

Abstract
Recognition of immunoglobulin G (IgG) by surface receptors for the Fc domain of immunoglobulin G (Fcγ), FcγRs, can trigger both humoral and cellular immune responses. Two human cytomegalovirus (HCMV)-encoded type I transmembrane receptors with Fcγ-binding properties (vFcγRs), gp34 and gp68, have been identified on the surface of HCMV-infected cells and are assumed to confer protection against IgG-mediated immunity. Here we show that Fcγ recognition by both vFcγRs occurs independently of N-linked glycosylation of Fcγ, in contrast with the properties of host FcγRs. To gain further insight into the interaction with Fcγ, truncation mutants of the vFcγR gp68 ectodomain were probed for Fcγ binding, resulting in localization of the Fcγ binding site on gp68 to residues 71 to 289, a region including an immunoglobulin-like domain. Gel filtration and biosensor binding experiments revealed that, unlike host FcγRs but similar to the herpes simplex virus type 1 (HSV-1) Fc receptor gE-gI, gp68 binds to the CH2-CH3 interdomain interface of the Fcγ dimer with a nanomolar affinity and a 2:1 stoichiometry. Unlike gE-gI, which binds Fcγ at the slightly basic pH of the extracellular milieu but not at the acidic pH of endosomes, the gp68/Fcγ complex is stable at pH values from 5.6 to pH 8.1. These data indicate that the mechanistic details of Fc binding by HCMV gp68 differ from those of host FcγRs and from that of HSV-1 gE-gI, suggesting distinct functional and recognition properties.
Both alpha- and betaherpesviruses encode proteins that recognize the Fc region of immunoglobulin G (IgG) molecules (Fcγ) (7, 18, 33). The viral Fcγ receptor (vFcγR) in alphaherpesviruses is a heterodimer of the two transmembrane proteins gE and gI, which are found in the viral envelope and on the surface of infected cells (5, 15, 24, 30, 54). Studies with herpes simplex virus type 1 (HSV-1) have shown that simultaneous binding of human anti-HSV IgG to both an HSV antigen with its Fab arms and to gE-gI with its Fcγ region, a phenomenon referred to as antibody bipolar bridging, protects the virus and infected cells from IgG-mediated immune responses (14, 16, 36, 51). gE-gI-mediated endocytosis of anti-HSV IgG/HSV antigen complexes followed by degradation of anti-HSV IgG has also been proposed based on the finding that gE-gI binds Fcγ at pH 7.4 but does not bind at pH 6.0 (47). Biochemical and structural analyses of gE-gI binding to Fcγ revealed that gE-gI interacts with the Fcγ CH2-CH3 interdomain junction with a stoichiometry of two molecules of gE-gI per Fcγ (47, 48). This symmetric interaction with Fcγ, which is a twofold symmetric homodimer in which each polypeptide chain contains an N-terminal hinge followed by the CH2 and CH3 domains, is analogous to that previously identified for protein A (11), protein G (43), rheumatoid factor (9), and FcRn (32). Each of these proteins recognizes the CH2-CH3 interdomain interface, which contains a six-residue consensus Fcγ binding site (12). In contrast, host Fcγ receptors (FcγRs; FcγRI, FcγRIIa, FcγRIIb, and FcγRIII) bind Fcγ with 1:1 stoichiometry in an asymmetric manner, contacting residues in the CH2 domain and in the CH1-CH2 hinge, which connects the Fab to Fcγ (39, 45).
Even though Fcγ binding activity has long been reported for cells infected with the betaherpesvirus human cytomegalovirus (HCMV), the effects of Fcγ binding are unknown (17, 19, 26, 41, 42, 53). HCMV vFcγRs gp34 and gp68 were recently demonstrated to be encoded by independent genes, TRL11/IRL11 (3, 29) and UL119-UL118 (3), respectively. Both vFcγRs, gp34 and gp68, were shown to be cell surface proteins that bind to Fcγ (3, 29). gp34 and gp68 share binding properties with gE-gI, the HSV-1 vFcγR, in that each is specific for human IgG but not human IgA or IgM. The HCMV vFcγRs, however, bind all four human IgG subclasses (IgG1, IgG2, IgG3, and IgG4) (2, 3), whereas gE-gI does not bind IgG3 (22, 55). gp34 and gp68 differ in their specificities for IgG from various mammal species, with gp68 being more restrictive than gp34 (3). Although gp34, gp68, gE-gI, and fcr-1/m138, the mouse cytomegalovirus-encoded FcγR (50), exhibit Fcγ binding activity, they do not share sequence homology. Thus, each vFcγR likely binds the Fc region of IgG by a different set of interactions.
To facilitate an understanding of how HCMV vFcγRs recognize IgG, we expressed and purified the ectodomains of gp34 and gp68 and characterized the interaction between gp68 and Fcγ. We show that both vFcγRs recognize Fcγ in a manner independent of N-linked glycosylation of the Fcγ CH2 domain. The gp34 ectodomain was unsuitable for biochemical characterization because of aggregation. However, we used the gp68 ectodomain to demonstrate that the Fcγ binding region is contained within gp68 residues 71 to 289, a region that includes a predicted immunoglobulin-like domain, and that gp68 interacts with the Fcγ CH2-CH3 interdomain junction with a nanomolar affinity and a stoichiometry of two molecules of gp68 per Fcγ dimer.
MATERIALS AND METHODS
Cells.
African green monkey CV-I (ATCC CCL-70) and human tk−143 (ATCC CRL-8303) cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum, penicillin, streptomycin, and 2 mM glutamine.
Viruses and plasmids.
HSV-1 was propagated and the virus titer was determined on Vero cells grown in Dulbecco modified Eagle medium containing 10% fetal calf serum, penicillin, streptomycin, and 2 mM glutamine. The recombinant vaccinia virus (rVV) gp68 expressing the FLAG epitope-tagged gp68 has been described previously (3). The coding sequences of TRL11 (gp34) and UL119-UL118 (gp68) were amplified from plasmids p7.5kTRL11FLAG (3) and p7.5kUL119-118FLAG (3), respectively, using primer pairs 5′-GTCTAGGGATCCATGCAGACCTACAGCACCCC-3′ and 5′-GCTTAAGAATTCCTACTGTAAATCCCCGTCCACCG-3′ and 5′-GACTTAGATCTACATGTGTTCCGTACTGGCG-3′ and 5′-GGAAGAATTCTACCACTGCTTGAAGTAGGGCACCG-3′, respectively. The PCR fragments were cloned into the vaccinia virus recombination vector p7.5k131a via BamHI and EcoRI and BglII and EcoRI, respectively (recognition sites are underlined). C-terminal truncation mutants of gp68 were generated using the forward primer 5′-GACTTAGATCTACATGTGTTCCGTACTGGCG-3′ and the reverse primers 5′-GAAACTAGTGTCCTCGAACAGCGGGTCGCTC-3′ [gp68 (26-292), encoding residues 26 to 292], 5′-GAAACTAGTCCGTTGTCCGTTATACGTCACG-3′ [gp68 (26-251), encoding residues 26 to 251] and 5′-GAAACTAGTCACGCGGACCCGCATCGTG-3′ [gp68 (26-206), encoding residues 26 to 206] (where residue 1 is the N-terminal Met of the immature protein and residues 1 to 25 are predicted to define the signal peptide). The PCR fragments were cloned using BglII and SpeI (recognition sites are underlined) into the vaccinia virus recombination vector p7.5k131a-FLAG containing the FLAG coding sequence after a SpeI site, resulting in vaccinia virus recombination vectors that encode C-terminally FLAG-tagged truncation mutants of gp68 [gp68 (26-292), gp68 (26-251), and gp68 (26-206)]. For the gp68 (71-292) mutant, the 5′-end region (nucleotides [nt] 1 to 84) and the 3′-end region (nt 213 to 876) were separately amplified using primer pairs 5′-GACTTAGATCTACATGTGTTCCGTACTGGCG-3′, 5′-CGCGCTAGCGCTTGTGGTGCTACTTTTC-3′ and 5′-GCGGCTAGCACGACGCAGAAAGAGGGG-3′, 5′-GAAACTAGTGTCCTCGAACAGCGGGTCGCTC-3′, respectively, digested with NheI (recognition sites are underlined), and ligated. After PCR amplification using primer pair 5′-GACTTAGATCTACATGTGTTCCGTACTGGCG-3′ and 5′-CGCGAATTCTACATGTAGGTCACGTACAAAAG-3′, the product was digested by BglII and EcoRI (recognition sites are underlined) and ligated into the BamHI and EcoRI sites of the V5/six-His-tag coding vector pGene/V5-His B (Invitrogen, Karlsruhe, Germany). The resulting DNA encoding gp68 (71-292) with a C-terminal V5/six-His tag was subcloned into the vaccinia virus recombination vector p7.5k131a. The human high-affinity Fcγ receptor Ia (CD64; GenBank accession no. NP000557) was cloned from CD64 cDNA (1) into p7.5k131a, using primer pair 5′-CACAGGATCCATGTGGTTCTTGACAACTC-3′ and 5′-AGCTGAGCTCCTACGTGGCCCCCTGGGGCTC-3′ and restriction enzymes BamHI and SacI (recognition sites are underlined). The human medium-affinity Fcγ receptor IIa (CD32; GenBank accession no. NP067674) was cloned from CD32 cDNA into p7.5k131a using primer pair 5′-GAGAAGATCTATGTCTCAGAATGTATGTCCC-3′ and 5′-AGCTGAGCTCTTAGTTATTACTGTTGACATG-3′ and restriction enzymes BglII and SacI (recognition sites are underlined).
