

Abstract
We aimed to identify genomic markers in hepatitis B virus (HBV) that are associated with hepatocellular carcinoma (HCC) development by comparing the complete genomic sequences of HBVs among patients with HCC and those without. One hundred patients with HBV-related HCC and 100 age-matched HBV-infected non-HCC patients (controls) were studied. HBV DNA from serum was directly sequenced to study the whole viral genome. Data mining and rule learning were employed to develop diagnostic algorithms. An independent cohort of 132 cases (43 HCC and 89 non-HCC) was used to validate the accuracy of these algorithms. Among the 100 cases of HCC, 37 had genotype B (all subgenotype Ba) and 63 had genotype C (16 subgenotype Ce and 47 subgenotype Cs) HBV infection. In the control group, 51 had genotype B and 49 had genotype C (10 subgenotype Ce and 39 subgenotype Cs) HBV infection. Genomic algorithms associated with HCC were derived based on genotype/subgenotype-specific mutations. In genotype B HBV, mutations C1165T, A1762T and G1764A, T2712C/A/G, and A/T2525C were associated with HCC. HCC-related mutations T31C, T53C, and A1499G were associated with HBV subgenotype Ce, and mutations G1613A, G1899A, T2170C/G, and T2441C were associated with HBV subgenotype Cs. Amino acid changes caused by these mutations were found in the X, envelope, and precore/core regions in association with HBV genotype B, Ce, and Cs, respectively. In conclusion, infections with different genotypes of HBV (B, Ce, and Cs) carry different genomic markers for HCC at different parts of the HBV genome. Different HBV genotypes may have different virologic mechanisms of hepatocarcinogenesis.
Chronic infection by the hepatitis B virus (HBV) causes an increased risk of hepatocellular carcinoma (HCC) of more than 100-fold (2). The relationship between HBV genotype and viral mutation with hepatocarcinogenesis is controversial. A case-control study from Taiwan suggested that genotype C HBV is more closely associated with cirrhosis and HCC in those who are older than 50 years, whereas genotype B is more common in patients with HCC who are less than 50 years old (18). Our previous cohort study of 426 cases of chronic hepatitis B patients also revealed a higher risk of HCC and liver cirrhosis in genotype C infection (5). On the other hand, reports from Japan and China did not confirm the higher malignant potential of genotype C HBV (27, 33).
Recently studies reported that the prevalence of basal core promoter mutants (A1762T and G1764A) is associated with more aggressive progression of liver disease and development of HCC (19, 30, 33). Several HBV genes, including truncated pre-S2/S and X genes, have been found in hepatoma tissue (15, 16, 23). Another hot spot mutation in the core promoter region is the G1896A and G1899A mutation (12, 30). HBV DNA integration into the host genome may allow persistence of the viral genome in the host and alteration of cell kinetics and cellular metabolism (3, 4, 29). Whether certain mutations of the HBV genes facilitate the integration of the viral genome and virus-host interaction is not known.
Two major reasons for discrepant results from various studies are (i) the small numbers of patients involved in these studies and (ii) the fact that most studies focus on a particular portion of the HBV genome (22). The aim of the present study was to identify markers in the HBV genome for HCC development by studying the complete genomic sequence of HBV among patients with HCC compared to age-matched individuals presenting with the infection but no HCC development.
(Part of this work has been presented at Digestive Diseases Week, 14 to 19 May 2005, Chicago, IL.)
MATERIALS AND METHODS
Patients.
We conducted a case control-study of 100 patients with HBV-related HCC and 100 age-matched HBV-infected patients as controls. All patients who presented with HBV-related HCC to the Joint Hepatoma Clinic, Prince of Wales Hospital, Hong Kong, from July 1999 to December 2000 were studied. HCC was diagnosed by histology or a combination of ultrasonography, computerized tomography or magnetic resonance imaging, and/or hepatic angiography. Age-matched patients with no evidence of HCC in the control group were selected from a cohort of chronic hepatitis B patients recruited from the liver clinics of the same hospital (5). The control cohort was recruited in a similar time frame as the cases (from December 1997 to July 2000). They were prospectively followed up until June 2003 with regular ultrasound and alfa-fetoprotein surveillance to confirm the absence of HCC. Serum samples from all patients were stored at −80°C. Informed consent was obtained from all patients, and ethics committee approval was obtained. An independent cohort of patients with known HBV infection (HBsAg positive) with or without HCC was studied to validate the findings of the case-control study.
DNA extraction, amplification, sequencing, and determination of genotype.
HBV DNA was extracted from 100 μl of serum using the QIAamp DNA blood mini kit (Qiagen GmBH, Hilden, Germany) according to the manufacturer's instructions. To obtain the full-length HBV DNA sequence, we performed seminested PCR to amplify three overlapping fragments of the HBV genome. For each fragment, 5 μl of the extracted DNA was used with Taq DNA polymerase (Amersham Biosciences, Uppsala, Sweden) and Pfu DNA polymerase (Promega, Madison, WI) in the first-round PCR and with Taq DNA polymerase alone in the second-round PCR. The final PCR product was examined on a 1.0% agarose-ethidium bromide gel run in 1× Tris-borate-EDTA buffer.
