

Abstract
We describe a method for the isolation of conditional knockouts of essential multiply spliced genes in which the entire body of the gene downstream of the ATG start codon is left untouched but can be switched off rapidly and completely by adding tetracycline to the culture medium. The approach centers on a “promoter-hijack” strategy in which the gene's promoter is replaced with a minimal promoter responsive to the tetracycline-repressible transactivator (tTA). Elsewhere in the genome, a cloned fragment of the gene's promoter is used to drive expression of a tTA. Thus, the gene is essentially regulated by its own promoter but through the intermediary tTA. Using this strategy, we generated a conditional knockout of chromokinesin KIF4A, an important mitotic effector protein whose mRNA is multiply spliced and whose cDNA is highly toxic when overexpressed in cells. We used chicken DT40 cells, but the same strategy should be applicable to ES cells and, eventually, to mice.
Keywords: DT40, kif4, knockout, method, mitosis
Gene knockouts and RNAi are commonly used to probe gene function in vertebrate cells (1). In knockouts, mRNA production is shut down at its source, whereas in RNAi methods, mRNAs are produced but continuously destroyed. Knockouts are more laborious to obtain but have three potential advantages. First, protein depletion in conditional knockouts is more complete than typically achieved with RNAi. Second, phenotypes produced under conditions of stable RNAi can potentially change if mRNA levels vary because of changes in transcription rates or the efficiency of RNAi. Third, gene knockouts avoid misleading off-target effects that can occur in RNAi studies (2).
Chicken DT40 B lymphocytes are advantageous for gene-targeting studies because of their rapid generation time (9 h) and high frequency of homologous recombination, >80% for certain loci (3). In some instances, multiple knockouts of nonessential genes have been isolated (4–8). DT40 conditional knockouts are particularly useful for functional analysis of essential housekeeping genes, especially where early embryonic lethality of mouse knockouts has made such analyses impractical. DT40 knockouts have contributed to our understanding of Ig gene conversion, DNA damage and repair pathways, signaling pathways, apoptotic execution, chromatin structure, kinetochore assembly and function, and mitotic chromatin condensation (for reviews, see ref. 9).
Unfortunately, obtaining conditional knockouts of essential genes in DT40 cells is not yet routine. Previously, such knockouts have involved deletion of all or a portion of the target gene while keeping the cell alive via expression of an exogenous cDNA under control of a tetracycline-regulated promoter. However, knockouts may be unobtainable for several reasons. (i) Overexpression of the exogenous cDNA may be toxic. (ii) Many gene transcripts undergo alternative splicing, producing multiple variant protein isoforms. If more than one of these isoforms has an essential function, then expression of a single cDNA cannot rescue cell life. (iii) Expression at inappropriate times of the cell cycle may be toxic or ineffective for certain genes involved in cell-cycle regulation.
Here, we describe a “promoter-hijack” approach for obtaining conditional knockouts of essential cell cycle genes. The key to this method is that coding sequences of the gene are left intact so that any transcripts produced can undergo normal RNA processing. The gene is under control of its own or a compatible promoter, but this operates indirectly through an intermediary tetracycline-repressible transactivator (tTA). As a result, we can abruptly turn off transcription of the endogenous target gene by adding doxycycline to the culture medium. Here, this method is applied to chicken DT40 cells, but it should also be suitable for obtaining knockouts of essential genes in mouse ES cells and in mice.
Results
Chromokinesin KIF4A has multiple roles in mitotic chromosome structure, spindle assembly, and cytokinesis (10–15). To dissect these functions, we tried to obtain a conditional knockout of the kif4a gene in DT40 cells by conventional means [supporting information (SI) Fig. 5]. However, concerted attempts failed. Apparently, continuous overexpression of the kif4a cDNA is toxic, and expression of a single cDNA fails to provide full KIF4A function in DT40 cells. A conditional knockout of this challenging gene therefore required a different approach.
Use of Endogenous Promoters for Physiological Transgene Expression.