Generation of rVVs.
The construction of rVVs has been described elsewhere (49). Briefly, the gene of interest inserted in the vaccinia virus recombination vector p7.5k131a was transferred into the thymidine kinase open reading frame of the virus genome (strain Copenhagen). rVVs were selected with bromodeoxyuridine (100 μg/ml) using tk−143 cells.
Immunoprecipitation and immunoblotting.
For characterization of the binding of gp68 truncation mutants to Fcγ, CV-I cells were infected with rVVs (5 PFU/cell) for 14 h, washed twice with ice-cold phosphate-buffered saline (pH 7.2), and lysed on ice in 1% NP-40-lysis buffer (140 mM NaCl, 20 mM Tris pH 7.6, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 50 μM leupeptin, and 1 μM pepstatin A). One microgram of human Fcγ fragment (Rockland Immunochemicals, Gilbertsville, PA), 1 μg mouse anti-V5 antibody (Invitrogen, Karlsruhe, Germany), or 4 μg goat anti-FLAG antibody-coupled agarose (Bethyl Laboratories, Montgomery, TX) was added to the postnuclear supernatant for 1 h at 4°C. Fcγ and potentially associated proteins were precipitated using protein A- or protein G-Sepharose (GE Healthcare, Munich, Germany) overnight at 4°C. Pellets were washed three times with a low-salt buffer (150 mM NaCl, 2 mM EDTA, 10 mM Tris [pH 7.6], 0.2% NP-40), two times with a high-salt buffer (500 mM NaCl, 2 mM EDTA, 10 mM Tris [pH 7.6], 0.2% NP-40), and once with 10 mM Tris (pH 8.0). An aliquot of the precipitate was digested with endoglycosidase H (Endo H; Roche, Mannheim, Germany) for 14 h using 5 mU according to the manufacturer's instructions. Proteins were separated by use of 4 to 12% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane, and probed with mouse anti-FLAG M2 antibody (Sigma-Aldrich, Munich, Germany) or mouse anti-V5 antibody. For detection, a peroxidase-coupled goat anti-mouse antibody (Dianova, Hamburg, Germany) was visualized using the ECL Plus chemiluminescence system (GE Healthcare).
Immunoprecipitation analyses of metabolically labeled proteins were performed as described previously (3). In brief, proteins were labeled for 1 h with 35S-labeled Redivue Pro-Mix (GE Healthcare), followed by lysis and washing procedures as described above. Untreated human Fcγ (1 μg; Rockland), enzymatically deglycosylated human Fcγ, or mouse anti-human CD64 (2 μg; Ancell Corp., Bayport, MN) were incubated for 1 h at 4°C before protein A- or protein G-Sepharose was added for an additional hour at 4°C. Before wild-type Fc (wtFc) and nonbinding Fc (nbFc) were used for protein precipitations, Fcγ proteins were covalently coupled to cyanogen bromide-activated (CNBr) Sepharose (GE Healthcare) according to manufacturer's instructions. Postnuclear cell lysates were precleared first with mock coupled CNBr Sepharose before the Fcγ-coupled Sepharose was added for 1 h at 4°C. The immune complexes were dissociated in sample buffer and separated by 10% SDS-PAGE or 10 to 13% gradient SDS-PAGE. Dried gels were exposed to Kodak BioMaxMR films at −70°C for 1 to 5 days.
Deglycosylation of human Fcγ.
Twenty micrograms of human Fcγ (Rockland) was deglycosylated overnight with either 0.025 U Endo H (Roche) or peptide N-glycosidase F (Roche) according to the manufacturer's instructions. Controls were treated the same but without any enzyme. After dialysis against PBS (pH 7.2), precipitation of Fcγ was performed as described above. Deglycosylation of Fcγ was verified by a shift in migration on silver-stained SDS-PAGE gels (data not shown).
Expression and purification of soluble HCMV gp68 and HCMV gp34 proteins.
The gp68 ectodomain [gp68 (26-289)] and an N-terminally truncated form of the gp68 ectodomain lacking the first 42 amino acids of the predicted mature protein containing 25 potential O-GalNAc-glycosylation sites [gp68 (68-289)] were expressed in baculovirus-infected insect cells. DNA encoding gp68 (26-289) (residues 26 to 289) with a C-terminal Factor Xa-cleavable six-His tag was PCR amplified and subcloned between the BamHI and EcoRI sites of pAcGP67A (Pharmingen) in frame with the gp67 signal peptide. DNA encoding gp68 (68-289) (residues 68 to 289) was PCR amplified and subcloned in frame with the HSV-1 gI signal peptide and C-terminal factor Xa-cleavable six-His tag of an HSV-1 gI expression construct with a pAcUW51 (Pharmingen) backbone (47) using BstXI and AvrII. Recombinant baculovirus stocks were generated by cotransfection of the expression plasmids with linear wild-type baculovirus DNA in Hi5 insect cells (Invitrogen). Supernatants of baculovirus-infected Hi5 cells containing gp68 (26-289) or gp68 (68-289) were buffer exchanged into Ni-binding buffer (40 mM Tris pH 8, 300 mM NaCl, 10 mM imidazole) and passed over a Ni-nitrilotriacetic acid agarose column (Qiagen). gp68 (26-289) and gp68 (68-289) were eluted in the same buffer containing 250 mM imidazole and further purified by size-exclusion chromatography on a Superdex 75 HiLoad 16/60 column (GE Healthcare) that was equilibrated in 20 mM HEPES (pH 7.8)-150 mM NaCl. Analysis of the peak fractions on a 12% SDS-PAGE gel showed bands migrating with an apparent molecular mass of 55 and 35 kDa for gp68 (26-289) and gp68 (68-289), respectively (see Fig. 4A). The gp34 ectodomain [gp34 (24-182)] was overexpressed in CHO cells. DNA encoding gp34 (24-182) (residues 24 to 182, where residue 1 is the N-terminal Met of the immature protein and residues 1 to 23 are predicted to define the signal peptide) was PCR amplified with a C-terminal factor Xa-cleavable six-His tag and subcloned in frame with the rat IgG2a signal sequence into pBJ5-GS (31), a mammalian expression vector that carries the glutamine synthetase gene as a means of selection and amplification in the presence of the drug methionine sulfoximine (6). Stable cell lines expressing gp34 (24-182) were generated by Lipofectamine 2000 (Invitrogen) transfection of the expression plasmid in CHO cells. gp34 (24-182) was purified from CHO cell supernatants on a human IgG-Sepharose column (GE Healthcare) by washing them with 40 mM Tris (pH 8)-300 mM NaCl and eluting them with 50 mM diethylamine (pH 11.5) that was immediately neutralized with 1 M Tris (pH 7), followed by a HiTrap chelating high-performance column (GE Healthcare) eluted with 40 mM Tris (pH 8)-300 mM NaCl-250 mM imidazole. The aggregation state of gp34 (24-182) was analyzed on a Superdex 75 HR 10/30 column (GE Healthcare).
