

Abstract
Sooty mangabeys (SMs) naturally infected with simian immunodeficiency virus (SIV) do not develop AIDS despite high levels of virus replication. At present, the mechanisms underlying this disease resistance are poorly understood. Here we tested the hypothesis that SIV-infected SMs avoid immunodeficiency as a result of virus replication occurring in infected cells that live significantly longer than human immunodeficiency virus (HIV)-infected human cells. To this end, we treated six SIV-infected SMs with potent antiretroviral therapy (ART) and longitudinally measured the decline in plasma viremia. We applied the same mathematical models used in HIV-infected individuals and observed that SMs naturally infected with SIV also present a two-phase decay of viremia following ART, with the bulk (92 to 99%) of virus replication sustained by short-lived cells (average life span, 1.06 days), and only 1 to 8% occurring in longer-lived cells. In addition, we observed that ART had a limited impact on CD4+ T cells and the prevailing level of T-cell activation and proliferation in SIV-infected SMs. Collectively, these results suggest that in SIV-infected SMs, similar to HIV type 1-infected humans, short-lived activated CD4+ T cells, rather than macrophages, are the main source of virus production. These findings indicate that a short in vivo life span of infected cells is a common feature of both pathogenic and nonpathogenic primate lentivirus infections and support a model for AIDS pathogenesis whereby the direct killing of infected cells by HIV is not the main determinant of disease progression.
Human immunodeficiency virus (HIV) arose in the human population through multiple episodes of cross-species zoonotic transmission of primate lentiviruses infecting African nonhuman primates and collectively defined simian immunodeficiency viruses (SIVs) (14). Of these viruses, SIVcpz is the source of the HIV type 1 (HIV-1) pandemic, while SIVsmm, which naturally infects sooty mangabeys (SMs; Cercocebus atys), is the source of the HIV-2 epidemic. Importantly, SIVsmm is also the origin of SIVmac viruses, whose infection of rhesus macaques (RMs; Maccaca mulata) is the best-studied animal model for AIDS pathogenesis and vaccines (19).
SIVsmm infection of SMs is common in the wild and in captivity and is typically nonpathogenic, with the majority of animals maintaining healthy peripheral CD4+ T-cell counts despite chronic high levels of virus replication (8, 41, 47, 52). The nonpathogenicity of SIV infection of SMs is confirmed by the fact that there has been only one report of classic AIDS in a naturally infected animal (23). Importantly, the absence of disease progression despite high levels of virus replication has also been shown in other monkey species naturally infected with SIV (5, 18, 36). These findings are in stark contrast to the observation that, in HIV-infected individuals, high viral loads predict faster disease progression (25, 26). Despite intense studies by us and others (2, 3, 8, 10, 30, 35, 45, 47, 52, 53), it is still unclear why SIV infection is nonpathogenic in SMs; however, it is increasingly recognized that a better understanding of the mechanisms underlying this lack of disease will provide important clues as to the pathogenesis of AIDS in HIV-infected individuals (46, 50, 51).
A possible explanation for the preserved CD4+ T-cell homeostasis and absence of AIDS in SIV-infected SMs with high viral loads is a reduced in vivo virus cytopathicity, which would result in a significantly longer average life span of infected cells. The in vivo life span of infected cells during chronic HIV infection was first assessed in seminal studies in which HIV-infected patients were treated with potent antiretroviral therapy (ART) (17, 39, 55). In these as well as more recent studies (24, 38), the analysis of viremia changes post-ART show a rapid initial decline that is thought to reflect the loss of short-lived virus-producing cells (most likely activated CD4+ T cells), followed by a slower decline that is suggestive of the loss of a population of longer-lived virus-producing cells (possibly resting CD4+ T cells or macrophages). Based on these studies, it was concluded that the bulk of HIV replication (92 to 99%) occurs in recently infected cells that die soon after infection, with an average life span on the order of 1 day after the start of viral production, and only 1 to 8% of virus production is derived from long-lived cells with an average life span on the order of 1 to 4 weeks (38).
In the current study, we used a similar experimental strategy to measure the in vivo life span of virus-infected cells in SMs. We treated six animals with 9-R-(2-phosphonomethoxypropyl)adenine (PMPA; tenofovir) and beta-2,3-dideoxy-3-thia-5-fluorocytidine (FTC; emtricitabine) and analyzed the slope of the decline in viremia after ART using the same mathematical model previously applied to HIV infection (38). As antiretroviral therapy consisting of reverse transcriptase inhibitors such as PMPA and FTC does not affect preformed virus or the ability of previously infected cells to continue to produce new virions, this experimental system allows a reliable quantitation of the in vivo turnover of virions and infected cells, thus providing indirect information on what cell type(s) supports virus replication in this nonpathogenic model of infection. Consistent with a previous unpublished experiment conducted by Grant, Staprans, and Feinberg using PMPA monotherapy, we found that SMs naturally infected with SIV present the same two-phase decay of viremia following ART observed in HIV-infected individuals and SIV-infected RMs, with the bulk of virus replication sustained by short-lived cells. These data suggest that the absence of disease in SMs is unlikely to be related to reduced intrinsic virus cytopathicity and suggest a key role for species-specific host factors in determining the outcome of a primate lentiviral infection.
MATERIALS AND METHODS
Animals.
Six SMs naturally infected with SIV were treated with PMPA and FTC for 42 days. Both drugs were given subcutaneously at a dose of 30 mg/kg of body weight/day. PMPA and FTC were provided by Gilead Sciences. Animals were maintained according to NIH guidelines. These studies were approved by the Institutional Animal Care and Use Committees of Emory University and the University of Pennsylvania.
Viral Load.
Viral quantification was performed on plasma from peripheral blood as previously described (45).
Lymph node biopsy.
Lymph node biopsies were collected once at pretreatment (baseline) and at days 2, 21, and 63 post-ART. Animals were anesthetized with ketamine-Telazol, the skin over the axillary or inguinal lymph node was prepared for surgery, a small incision was made, and blunt dissection was performed to remove the node. Lymph nodes were homogenized and passed through a 70-μm cell strainer to obtain lymphocytes or paraformaldehyde fixed for in situ hybridization to detect SIV RNA and immunohistochemical staining for CD3 and Ki67.
Flow cytometry.