Efficient rescue of cells bearing a conditional knockout of condensin subunit smc2 with a wild-type smc2 cDNA requires that cDNA expression be regulated by a fragment of the smc2 promoter (16). We therefore tested whether stable expression of the kif4a cDNA could be achieved by using its own or another cellular promoter. The following promoter fragments were cloned: kif4apro (6.0 kb; see Fig. 2a), incenppro (5.7 kb), smc2pro (3.8 kb), and survivinpro (2.0 kb). (See SI Methods.) We used these to drive expression of differing tTA variants, testing the strength of each promoter/tTA pair by determining its ability to drive the expression of a GFP-lamin A reporter under control of a minimal tTA-responsive promoter (tetO) (17).
Fig. 2.

Promoter-hijack approach for generating conditional knockout cell lines. (a) The promoter-hijack knockout strategy. +, wild-type allele; ×, promoter-hijack vector-targeted allele. Tables show targeting efficiencies for each vector into the first and second alleles. For the second targeting, three different vectors were transfected into heterozygotes in which the first allele had been targeted by promoter-hijack vector in the presence or absence of pdk11 expression vector. (b) Physical map of the kif4a locus, showing the promoter-hijack targeting constructs and probe for Southern blotting. (c) Southern blotting revealed a polymorphism 5′ to the kif4a gene (left-most lane). Both alleles were targeted by the same targeting vector, yielding two Southern blotting patterns, depending on which allele was targeted first, and confirming correct targeting of the locus. I–IV correspond to each of the steps described in a. (d) Physical map of the kif4A locus showing the disruption constructs and probe for Southern blotting.
Cells were analyzed by flow cytometry 24 h after transient transfection with tTA and reporter constructs (Fig. 1a). The median GFP signal within the GFP-lamin A-expressing population provided an indication of promoter strength. This analysis revealed that cloned fragments of the kif4a, incenp, and survivin promoters are functional, albeit weaker than the CMV promoter (Fig. 1b). Surprisingly, the cloned smc2 promoter was stronger than the CMV promoter in this assay.
Fig. 1.

The kif4a promoter is much weaker than the cytomegalovirus (CMV) promoter. (a) Examples of the flow cytometry analysis. Medians for GFP-positive cells (transfected cells) are shown. (b) Comparative analysis of different promoters by using flow cytometry after transient transfection of tetO-GFP lamin A and various tTA vectors. Median values (as in a) are normalized to 1.0 for CMV-tTA2. Error bars represent SD (n ≥ 3).
Multiple Splice Variants of KIF4A Are Expressed in DT40 Cells.
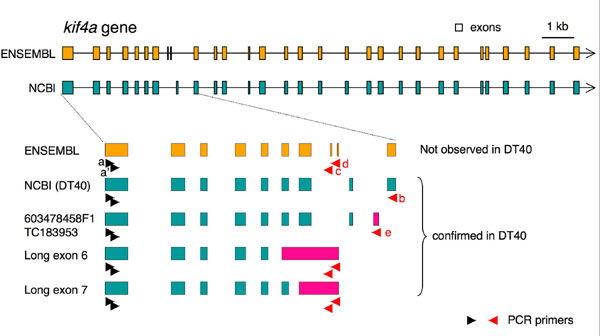
We obtained cell lines with relatively stable KIF4A expression when the cDNA was driven indirectly by its own promoter (see SI Fig. 6a). However, we still failed to obtain a conditional knockout of kif4a in these cells, suggesting that multiple splice variants of KIF4A may be required for cell survival. The kif4a gene contains either 30 exons (Ensembl database) or 29 exons [National Center for Biotechnology Information (NCBI) database], with predicted differences around exon 8 and 9 (see SI Fig. 7). Our cDNA matched the latter.
A search of chicken databases revealed ESTs confirming the alternative splicing of kif4a. For example, two ESTs (603478458F1, TC183953) contain an extra exon located between canonical exons 9 and 10. The presence of this exon was confirmed by RT-PCR of DT40 polyA (+) RNA (SI Fig. 7). No ESTs were found encoding exons 8 and 9 predicted by the Ensembl database, and transcripts of those putative exons were not detected by RT-PCR. Instead, unexpected isoforms with an extended exon 6 or 7 were detected in DT40 cells by using specific primers corresponding to either exons 8 or 9 (SI Fig. 7). Altogether, this preliminary analysis revealed that at least four isoforms of KIF4A are expressed in DT40 cells.