FIG. 4.
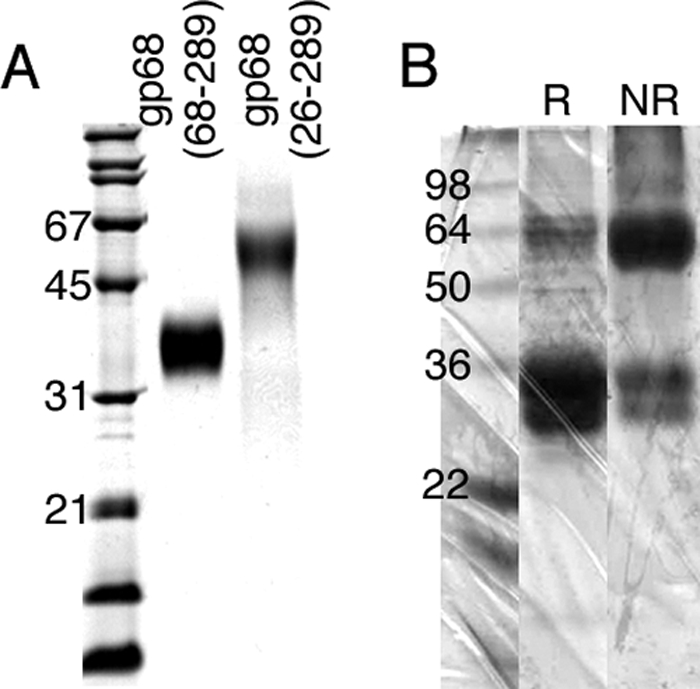
SDS-PAGE analyses of purified recombinant gp68 and gp34 ectodomains. (A) gp68 (26-289) and gp68 (68-289); (B) gp34 (24-182) under reducing (R) and nonreducing (NR) conditions.
Gel filtration binding assay.
wtFc, heterodimeric Fc (hdFc), and nbFc, expressed in CHO cells and purified as described previously (47), were each mixed with gp68 (68-289) in 2:1 [104 μM gp68 (68-289), 52 μM Fc] and/or 1:1 [52 μM gp68 (68-289), 52 μM Fc] molar ratios in 50-μl total volumes and injected onto a Superdex 200 10/30 column (GE Healthcare) in 20 mM HEPES (pH 7.8)-150 mM NaCl. wtFc, hdFc, and nbFc were each examined alone at a concentration of 52 μM; gp68 (68-289) was examined alone at a concentration of 52 μM. A 1:1 molar ratio of gp68 (68-289) and hdFc was also run under acidic conditions in 50 mM sodium citrate (pH 5.6)-150 mM NaCl-1 mM EDTA.
Biosensor studies.
Surface plasmon resonance studies were performed using a BIAcore 2000 instrument. For the affinity analyses of gp68 binding to wtFc and hdFc, purified recombinant wtFc, hdFc, and nbFc were each immobilized on a research-grade CM5 biosensor chip (Biacore) using primary amine coupling as described in the Biacore manual. gp68 (26-289) and gp68 (68-289) were each injected over a chip where one flow cell was mock coupled with buffer only and the other three flow cells were coupled with either wtFc, hdFc, or nbFc at low coupling densities (∼100 to 200 resonance units ([RU]). Threefold dilutions of gp68 (26-289) and gp68 (68-289) were made starting at 6,000 nM and ending at 8 nM and assayed in 50 mM HEPES (pH 7.8 or 8.1), 150 mM NaCl, 3 mM EDTA, and 0.005% (vol/vol) P-20 surfactant at a flow rate of 50 μl/min. The signal returned to baseline after approximately 30 min, indicating complete dissociation of the gp68/Fcγ complex and thus regeneration of the Fcγ-coupled biosensor surface. Kinetic binding data were analyzed with both mono- and bivalent ligand models using Clamp (35), resulting in equilibrium dissociation constants (KDs) for either a single-binding event, KD, or the first and second binding events, KD1 and KD2, as described previously (47). For the comparison of gp68 binding to hdFc at pH 6.0 and pH 8.1, gp68 (68-289) was immobilized using primary amine coupling, as described above, and threefold dilutions of hdFc were injected at pH 8.1 (50 mM HEPES [pH 8.1], 150 mM NaCl, 3 mM EDTA, 0.005% [vol/vol] P-20 surfactant) or pH 6.0 (50 mM sodium phosphate [pH 6.0], 150 mM NaCl, 3 mM EDTA, 0.005% [vol/vol] P-20 surfactant) at a flow rate of 5 μl/min. The KD was determined at each pH by fitting the equilibrium binding response as a function of hdFc concentration to a single-site binding model.
RESULTS
Intact binding of the HCMV FcγRs gp34 and gp68 to deglycosylated Fcγ.
The Fcγ is a homodimer formed by two N-linked glycopeptide chains, each of which contains two immunoglobulin constant domains, CH2 and CH3. The chains form a covalent dimer through interchain disulfide bonds in the N-terminal hinge region. In addition, the CH3 domains pair through protein-protein interactions, and the CH2 domains interact through N-linked oligosaccharide chains that are attached to Asn297. It is well established that glycosylation of Fcγ is essential for recognition and activation of host FcγRs (21), which recognize the CH1-CH2 hinge and CH2 domain of Fcγ (45). As a consequence, removal of the N-glycan results in a loss of FcγR binding (44, 52). By contrast, carbohydrate depletion of IgG does not prevent binding by the Staphylococcus aureus Fc receptor protein A (37), which contacts the interface between the CH2 and CH3 domains (11). To test whether the interaction of the HCMV FcγRs gp34 and gp68 with Fcγ is sensitive to the presence of carbohydrate on Fcγ, we compared the binding levels of the vFcγRs to enzymatically deglycosylated Fcγ and to glycosylated Fcγ. The HCMV FcγRs and a control protein, the host FcγRI (CD64), which binds glycosylated but not deglycosylated Fcγ (44), were expressed by rVVs and precipitated from lysates following metabolic labeling of cells using both forms of Fcγ. A lysate of CV-I cells infected with wild-type VV (wtVV) (Fig. 1, lanes 1 to 5), precipitation of gp34, gp68, and CD64 with untreated Fcγ (Fig. 1, lanes 1, 6, 11, and 21), and immunoprecipitation of CD64 using a specific antibody (Fig. 1, lane 16) were used as controls for binding. As expected, the binding capacity of deglycosylated Fcγ to CD64 was strongly impaired (Fig. 1, lanes 17 and 18). In contrast, gp34 and gp68 exhibited strong and unaltered binding activities to deglycosylated Fcγ in comparison to glycosylated, mock-treated Fcγ (Fig. 1, lanes 7 to 10 and 12 to 15). These results exclude direct binding of HCMV gp34 and gp68 to the Asn297-linked glycan and indicate a conformational requirement for Fcγ recognition different from that of host FcγRs (27, 44). Moreover, gp34 and gp68 exhibit a glycan-independent binding mode to Fcγ like the Staphylococcus aureus protein A, although neither HCMV protein interferes with Fcγ binding to protein A (3).