Immunophenotyping using multicolor flow cytometry was performed on mononuclear cells isolated from peripheral blood (collected by venipuncture) and lymph nodes. Quantitation of CD3+ CD4+ and CD3+ CD8+ T cells was performed, and the percentage of individual T-cell subsets (naïve, effector, and memory) was determined by the expression of CD28 and CD95. The activation and proliferation state of T cells was monitored by the expression of HLADR, CD25, CD69, and Ki67. Antibodies used in this study were anti-CD4-peridinin chlorophyll protein (PerCP) (clone L200), anti-CD8-Pacific Blue (clone RPA-T8), anti-CD25-phycoerythrin-Cy7 (clone 2A3), Ki67-fluorescein isothiocyanate (clone B56), anti-CD3-Alexa 700 (clone SP34-2), anti-CD69-PerCP (clone L78), anti-CD95-allophycocyanin (clone DX2), and anti-HLA-DR-PerCP (clone G46-6) (all from BD PharMingen, San Diego, CA) and anti-CD28-phycoerythrin-Cy7 (clone 28.2) from eBioscience, San Diego, CA. Peripheral blood mononuclear cells (PBMC) were analyzed by seven-color fluorescent antibody staining to determine the percentage and absolute number of specific cell subpopulations. Flow cytometric acquisition and analysis of samples were performed on at least 10,000 acquired events, gated on lymphocytes, on an LSRII flow cytometer driven by the FACSDiVa software. Data analysis was performed using the FlowJo software (Treestar, Inc., Ashland, OR).
In situ hybridization and immunohistochemistry.
In situ hybridization was performed as previously described (11, 22). Immunohistochemical staining was performed using the biotin-free Double Vision polymer detection system (Biocare Medical) according to the manufacturer's instructions. In brief, tissue sections (5 μm) were mounted on glass slides, dewaxed, and rehydrated with double-distilled water. Antigen retrieval was performed by heating sections in 1× DIVA Decloacker reagent (Biocare Medical) in a 95°C water bath for 30 min followed by cooling to room temperature. Tissues were blocked with blocking reagent (Biocare Medical) for 1 h at room temperature. Endogenous peroxidase was blocked with 3% (vol/vol) H2O2 in phosphate-buffered saline (pH 7.4). Primary antibodies were diluted in 10% blocking reagent in TNB (0.1 M Tris-HCl pH 7.5, 0.15 M NaCl, and 0.5% blocking reagent [NEN]) and incubated overnight at 4°C. Mouse anti-Ki67 was resolved with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA) first, and rabbit monoclonal anti-CD3 was resolved second using Vulcan Fast Red (Biocare Medical). Sections were dehydrated, mounted in Permount (Fisher Scientific), and examined by light microscopy using an Olympus BX60 upright microscope with the following objectives: 10× (0.3 numerical aperture) and 40× (0.75 numerical aperture). Light micrographs were taken using a Spot color mosaic camera (model 11.2) using Spot acquisition software version 4.5.9 (Diagnostic Instruments, Inc.). Primary antibodies used for immunohistochemical experiments were mouse anti-human Ki67 (clone MIB-1; DakoCytomation) and rabbit monoclonal anti-human CD3 (clone SP7; Neomarkers). Isotype-matched negative control antibodies were used in every experiment and uniformly showed no staining (data not shown).
ELISPOT.
Enzyme-linked immunospot (ELISPOT) assays were performed as previously described (53). Gamma interferon (IFN-γ) and interleukin-2 (IL-2) ELISPOT kits for humans that are cross-reactive with monkeys were obtained from Mabtech (Nacka, Sweden). ELISPOT assays were performed on unfractionated PBMC suspended in R-10 medium supplemented with 50 μM 2-beta-mercaptoethanol. Sterile 96-well polyvinylidene difluoride ELIIP10SSP plates (Millipore, Bedford, MA) coated with anti-cytokine monoclonal antibody were plated with cells at concentrations ranging between 300,000 and 500,000 cells/well for SIV-specific stimulation. Positive controls included stimulation with concanavalin A (5 μg/ml; Sigma, St. Louis, MO) or staphylococcal enterotoxins A and B (100 ng/ml each; Sigma, St. Louis, MO). After overnight stimulation with SIV peptide pools at 37°C in a 5% CO2 incubator, cells were removed by extensive washing and incubated for 2 h at room temperature with biotinylated detector monoclonal antibody (Mabtech). Spots were developed by successive incubation with streptavidin-alkaline phosphatase followed by the substrate nitroblue tetrazolium-5-bromo-4-chloro-3 indolyl phosphate buffer (Bio-Rad Laboratories, Hercules, CA). Spots were counted on a KS ELISPOT automated reader system (Carl Zeiss, Inc., Thornwood, NY) using KS ELISPOT 4.2 software (performed by ZellNet Consulting, Inc., Fort Lee, NJ). Frequencies of responding cells obtained after subtracting background spots in medium-containing wells were expressed as spot-forming cells per million PBMC. ELISPOT responses of >50 spot-forming cells per million PBMC were considered positive.
Statistical analysis.
Friedman's test was used to determine the significance of the observed differences of specific immunological markers, with Dunn's multiple comparison posttest identifying statistically significant differences in the mean percentage before, during, and after ART (α < 0.05). Correlations were performed using either the Pearson correlation or the Spearman ranked correlation, depending on normality with an α of 0.05. Graphical and statistical analyses were performed using GraphPad Prism software version 4.0b.
Mathematical modeling.
Mathematical analyses to evaluate the life span of both short- and long-lived productively infected CD4+ T cells was performed by fitting the viral load decays in each animal on ART to the two-phase decay model as previously described (38). Briefly, the logarithm of the predicted viral load was fit to the logarithm of the viral load data using nonlinear least squares regression based on a Levenberg-Marquardt algorithm. For each animal, we estimated the pharmacological delay, t, the lumped parameter NkT0,d, and when a second phase was present, m. When a second phase was not present (monkeys FDv and FUs), m was set to 0 and NkT0 was set equal to c, as this is a necessary condition for the attainment of a viral set point in a one-phase decay model (39). As done previously, we assumed that the viral load in each animal had attained its set point before therapy was initiated. Further, we assumed that the efficacy of therapy was 100%, and thus we only obtained minimal estimates of the true infected cell life spans. The fraction of viral production from long-lived cells was estimated by extrapolating the second-phase decay curve back to time zero and calculating the fraction of baseline viral load that this represented.
RESULTS
Antiretroviral therapy suppresses viral replication in SMs chronically infected with SIV.