For fragment A, PCR was carried out with P1 and P2 primers with a 5-min initial denaturation at 95°C, followed by 10 cycles of amplification (94°C for 36 s, 60°C for 36 s, and 72°C for 2.5 min), then 30 cycles of amplification (94°C for 36 s, 50°C for 36 s, and 72°C for 2.5 min), and a 7-min final extension at 72°C. The sequences of all primers used for PCR and sequencing in this study are shown in Table 1. The PCR product was further amplified in a seminested PCR with P1 and P3. PCR was carried out with a 5-min initial denaturation at 95°C, followed by 10 cycles of amplification (94°C for 36 s, 60°C for 36 s, and 72°C for 2 min), then 30 cycles of amplification (94°C for 36 s, 52°C for 36 s, and 72°C for 2 min), and a 7-min final extension at 72°C. For fragment B, PCR was carried out with the P4 and P5 primers, and the PCR product was further amplified in a seminested PCR with the P5 and P6 primers. For fragment C, PCR was carried out with the P7 and P9 primers. The PCR product was further amplified in a seminested PCR with the P8 and P9 primers. Both strands of PCR products were directly sequenced with the DYEnamic ET Dye Terminator cycling sequencing kit for MegaBACE (Amersham Biosciences, Piscataway, NJ).
TABLE 1.
Primers used for PCR amplification and sequencing of the HBV genome
| Function and primer | Nucleotide sequence (5′→3′) | Position | Direction |
|---|---|---|---|
| PCR | |||
| P1 | TTTTTCACCTCTGCCTAATCA | 1821-1841 | Sense |
| P2 | CCCTAGAAAATTGAGAGAAGTC | 262-283 | Antisense |
| P3a | CCACTGCATGGCCTGAGGATG | 3193-3213 | Antisense |
| P4 | GCCTCATTTTGTGGGTCACCATA | 2801-2824 | Sense |
| P5 | TTCTTTGACATACTTTCCA | 979-997 | Antisense |
| P6a | TTGGGGTGGAGCCCTCAGGCT | 3070-3090 | Sense |
| P7a | TTGGCCAAAATTCGCAGTC | 300-318 | Sense |
| P8a | CCCCACTGTTTGGCTTTCAG | 714-734 | Sense |
| P9a | GTTGATAAGATAGGGGCATTTGGTGG | 2299-2325 | Antisense |
| Sequencing | |||
| S1 | CTCCGGAACATTGTTCACCT | 2031-2050 | Sense |
| S2 | AAGGTGGGAAACTTTACTGGGC | 2469-2490 | Sense |
| S3 | GCTGACGCAACCCCCACTGG | 1186-1205 | Sense |
| S4 | TCGCATGGAGACCACCGTGA | 1604-1623 | Sense |
| S5 | GGCAAAAACGAGAGTAACTC | 1940-1959 | Antisense |
| S6 | GGGTCGTCCGCGGGATTCAG | 1441-1460 | Antisense |
| S7 | GACATACTTTCCAATCAATAGG | 970-991 | Antisense |
| S8 | GAAGATGAGGCATAGCAGCAGG | 411-433 | Antisense |
| S9 | CATGCTGTAGCTCTTGTTCC | 2831-2850 | Antisense |
Primer also used for sequencing.
Molecular evolutionary analyses.
All HBV genomic sequences in this study and typical genome sequences of different genotypes were multiply aligned using CLUSTALW (28) version 1.83 and corrected manually by visual inspection. The numbering of HBV nucleotides started at the EcoRI cleavage site. Genetic distances were estimated by Kimura's two-parameter method (20), and the phylogenetic trees were constructed by the neighbor-joining method (24). The reliability of the pairwise comparison and phylogenetic tree analysis was assessed by bootstrap resampling (13) with 1,000 replicates. Phylogenetic and molecular evolutionary analyses were done using MEGA version 3.0 (21). HBV genotypes and subgenotypes were determined by comparison with 122 full genome sequences downloaded from GenBank.
Data mining framework.
The data mining framework is shown in Fig. 1. The process involved seven modules. After the molecular evolutionary analyses, the data were passed to the clustering module to check whether clusters existed, based on the phylogenetic tree analysis. These clusters are possible genotypes or subgroups possessing differences in some nucleotides which do not have any effects on the classification of HCC. If clusters were found, each cluster was analyzed separately for potential genetic marker sites. While genotype B HBV appeared to be a homogenous group, the phylogenetic tree results showed that there exist two subgroups (clusters) in genotype C among the HBV strains collected (Fig. 2) (9). All three (sub)groups (B, Cs, and Ce) were analyzed separately in the learning and classification parts.
FIG. 1.

Flow diagram for the data mining and classification process.
FIG. 2.

Phylogenetic tree of the full-genome sequencing of HBV in the case-control study. All patients were infected with either genotype B or C HBV. Two subgenotypes (Ce and Cs) could be identified in genotype C HBV due to a more than 4% difference in the entire HBV sequence.