A Promoter-Hijack Strategy for Conditional Shutoff of the KIF4A Gene.
We developed a strategy designed to allow the conditional expression of all KIF4A splice variants at near physiological levels (Fig. 2a). Briefly, the idea was to “hijack” the gene promoter and use it to drive the tTA, which would then drive expression of the endogenous kif4a locus instead of a cDNA.
A targeting vector was constructed to replace 7.5 kb of the 5′ untranslated region (UTR) from the kif4a gene with a minimal tet-responsive promoter and puromycin resistance cassette flanked by Lox P sites (Fig. 2b). Integration of this vector does not disturb sequences downstream of the ATG. This vector was transfected into wild-type cells, with a targeting efficiency of 47% (9/19) (Fig. 2a). Southern blot analysis of wild-type cells with a 5′ probe revealed a polymorphism in the extreme 5′ UTR of the kif4a gene in DT40 cells. Both alleles were targeted by the same promoter-hijack targeting vector (Fig. 2c).
Excision of the lox P puromycin cassette from the correctly targeted promoter-hijack vector by transient transfection of a plasmid encoding Cre-recombinase yielded heterozygotes with the minimal tTA-responsive promoter (tetO) inserted just in front of the ATG. These cells were then transfected with kif4apro-tTA2 either alone or in combination with a vector driving constitutive expression of upstream gene pdk11 (see next section). Heterozygous clones with one untouched kif4a allele and one promoter-hijack allele driven by kif4apro-tTA2 were tested for tTA activity by using a GFP-lamin A cDNA reporter, as in Fig. 1. Regulated expression of the targeted kif4a locus in these clones was confirmed by growth in the presence or absence of doxycycline (dox) for 24 h and determination of the KIF4A protein levels by immunoblotting (SI Fig. 6b). In clones chosen for the second targeting, KIF4A expression was moderately elevated when one allele was driven by kif4apro-tTA2, but the protein fell to wild-type levels after 1 day with dox.
Rescue of Putative Upstream Genes.
The 7,463 kb deleted by the kif4a promoter-hijack vector included the putative kif4a promoter and coding region of pdk11 but also the promoter for ENSGALG00000004206 and coding region of ENSGALG00000004202, two predicted genes (Fig. 2b) that could possibly be essential for life. An EST of pdk11 is present in a chicken intestinal lymphocyte library, but we could not detect pdk11 expression in DT40 cells by RT-PCR (data not shown).
We used two strategies to minimize potential effects of the promoter-hijack vector on these genes. First, we knocked-out the second kif4a allele by using a targeting vector that disrupts the kif4a ORF but leaves the three upstream genes undisturbed. This vector inserts a puromycin resistance cassette into a BamHI site in exon 3 of kif4a, thereby permitting the transcription of mRNA encoding the first 167 aa of KIF4A (Fig. 2d). We also constructed a vector driving ectopic expression of the predicted pdk11 ORF because we could not exclude that the disruption vector targeting might perturb upstream elements of the pdk11 promoter.
KIF4A Knockout Cells Obtained by the Promoter-Hijack Strategy.
Using the promoter-hijack strategy, we obtained seven independent kif4a conditional knockout cell lines (Fig. 2a). The kif4a promoter-hijack, disruption, or kinesin motor deletion vectors were transfected into four independent kif4a heterozygous lines with or without an ectopic constitutive pdk11 expression vector. After 1–2 weeks of culture under selective conditions, candidate clones were transferred to replicate plates in new media and cultured for 3–6 days in the presence or absence of 0.5 μg/ml dox. Clones exhibiting retarded growth or death in the presence of dox were further analyzed by Southern blotting with an external 5′ probe to confirm correct targeting (Fig. 2c).
We obtained conditional kif4a knockout clones by using four different combinations of vectors (Fig. 2a). Importantly, one kif4a knockout clone was isolated in which both alleles were targeted by the promoter-hijack vector without ectopic expression of PDK11. All clones showed essentially identical phenotypes, therefore pdk11 and the two predicted upstream genes are not essential in DT40 cells.