Six SMs naturally infected with SIV with viral loads ranging between 1.69 × 103 and 1.33 × 105 copies/ml of plasma were treated for 42 days with a potent antiretroviral regimen that included two reverse transcriptase inhibitors, i.e., PMPA and FTC, that were administered subcutaneously at a dose of 30 mg/kg/day. Treatment was well tolerated in all animals, with no discernible side effects. As shown in Fig. 1, this antiretroviral regimen induced a rapid and significant decline of the plasma viral load in all six SIV-infected SMs, with four of six animals suppressing viral replication below the limit of detection (i.e., 160 copies/ml of plasma) and the remaining two showing a >2-log decline in viremia after 17 days of therapy. Viral replication was also inhibited in lymph nodes (data not shown). Interestingly, interruption of therapy caused a rapid rebound in SIV viral load, the kinetics of which were remarkably similar to those observed during acute SIV infection of SMs (13, 45).
FIG. 1.

Antiretroviral therapy suppressed viral replication in SIV-infected SMs. (A) Plasma viral load of six SMs chronically infected with SIV, designated FAy, FCy, FDv, FEy, FUs, and FVs, before, during (shaded), and after ART. SMs were treated with PMPA and FTC for 42 days.
In all, these data indicate that natural SIV infection of SMs is similar to HIV infection of humans in that suppression of virus replication by ART consisting of reverse transcriptase inhibitors results in a rapid and profound decline of plasma viremia, thus suggesting that the bulk of virus replication occurs in short-lived infected cells. As shown below, mathematical modeling can be used to analyze the changes in viral load induced by ART and to estimate the average in vivo turnover of virus and infected cells, as well as the relative contribution of short- versus long-lived infected cells.
Immunological changes induced by antiretroviral therapy in SMs naturally infected with SIV.
Pathogenic HIV infection of humans and SIVmac infection of RMs are associated with progressive depletion of CD4+ T cells and high levels of lymphocyte activation and proliferation (16, 21, 27, 28, 32, 34, 42-44). In these pathogenic infections, inhibition of viral replication by ART induces a series of profound immunological changes in the infected hosts, consisting mainly of (i) increased CD4+ T-cell counts and (ii) a decreased fraction of activated and/or proliferating T cells (7).
In HIV-infected humans, the ART-induced increase of the pool of circulating CD4+ T cells shows a typically biphasic trend. The initial increase involves mainly memory cells and is the result of both the interruption of virus-induced killing and the rapid redistribution of cells that were previously trapped in lymphoid tissues (6). The second, slower phase of CD4+ T-cell increase may last up to several years, involves both naïve and memory CD4+ T cells, and is correlated with the presence of lower levels of immune activation (9, 12, 15, 48). In our group of SMs naturally infected with SIV and treated with ART, we observed only minor fluctuations in the number of circulating CD4+ T cells, with five out of six animals showing a modest increase in CD4 T-cell counts during the first week of therapy (Fig. 2A). Further fluctuations in the fraction of CD4+ and CD8+ T cells (measured as the percentage of the total CD3+ T-cell population) were observed in both the blood and lymph nodes during the follow-up post-initiation of ART; however, none of these changes was statistically significant (Fig. 2B and C). Minor fluctuations were also observed in the percentages of naïve (CD28+ CD95−), memory (CD28+ CD95+), and effector (CD28− CD95+) CD4+ T cells (data not shown). The lack of CD4+ T-cell increase in ART-treated SMs naturally infected with SIV was not entirely unexpected, given that these animals had relatively high baseline levels of CD4+ T cells. Interestingly, interruption of therapy was not associated with a significant decrease in the percentage or number of CD4+ T cells (Fig. 2), confirming that the level of virus replication does not appear to determine the size of the circulating CD4+ T-cell pool during natural SIV infection of SMs (47, 52).
FIG. 2.

Modest increases in CD4+ T cells in SMs treated with ART. (A) Plasma viral load is on the right y axis, and the CD4+ T-cell count is on the left y axis for each of the SMs before, during (shaded), and after ART. (B) Average fraction of CD3+ CD4+ T cells in peripheral blood 42 days before the initiation of ART, after 6 weeks of continuous ART, and 21 days after the termination of ART. Friedman's test demonstrated that the means were not significantly different. (C) Average fraction of CD3+ CD4+ (red) and CD3+ CD8+ (blue) T cells in peripheral blood (left) and lymph nodes (right) before, during (shaded), and after ART.
In HIV-infected individuals, suppression of virus replication is associated with a rapid decrease in T-cell turnover and reduced expression of markers of T-cell activation (1, 15, 16, 28). To first assess the ART-induced changes in the T-cell turnover, we sequentially measured the expression of the proliferation marker Ki67 on T cells of our group of SMs. As shown in Fig. 3A, ART induced relatively minor changes on the number of proliferating CD4+ T cells. Interruption of therapy was associated with an increase in the number and percentage of proliferating CD4+ T cells that reached levels higher than those observed prior to treatment in five of six animals (Fig. 3A and B). Since this increase in CD4+ Ki67+ T cells is temporally associated with the rebound of viremia, it is unclear if increased SIV replication (i) induces proliferation of SIV-specific CD4+ T cells or, alternatively, (ii) if the increased frequency of activated CD4+ T cells actually contributes to the finding that, after interruption of ART, virus replication transiently reaches levels even higher than the pretherapy set point (Fig. 1). In all our animals, we observed that ART induced a decline in the level of proliferating CD8+ T cells, which was then followed by a significant increase of these cells after termination of therapy (Fig. 3B). Importantly, the increase in CD8+ T-cell proliferation at the time of treatment interruption was only transient and likely represented a self-limiting response to the increased viral burden, as the return to the pretherapy set point viremia was associated with the decline of CD8+ T-cell proliferation to pretreatment levels. Immunohistochemical analysis of Ki67+ staining in lymph node tissue before, during, and after therapy confirmed the changes in the level of proliferating T cells observed by flow cytometry (Fig. 3C).
FIG. 3.

Reduced T-cell proliferation during ART treatment in SMs. (A) Plasma viral load is on the right y axis, and the number of CD4+ KI67+ T cells is on the left y axis for each of the SMs before, during (shaded), and after ART. (B) Percentage of CD4+ KI67+ (left) and CD8+ KI67+ (right) T cells in the peripheral blood of each of the SIV-infected SMs before, during (shaded), and after ART. The average fractions of CD4+ KI67+ T cells (left) and CD8+ KI67+ T cells (right) in peripheral blood 42 days before the initiation of ART, after 6 weeks of continuous ART, and 21 days after the termination of ART are shown. Friedman's test was used to compare the differences in the means. Statistically significant differences were observed in the percentage of CD8+ KI67+ T cells (α = 0.012); thus, Dunn's comparison posttest was performed (depicted by an asterisk). (C) Immunohistochemical analysis of Ki67 (brown) and CD3 (pink) staining in lymph node tissue before, during, and after therapy.
