Google Scholar]
- 30.van der Horst G, Farih-Sips H, Lowik CW, Karperien M. Bone. 2003;33:899–910. doi: 10.1016/j.bone.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. Cell Metab. 2006;3:25–34. doi: 10.1016/j.cmet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Krishnan V, Pereira FA, Qiu Y, Chen CH, Beachy PA, Tsai SY, Tsai MJ. Science. 1997;278:1947–1950. doi: 10.1126/science.278.5345.1947. [DOI] [PubMed] [Google Scholar]
- 33.Krishnan V, Elberg G, Tsai MJ, Tsai SY. Mol Endocrinol. 1997;11:1458–1466. doi: 10.1210/mend.11.10.9992. [DOI] [PubMed] [Google Scholar]
- 34.Chen JK, Taipale J, Cooper MK, Beachy PA. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tontonoz P, Hu E, Spiegelman BM. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 36.Cooney AJ, Tsai SY, O'Malley BW, Tsai MJ. Mol Cell Biol. 1992;12:4153–4163. doi: 10.1128/mcb.12.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooney AJ, Leng X, Tsai SY, O'Malley BW, Tsai MJ. J Biol Chem. 1993;268:4152–4160. [PubMed] [Google Scholar]
- 38.Kliewer SA, Umesono K, Heyman RA, Mangelsdorf DJ, Dyck JA, Evans RM. Proc Natl Acad Sci USA. 1992;89:1448–1452. doi: 10.1073/pnas.89.4.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran P, Zhang XK, Salbert G, Hermann T, Lehmann JM, Pfahl M. Mol Cell Biol. 1992;12:4666–4676. doi: 10.1128/mcb.12.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leng X, Cooney AJ, Tsai SY, Tsai MJ. Mol Cell Biol. 1996;16:2332–2340. doi: 10.1128/mcb.16.5.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai SY, Tsai MJ. Endocr Rev. 1997;18:229–240. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 42.Eubank DW, Duplus E, Williams SC, Forest C, Beale EG. J Biol Chem. 2001;276:30561–30569. doi: 10.1074/jbc.M103019200. [DOI] [PubMed] [Google Scholar]
- 43.El-Jack AK, Hamm JK, Pilch PF, Farmer SR. J Biol Chem. 1999;274:7946–7951. doi: 10.1074/jbc.274.12.7946. [DOI] [PubMed] [Google Scholar]
- 44.Hoshizaki DK, Blackburn T, Price C, Ghosh M, Miles K, Ragucci M, Sweis R. Development. 1994;120:2489–2499. doi: 10.1242/dev.120.9.2489. [DOI] [PubMed] [Google Scholar]
- 45.Hayes SA, Miller JM, Hoshizaki DK. Development. 2001;128:1193–1200. doi: 10.1242/dev.128.7.1193. [DOI] [PubMed] [Google Scholar]
- 46.Abel T, Michelson AM, Maniatis T. Development. 1993;119:623–633. doi: 10.1242/dev.119.3.623. [DOI] [PubMed] [Google Scholar]
- 47.Sagami I, Tsai SY, Wang H, Tsai MJ, O'Malley BW. Mol Cell Biol. 1986;6:4259–4267. doi: 10.1128/mcb.6.12.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ladias JA, Karathanasis SK. Science. 1991;251:561–565. doi: 10.1126/science.1899293. [DOI] [PubMed] [Google Scholar]
- 49.Xu Z, Kandror KV. J Biol Chem. 2002;277:47972–47975. doi: 10.1074/jbc.C200486200. [DOI] [PubMed] [Google Scholar]
- 50.Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eguchi J, Yan QW, Schones DE, Kamal M, Hsu CH, Zhang MQ, Crawford GE, Rosen ED. Cell Metab. 2008;7:86–94. doi: 10.1016/j.cmet.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}