FIG. 1.

Consequences of Fcγ deglycosylation for HCMV FcγRs (A) and CD64 (FcγRI) (B) binding. CV-I cells were infected with rVV or wtVV at a multiplicity of infection of 5 for 14 h. [35S]methionine-labeled cells were lysed in 1% NP-40 lysis buffer, and precipitation of proteins was performed with either enzymatically deglycosylated Fcγ (F, peptide N-glycosidase F treated; H, Endo H treated), mock-treated Fcγ (cF or cH; no enzyme added), untreated Fcγ (−), or anti-CD64 antibody followed by protein A- or protein G-Sepharose. Precipitated proteins were separated by SDS-10 to 13% PAGE. Partially glycosylated forms of gp34 and gp68 (3) are indicated by asterisks. α, anti-.
Amino acids 71 to 292 of HCMV gp68 are required for Fcγ binding.
The HCMV UL119-UL118-encoded FcγR gp68 is a type I transmembrane protein with an N-terminal hydrophobic signal peptide (comprising the first 25 to 28 residues) and carrying multiple predicted N- and O-linked glycans within the mature ectodomain (Fig. 2A). An immunoglobulin-like domain in good agreement with the consensus sequence for variable-like domains (40) is predicted for gp68 residues 91 to 190. The putative immunoglobulin-like domain includes positionally conserved cysteines, Cys116 and Cys169 (3), and showed its highest degree of sequence identity to the third domain of FcγRI (CD64) (20% amino acid identity and 31% amino acid similarity). To test whether the region including the immunoglobulin-like domain is involved in Fcγ recognition and to define the minimal region of gp68 required for Fcγ binding, soluble gp68 truncation mutants, each lacking the predicted transmembrane and cytoplasmic regions of the protein, were constructed. The following gp68 truncation mutants were expressed with a C-terminal FLAG epitope (Fig. 2A), which allowed for verification of protein expression by immunoprecipitation with FLAG-specific antibodies: gp68 (26-292) (the complete ectodomain), gp68 (26-251) (mutant lacking 41 C-terminal residues of the ectodomain), and gp68 (21-206) (mutant lacking 84 C-terminal residues of the ectodomain). A further deletion mutant was constructed to remove residues at the N terminus of the ectodomain predicted to contain 25 potential O-glycans. This mutant, referred to as gp68 (71-292), was constructed by fusing the N-terminal signal sequence of gp68 to residues 71 to 292 followed by C-terminal V5/six-His epitopes. All gp68-derived mutants were expressed by rVVs, and proteins with Fcγ-binding properties were precipitated from cell lysates with Fcγ or epitope tag-specific antibodies, separated by SDS-PAGE, and detected by immunoblotting using epitope-specific IgG (Fig. 2B). Lysates from cells infected with wtVV or mock-infected cells were used as controls. Epitope-specific antibodies immunoprecipitated mutant proteins that exhibited the expected molecular masses (Fig. 2B, left panel), as confirmed by deglycosylation using Endo H (Fig. 2B, right panel). Binding to Fcγ, however, was restricted to gp68 (26-292), containing the complete gp68 ectodomain, and gp68 (71-292), which lacked the N-terminal O-glycosylated subdomain (Fig. 2B). Removal of residues 206 to 292 abrogated Fcγ binding, pointing to an indispensable contribution of this region of the gp68 ectodomain, which is located C terminally from the putative immunoglobulin-like domain, in the Fcγ interaction.
FIG. 2.

Schematic representation of gp68 truncation mutants and their Fc binding properties. (A) The immature full-length gp68 (gp68FL) contains a predicted ∼25 residue N-terminal signal peptide (indicated by an open rectangle) followed by an ∼268-residue ectodomain (residues 26 to 293), 21-residue transmembrane region (residues 294 to 314), and 31-residue cytoplasmic tail (residues 315 to 345). All gp68 mutants are secreted proteins that lack the transmembrane and cytosolic domains and contain a C-terminal FLAG or V5/six-His tag as indicated. The predicted molecular mass of each deglycosylated and glycosylated HCMV gp68 mutant is indicated on the right. TM, transmembrane region; signal, signal peptide; Ig-like domain, immunoglobulin-like domain. (B) The truncated forms of gp68 were expressed by rVVs and precipitated from cell lysates either with human Fcγ and protein A-Sepharose or with goat anti-FLAG antibody coupled to agarose or with mouse anti-V5 antibodies and protein G-Sepharose. Cell lysates from CV-I cells infected with wtVV or mock infected were used as controls. An aliquot of the precipitate was deglycosylated by Endo H treatment prior to separation of the proteins by gradient SDS-PAGE. After transfer to a nitrocellulose membrane, proteins were detected using an anti-FLAG M2 or anti-V5 antibody. Bands of Fcγ due to cross-reactivity of the secondary anti-FLAG M2 antibody are indicated by asterisks. WB, Western blot analysis; IP, immunoprecipitation; α, anti-.
Fcγ binding characteristics of gp68 differ from those of host CD64 (FcγRI) and CD32 (FcγRII).
The binding interface of Fcγ bound to the host CD16 (FcγRIII), defined by crystallization studies, is formed by domains 1 and 2 of CD16 and the lower hinge region between the antibody Fab and Fcγ in conjunction with a contact site on the CH2 domain of Fcγ, resulting in a 1:1 stoichiometry (45). A common set of residues on Fcγ is involved in binding FcγRs, although CD64 and CD32 also interact with Fcγ residues outside of the common set (44). In contrast, biochemical and structural analyses of gE-gI binding to Fcγ revealed that gE-gI interacts with the Fcγ CH2-CH3 interdomain junction with a stoichiometry of two molecules of gE-gI per Fcγ (47, 48). We previously demonstrated that HSV-1 gE-gI does not bind to nbFc (a nonbinding mutant Fcγ in which each Fcγ subunit contained six point mutations in the CH2-CH3 domain interface) but retains normal binding to a recombinant wtFc (47), consistent with crystallographic studies of a 2:1 gE-gI/Fcγ complex (48). To analyze the mode of interaction of the cytomegaloviral FcγR gp68 to Fcγ, we compared the binding levels of wtFc and nbFc to gp68, the host FcγRs CD32 and CD64, and HSV-1 gE-gI, respectively (Fig. 3). As expected, mutation of both chains in the CH2-CH3 interdomain hinge of the nbFc molecule caused a loss of binding to HSV-1 gE-gI but not to CD32 and CD64 (44, 47, 48). The impaired interaction of CD32 to nbFc resulted from utilization of additional residues outside of the common FcγR binding site in the hinge region/CH2 domain (44). Interestingly, nbFc did not precipitate gp68, suggesting a binding mode similar to that of the herpesviral FcγR gE-gI.
FIG. 3.

Fcγ binding characteristics of gp68 differ from those of host CD64 (FcγRI) and CD32 (FcγRII). CV-I cells were infected with rVVs expressing gp68 and the host Fcγ receptors CD32 and CD64 or with HSV-1 strain F at a multiplicity of infection of 4 for 14 h before metabolic labeling (as described in the legend to Fig. 1). Proteins were precipitated using either wtFc or nbFc coupled to CNBr-activated Sepharose. Dissociated immune complexes were separated by 10% SDS-PAGE. IP, immunoprecipitation.
Expression and purification of soluble HCMV gp68 and HCMV gp34 proteins.