To further characterize in our cohort of SIV-infected SMs the impact of ART on the level of immune activation, we longitudinally assessed the expression of the markers CD25, CD69, and HLA-DR on T cells. As shown in Fig. 4A and B, we observed that ART induced a modest decrease in the fraction of CD4+ CD25+, CD8+ CD25+, and CD8+ CD69+ T cells in blood. A similar nonsignificant trend toward a reduced expression of activation markers on T cells during therapy was also observed in lymph nodes (Fig. 4C). Importantly, no correlation was found, at any time point during the follow-up period, between the ART-induced changes in CD4+ T-cell count and the fraction of activated CD4+ or CD8+ T cells (data not shown). Coincident with the increase of viral replication observed upon the termination of ART, we observed a statistically significant increase (P < 0.001) in the fraction of activated CD8+ CD25+ T cells (Fig. 4B).
FIG. 4.

Minor reductions in levels of activated T cells in ART treated SIV-infected SMs. (A) Average fraction of CD4+ T cells (left) expressing CD25+ (red) and CD69+ (blue), as well as the average fraction of CD8+ T cells (right) expressing CD25+ (red) and CD69+ (blue) in peripheral blood before, during (shaded), and after ART. (B) Mean percentage of CD4+ CD25+, CD4+ CD69+ (left), and CD8+ CD25+, CD8+ CD69+ (right) T cells in peripheral blood 42 days before the initiation of ART, after 6 weeks of continuous ART, and 21 days after the termination of ART. Friedman's test was used to compare the differences in the means. Statistically significant differences were observed in the percentage of CD8+ CD25+ T cells (α = 0.0055); thus, Dunn's comparison posttest was performed (depicted by an asterisk).
To determine the effect of inhibiting virus replication on the level of circulating SIV-specific T cells, IFN-γ and IL-2 ELISPOT responses to pools of peptides representing the entire SIVmac239 proteome were measured before, during, and after ART as previously described (53). While IFN-γ ELISPOT responses were detected in all six SMs at baseline (Fig. 5), suppression of virus replication was associated, in five out of six animals, with a transient 2- to 13-fold decline in IFN-γ ELISPOT responses (Fig. 5). At day 84 and 150, i.e., 6 and 15 weeks after stopping ART, we observed an increase of the SIV-specific T-cell response that returned to levels similar to baseline. IL-2 ELISPOT responses to SIV peptides were lower in magnitude compared to the IFN-γ ELISPOT responses but showed a similar kinetic pattern during the course of ART (data not shown). The trend for decline in ELISPOT responses during suppression of viremia followed by a rebound when virus replication resumed suggests that these changes in SIV-specific T-cell responses were antigen driven. The transient increase in proliferating and activated CD8+ T lymphocytes soon after stopping ART raises the possibility that an increase in frequency of SIV-specific CD8+ T lymphocytes coincided with control of rebound viremia to pre-ART set point levels. However, in the absence of ELISPOT data at more frequent time points soon after stopping ART, it is not possible to definitively ascertain the temporal relationship between viremia control and the SIV-specific T-lymphocyte response.
FIG. 5.

SIV-specific IFN-γ ELISPOT responses in SMs. IFN-γ ELISPOT responses in six SMs naturally infected with SIV were measured by peptide stimulation before, during, and after treatment with PMPA and FTC. The magnitudes of IFN-γ ELISPOT responses to SIV proteins are shown on the y axis, and individual time points are indicated on the x axis. Colored bars depict the frequency of the IFN-γ ELISPOT response to each tested SIV protein. Numbers on the tops of columns denote the number of SIV proteins testing positive.
Taken together, these data indicate that short-term suppression of viral replication induces only minor changes in the level of CD4+ T cells, immune activation, and SIV-specific T-cell responses in SMs naturally infected with SIV.
SIV infection of SMs is characterized by rapid virus turnover and a short in vivo life span of infected cells.
In a series of influential studies, the kinetic analysis of the ART-induced decline of viremia has provided important insight into the turnover of virions and infected cells during HIV infection (17, 38, 39, 55). These early studies estimated the average life span of free virions (<6 h) and productively infected cells (<1.6 days), as well as the fraction of virus replication occurring in short-lived (90 to 99%) versus long-lived (1 to 10%) cells (38, 39), while more recent studies have suggested that free virions are cleared with a half-life of <1 h and infected cells live ∼1 day while producing virus (24, 40).
In SMs naturally infected with SIV, a more limited impact of virus replication on the homeostasis of the CD4+ T-cell pool could theoretically be explained by either a longer life span of short-lived productively infected cells (likely, activated CD4+ T cells) or an increased fraction of virus replication occurring in long-lived cells (possibly macrophages). To this end, we applied to the data generated from our group of ART-treated SIV-infected SMs the “long-lived infected cell model” (38), which accounts for the biphasic decay of plasma viremia and allowed us to estimate both the life span of productively infected cells and the fraction of virus produced by short- versus long-lived cells. Under 100% effective therapy, plasma viral load, V, is predicted to decay from its baseline value, V0, according to the formula shown below, where δ is the decay rate of short-lived productively infected CD4+ T cells, μ is the decay rate of infected long-lived cells, N is the burst size, k is the rate constant for infection of short-lived cells, T0 is the baseline CD4+ T-cell count, c is the clearance rate of free virus, assumed to be equal to its previously estimated value of 23 days−1, τ is the pharmacological delay, and t is the time on therapy (38).
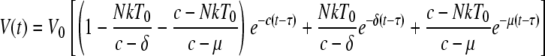 |
Fitting viral load decay data to this formula allows one to obtain minimal estimates of δ and μ and to calculate the fraction of virus produced by short- and long-lived infected cells (38). For animals FDv and FUs, there was only one phase of viral decay that was evident before the viral load became undetectable. For these monkeys, we set μ at 0 and made estimates using the one-phase viral decay model (39).
We determined that treatment with PMPA and FTC of SIV-infected SMs resulted in a biphasic decline in viral load, with the rapid phase of viremia decline lasting <7 days (Fig. 6A). This decline accounted for 92 to 99% of viral replication in all animals. From the rate of decline we estimated that on average the half-life of productively infected cells is at most 1.1 ± 0.25 days (mean ± standard error). The second phase of decline lasted for 14 days (Fig. 6A) and is due to virus production from longer-lived cells with a half-life of at most 15.2 ± 15.7 days. Lastly, we estimated that for the four SMs in which a two-phase decay was apparent, an average 3% of virus production is from long-lived infected cells, with a range of 92 to 99% of replication occurring in short-lived cells. The rate of viral decay and estimated half-life of infected cells in each individual SM is shown in Fig. 6B. In all, this analysis shows that natural SIV infection of SMs is similar to pathogenic HIV and SIV infections of humans and macaques in that it is characterized by rapid viral turnover and the vast majority of viral replication is occurring in short-lived infected cells, with an average in vivo life span strikingly close to that estimated in pathogenic HIV and SIV infections (38). These findings clearly indicate that the preserved homeostasis of CD4+ T cells and AIDS resistance of SMs naturally infected with SIV are unlikely to be related to a longer life span of infected cells or an increased fraction of virus coming from long-lived infected cells.