Two of the kif4a knockout clones were selected for further analysis. In those clones, both alleles were targeted by the promoter-hijack vector, either in the absence (x/x1) or presence (x/x2) of ectopic PDK11 expression.
Characterization KIF4A Conditional Knockout Cells.
In the absence of dox, the doubling time of the kif4a knockout cell line was 11.3 h for kif4a x/x1 and 12.9 h for x/x2. This time compares to 8.9 h for wild type and 10.4 h for kif4a heterozygotes expressing kif4apro-tTA2. The apparent increased doubling time reflects a background of cell death in these lines. The growth of kif4a knockout cells slowed dramatically within 24 h of dox addition (Fig. 3a). This slowing was accompanied by an increase in cell death as measured by annexin V-PE staining (Fig. 3b).
Fig. 3.

Initial characterizations of kif4a knockout cell lines. (a) Growth was inhibited within 24 h after the addition of dox. (b) Percentage of apoptotic cells increased within 24 h after the addition of dox. (c) mRNA was shut off within 8 h after the addition of dox. Wild-type level is 1.0. (d) KIF4A protein almost disappeared within 24 h after the addition of dox. α-Tubulin was used as a loading control. (a–c) Error bars represent SD (n ≥ 3).
Quantitative RT-PCR revealed that kif4a mRNA levels decreased significantly within 2 h after adding dox and almost disappeared within 8 h (Fig. 3c). Concomitantly, KIF4A protein, which was originally 3- to 5-fold over-expressed compared with the wild type, fell within 24 h to <5% of wild-type levels (data not shown) and became undetectable by immunoblotting at 48 h after addition of dox (Fig. 3d).
Expression of the canonical (NCBI) kif4a cDNA failed to support the growth of kif4a knockout cells after dox addition. Although mRNA expression was confirmed by RT-PCR in one clone, this became undetectable after 1 week in culture even in the presence of dox, where KIF4A expression is required to keep cells alive. This finding explains the failure of the conventional knockout strategy for kif4a.
We modified the promoter-hijack targeting vector to insert sequences encoding GFP and a triple-affinity purification (TrAP) tag (Fig. 4a) in front of the ATG of the endogenous kif4a allele in a heterozygous cell line expressing kif4apro-tTA2. Tagging endogenous genes by knockin is done routinely in yeasts but rarely in vertebrate cells (18, 19). In these cells, all KIF4A isoforms expressed from the targeted allele are tagged with GFP. Immunoblotting confirmed that TrAPGFP-KIF4A was expressed at levels comparable to endogenous KIF4A (Fig. 4b). The tagged protein colocalized with endogenous KIF4A (Fig. 4 c and d), and we could isolate both tagged and endogenous KIF4A from cell lysates by using streptavidin beads (Fig. 4e). This finding suggested that the tagged protein forms a complex with the endogenous protein. Live imaging of the tagged protein revealed the dynamic localization of KIF4A during mitotic progression (Fig. 4f and SI Movie 1). During prometaphase and metaphase, TrAPGFP-KIF4A is concentrated along the chromosome axis with a diffuse pool in the cytosol. During anaphase, a fraction of TrAPGFP-KIF4A transfers to the central spindle, then concentrates at the midzone and midbody from telophase to cytokinesis. TrAPGFP-KIF4A protein will be a useful tool for future detailed phenotypic and biochemical analyses of KIF4A function.
Fig. 4.