Two soluble versions of gp68 were expressed in baculovirus-infected insect cells: the gp68 ectodomain [gp68 (26-289)] and an N-terminally truncated form of the gp68 ectodomain [gp68 (68-289); similar to gp68 (71-292) expressed using rVV] that lacks the first 42 amino acids of the predicted mature protein containing 25 potential O-GalNAc-glycosylation sites (25). Analysis of the purified proteins on a 12% SDS-PAGE gel showed bands migrating with an apparent molecular mass of 55 and 35 kDa for gp68 (26-289) and gp68 (68-289), respectively (Fig. 4A).
The gp34 ectodomain [gp34 (24-182)] was expressed as a secreted protein in CHO cells. Analysis of purified gp34 (24-182) on a 12% SDS-PAGE gel under nonreducing conditions revealed that ∼10% of the protein migrated with an apparent molecular mass of 30 kDa and ∼90% migrated with an apparent molecular mass of 60 kDa. By contrast, under reducing conditions, most of the protein migrated with an apparent molecular mass of 30 kDa (Fig. 4B). Thus, a disulfide-linked gp34 homodimer [gp34 (24-182) contains 5 cysteines] may be present in the nonreducing environment of the extracellular milieu when the intact protein is present at the cell surface. gp34 (24-182) eluted as a very broad peak on a size exclusion column (data not shown), suggesting that our gp34 (24-182) preparation contained multiple aggregated species and was not suitable for further biochemical characterization.
The HCMV gp68 ectodomain binds the CH2-CH3 interdomain interface of human Fcγ.
We previously used wtFc, nbFc, and a heterodimeric Fcγ containing one wtFc and one nbFc chain (hdFc) to demonstrate that gE-gI binds Fcγ with a 2:1 stoichiometry (47). To address whether gp68 binds to the CH2-CH3 interdomain interface region on Fcγ and to determine the gp68/Fcγ binding stoichiometry, we designed a gel filtration assay to compare the elution profiles of gp68 (68-289) mixed with wtFc, hdFc, or nbFc, which were expressed in CHO cells and purified as previously described (47). If the interaction interface between gp68 (68-289) and Fcγ encompasses the Fcγ CH2-CH3 interdomain interface, mixtures of gp68 (68-289) with wtFc, hdFc, and nbFc will have different elution profiles when run on a gel filtration column. Otherwise, if the residues in the Fcγ CH2-CH3 interdomain interface do not significantly contribute to the gp68 (68-289)/Fcγ interface, as in the Fcγ/FcRγIII complex (45), mixtures of gp68 (68-289) with each of the Fcγ variants, which differ in residues located in the CH2-CH3 interdomain interface region of one or both subunits, will have similar elution profiles.
In the gel filtration assay, gp68 (68-289) eluted at 15.7 ml, consistent with an ∼35-kDa monomeric protein, and wtFc, nbFc, and hdFc eluted at 14.6, 14.4, and 14.2 ml, respectively, consistent with dimeric Fcγ differing by one or two six-His tags (Fig. 5A). When gp68 (68-289) was mixed with nbFc and loaded onto the gel filtration column, the only peaks observed were at 15.7 and 14.2 ml, identical to the elution volumes for gp68 (68-289) and nbFc alone (Fig. 5A). The elution profile for a mixture of gp68 (68-289) with hdFc, however, contained a new peak at 13.0 ml (Fig. 5A). Likewise, elution profiles of gp68 (68-289) with wtFc also contained additional species that eluted at 12.5 and 12.3 ml depending on the molar ratio (Fig. 5A). The additional peaks observed for gp68 (68-289) in the presence of either wtFc or hdFc in comparison to any of the proteins alone demonstrate that gp68 (68-289) binds wtFc and hdFc, consistent with the binding of gp68 (71-292) to Fcγ, whereas the similarity between the elution profiles of either gp68 (68-289) and nbFc alone and the mixture of gp68 (68-289) and nbFc indicates that gp68 (68-289) does not bind nbFc. Thus, gp68 (68-289) recognized wtFc by using at least some of the six residues in the Fc CH2-CH3 interdomain interface that were altered to create nbFc. Furthermore, binding of gp68 (68-289) to hdFc suggests that the gp68 (68-289) binding site on Fcγ is likely contained within a single Fc CH2-CH3 interdomain interface instead of being composed of residues located in the CH2-CH3 interdomain interface of both subunits of the Fcγ dimer.
FIG. 5.

Gel filtration assay revealing that one molecule of gp68 binds each CH2-CH3 interdomain interface of Fcγ. Elution profiles are shown for mixtures of gp68 (68-289) with wtFc, hdFc, and nbFc, which contain zero, one, and two CH2-CH3 mutant Fcγ chains (see text), respectively, at pH 7.8 (A) and pH 5.6 (B). The elution volumes of gp68, wtFc, hdFc, and nbFc, and 1:1 and 2:1 [gp68 (68-289)/Fcγ] complexes are indicated with dotted lines. mAU, milli-absorbance units.
Two HCMV gp68 molecules bind to each wtFc.
To determine the stoichiometry of the gp68 (68-289)/Fcγ interaction, we analyzed the elution profiles of different molar ratios of gp68 (68-289) and wtFc or hdFc, which contain two or one potential binding sites, respectively. A 1:1 molar ratio of gp68 (68-289) to wtFc eluted as two peaks: one at 12.5 ml, corresponding to a new species with a higher molecular mass than either protein alone, and one at 14.5 ml, corresponding to excess wtFc (∼50% of the initial wtFc) (Fig. 5A). When the molar ratio was increased to 2:1, the major peak shifted to 12.3 ml, the excess wtFc peak disappeared, and there was a slight excess of gp68 (68-289) (<10% of the total sample) (Fig. 5A), all of which indicate a stoichiometry of two gp68 (68-289) molecules binding to one wtFc dimer. In addition, the elution profile for a sample containing a 2:1 molar ratio of gp68 (68-289) to hdFc contained a major peak at 13.0 ml, likely corresponding to a 1:1 gp68 (68-289)/hdFc complex, and a minor peak at 15.7 ml, corresponding to excess gp68 (68-289) [∼50% of the initial gp68 (68-289)] (Fig. 5A). Thus, a comparison of the binding characteristics of gp68 (68-289) to wtFc with those to hdFc and nbFc, two CH2-CH3 interdomain interface variants, using a gel filtration chromatography assay demonstrated that one molecule of gp68 (68-289) binds to the CH2-CH3 interdomain interface of each wtFc subunit, similar to the binding of HSV-1 gE-gI to wtFc (47). In addition, the fact that the gp68 (68-289)/wtFc complex migrated only slightly slower than the 1:1 gp68 (68-289)/hdFc complex suggested that the gp68 (68-289)/wtFc complex does not contain higher-order multimers of 2:1 vFcγR/Fcγ complexes.
Biosensor binding experiments support a bivalent binding model with nanomolar affinities.