FIG. 6.

SMs have a short in vivo life span of SIV-infected cells. (A) Plasma concentrations of SIV RNA (in copies/ml) for the six SIV-infected animals after treatment with ART was begun on day zero. (B)The two-phase decay model of Perelson et al. (38) was fit using nonlinear least-squares regression to the viral load data (circles) for each animal (FAy, FDv, FUs, FCy, FEy, and FVs), and the death rates, d (of short-lived infected cells) and m (of long-lived cells) were estimated as described in Materials and Methods. The solid line shows the best-fit theoretical curve. The half-lives of short- and long-lived infected cells were computed from the best-fit parameters as the ln 2/d and ln 2/m, where ln 2 is the natural logarithm of 2 (0.693).
DISCUSSION
One of the most intriguing observations in HIV/AIDS research is that, in striking contrast to HIV-infected humans, natural hosts of SIV remain persistently asymptomatic and usually maintain normal CD4+ T-cell counts, despite chronic high levels of virus replication and relatively low cellular immune responses to SIV (5, 8, 10, 18, 36, 41, 47). The observation that nonpathogenic SIV infections of natural hosts are associated with high viremia provides an obvious challenge to the paradigm that the level of virus replication is the main (or even the sole) determinant of progression to AIDS during pathogenic HIV infection in humans and SIVmac infection in Asian macaques (17, 26, 31, 49, 54, 55).
Our early studies of SMs naturally infected with SIV led us to hypothesize that the absence of generalized immune activation is a mechanism that favors the preservation of CD4+ T-cell homeostasis in these animals (47). Consistent with this hypothesis are the results of studies in which SMs and RMs were experimentally infected with uncloned SIVsmm, resulting in high levels of virus replication in both species but with only RMs developing chronic immune activation and progressing to simian AIDS (13, 29, 30, 45). While these earlier studies, as well as similar studies performed in mandrills and African green monkeys (5, 20, 33, 36), clearly identified a consistent association between nonpathogenicity and low levels of T-cell activation and apoptosis, the contribution of these findings to the absence of disease progression is still unclear.
To explain the lack of disease in natural hosts for SIV, and particularly in SMs, it could be hypothesized that, in these animals, the high level of virus replication is less damaging to the CD4+ T-cell pool because either (i) infected CD4+ T cells survive the infection for a longer period of time, thus producing more virions on a per cell basis, or (ii) a higher fraction of virus replication is supported by long-lived productively infected cells, such as macrophages. In both scenarios, the infection will be associated at any given time with fewer CD4+ T cells being infected and killed by the virus, thus favoring the overall maintenance of their homeostasis. To test this hypothesis we have applied to SMs the same experimental approach and mathematical modeling previously used to assess the turnover of virus and infected cells in HIV-infected humans and SIV-infected RMs (38, 39). With this kinetic analysis of the ART-induced decline of viremia, it is possible to assess the fractions of short- and long-lived cells that are producing virus and provide estimates of their life spans (37, 38). For example, if we observed a biphasic viral load decline with a 1- to 2-log first-phase decline but with a slope less steep than observed in HIV-infected humans or SIV-infected RMs, this would suggest that, while in SIV-infected SMs the bulk of viral replication also takes place in short-lived cells (presumably, activated CD4+ T cells), these cells tend to live longer in SMs than in HIV-infected humans and SIV-infected RMs. Alternatively, the finding of a transition to the phase of slow decline occurring sooner in SIV-infected SMs would suggest that, in these animals, the longer-lived population of infected cells (likely macrophages) is responsible for a significantly larger fraction of the viral production than in HIV-infected humans and SIV-infected RMs. However, neither of these two potential experimental outcomes was observed in this study.
The current study indicates that all six ART-treated SMs naturally infected with SIV experienced a rapid decline of plasma viremia, with four of the six animals showing a decline of plasma viral load below the limit of detection and the other animals exhibiting a 2- to 3-log decrease. Our mathematical modeling of these data suggested that, during natural SIV infection of SMs, the average in vivo estimated half-life of short-lived virus-producing cells is approximately 1.1 days, with 92 to 99% of virus replication occurring in these cells. Because drug therapy is presumably not 100% effective, the true half-life of infected cells is probably shorter than 1.1 days. Thus, these data are very similar to the findings for HIV infection for the productively infected CD4+ T-cell half-life of 0.7 days (24) with 92 to 99% of virus production from these short-lived cells (38). In these latter studies, the estimate of a 0.7-day half-life was obtained under a very potent three-drug regimen, which should be significantly closer to 100% effective than the two-drug regimen used here. Similarly, estimates of the half-life of productively infected cells in SIV infection of macaques using one drug, PMPA, ranged between 0.7 and 1.4 days with an average of about 1 day (31), while a more recent study using quadruple ART found a mean half-life of 0.47 days (range, 0.37 to 0.50) (4). The interpretation of these data is that SMs naturally infected with SIV are similar to HIV-infected humans and SIV-infected RMs in terms of the short in vivo life span of the vast majority of productively infected cells, which in turn suggests that SIVsmm is equally cytopathic in vivo for SM infected cells as are HIV for human cells and SIVmac for macaque cells. Collectively, these data are consistent with those obtained in an unpublished study by Grant, Staprans, and Feinberg in which SMs naturally infected with SIV were treated with PMPA monotherapy. Interestingly, a short in vivo life span of infected cells was also observed using the same experimental system but in a different natural SIV host, African green monkeys experimentally infected with SIVagm-sab treated with PMPA and FTC (36a).
It should be noted that a short in vivo life span of productively infected cells does not necessarily imply intrinsically high viral cytopathicity, as this finding could also be explained by the fact that activated CD4+ T cells are highly susceptible to activation-induced cell death (and thus are destined to die regardless of their infection) or, alternatively, by the fact that productively infected CD4+ T cells may be killed by SIV-specific cytotoxic T lymphocytes (CTL). As such, our experiments and mathematical modeling do not rule out the possibility that the similarly short life span of infected cells observed in pathogenic and nonpathogenic models of primate lentiviral infection may reflect different mechanisms of death of the infected cells. However, even if the exact mechanisms (i.e., virus direct cytopathicity versus activation-induced cell death versus CTL-mediated lysis) determining the life span of infected cells in SMs naturally infected with SIV remain unclear, our data clearly indicate that the preservation of CD4+ T-cell homeostasis and AIDS resistance of SIV-infected SMs is unlikely to be simply due to a longer life span of infected cells.