For each cluster, the data were divided into training and testing sets. The training samples were then passed to the feature selection module to find the useful features (potential marker sites) for classification. The feature selection was based on the information gains of each aligned site. The details of the information gain calculation are given in the appendix. The main purpose of feature selection was to reduce the number of features used in classification while maintaining acceptable classification accuracy. The sites were then ranked according to their respective information gains, which can reflect their potentials to distinguish between the control and the HCC groups. The ranked and computed information gain of each aligned site can be displayed with the aligned sequences by our viewer tools. The top 10, 20, 30, 40, and 50 ranked (information gain) sites were included as the selected features for the classifier learning module and preprocessing modules in turn to see which one gave the best result.
The selected features were extracted and passed to the classifier learning module, wherein a rule-based classifier was learned. Rule learning tries to learn rules from a set of training data (samples). It can be modeled as a search problem of finding the best rules that classify the training examples with minimum classification error. Generic genetic programming (31), which is a type of evolutionary algorithm (1), was adopted as our search and optimization algorithm to learn the rules. The testing data were then transferred to the preprocessing module with the marker sites selected by the feature selection module. The testing data were preprocessed, and only the part relevant to the selected sites was kept. This part of the testing data was then used for prediction evaluation in the classification module.
Prediction results were output from the classification module. They were then verified by the actual classes given in the testing samples. If the verification results were unsatisfactory, the process was repeated, starting from the features selection.
In the final validation module, when a reasonable classifier was obtained, the classifier could be further validated by testing with previously unanalyzed validation samples.
Statistical analysis.
In the case-control study with 100 HCC cases and 100 non-HCC age-matched controls, 90% of the samples were selected randomly as the training set and the remaining 10% formed the testing set in each experiment. For each data set, the experiment was repeated 10 times by picking different training sets. For each learning and evaluation experiment, sensitivity and specificity as defined below were estimated as the fitness or performance indicators of the classification rules. The average sensitivity and specificity of the testing set in the case-control study and of the validation cohort were determined. The 95% confidence intervals (CIs) of the sensitivity and specificity as well as the likelihood ratios were determined based on the performance of the algorithms on the entire data set. The odds ratios (ORs) and 95% CIs for HCC among patients with different numbers of HCC-related mutations were also calculated. When any zero cell occurred in the two-by-two contingency table, we added 0.5, based on the Haldane correction (14), to all of the cells in the calculation of ORs and 95% CIs. The statistical significance was examined at the conventional level of 0.05 by analysis of variance, the chi-square test, or Fisher's exact test as appropriate.
RESULTS
Case-control study.
The demographic characteristics, clinical diagnoses, and HBV genotypes of the HCC and control groups are listed in Table 2. Among the 100 cases of HCC, there were 37 cases who had genotype B HBV and 63 cases who had genotype C HBV. In the control group, 51 cases were infected with HBV genotype B HBV and 49 cases with genotype C HBV. There was a significant male preponderance in both the HCC and control groups for both genotypes. Sixty-seven percent of cases in the HCC group had cirrhosis, compared to only 13% in the control group. The percentages of patients with cirrhosis in the genotype B subgroup (35.1%) and genotype C subgroup (31.7%) were quite similar.
TABLE 2.
Demographic characteristics, clinical diagnoses, HBeAg/anti-HBe status, and genotypes of HBV for the HCC and control groups
| Parameter (unit) | Valuea for the indicated group and HBV genotype (n)
|
|||||
|---|---|---|---|---|---|---|
| HCC (100)
|
Non-HCC (control) (100)
|
|||||
| B (37) | Ce (16) | Cs (47) | B (51) | Ce (10) | Cs (39) | |
| Age (yr) | 51 ± 14 | 54 ± 14 | 50 ± 11 | 52 ± 13 | 48 ± 10 | 47 ± 13 |
| Males | 35 (95) | 13 (81) | 39 (83) | 42 (82) | 6 (60) | 25 (64) |
| Hemoglobin (g/dl) | 12.8 ± 1.6 | 12.8 ± 1.8 | 13.0 ± 2.7 | 14.9 ± 1.0 | 14.0 ± 2.0 | 15.0 ± 1.2 |
| White cells (109/liter) | 7.7 ± 3.2 | 6.3 ± 3.4 | 6.6 ± 2.6 | 6.5 ± 2.0 | 6.0 ± 2.0 | 7.0 ± 2.6 |
| Platelet (109/liter) | 202 ± 117 | 201 ± 128 | 204 ± 96 | 166 ± 60 | 153 ± 68 | 163 ± 60 |
| International normalized ratio | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.2 | 1.0 ± 0.1 |
| Creatinine (mmol/liter) | 89 ± 23 | 92 ± 17 | 91 ± 20 | 89 ± 20 | 77 ± 17 | 82 ± 18 |
| Albumin (g/liter) | 33 ± 6 | 32 ± 4 | 33 ± 5 | 38 ± 4 | 37 ± 4 | 38 ± 5 |
| Alanine transaminase (IU/liter) | 103 ± 90 | 82 ± 54 | 101 ± 102 | 94 ± 36 | 81 ± 21 | 98 ± 49 |
| Alkaline phosphatase (IU/liter) | 227 ± 124 | 239 ± 154 | 212 ± 145 | 94 ± 36 | 81 ± 21 | 98 ± 49 |
| Bilirubin (μmol/liter) | 32 ± 58 | 30 ± 23 | 27 ± 48 | 10 ± 7 | 12 ± 9 | 12 ± 13 |
| Liver cirrhosis present | 24 (65) | 14 (88) | 29 (62) | 6 (11) | 2 (20) | 6 (15) |
| Anti-HCV present | 0 | 0 | 1 | 0 | 0 | 0 |
Continuous variables are expressed as means ± standard deviations. Categorical variables are expressed as number (percentage).