Integration of TrAPGFP in the kif4a genomic locus and application of the cell line. (a) Schematic presentation of TrAPGFPkif4a. (b) TrAPGFP-KIF4A is expressed at similar to wild-type cell level. (c–c‴) Four-percent paraformaldehyde-fixed cells. TrAPGFP-KIF4A localizes axially on chromosomes and diffusely in cytosol during prometaphase. Note that KIF4A-specific antibodies did not show specific staining in prometaphase cells under these fixation conditions. DNA was stained with DAPI. (d–d‴) Cold methanol/acetic acid-fixed cells. TrAPGFP-KIF4A colocalized with endogenous KIF4A on chromosome axes. Note that TrAPGFP-KIF4A was stained by using anti-SBP antibody because the GFP signal was lost after this fixation. (Scale bar: 5 μm.) (e) One-step crude purification of TrAPGFP-KIF4A from lysates of cycling cells with streptavidin beads revealed that TrAPGFP-KIF4A protein is complexed with endogenous KIF4A protein. Equivalent amounts of cells were loaded in each lane unless otherwise indicated. (f) Stills from SI Movie 1of mitosis in a TrAPGFP-KIF4A integrant cell line. TrAPGFP-KIF4A protein localizes on chromosome axes and diffusely in cytosol throughout mitosis. During anaphase, part of KIF4A transfers to the central spindle (filled arrow) and later concentrates at the midzone and midbody. A vestigial midbody from a previous division is labeled with the empty arrow. Because two movies following the same cell continuously were fused, two “time = 0 points” are shown. The first, from the beginning to the onset of anaphase, shows one focal plane. The second, from the onset of anaphase to late cytokinesis, is a projection of 10 focal planes. Time interval, 1 min.
Discussion
The “promoter-hijack” approach described here has enabled us to obtain a conditional knockout of kif4a, a gene that had previously been refractory to the conditional knockout approach in DT40 cells. This method has also enabled us to isolate a conditional knockout of incenp, which had eluded us despite several years of effort. Detailed phenotypic analyses of the Kif4A and incenp knockouts will be the subject of future communications (K.S., H.O., Zhenjie Xu, S.R., and W.C.E., unpublished data).
The key innovation of our promoter-hijack approach is to retain at least one allele in which the coding region, including introns and the 3′ UTR of the gene, remains intact, while replacing its promoter with a minimal promoter responsive to the tTA transcriptional activator. By cloning a fragment of the endogenous gene promoter and using that to drive expression of the tTA, we achieve a situation wherein the gene produces its normal pre-mRNA transcript (including both introns and exons) essentially under the control of its own promoter but through the intermediary tTA. This method enables transcription from the endogenous gene locus to be shut off extremely rapidly and efficiently by adding dox to the culture medium. Another advantage of regulating the expression of the endogenous gene is that the transcript is produced at its normal location within the nucleoplasm. Thus, any potential spatial regulation of transcript dynamics or processing is preserved. Of course, the final levels of gene expression will be affected by factors including the copy number and half-life of the transfected tTA.
The “promoter-hijack” strategy does not require cloning and expression of the cDNA for the gene being studied. The method is therefore applicable to cell cycle-regulated, alternatively spliced genes as well as very large genes for which cDNA cloning can be problematic. For example, use of the promoter-hijack vector to target one allele of incenp followed by disruption of the other allowed us to isolate a conditional allele of the endogenous INCENP locus, which is flanked upstream by a nearby essential gene or essential noncoding element. Therefore, the promoter-hijack strategy may be applicable to most if not all essential genes.
Although used less widely than ES cells for gene targeting, chicken DT40 cells offer several advantages for studies of housekeeping genes. With a doubling time of 9 h, stable DT40 cell lines expressing particular isoforms, mutants, or tagged proteins in the knockout background can be established within weeks by introduction of a cDNA or by genomic targeting. The ability to complement null mutations can provide evidence that tagged proteins are biologically active and enable a definitive mutational analysis of the protein being studied. Rapid generation time, growth in suspension, and ability to synchronize the cells by centrifugal elutriation in the absence of drugs (20) render the DT40 system highly suitable for biochemical analysis of wild-type and mutant cells. Furthermore, although these nonadherent B-cells require a high-end microscope for optimal phenotypic analysis, they can be used for high-resolution live-cell analysis. Indeed, all of the isoforms of KIF4A can be visualized and isolated as a result of the knockin of sequences encoding a TrAPGFP tag into the endogenous locus at the 5′ end of the transcription unit. With the ability to readily isolate conditional alleles of essential housekeeping genes described here and with the availability of a wide range of shared reagents and protocols (9), DT40 cells will continue to grow in utility as a model genetic system.