The current study also gave us the opportunity to study, in SMs naturally infected with SIV, the kinetics of viral replication after cessation of antiretroviral therapy. We observed that in all animals a rapid rebound of viremia followed the interruption of treatment, with kinetics characterized by a peak of virus replication occurring at 1 to 2 weeks postinterruption and a subsequent decline to a set point level that was strikingly similar to that observed prior to therapy. It should be noted that the kinetics of viral rebound and reestablishment of the viral set point is remarkably similar to the kinetics of viral replication observed during the first weeks of acute SIVsmm infection (13), which may reflect target cell availability, an influence of SIV-specific CTL, or a combination of the two.
In most HIV-infected individuals, the suppression of virus replication by highly active ART results in an increase in CD4+ T-cell counts as well as a significant decrease in the levels of immune activation (7). In this cohort of SMs naturally SIV infected, we observed only a modest, nonsignificant increase in the number of circulating CD4+ T cells at the time of ART-induced suppression of virus replication, with no detectable decline of CD4+ T-cell counts at the time of therapy interruption. With respect to the level of immune activation, we observed that ART induced a minor decrease in the levels of T-cell proliferation (measured by Ki67 expression) and activation (measured by CD25, CD69, and HLA-DR expression) in SIV-infected SMs. Interestingly, an increase in the number of CD4+ Ki67+ and CD8+ Ki67+ T cells was observed after therapy interruption and coincident with the rebound of virus replication. Taken together, these data indicate that, in SMs naturally SIV infected, suppression of virus replication by ART had a limited impact on both CD4+ T cells and the prevailing level of T-cell activation and proliferation. This observation is consistent with the observation that, in SMs naturally SIV infected, chronic virus replication does not translate into peripheral CD4+ T-cell depletion and does not induce high levels of immune activation. Indirectly, the presented data also support the hypothesis that, in HIV-infected patients, the ability of ART to reduce the immune activation, rather than its suppression of viral replication, dictates the overall pace of CD4+ T-cell recovery (15).
In summary, the presented data indicate that natural SIV infection of SMs is associated with the short in vivo life span of productively infected cells and suggest that, in these animals, the bulk of virus replication occurs in activated CD4+ T cells. As such, these data rule out that a prolonged survival of infected cells and/or a reduced intrinsic cytopathicity of SIVsmm for SM cells is the key mechanism underlying the preservation of CD4+ T cells and resistance to AIDS in these animals.
Acknowledgments
We thank Elizabeth Strobert and James Else, as well as Stephanie Ehnert and Chris Souder, for their assistance with the animal studies conducted at the Yerkes National Primate Research Center and the research staff of the Emory Center for AIDS Research Virology Core, particularly Benton Lawson and Seema Garg, for viral quantification. We are thankful to Ann Chahroudi for critical review of the manuscript.
S.N.G. was supported by NRSA fellowship 1F31A1066400-01A1. Portions of this work were done under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. This work was supported by NIH grants AI52755, AI66996, and HL75766 (to G.S.), AI28433 and RR06555 (to A.S.P.), AI60451 (to D.L.S.), AI65325 (to C.A.), AI64066 (to I.P.), R01-AI49809 (to A.K.), and RR-00165 (Yerkes National Primate Research Center).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Published ahead of print on 23 January 2008.
REFERENCES
- 1.Autran, B., G. Carcelain, T. S. Li, C. Blanc, D. Mathez, R. Tubiana, C. Katlama, P. Debre, and J. Leibowitch. 1997. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 277112-116. [DOI] [PubMed] [Google Scholar]
- 2.Bostik, P., A. E. Mayne, F. Villinger, K. P. Greenberg, J. D. Powell, and A. A. Ansari. 2001. Relative resistance in the development of T cell anergy in CD4+ T cells from simian immunodeficiency virus disease-resistant sooty mangabeys. J. Immunol. 166506-516. [DOI] [PubMed] [Google Scholar]
- 3.Bostik, P., E. S. Noble, A. E. Mayne, L. Gargano, F. Villinger, and A. A. Ansari. 2006. Central memory CD4 T cells are the predominant cell subset resistant to anergy in SIV disease resistant sooty mangabeys. AIDS 20181-188. [DOI] [PubMed] [Google Scholar]
- 4.Brandin, E., R. Thorstensson, S. Bonhoeffer, and J. Albert. 2006. Rapid viral decay in simian immunodeficiency virus-infected macaques receiving quadruple antiretroviral therapy. J. Virol. 809861-9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broussard, S. R., S. I. Staprans, R. White, E. M. Whitehead, M. B. Feinberg, and J. S. Allan. 2001. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J. Virol. 752262-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bucy, R. P., R. D. Hockett, C. A. Derdeyn, M. S. Saag, K. Squires, M. Sillers, R. T. Mitsuyasu, and J. M. Kilby. 1999. Initial increase in blood CD4+ lymphocytes after HIV antiretroviral therapy reflects redistribution from lymphoid tissues. J. Clin. Investig. 1031391-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carcelain, G., P. Debre, and B. Autran. 2001. Reconstitution of CD4+ T lymphocytes in HIV-infected individuals following antiretroviral therapy. Curr. Opin. Immunol. 13483-488. [DOI] [PubMed] [Google Scholar]
- 8.Chakrabarti, L. A., S. R. Lewin, L. Zhang, A. Gettie, A. Luckay, L. N. Martin, E. Skulsky, D. D. Ho, C. Cheng-Mayer, and P. A. Marx. 2000. Normal T cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J. Virol. 741209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dion, M. L., R. Bordi, J. Zeidan, R. Asaad, M. R. Boulassel, J. P. Routy, M. M. Lederman, R. P. Sekaly, and R. Cheynier. 2007. Slow disease progression and robust therapy-mediated CD4+ T cell recovery are associated with efficient thymopoiesis during HIV-1 infection. Blood 1092912-2920. [DOI] [PubMed] [Google Scholar]