Validation cohort.
The validation samples came from a serum bank of patients with known HBV infection (HBsAg positive) with or without HCC. This was an independent cohort of 132 cases, including 43 patients with HCC (18 patients with genotype B and 25 with genotype C HBV infection) and 89 non-HCC subjects (41 with genotype B and 48 with genotype C HBV infection). There was no overlap between this validation cohort and the test cohort described above.
Subgenotype prevalence in subjects.
Genotype B HBV appeared to be a homogenous group, and all belonged to subgenotype Ba (26). However, the phylogenetic tree results showed that there existed two subgroups, namely, Ce (found predominantly in East Asia) and Cs (found predominantly in Southeast Asia), in genotype C among the HBV strains collected (Fig. 2). This is in concordance with our previous phylogeny with published full-length sequences in GenBank (9).
The clinical characteristics of patients with genotype B and subgenotypes of genotype C are shown in Table 2. No significant difference in age (P = 0.46), gender (P = 0.06), or presence of HCC (P = 0.11) was observed between patients with genotype B and subgenotype C. The proportions of cirrhosis in HCC patients with HBV genotype B, subgenotype Ce, and subgenotype Cs were 65%, 88%, and 62%, respectively. The risk of cirrhosis and HCC for subgenotype Ce was higher than for the others, but this result did not show a statistically significant difference (P = 0.16%). These percentages of cirrhosis were much higher than the proportions of cirrhosis in control patients with HBV genotype B (P < 0.001), subgenotype Ce (P < 0.001), or subgenotype Cs (P < 0.001).
HCC-related mutations.
Among HCC patients with genotype B HBV, mutations in the following sites were commonly found: A1762T (81.1%) and G1764A (81.1%), C1165T (18.9%), T2712C/A/G (70.3%), and A/T2525C (21.6%). The mutations at these nucleotide positions in the HCC and control groups are shown in Table 3. In the group with HBV subgenotype Ce, the mutations T31C (37.5%), T53C (37.5%), and A1499G (62.5%) were associated with HCC development: (Table 3). In the group with HBV subgenotype Cs, the mutations G1613A (38.3%), G1899A (27.7%), T2170C/G (34.0%), and T2441C (21.3%) were associated with HCC development (Table 3). Combining the patients from the case-control study and the independent validation cohort, the presence of an increasing number of HCC-related mutations in each HBV genotype/subtype was associated with an increased risk of HCC (Table 4). All mutations associated with HCC development had amino acid changes in at least one of the four open reading frame of HBV (Table 5). Amino acid changes in the X region were found only in genotype B HBV. Envelope region amino acid changes were found in HBV subgenotype Ce, whereas precore/core region amino acid changes were found in HBV subgenotype Cs.
TABLE 3.
Mutations in different genotypes associated with HCC development in the case-control study (100 patients with HCC and 100 control patients)
| Genotype | Nucleotide
|
% in group
|
|||||
|---|---|---|---|---|---|---|---|
| Position | Wild type | Mutant | Control
|
HCC
|
|||
| Wild type | Mutant | Wild type | Mutant | ||||
| B | 1762 | A | T | 57.1 | 42.9 | 18.9 | 81.1 |
| 1764 | G | A | 57.1 | 42.9 | 18.9 | 81.1 | |
| 1165 | C | T | 93.9 | 6.1 | 81.1 | 18.9 | |
| 2712 | T | C/A/G | 63.3 | 36.7 | 29.7 | 70.3 | |
| 2525 | A, T | C | 87.8 | 12.2 | 78.4 | 21.6 | |
| Ce | 31 | T | C | 100 | 0 | 62.5 | 37.5 |
| 53 | T | C | 90 | 10 | 62.5 | 37.5 | |
| 1499 | A | G | 80 | 20 | 37.5 | 62.5 | |
| Cs | 1613 | G | A | 83.8 | 16.2 | 61.7 | 38.3 |
| 1899 | G | A | 97.3 | 2.7 | 62.3 | 27.7 | |
| 2170 | T | C/G | 91.9 | 8.1 | 66.0 | 34.0 | |
| 2441 | T | C | 100.0 | 0.0 | 78.7 | 21.3 | |
TABLE 4.