Binding of the HCMV gp68 ectodomain variants to Fcγ were further characterized using a surface plasmon resonance assay, in which wtFc (“ligand”) was immobilized and gp68 (26-289) or gp68 (68-289) (“analyte”) was injected. A comparison of the gp68 (26-289) and gp68 (68-289) binding curves with those predicted by mono- and bivalent ligand models showed that the interactions between gp68 (26-289) and wtFc and between gp68 (68-289) and wtFc were best described by a bivalent ligand model (Fig. 6A), consistent with the gel filtration studies that identified two gp68 binding sites on wtFc. Using a bivalent ligand model, we determined KDs for the first and second binding events (KD1 and KD2) in which gp68 interacts with Fc as follows: for gp68 (26-289) binding to immobilized wtFc, KD1 equals 470 nM and KD2 equals 1,600 nM; for gp68 (68-289) binding to immobilized wtFc, KD1 equals 140 nM and KD2 equals 240 nM (Fig. 6A). The finding that gp68 (68-289) binds wtFc with affinities that are three- to sixfold tighter than those of the gp68 (26-289)/wtFc interaction suggests that the N-terminal 42 residues of the mature gp68 protein and their likely associated O-linked glycans are not critical for recognition of Fc, consistent with the Fcγ pulldown experiments that indicated similar binding levels of Fcγ with gp68 (26-292) and with gp68 (71-292) (Fig. 2B). The KD values derived from an analysis of kinetic binding data of gp68 to immobilized hdFc were similar to those for gp68 binding to the highest-affinity site on wtFc [gp68 (26-289), KD = 300 nM; gp68 (68-289), KD = 85 nM] (Fig. 6B), confirming that the single binding site on hdFc mimics each of the gp68 binding sites on wtFc. As expected, binding of the gp68 ectodomains to 1 μM nbFc was not detectable in this assay (data not shown). Previous measurements of binding of human Fcγ to acetone-fixed HCMV-infected human embryonic lung fibroblasts estimated a KD of 5 nM (2); however, this value represents a macroscopic KD that includes avidity effects due to cross-linking of IgG by gp68 proteins, gp34 proteins, and complexes containing one of each vFcγR.
FIG. 6.

Surface plasmon resonance binding data for gp68 (26-289) and gp68 (68-289) binding to Fcγ. Sensorgrams for binding of injected gp68 (26-289) or gp68 (68-289) at various concentrations (6,000 nM, 2,000 nM, 667 nM, 222 nM, 74 nM, 25 nM, and 8 nM) at pH 7.8 over a wtFc surface (∼200 RU of primary amine-coupled wtFc) (A) or a hdFc surface (∼100 RU of primary amine-coupled hdFc) (B) are shown in black. Fits for a bivalent ligand binding model, which assumes two independent binding sites on Fcγ, and for a single-site binding model are shown as blue lines, and affinities are indicated, with KD1 and KD2 corresponding to the first and second binding events, respectively, for the bivalent ligand model. Residuals are shown below each fit. (C) Equilibrium binding responses for hdFc injected over gp68 (68-289) at pH 8.1 (triangles) or pH 6.0 (squares) are shown at various injection concentrations. Data are plotted as percent binding at equilibrium (normalized for maximal binding) for each injection of hdFc and fit to a single-binding-site isotherm. The derived KDs are 60 nM at pH 6.0 and 300 nM at pH 8.1. Four independent determinations of the KD at either pH 7.8 or 8.1 ranged from 85 nM to 300 nM; thus, the affinity of gp68 binding to Fc is similar, or even slightly tighter, at pH 6.0 than at pH 8.1.
The interaction between HCMV gp68 and Fcγ is stable at acidic pH.
The interaction between HSV-1 gE-gI and Fcγ is sharply pH dependent, with a 10-fold decrease in affinity between pH 7.4 and pH 6.6 and unmeasurable binding at pH values of ≤6.2 as determined by a Biacore assay (47). The unusual pH sensitivity of the gE-gI/Fcγ interaction allowed for easy regeneration of gE-gI-coupled biosensor chips after each injection of Fcγ at acidic pH (pH 5.0) (47). However, when we tried to set up a similar Biacore binding assay for gp68/Fcγ, acidic pH did not disrupt the interaction between Fcγ and gp68, and regeneration of the biosensor surface could be achieved only with long dissociation times (>30 min). To verify the lack of a sharp pH dependence for the gp68/Fcγ interaction, we repeated the gel filtration binding assay at acidic pH and measured the KD at acidic pH using a surface plasmon resonance assay. In the gel filtration assay at pH 5.6, a 1:1 molar ratio of gp68 (68-289)/hdFc eluted from the column as expected for a 1:1 complex (Fig. 5B), demonstrating the stability of the gp68/Fcγ complex in solution at pH 5.6. Further assessment of the interaction at acidic pH by surface plasmon resonance showed that the affinity for binding of gp68 (68-289) to hdFc at pH 6 (KD = 60 nM) was similar to, and possibly even tighter than, the affinities observed at pH 7.8 or pH 8.1 (KD = 85 to 300 nM) (Fig. 6C). Thus, the gp68/Fcγ complex is stable between pH 5.6 and 8.1, in contrast to the gE-gI/Fcγ complex.
DISCUSSION
Structural requirements for gp68 Fcγ binding.
In this study we have investigated the mode of HCMV vFcγR binding to Fcγ and found distinct differences in comparison with the binding of host FcγRs, on the one hand, and a prototypic vFcγR, HSV gE-gI, on the other hand. Our initial finding revealed a fully maintained binding of gp34 and gp68 to deglycosylated Fcγ lacking the N-linked glycans that are attached to Asn297. This finding was not expected based on the following several facts: (i) the existing, albeit limited, sequence relatedness of gp68 and gp34 with host FcγRs (3) but not with herpesviral or microbial Fc binding proteins; (ii) the antagonistic effect of gp34 and gp68 on the activation of distinct host FcγRs by immune complexes (E. Corrales-Aguilar and H. Hengel, unpublished data); and (iii) the lack of gp34 and gp68 competition with Staphylococcus aureus protein A for Fcγ binding, which contrasts to HSV gE-gI (8, 23). Removal of the N-linked glycans results in conformational changes in the CH2 domain and increased internal disorder of the Fcγ affecting the interface between Fcγ and host FcγRs (27, 56). Based on the fact that gp34 and gp68 binding was insensitive to deglycosylation-induced changes of the Fcγ, we hypothesized that a contact site distinct from the CH2-proximal hinge region mediating binding of the host FcγRII and FcγIII (45, 57) is more likely recognized by the vFcγRs. This assumption prompted us to take advantage of recombinant Fcγ mutants of the CH2-CH3 interdomain interface, which had been used to map the site recognized by HSV gE-gI (47). Our results demonstrated that Fcγ binding of gp68, like the binding of gE-gI, is affected by mutations at the CH2-CH3 domain interface, suggesting that gp68 contacts this region of Fcγ. Consistent with this suggestion, gp68 formed a 2:1 complex with Fcγ. Although gp68 targets a similar binding site on Fcγ, several findings support the notion that gp68 adopts an Fc binding mode that differs from that of HSV gE-gI, as follows: (i) stability of the gp68/Fcγ complex at pH 6, (ii) a maintained strong binding to the IgG3 subclass (3), (iii) a lack of competition with Staphylococcus aureus protein A, and (iv) effects on IgG effector functions that differ from those exerted by HSV gE (H. Reinhard, E. Corrales-Aguilar, and H. Hengel, unpublished data).
Despite their selective binding to IgG with high affinities, the herpesviral FcγRs HSV gE-gI and mouse cytomegalovirus (MCMV) m138/fcr-1 also exhibit IgG-independent functions. HSV gE and gI play an important role in virus spread from cell to cell (4, 13), and MCMV m138/fcr-1 attenuates NKG2D-mediated NK cell responses, resulting in a severely attenuated replication of gene deletion mutants in vivo (10, 28). By analogy, gp68 may also have additional functions that are unrelated to Fcγ binding.
pH dependence of the gp68/Fcγ interaction.
Although HCMV gp68 and HSV-1 gE-gI share the same or an overlapping binding site on Fcγ, the finding that the gp68/Fcγ interaction is stable at pH values between 5.6 and 8.1 but that gE-gI binds only at neutral or basic pH raises the question whether the downstream events after Fc binding differ for gp68 and gE-gI. Binding of gE-gI to anti-HSV IgG/antigen complexes in an antibody bipolar bridged complex has been shown to abrogate IgG-mediated immune responses (14, 16, 36, 51). gE-gI may also contribute to immune evasion by transporting free IgG and antibody bipolar bridged complexes into endosomes, where the gE-gI/IgG complex would dissociate in the ∼pH 6 environment of early endosomes, and IgG-antigen complexes would be targeted for proteolytic degradation while gE-gI would be recycled back to the cell surface (47). By contrast, IgG is predicted to remain bound to gp68 in early endosomes. Since both gp34 and gp68 are relatively short-lived proteins that are rapidly endocytosed and eventually transported to lysosomes and degraded (3), they could transport bound IgG to lysosomes, where proteases could digest both gp68 and the bound IgG.