The promoter-hijack strategy should be applicable to any cell line for which gene-targeting procedures are established, including human cell lines (21, 22) and mouse ES cells. It may therefore be possible to generate conditional knockouts for essential mouse genes that could be rapidly switched off throughout the entire animal by administration of dox.
Methods
Materials.
Unless otherwise noted, all chemicals and primers were purchased from Sigma–Aldrich, all enzymes required for subcloning were obtained from New England BioLabs, and all cell culture products were purchased from Gibco BRL/Life Technologies, a division of Invitrogen. Antibodies used for immunoblotting/indirect immunofluorescence analysis were rabbit anti-KIF4A at 1:500 (IB, IF); mouse anti-tubulin B512 (Sigma) at 1:4,000 (IB), 1:1,000 (IF); anti-SBP 1:250 (IB), 1:20 (IF).
Cell Culture, Transfections, and Selections.
Ten million DT40 cells (grown in RPMI medium 1640 plus 10% FBS, 1% chicken serum, penicillin, and streptomycin) were suspended in 0.5 ml of Optimem per cuvette (0.4 mm; Bio-Rad). Ten to 20 μg of linearized DNA was added to each cuvette and kept on ice for 5 min before and after electroporation (300 V; 950 microfarads in a GenePulser; Bio-Rad). Cells were transferred to 96-well plates, either directly or after 24 h in selective media (puromycin at 0.5 μg/ml and blasticidin at 25 μg/ml from Calbiochem; Histidinol at 1.5 mg/ml from Sigma; or G418 at 1.5 mg/ml from Invitrogen). For Cre-recombinase transient transfection, 1.0 × 107 DT40 cells were rinsed with PBS and then suspended in 0.1 ml of nucleofector solution V per cuvette. Two to 10 μg of DNA was added to each cuvette and electroporated (Program B-30, Nucleofector II; Amaxa). After 24 h, cells were transferred to 96-well dishes containing nonselective media. Clones were transferred to 24-well dishes containing selective (puromycin 0.5 μg/ml) and nonselective media, and those that died in selective media were chosen for further study. Excision of the puromycin resistance cassette was confirmed by Southern blotting.
Construction of TrAPGFP.
The plasmids (pTrAP) containing a His tag, streptavidin-binding peptide (SBP), and S-tag were designed in our laboratory. Primers used to generate the TrAP tag are described in the SI Methods.
Construction of Endogenous Promoters and Flow Cytometry Analysis.
All of the promoters were generated from a phage DNA corresponding to each gene and cloned into BsrGI/EcoRI sites of tTA2, -3, and -4 (Clontech).
Two to 10 micrograms of tTA and pUHD10.3 GFP-lamin A plasmids were cotransfected into DT40 wild-type cells by electroporation, and these cells were harvested after 24 h and analyzed by flow cytometry (Becton Dickinson). The median of GFP-positive cells was used as an indication of promoter strength. Primers used to generate the promoters are described in the SI Methods.
TrAPGFP KIF Isolation from Cell Lysate.
Exponentially growing cells were collected by centrifugation and lysed in lysis buffer [50 mM Tris·HCl (pH 7.4), 50 mM NaCl, 0.5% Nonidet P-40, 3 mM CaCl2, 30 μg/ml RNase A, 40 μg/ml Micrococcal DNase, Aprotinin, 1 μg/ml each Chymostatin, Leupeptin, Antipain, Pepstatin A, and 1 mM phenylmethylsulfonylfluoride] for 40 min on ice. Ten millimolar EDTA was added and NaCl up to 150 mM. The lysate was centrifuged at 4°C for 20 min, and Streptavidin agarose beads (Pierce) were mixed with the cleared lysate (supernatant) for 1 h at 4°C. The beads were washed with wash buffer [50 mM Tris·HCl (pH 7.4), 50 mM NaCl, 0.5% Nonidet P-40] three times and then eluted with [50 mM Tris·HCl (pH 7.4), 50 mM NaCl, 0.5% Nonidet P-40, 4 mM biotin]. The remaining beads were boiled in sample buffer.