ORs for HCC with different number of mutations in different HBV genotypes/subtypes
| Genotype and no. of mutations | Case-control study
|
Validation cohort
|
||||||
|---|---|---|---|---|---|---|---|---|
| No. in group
|
OR (95% CI) | P | No. in group
|
OR (95% CI) | P | |||
| HCC | Non-HCC | HCC | Non-HCC | |||||
| B | ||||||||
| 0 | 3 | 16 | Referent | 1 | 23 | Referent | ||
| 1 | 0 | 6 | 0.36 (0.02-8.04)a | 0.55 | 1 | 2 | 11.50 (0.50-261.97) | 1.00 |
| 2 | 7 | 13 | 2.87 (0.62-13.37) | 0.27 | 7 | 5 | 32.20 (3.20-323.67) | 0.0006 |
| 3 | 21 | 12 | 9.33 (2.25-38.71) | 0.001 | 7 | 10 | 16.10 (1.74-148.68) | <0.0001 |
| 4 | 6 | 4 | 8.00 (1.37-46.81) | 0.03 | 1 | 1 | 13.65 (2.31-84.43) | 0.0009 |
| 5 | 0 | 0 | Undefined | 1 | 0 | 47.00 (1.28-1722.22)a | 0.11 | |
| Ce | ||||||||
| 0 | 3 | 7 | Referent | 0 | 7 | Referent | ||
| 1 | 6 | 3 | 4.67 (0.67-32.36) | 0.18 | 4 | 0 | 135.00 (2.26-8069.03)a | 0.001 |
| 2 | 5 | 0 | 23.57 (1.00-556.11)a | 0.03 | 3 | 1 | 35.00 (1.12-1094.80)a | 0.0008 |
| 3 | 2 | 0 | 10.71 (0.40-287.84) | 0.15 | 1 | 0 | 45.00 (0.61-3297.13)a | 0.02 |
| Cs | ||||||||
| 0 | 12 | 29 | Referent | 2 | 27 | Referent | ||
| 1 | 18 | 9 | 4.83 (1.70-13.75) | 0.002 | 7 | 9 | 10.50 (1.84-60.01) | <0.0001 |
| 2 | 12 | 1 | 29.00 (3.38-248.49) | <0.0001 | 3 | 4 | 10.13 (1.27-80.61) | <0.0001 |
| 3 | 5 | 0 | 25.96 (1.33-505.87)a | 0.005 | 4 | 0 | 99.00 (4.05-2418.57)a | <0.0001 |
| 4 | 0 | 0 | Undefined | 0 | 0 | Undefined | ||
Haldane's approximation was used for estimation of the OR and 95% CI.
TABLE 5.
Amino acid mutations in different genotypes associated with HCC development
| Genotype | Nucleotide
|
Amino acid in region:
|
|||||
|---|---|---|---|---|---|---|---|
| Position | Wild type | Mutant | Surface | Polymerase | Precore/core | X | |
| B | 1165 | C | T | RNase H, P2S | |||
| 1762 | A | T | K130M | ||||
| 1764 | G | A | V131I | ||||
| 2525 | A, T | C | TP, K/N73N | ||||
| 2712 | T | C/A/G | TP, Y136H/N/D | ||||
| Ce | 31 | T | C | Pre-S1, D134 (no change) | Spacer, S137P | ||
| 53 | T | C | Pre-S1, F141L | Spacer, L144P | |||
| 1499 | A | G | RNase H, H113R | P43 (no change) | |||
| Cs | 1613 | G | A | RNase H, R151K | E80 (no change) | ||
| 1899 | G | A | Precore, G29D | ||||
| 2170 | T | C/G | Core, N90N/K | ||||
| 2441 | T | C | N45 (no change) | Core, S181P | |||
Diagnostic algorithm.
Using the method of rule learning using evolutionary algorithms, the following algorithms for risk estimation was established. The classification rules for genotype B were as follows:
IF A1762G1764 and T1165, then HCC
IF T1762A1764 and ACG2712, then HCC
IF T1762A1764 and T2712 and C2525, then HCC
ELSE, non-HCC.
Using this algorithm, the sensitivity (95% CI) and specificity (95% CI) of diagnosing HCC in the testing cohort were 0.75 (0.61 to 0.89) and 0.66 (0.53 to 0.79), respectively, and those in the validation cohort were 0.72 (0.51 to 0.93) and 0.73 (0.59 to 0.87), respectively. The positive and negative likelihood ratios (95% CIs) for the performance of the algorithm in the testing cohort were 2.21 (1.27 to 3.14) and 0.38 (0.15 to 0.60), respectively, and those in the validation cohort were 2.67 (1.12 to 4.21) and 0.38 (0.09 to 0.68), respectively.
The classification rules for the Ce cluster of genotype C were as follows:
IF C31 OR C53 OR G1499, then HCC
ELSE, non-HCC.
Using this algorithm, the sensitivity (95% CI) and specificity (95% CI) of diagnosing HCC in the testing cohort were 0.75 (0.54 to 0.96) and 0.70 (0.42 to 0.98), respectively, and those in the validation cohort were 1.00 (not available) and 0.75 (0.47 to 1.00), respectively. The positive and negative likelihood ratios (95% CIs) for the performance of the algorithm in the testing cohort were 2.50 (0.03 to 4.97) and 0.36 (0.02 to 0.69), respectively, and those in the validation cohort were 4.00 (0.00 to 8.53) and 0.00 (not available), respectively.
The classification rules for the Cs cluster of genotype C were as follows:
IF A1613 OR A1899 OR CG2170 OR C2441, then HCC
ELSE, control.
Using this algorithm, the sensitivity (95% CI) and specificity (95% CI) of diagnosing HCC in the testing cohort were 0.72 (0.59 to 0.85), 0.72 (0.58 to 0.86), respectively, and those in the validation cohort were 0.88 (0.73 to 1.00) and 0.63 (0.48 to 0.78), respectively. The positive and negative likelihood ratios (95% CIs) for the performance of the algorithm in the testing cohort were 2.57 (1.20 to 3.94) and 0.39 (0.20 to 0.58), respectively, and those in the validation cohort were 2.38 (1.32 to 3.44) and 0.19 (0.00 to 0.44), respectively.