MCMV m138/fcr-1 glycoprotein is another surface-resident vFcγR that trafficks to endolysosomal compartments. In addition to its Fcγ binding capabilities, m138/fcr-1 down-modulates CD80 (34), the NKG2D ligand H60, and murine UL16-binding protein-like transcript (MULT-1) (28). The down-regulation of MULT-1 is achieved by m138/fcr-1-mediated interference of the recycling of surface MULT-1, leading to its subsequent proteolytic degradation in a lysosomal compartment (28). Interestingly, both effects, i.e., MULT-1 removal from the cell surface and Fcγ binding, are mediated by the same N-terminal portion of the fcr-1 ectodomain (Ig1), which is one of three putative immunoglobulin-like domains (28). Taken together, it is tempting to speculate that herpesviral FcγRs in general are lysosomotropic proteins shuttling target proteins and IgG-immune complexes from the cell surface to distinct endolysosomal compartments. Moreover, vFcγRs may function to deliver their cargo to endosomal compartments where they can be processed and loaded onto major histocompatibility complex (MHC) molecules, usually class II MHC molecules but also possibly class I MHC molecules involved in cross-presentation provided that the MHC molecules escaped from downregulation by HCMV (20, 38). Thus, the observed pH differences of Fcγ binding and release from vFcγRs may reflect adaptation to specific pH values prevailing at certain sites along the increasingly acidic milieu of the endolysosomal route and therefore depend on vFcγR destination.
In conclusion, this study provides evidence that HCMV gp68 has found a means to bind Fcγ that differs from those of both host FcγRs and vFcγRs. In this context it should be mentioned that the gp68 binding site at the CH2-CH3 interdomain junction is distant from the FcγR binding sites involving the hinge between the Fcγ and Fab domains and the upper portion of the CH2 domain (45, 46). Since gp68, like HSV-1 gE-gI, is able to antagonize host FcγR-dependent responses (H. Reinhard, E. Corrales-Aguilar, and H. Hengel, unpublished data), these vFcγRs must have evolved binding interactions that allow them to simultaneously contact the CH2-CH3 interdomain interface and to block the host FcγR binding site. The structural basis for these interactions remains an intriguing problem to be solved.
Acknowledgments
We thank Peter Snow and Inder Nangiana for insect cell expression of gp68 proteins, Brian Seed for the CD64 and CD32 cDNAs, Manuela Fiedler and Philipp Lacher for constructing rVVs, and Eugenia Corrales-Aguilar for critical reading of the manuscript.
This work was supported by a Leukemia and Lymphoma Society Postdoctoral Fellowship (E.R.S.), a Max Planck Research Award (P.J.B.), the National Institutes of Health (2 R37 AI041239-06A1 to P.J.B.), DFG grant He 2526/6-2, and EU grant FP6-037517.
Footnotes
Published ahead of print on 23 January 2008.
REFERENCES
- 1.Allen, J. M., and B. Seed. 1989. Isolation and expression of functional high-affinity Fc receptor complementary DNAs. Science 243378-381. [DOI] [PubMed] [Google Scholar]
- 2.Antonsson, A., and P. J. Johansson. 2001. Binding of human and animal immunoglobulins to the IgG Fc receptor induced by human cytomegalovirus. J. Gen. Virol. 821137-1145. [DOI] [PubMed] [Google Scholar]
- 3.Atalay, R., A. Zimmermann, M. Wagner, E. Borst, C. Benz, M. Messerle, and H. Hengel. 2002. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcγ receptor homologs. J. Virol. 768596-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balan, P., N. Davis-Poynter, S. Bell, H. Atkinson, H. Browne, and T. Minson. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 751245-1258. [DOI] [PubMed] [Google Scholar]
- 5.Baucke, R. B., and P. G. Spear. 1979. Membrane proteins specified by herpes simplex viruses. V. Identification of an Fc-binding glycoprotein. J. Virol. 32779-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bebbington, C. R., and C. C. G. Hentschel. 1987. The use of vectors based on gene amplification for the expression of cloned genes in mammalian cells, p. 163-188. In D. M. Glover (ed.), DNA cloning: a practical approach. IRL Press, Oxford, United Kingdom.
- 7.Budt, M., H. Reinhard, A. Bigl, and H. Hengel. 2004. Herpesviral Fcgamma receptors: culprits attenuating antiviral IgG? Int. Immunopharmacol. 41135-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman, T. L., I. You, I. M. Joseph, P. J. Bjorkman, S. L. Morrison, and M. Raghavan. 1999. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J. Biol. Chem. 2746911-6919. [DOI] [PubMed] [Google Scholar]
- 9.Corper, A. L., M. K. Sohi, V. R. Bonagura, M. Steinitz, R. Jefferis, A. Feinstein, D. Beale, M. J. Taussig, and B. J. Sutton. 1997. Structure of human IgM rheumatoid factor Fab bound to its autoantigen IgG Fc reveals a novel topology of antibody-antigen interaction. Nat. Struct. Biol. 4374-381. [DOI] [PubMed] [Google Scholar]
- 10.Crnkovic-Mertens, I., M. Messerle, I. Milotic, U. Szepan, N. Kucic, A. Krmpotic, S. Jonjic, and U. H. Koszinowski. 1998. Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 721377-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deisenhofer, J. 1981. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 202361-2370. [PubMed] [Google Scholar]
- 12.DeLano, W. L., M. H. Ultsch, A. M. de Vos, and J. A. Wells. 2000. Convergent solutions to binding at a protein-protein interface. Science 2871279-1283. [DOI] [PubMed] [Google Scholar]
- 13.Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J. Rainbow, and D. C. Johnson. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68834-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubin, G., E. Socolof, I. Frank, and H. M. Friedman. 1991. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J. Virol. 657046-7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favoreel, H. W., H. J. Nauwynck, P. Van Oostveldt, T. C. Mettenleiter, and M. B. Pensaert. 1997. Antibody-induced and cytoskeleton-mediated redistribution and shedding of viral glycoproteins, expressed on pseudorabies virus-infected cells. J. Virol. 718254-8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank, I., and H. M. Friedman. 1989. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J. Virol. 634479-4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frey, J., and B. Einsfelder. 1984. Induction of surface IgG receptors in cytomegalovirus-infected human fibroblasts. Eur. J. Biochem. 138213-216. [DOI] [PubMed] [Google Scholar]
- 18.Friedman, H. M. 2003. Immune evasion by herpes simplex virus type 1, strategies for virus survival. Trans Am. Clin. Climatol Assoc. 114103-112. [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa, T., E. Hornberger, S. Sakuma, and S. A. Plotkin. 1975. Demonstration of immunoglobulin G receptors induced by human cytomegalovirus. J. Clin. Microbiol. 2332-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hengel, H., W. Brune, and U. H. Koszinowski. 1998. Immune evasion by cytomegalovirus—survival strategies of a highly adapted opportunist. Trends Microbiol. 6190-197. [DOI] [PubMed] [Google Scholar]
- 21.Jefferis, R., and J. Lund. 2002. Interaction sites on human IgG-Fc for FcγR: current models. Immunol. Lett. 8257-65. [DOI] [PubMed] [Google Scholar]