- 10.Dunham, R., P. Pagliardini, S. Gordon, B. Sumpter, J. Engram, A. Moanna, M. Paiardini, J. N. Mandl, B. Lawson, S. Garg, H. M. McClure, Y. X. Xu, C. Ibegbu, K. Easley, N. Katz, I. Pandrea, C. Apetrei, D. L. Sodora, S. I. Staprans, M. B. Feinberg, and G. Silvestri. 2006. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 108209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estes, J. D., Q. Li, M. R. Reynolds, S. Wietgrefe, L. Duan, T. Schacker, L. J. Picker, D. I. Watkins, J. D. Lifson, C. Reilly, J. Carlis, and A. T. Haase. 2006. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J. Infect. Dis. 193703-712. [DOI] [PubMed] [Google Scholar]
- 12.Goicoechea, M., D. M. Smith, L. Liu, S. May, A. R. Tenorio, C. C. Ignacio, A. Landay, and R. Haubrich. 2006. Determinants of CD4+ T cell recovery during suppressive antiretroviral therapy: association of immune activation, T cell maturation markers, and cellular HIV-1 DNA. J. Infect. Dis. 19429-37. [DOI] [PubMed] [Google Scholar]
- 13.Gordon, S. N., N. R. Klatt, S. E. Bosinger, J. M. Brenchley, J. M. Milush, J. C. Engram, R. M. Dunham, M. Paiardini, S. Klucking, A. Danesh, E. A. Strobert, C. Apetrei, I. V. Pandrea, D. Kelvin, D. C. Douek, S. I. Staprans, D. L. Sodora, and G. Silvestri. 2007. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J. Immunol. 1793026-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn, B. H., G. M. Shaw, K. M. De Cock, and P. M. Sharp. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287607-614. [DOI] [PubMed] [Google Scholar]
- 15.Hazenberg, M. D., J. W. Stuart, S. A. Otto, J. C. Borleffs, C. A. Boucher, R. J. de Boer, F. Miedema, and D. Hamann. 2000. T cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95249-255. [PubMed] [Google Scholar]
- 16.Hellerstein, M. K., R. A. Hoh, M. B. Hanley, D. Cesar, D. Lee, R. A. Neese, and J. M. McCune. 2003. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J. Clin. Investig. 112956-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373123-126. [DOI] [PubMed] [Google Scholar]
- 18.Holzammer, S., E. Holznagel, A. Kaul, R. Kurth, and S. Norley. 2001. High virus loads in naturally and experimentally SIVagm-infected African green monkeys. Virology 283324-331. [DOI] [PubMed] [Google Scholar]
- 19.Johnson, P. R., and V. M. Hirsch. 1992. SIV infection of macaques as a model for AIDS pathogenesis. Int. Rev. Immunol. 855-63. [DOI] [PubMed] [Google Scholar]
- 20.Kornfeld, C., M. J. Ploquin, I. Pandrea, A. Faye, R. Onanga, C. Apetrei, V. Poaty-Mavoungou, P. Rouquet, J. Estaquier, L. Mortara, J. F. Desoutter, C. Butor, R. Le Grand, P. Roques, F. Simon, F. Barre-Sinoussi, O. M. Diop, and M. C. Muller-Trutwin. 2005. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J. Clin. Investig. 1151082-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs, J. A., R. A. Lempicki, I. A. Sidorov, J. W. Adelsberger, B. Herpin, J. A. Metcalf, I. Sereti, M. A. Polis, R. T. Davey, J. Tavel, J. Falloon, R. Stevens, L. Lambert, R. Dewar, D. J. Schwartzentruber, M. R. Anver, M. W. Baseler, H. Masur, D. S. Dimitrov, and H. C. Lane. 2001. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J. Exp. Med. 1941731-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, Q., L. Duan, J. D. Estes, Z. M. Ma, T. Rourke, Y. Wang, C. Reilly, J. Carlis, C. J. Miller, and A. T. Haase. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 4341148-1152. [DOI] [PubMed] [Google Scholar]
- 23.Ling, B., C. Apetrei, I. Pandrea, R. S. Veazey, A. A. Lackner, B. Gormus, and P. A. Marx. 2004. Classic AIDS in a sooty mangabey after an 18-year natural infection. J. Virol. 788902-8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio, A. S. Perelson, and D. D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T cell decay in vivo. J. Virol. 775037-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellors, J. W., A. Munoz, J. V. Giorgi, J. B. Margolick, C. J. Tassoni, P. Gupta, L. A. Kingsley, J. A. Todd, A. J. Saah, R. Detels, J. P. Phair, and C. R. Rinaldo, Jr. 1997. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann. Intern. Med. 126946-954. [DOI] [PubMed] [Google Scholar]
- 26.Mellors, J. W., C. R. Rinaldo, Jr., P. Gupta, R. M. White, J. A. Todd, and L. A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 2721167-1170. [DOI] [PubMed] [Google Scholar]
- 27.Mohri, H., S. Bonhoeffer, S. Monard, A. S. Perelson, and D. D. Ho. 1998. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science 2791223-1227. [DOI] [PubMed] [Google Scholar]
- 28.Mohri, H., A. S. Perelson, K. Tung, R. M. Ribeiro, B. Ramratnam, M. Markowitz, R. Kost, A. Hurley, L. Weinberger, D. Cesar, M. K. Hellerstein, and D. D. Ho. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 1941277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muthukumar, A., A. Wozniakowski, M. C. Gauduin, M. Paiardini, H. M. McClure, R. P. Johnson, G. Silvestri, and D. L. Sodora. 2004. Elevated interleukin-7 levels not sufficient to maintain T cell homeostasis during simian immunodeficiency virus-induced disease progression. Blood 103973-979. [DOI] [PubMed] [Google Scholar]
- 30.Muthukumar, A., D. Zhou, M. Paiardini, A. P. Barry, K. S. Cole, H. M. McClure, S. I. Staprans, G. Silvestri, and D. L. Sodora. 2005. Timely triggering of homeostatic mechanisms involved in the regulation of T cell levels in SIVsm-infected sooty mangabeys. Blood 1063839-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak, M. A., A. L. Lloyd, G. M. Vasquez, T. A. Wiltrout, L. M. Wahl, N. Bischofberger, J. Williams, A. Kinter, A. S. Fauci, V. M. Hirsch, and J. D. Lifson. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 717518-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okoye, A., M. Meier-Schellersheim, J. M. Brenchley, S. I. Hagen, J. M. Walker, M. Rohankhedkar, R. Lum, J. B. Edgar, S. L. Planer, A. Legasse, A. W. Sylwester, M. Piatak, Jr., J. D. Lifson, V. C. Maino, D. L. Sodora, D. C. Douek, M. K. Axthelm, Z. Grossman, and L. J. Picker. 2007. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J. Exp. Med. 2042171-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onanga, R., S. Souquiere, M. Makuwa, A. Mouinga-Ondeme, F. Simon, C. Apetrei, and P. Roques. 2006. Primary simian immunodeficiency virus SIVmnd-2 infection in mandrills (Mandrillus sphinx). J. Virol. 803301-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orendi, J. M., A. C. Bloem, J. C. Borleffs, F. J. Wijnholds, N. M. de Vos, H. S. Nottet, M. R. Visser, H. Snippe, J. Verhoef, and C. A. Boucher. 1998. Activation and cell cycle antigens in CD4+ and CD8+ T cells correlate with plasma human immunodeficiency virus (HIV-1) RNA level in HIV-1 infection. J. Infect. Dis. 1781279-1287. [DOI] [PubMed] [Google Scholar]