DISCUSSION
In this study, we demonstrated that certain genotypes (and subgenotypes) and mutations are associated with development of hepatic carcinogenesis. There seems to be a stratified risk of HCC, with each genotype (or subgenotype) being associated with a certain pattern of mutations. The significance of these genotypes and mutations was verified by use of an independent cohort which was composed of both HCC and non-HCC patients. Using these algorithms, the sensitivity of identifying a high-risk case ranged from 72% to 75% and the specificity ranged from 66% to 72%. Although the use of these algorithms had only moderate discriminatory capability to predict HCC (positive likelihood ratio of 2.21 to 2.57 and negative likelihood ratio of 0.36 to 0.39), our data suggested that different HBV genotypes and subgenotypes might have different predominant carcinogenic mechanisms.
The issue of HBV genotypes has been debated due to discrepant results in previous studies from different countries (18, 27). These differences may be explained by a distinct distribution of HBV subgenotypes in different geographical regions. In most Asian countries, only subgroup Ba of HBV is found, while the majority of Japanese patients with HBV have subgroup Bj (9). Genotype C HBV has a higher risk of HCC than genotype B HBV, which is probably related to a delayed HBeAg seroconversion, more active hepatitis, and a higher prevalence of basal core promoter mutations (5, 19, 32). Among genotype C HBV, there were also differences in the disease activity associated with different subgenotypes (6). Recently, we have shown that subgenotype Ce HBV was associated with the highest risk of HCC independent of other risk factors, including high HBV DNA levels and liver cirrhosis, among a longitudinal cohort of 1,006 chronic hepatitis B patients followed up for 7.7 years (10). The proportion of HCC in patients with subtype adw was found to be higher than that in patients with subtype adr (25). Going beyond attributing HCC to a specific genotype, this study suggests that different genotypes of HBV are associated with different mutations of the viral genome and thus may have separate mechanisms of hepatic carcinogenesis.
The basal core promoter mutant (T1762/A1764) is found to parallel the progression of liver disease and increases the risk of HCC for both genotype B and C HBV (19, 33). In common with previous studies, we also found mutation at codon 1762/1764 to be associated with HCC in genotype B HBV infection. The reason why 1762/1764 mutations were not identified as a marker for HCC in genotype C HBV was related to the high prevalence of mutations at these sites even among the non-HCC patients (8). However, this phenomenon may also mean that a selection pressure on the basal core promoter/X region of the HBV genome in genotype B HBV is associated with the development of HCC. The HCC-associated mutations selected by HBV subgenotype Ce are located in the envelope region, while those selected by HBV subgenotype Cs are located in the precore/core region. These findings offer additional support for the presence of various virologic mechanisms of hepatocarcinogenesis by different HBV genotypes/subgenotypes. The functions of these mutations and their gene products need further investigation.
HBV DNA appears to integrate into host DNA at different sites, exerting direct and indirect effects on the host genes (7). It has also been postulated that the integrated HBV genes can activate cellular genes remote from the site of HBV DNA integration, thereby influencing cellular proliferation and differentiation. This transactivation effect could be mediated through different signal transduction pathways. Identification of HCC-related mutations is only the first step in understanding the viral mechanism of hepatic carcinogenesis. Functional genomic studies of these mutations would have to be carried out in the future to elucidate the effects of these mutations on cell growth and death of hepatocytes.
There are several limitations in this study. First, although patients in the control group were age matched with those in the HCC group, the possibility of developing malignancy in the future cannot be denied. As there is no matching in the disease severity and liver cirrhosis, the HCC-related mutations may have an indirect effect on HCC development through increasing hepatic inflammation and liver cirrhosis. When the algorithms were tested with the independent validation cohort, a very high sensitivity and a satisfactory specificity were reported for both genotype B and C subgenotypes. Second, although this is by far the largest cohort of HCC and non-HCC cases to have full-length viral genomic analysis of HBV compared to previous studies (17, 22), the sample size is still relatively small. The 95% CIs for the sensitivity and specificity of the genomic algorithms are still wide. In the future, laboratory methods to detect these mutants in a more robust manner than does full-genome sequencing are needed to facilitate a larger-scale validation study. A larger cohort, preferably from a different geographic location, would also be needed to validate the generalization of our results. Third, we can only study patients with genotype B and subgenotypes Ce and Cs of HBV. We cannot study genotype A HBV and genotype D HBV, which are prevalent in Europe and Africa, because of our geographic limitations. Moreover, as most Hong Kong residents are immigrants from China, we did not have the information on the place where the ancestors of the patients acquired the infection. We believe that most of our patients originated from southern China, where HBV subgenotype Cs is more prevalent than subgenotype Ce. However, the methodology adopted in this study could be used in countries with other HBV genotypes for mining of HBV-related mutations. Finally, we have not worked out the functionality of these mutated codons and why they might lead to development of HCC. More work is required to elucidate the virologic and host responses to mutations. We cannot draw a conclusion on the causal relationship between these HBV mutations and HCC.