Supplementary Material
ACKNOWLEDGMENTS.
We thank T. Hori and T. Fukagawa (National Institute of Genetics, Mishima, Japan) for technical advice, and N. Cobbe, J. Paulson, and L. Harrington for comments on the manuscript. This work is supported by The Wellcome Trust, of which W.C.E. is a Principal Research Fellow.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0712083105/DC1.
References
- 1.Hudson DF, Morrison C, Ruchaud S, Earnshaw WC. Reverse genetics of essential genes in tissue-culture cells: “Dead cells talking.”. Trends Cell Biol. 2002;12:281–287. doi: 10.1016/s0962-8924(02)02281-x. [DOI] [PubMed] [Google Scholar]
- 2.Svoboda P. Off-targeting and other non-specific effects of RNAi experiments in mammalian cells. Curr Opin Mol Ther. 2007;9:248–257. [PubMed] [Google Scholar]
- 3.Buerstedde J-M, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 4.Sugawara H, Kurosaki M, Takata M, Kurosaki T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 1997;16:3078–3088. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirano S, et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 2005;24:418–427. doi: 10.1038/sj.emboj.7600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ageichik AV, Samejima K, Kaufmann SH, Earnshaw WC. Genetic analysis of the short splice variant of the inhibitor of caspase-activated DNase (ICAD-S) in chicken DT40 cells. J Biol Chem. 2007;282:27374–27382. doi: 10.1074/jbc.M704307200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruchaud S, et al. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 2002;21:1967–1977. doi: 10.1093/emboj/21.8.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada M, Hori T, Fukagawa T. The DT40 system as a tool for analyzing kinetochore assembly. In: Buerstedde JM, Takeda S, editors. Reviews and Protocols in DT40 Research, Subcellular Biochemistry. Vol 40. New York: Springer; 2006. pp. 91–106. [DOI] [PubMed] [Google Scholar]
- 9.Buerstedde JM, Takeda S, editors. Reviews and Protocols in DT40 Research, Subcellular Biochemistry. Vol 40. New York: Springer; 2006. [Google Scholar]
- 10.Mazumdar M, Sundareshan S, Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J Cell Biol. 2004;166:613–620. doi: 10.1083/jcb.200401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurasawa Y, Earnshaw WC, Mochizuki Y, Dohmae N, Todokoro K. Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. EMBO J. 2004;23:3237–3248. doi: 10.1038/sj.emboj.7600347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu C, Jiang W. Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proc Natl Acad Sci USA. 2005;102:343–348. doi: 10.1073/pnas.0408438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Midorikawa R, Takei Y, Hirokawa N. KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell. 2006;125:371–383. doi: 10.1016/j.cell.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 14.D'Avino PP, et al. Recruitment of Polo kinase to the spindle midzone during cytokinesis requires the Feo/Klp3A complex. PLoS ONE. 2007;2:e572. doi: 10.1371/journal.pone.0000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castoldi M, Vernos I. Chromokinesin Xklp1 contributes to the regulation of microtubule density and organization during spindle assembly. Mol Biol Cell. 2006;17:1451–1460. doi: 10.1091/mbc.E05-04-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudson DF, Vagnarelli P, Gassmann R, Earnshaw WC. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev Cell. 2003;5:323–336. doi: 10.1016/s1534-5807(03)00199-0. [DOI] [PubMed] [Google Scholar]
- 17.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukagawa T, et al. CENP-H, a constitutive centromere component, is required for centromere targeting of CENP-C in vertebrate cells. EMBO J. 2001;20:4603–4617. doi: 10.1093/emboj/20.16.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arakawa H, et al. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillespie DA, Henriques C. In: Reviews and Protocols in DT40 Research, Subcellular Biochemistry. Buerstedde JM, Takeda S, editors. Vol 40. New York: Springer; 2006. pp. 359–361. [Google Scholar]
- 21.Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papi M, Berdougo E, Randall CL, Ganguly S, Jallepalli PV. Multiple roles for separase auto-cleavage during the G2/M transition. Nat Cell Biol. 2005;7:1029–1035. doi: 10.1038/ncb1303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}