- 35.Pandrea, I., C. Apetrei, S. Gordon, J. Barbercheck, J. Dufour, R. Bohm, B. Sumpter, P. Roques, P. A. Marx, V. M. Hirsch, A. Kaur, A. A. Lackner, R. S. Veazey, and G. Silvestri. 2007. Paucity of CD4+ CCR5+ T cells is a typical feature of natural SIV hosts. Blood 1091069-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandrea, I., R. Onanga, C. Kornfeld, P. Rouquet, O. Bourry, S. Clifford, P. T. Telfer, K. Abernethy, L. T. White, P. Ngari, M. Muller-Trutwin, P. Roques, P. A. Marx, F. Simon, and C. Apetrei. 2003. High levels of SIVmnd-1 replication in chronically infected Mandrillus sphinx. Virology 317119-127. [DOI] [PubMed] [Google Scholar]
- 36a.Pandrea, I., R. M. Ribeiro, R. Gautam, T. Gaufin, M. Pattison, M. Barnes, C. Monjure, C. Stoulig, J. Dufour, W. Cyprian, G. Silvestri, M. D. Miller, A. S. Perelson, and C. Apetrei. 2008. Simian immunodeficiency virus SIVagm dynamics in African green monkeys. J. Virol. 823713-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perelson, A. S. 2002. Modelling viral and immune system dynamics. Nat Rev. Immunol. 228-36. [DOI] [PubMed] [Google Scholar]
- 38.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387188-191. [DOI] [PubMed] [Google Scholar]
- 39.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 2711582-1586. [DOI] [PubMed] [Google Scholar]
- 40.Ramratnam, B., S. Bonhoeffer, J. Binley, A. Hurley, L. Zhang, J. E. Mittler, M. Markowitz, J. P. Moore, A. S. Perelson, and D. D. Ho. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 3541782-1785. [DOI] [PubMed] [Google Scholar]
- 41.Rey-Cuille, M. A., J. L. Berthier, M. C. Bomsel-Demontoy, Y. Chaduc, L. Montagnier, A. G. Hovanessian, and L. A. Chakrabarti. 1998. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J. Virol. 723872-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ribeiro, R. M., H. Mohri, D. D. Ho, and A. S. Perelson. 2002. In vivo dynamics of T cell activation, proliferation, and death in HIV-1 infection: why are CD4+ but not CD8+ T cells depleted? Proc. Natl. Acad. Sci. USA 9915572-15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenzweig, M., M. A. DeMaria, D. M. Harper, S. Friedrich, R. K. Jain, and R. P. Johnson. 1998. Increased rates of CD4+ and CD8+ T lymphocyte turnover in simian immunodeficiency virus-infected macaques. Proc. Natl. Acad. Sci. USA 956388-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sachsenberg, N., A. S. Perelson, S. Yerly, G. A. Schockmel, D. Leduc, B. Hirschel, and L. Perrin. 1998. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 1871295-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silvestri, G., A. Fedanov, S. Germon, N. Kozyr, W. J. Kaiser, D. A. Garber, H. McClure, M. B. Feinberg, and S. I. Staprans. 2005. Divergent host responses during primary simian immunodeficiency virus SIVsm infection of natural sooty mangabey and nonnatural rhesus macaque hosts. J. Virol. 794043-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silvestri, G., M. Paiardini, I. Pandrea, M. Lederman, and D. Sodora. 2007. Understanding the benign nature of SIV infection in natural hosts. J. Clin. Investig. 1173148-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silvestri, G., D. L. Sodora, R. A. Koup, M. Paiardini, S. P. O'Neil, H. M. McClure, S. I. Staprans, and M. B. Feinberg. 2003. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18441-452. [DOI] [PubMed] [Google Scholar]
- 48.Smith, K., E. Aga, R. J. Bosch, H. Valdez, E. Connick, A. Landay, D. Kuritzkes, B. H. Gross, I. R. Francis, J. M. McCune, H. Kessler, and M. Lederman. 2004. Long-term changes in circulating CD4 T lymphocytes in virologically suppressed patients after 6 years of highly active antiretroviral therapy. AIDS 181953-1956. [DOI] [PubMed] [Google Scholar]
- 49.Staprans, S. I., P. J. Dailey, A. Rosenthal, C. Horton, R. M. Grant, N. Lerche, and M. B. Feinberg. 1999. Simian immunodeficiency virus disease course is predicted by the extent of virus replication during primary infection. J. Virol. 734829-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stebbing, J., B. Gazzard, and D. C. Douek. 2004. Where does HIV live? N. Engl. J. Med. 3501872-1880. [DOI] [PubMed] [Google Scholar]
- 51.Stevenson, M. 2003. HIV-1 pathogenesis. Nat. Med. 9853-860. [DOI] [PubMed] [Google Scholar]
- 52.Sumpter, B., R. Dunham, S. Gordon, J. Engram, M. Hennessy, A. Kinter, M. Paiardini, B. Cervasi, N. Klatt, H. McClure, J. M. Milush, S. Staprans, D. L. Sodora, and G. Silvestri. 2007. Correlates of preserved CD4+ T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J. Immunol. 1781680-1691. [DOI] [PubMed] [Google Scholar]
- 53.Wang, Z., B. Metcalf, R. M. Ribeiro, H. McClure, and A. Kaur. 2006. Th-1-type cytotoxic CD8+ T-lymphocyte responses to simian immunodeficiency virus (SIV) are a consistent feature of natural SIV infection in sooty mangabeys. J. Virol. 802771-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson, A., J. Ranchalis, B. Travis, J. McClure, W. Sutton, P. R. Johnson, S. L. Hu, and N. L. Haigwood. 1997. Plasma viremia in macaques infected with simian immunodeficiency virus: plasma viral load early in infection predicts survival. J. Virol. 71284-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373117-122. [DOI] [PubMed] [Google Scholar]