- 22.Johansson, P. J., T. Hallberg, V. A. Oxelius, A. Grubb, and J. Blomberg. 1984. Human immunoglobulin class and subclass specificity of Fc receptors induced by herpes simplex virus type 1. J. Virol. 50796-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson, P. J., F. A. Nardella, J. Sjoquist, A. K. Schroder, and P. Christensen. 1989. Herpes simplex type 1-induced Fc receptor binds to the Cγ2-Cγ3 interface region of IgG in the area that binds staphylococcal protein A. Immunology 668-13. [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, D. C., M. C. Frame, M. W. Ligas, A. M. Cross, and N. D. Stow. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 621347-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Julenius, K., A. Molgaard, R. Gupta, and S. Brunak. 2005. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15153-164. [DOI] [PubMed] [Google Scholar]
- 26.Keller, R., R. Peitchel, J. N. Goldman, and M. Goldman. 1976. An IgG-Fc receptor induced in cytomegalovirus-infected human fibroblasts. J. Immunol. 116772-777. [PubMed] [Google Scholar]
- 27.Krapp, S., Y. Mimura, R. Jefferis, R. Huber, and P. Sondermann. 2003. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 325979-989. [DOI] [PubMed] [Google Scholar]
- 28.Lenac, T., M. Budt, J. Arapovic, M. Hasan, A. Zimmermann, H. Simic, A. Krmpotic, M. Messerle, Z. Ruzsics, U. H. Koszinowski, H. Hengel, and S. Jonjic. 2006. The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J. Exp. Med. 2031843-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lilley, B. N., H. L. Ploegh, and R. S. Tirabassi. 2001. Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J. Virol. 7511218-11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litwin, V., W. Jackson, and C. Grose. 1992. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J. Virol. 663643-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin, W. L., and P. J. Bjorkman. 1999. Characterization of the 2:1 complex between the class I MHC-related Fc receptor and its Fc ligand in solution. Biochemistry 3812639-12647. [DOI] [PubMed] [Google Scholar]
- 32.Martin, W. L., A. P. West, Jr., L. Gan, and P. J. Bjorkman. 2001. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol. Cell 7867-877. [DOI] [PubMed] [Google Scholar]
- 33.Michelson, S. 2004. Consequences of human cytomegalovirus mimicry. Hum. Immunol. 65465-475. [DOI] [PubMed] [Google Scholar]
- 34.Mintern, J. D., E. J. Klemm, M. Wagner, M. E. Paquet, M. D. Napier, Y. M. Kim, U. H. Koszinowski, and H. L. Ploegh. 2006. Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J. Immunol. 1778422-8431. [DOI] [PubMed] [Google Scholar]
- 35.Morton, T. A., and D. G. Myszka. 1998. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Methods Enzymol. 295268-294. [DOI] [PubMed] [Google Scholar]
- 36.Nagashunmugam, T., J. Lubinski, L. Wang, L. T. Goldstein, B. S. Weeks, P. Sundaresan, E. H. Kang, G. Dubin, and H. M. Friedman. 1998. In vivo immune evasion mediated by the herpes simplex virus type 1 immunoglobulin G Fc receptor. J. Virol. 725351-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nose, M., and H. Wigzell. 1983. Biological significance of carbohydrate chains on monoclonal antibodies. Proc. Natl. Acad. Sci. USA 806632-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ploegh, H. L. 1998. Viral strategies of immune evasion. Science 280248-253. [DOI] [PubMed] [Google Scholar]
- 39.Radaev, S., S. Motyka, W. H. Fridman, C. Sautes-Fridman, and P. D. Sun. 2001. The structure of a human type III Fcγ receptor in complex with Fc. J. Biol. Chem. 27616469-16477. [DOI] [PubMed] [Google Scholar]
- 40.Raghavan, M., and P. J. Bjorkman. 1996. Fc receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 12181-220. [DOI] [PubMed] [Google Scholar]
- 41.Rahman, A. A., M. Teschner, K. K. Sethi, and H. Brandis. 1976. Appearance of IgG (Fc) receptor(s) on cultured human fibroblasts infected with human cytomegalovirus. J. Immunol. 117253-258. [PubMed] [Google Scholar]
- 42.Sakuma, S., T. Furukawa, and S. A. Plotkin. 1977. The characterization of IgG receptor induced by human cytomegalovirus. Proc. Soc. Exp. Biol. Med. 155168-172. [DOI] [PubMed] [Google Scholar]
- 43.Sauer-Eriksson, A. E., G. J. Kleywegt, M. Uhlen, and T. A. Jones. 1995. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure 3265-278. [DOI] [PubMed] [Google Scholar]
- 44.Shields, R. L., A. K. Namenuk, K. Hong, Y. G. Meng, J. Rae, J. Briggs, D. Xie, J. Lai, A. Stadlen, B. Li, J. A. Fox, and L. G. Presta. 2001. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2766591-6604. [DOI] [PubMed] [Google Scholar]
- 45.Sondermann, P., R. Huber, V. Oosthuizen, and U. Jacob. 2000. The 3.2-A crystal structure of the human IgG1 Fc fragment-FcγRIII complex. Nature 406267-273. [DOI] [PubMed] [Google Scholar]
- 46.Sondermann, P., and V. Oosthuizen. 2002. X-ray crystallographic studies of IgG-Fc gamma receptor interactions. Biochem. Soc. Trans. 30481-486. [DOI] [PubMed] [Google Scholar]
- 47.Sprague, E. R., W. L. Martin, and P. J. Bjorkman. 2004. pH dependence and stoichiometry of binding to the Fc region of IgG by the herpes simplex virus Fc receptor gE-gI. J. Biol. Chem. 27914184-14193. [DOI] [PubMed] [Google Scholar]
- 48.Sprague, E. R., C. Wang, D. Baker, and P. J. Bjorkman. 2006. Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol. 4e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staib, C., I. Drexler, and G. Sutter. 2004. Construction and isolation of recombinant MVA. Methods Mol. Biol. 26977-100. [DOI] [PubMed] [Google Scholar]
- 50.Thale, R., P. Lucin, K. Schneider, M. Eggers, and U. H. Koszinowski. 1994. Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J. Virol. 687757-7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Vliet, K. E., L. A. De Graaf-Miltenburg, J. Verhoef, and J. A. Van Strijp. 1992. Direct evidence for antibody bipolar bridging on herpes simplex virus-infected cells. Immunology 77109-115. [PMC free article] [PubMed] [Google Scholar]
- 52.Walker, M. R., J. Lund, K. M. Thompson, and R. Jefferis. 1989. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing Fc gamma RI and/or Fc gamma RII receptors. Biochem. J. 259347-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westmoreland, D., S. St Jeor, and F. Rapp. 1976. The development by cytomegalovirus-infected cells of binding affinity for normal human immunoglobulin. J. Immunol. 1161566-1570. [PubMed] [Google Scholar]
- 54.Westmoreland, D., and J. F. Watkins. 1974. The IgG receptor induced by herpes simplex virus: studies using radioiodinated IgG. J. Gen. Virol. 24167-178. [DOI] [PubMed] [Google Scholar]
- 55.Wiger, D., and T. E. Michaelsen. 1985. Binding site and subclass specificity of the herpes simplex virus type 1-induced Fc receptor. Immunology 54565-572. [PMC free article] [PubMed] [Google Scholar]
- 56.Yamaguchi, Y., M. Nishimura, M. Nagano, H. Yagi, H. Sasakawa, K. Uchida, K. Shitara, and K. Kato. 2006. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim. Biophys. Acta 1760693-700. [DOI] [PubMed] [Google Scholar]
- 57.Zhang, Y., C. C. Boesen, S. Radaev, A. G. Brooks, W. H. Fridman, C. Sautes-Fridman, and P. D. Sun. 2000. Crystal structure of the extracellular domain of a human Fc gamma RIII. Immunity 13387-395. [DOI] [PubMed] [Google Scholar]