In conclusion, this study suggests that HBV genotypes B and C demonstrate different point mutations which might be associated with high risk of hepatic carcinogenesis. The difference in the locations of these mutations in the HBV genome may reflect the underlying mechanisms of hepatocarcinogenesis of the different HBV genotype/subgenotypes. The detection of these mutations has shown promising results in the association with a higher cancer risk. By combining this information with other clinical risk factors for HCC, including HBV DNA levels and liver cirrhosis status (10, 11), future clinical algorithms can be refined. It is possible that these diagnostic algorithms may shed light on which patients with chronic HBV infection require more frequent screening and surveillance for HCC development.
APPENDIX
The information gain of a feature (attribute) is the reduction in uncertainty (entropy) that results if the attribute is used for classification. Hence, the higher the information gain, the better. The following equation gives the entropy, E, of an attribute X with n values, X1 … Xn, where P(Xj) is the frequency of the value Xj: E(X) = 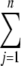 −P(Xj)log2P(Xj).
−P(Xj)log2P(Xj).
Specific to a typical DNA classification problem, we assumed that the data had M classes, C1 … CM. For each aligned site position, it has N possible nucleotides, V1 … VN. We defined Cm as the number of sequences in class Cm. Cmi is the number of sequences in class Cm whose character at the aligned site is Vi, which could be A, T, G, or C in our case. The remainder of X, R(X) was defined as follows:
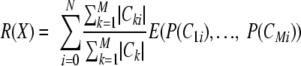 |
The information gain, IGj, of the aligned site j is the difference between the original information content E(C) of the data set and the amount of information needed to classify all the unclassified data left in the data set after applying site j for classification: IGj = E(C) − R(j).
The features were ranked by the information gains, and then the top-ranked features were chosen for classification. A site with higher information gain would contribute more discriminatory power to the classification such that more samples could be distinguished by this site.
Acknowledgments
This study was supported by a grant from the Innovation and Technology Fund (ITS/188/01) of the Hong Kong SAR to J.J.Y.S.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print on 23 January 2008.
REFERENCES
- 1.Angeline, P. 1993. Evolutionary algorithms and emergent intelligence. Ph.D. dissertation. The Ohio State University, Columbus, OH.
- 2.Beasley, R. P., L. Y. Hwang, C. C. Lin, and C. S. Chien. 1981. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet ii1129-1133. [DOI] [PubMed] [Google Scholar]
- 3.Brechot, C. 2004. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology 127S56-61. [DOI] [PubMed] [Google Scholar]
- 4.Chami, M., D. Ferrari, P. Nicotera, P. Paterlini-Brechot, and R. Rizzuto. 2003. Caspase-dependent alterations of Ca2− signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 27831745-31755. [DOI] [PubMed] [Google Scholar]
- 5.Chan, H. L. Y., A. Y. Hui, M. L. Wong, A. M. L. Tse, L. C. T. Hung, V. W. S. Wong, and J. J. Y. Sung. 2004. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 531494-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan, H. L. Y., C. H. Tse, E. Y. T. Ng, K. S. Leung, K. H. Lee, S. K. W. Tsui, and J. J. Y. Sung. 2006. Phylogenetic, virological and clinical characteristics of genotype C hepatitis B virus with TCC at codon 15 of the precore region. J. Clin. Microbiol. 44681-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan, H. L. Y., and J. J. Y. Sung. 2006. Hepatocellular carcinoma and hepatitis B virus. Semin. Liver Dis. 26153-161. [DOI] [PubMed] [Google Scholar]
- 8.Chan, H. L. Y., N. W. Y. Leung, M. Hussain, M. L. Wong, and A. S. F. Lok. 2000. Hepatitis B e antigen-negative chronic hepatitis B in Hong Kong. Hepatology 31763-768. [DOI] [PubMed] [Google Scholar]
- 9.Chan, H. L. Y., S. K. W. Tsui, C. H. Tse, E. Y. T. Ng, T. C. C. Au, L. Yuen, A. Bartholomeusz, K. S. Leung, K. H. Lee, S. Locarnini, and J. J. Y. Sung. 2005. Epidemiological and virological characteristics of two subgroups of genotype C hepatitis C virus. J. Infect. Dis. 1912022-2032. [DOI] [PubMed] [Google Scholar]
- 10.Chan, H. L. Y., C. H. Tse, F. Mo, J. Koh, V. W. S. Wong, G. L. H. Wong, S. L. Chan, W. Yeo, J. J. Y. Sung, and T. S. K. Mok. 2008. High viral load and hepatitis B virus subgenotype Ce are associated with increased risk of hepatocellular carcinoma. J. Clin. Oncol. 26177-182. [DOI] [PubMed] [Google Scholar]
- 11.Chen, C. J., H. I. Yang, J. Su, C. L. Jen, S. L. You, S. N. Lu, G. T. Huang, and U. H. Iloeje. 2006. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 29565-73. [DOI] [PubMed] [Google Scholar]
- 12.Cho, S. W., Y. J. Shin, K. B. Hahm, J. H. Jin, Y. S. Kim, J. H. Kim, and H. J. Kim. 1999. Analysis of the precore and core promoter DNA sequence in liver tissues from patients with hepatocellular carcinoma. J. Korean Med. Sci. 14424-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39783-791. [DOI] [PubMed] [Google Scholar]
- 14.Haldane, J. B. S. 1956. The estimation and significance of the logarithm of a ratio of frequencies. Ann. Hum. Genet. 20309-311. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh, Y. T., I. J. Su, H. C. Wang, W. W. Chang, H. Y. Lei, M. D. Lai, W. T. Chang, and W. Huang. 2004. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 252023-2032. [DOI] [PubMed] [Google Scholar]
- 16.Hwang, G. Y., C. Y. Lin, L. M. Huang, Y. H. Wang, J. C. Wang, C. T. Hsu, S. S. Yang, and C. C. Wu. 2003. Detection of the hepatitis B virus X protein (HBx) antigen and anti-HBx antibodies in cases of human hepatocellular carcinoma. J. Clin. Microbiol. 415598-5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajiya, Y., K. Hamasaki, K. Nakata, Y. Nakagawa, S. Miyazoe, Y. Takeda, K. Ohkubo, T. Ichikawa, K. Nakao, Y. Kato, and K. Eguchi. 2002. Full-length sequence and functional analysis of hepatitis B virus genome in a virus carrier: a case report suggesting the impact of pre-S and core promoter mutations on the progression of the disease. J. Viral Hepat. 9149-156. [DOI] [PubMed] [Google Scholar]
- 18.Kao, J. H., P. J. Chen, M. Y. Lai, and D. S. Chen. 2000. Hepatitis B genotypes correlate with clinical outcome in patients with chronic hepatitis B. Gastroenterology 118554-559. [DOI] [PubMed] [Google Scholar]
- 19.Kao, J. H., P. J. Chen, M. Y. Lai, and D. S. Chen. 2003. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carrier. Gastroenterology 124327-334. [DOI] [PubMed] [Google Scholar]
- 20.Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 15111-120. [DOI] [PubMed] [Google Scholar]
- 21.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings Bioinformatics 5150-163. [DOI] [PubMed] [Google Scholar]
- 22.Lin, X., G. S. Qian, P. X. Lu, L. Wu, and Y. M. Wen. 2001. Full-length genomic analysis of hepatitis B virus isolates in a patient progressing from hepatitis to hepatcellular carcinoma. J. Med. Virol. 64299-304. [PubMed] [Google Scholar]
- 23.Oon, C. J., W. N. Chen, K. T. Goh, S. Mesenas, H. S. Ng, G. Chiang, C. Tan, S. Koh, S. W. Teng, I. Toh, M. C. Moh, K. S. Goo, K. Tan, A. L. Leong, and G. S. Tan. 2002. Molecular characterization of hepatitis B virus surface antigen mutant in Singapore patients with hepatocellular carcinoma and hepatitis B virus carrier negative for HBsAg but positive for anti-HBs and anti-HBc. J. Gastroenterol. Hepatol. 17S491-S496. [DOI] [PubMed] [Google Scholar]
- 24.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4406-425. [DOI] [PubMed] [Google Scholar]
- 25.Sakai, T., K. Shiraki, H. Inoue, H. Okano, M. Deguchi, K. Sugimoto, S. Ohmor, K. Murata, H. Fujioka, K. Takase, Y. Tameda, and T. Nakano. 2002. HBV subtype as a marker of the clinical courser of chronic HBV infection in Japanese patients. J. Med. Virol. 68175-181. [DOI] [PubMed] [Google Scholar]
- 26.Sugauchi, F., H. Kumada, H. Sakugawa, M. Komatsu, H. Niitsuma, H. Watanabe, Y. Akahane, H. Tokita, T. Kato, Y. Tanaka, E. Orito, R. Ueda, Y. Miyakawa, and M. Mizokami. 2004. Two subtypes of genotype B (Ba and Bj) of hepatitis B virus in Japan. Clin. Infect. Dis. 381222-1228. [DOI] [PubMed] [Google Scholar]
- 27.Sumi, H., O. Yokosuka, N. Seki, M. Arai, F. Imazeki, T. Kurihara, T. Kanda, K. Fukai, M. Kato, and H. Saisho. 2003. Influence of hepatitis B virus genotypes on the progression of chronic liver disease. Hepatology 3719-26. [DOI] [PubMed] [Google Scholar]
- 28.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tralhao, J. G., J. Roudier, S. Morosan, C. Giannini, H. Tu, C. Goulenok, F. Carnot, F. Zavala, V. Joulin, D. Kremsdorf, and C. Brechot. 2002. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: an alternative mechanism of HBx-related pathogenesis. Proc. Natl. Acad. Sci. USA 996991-6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsubota, A., Y. Arase, F. Ren, H. Tanaka, K. Ikeda, and H. Kumada. 2001. Genotype may correlate with liver carcinogenesis and tumor characteristics in cirrhotic patients infected with hepatitis B virus subtype adw. J. Med. Virol. 65257-265. [DOI] [PubMed] [Google Scholar]
- 31.Wong, M. L., and K. S. Leung. 2000. Data mining using grammar based genetic programming and applications. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 32.Yu, M. W., S.-H. Yeh, P.-J. Chen, Y.-F. Liaw, C.-L. Lin, C.-J. Liu, W.-L. Shih, J.-H. Kao, D.-S. Chen, and C.-J. Chen. 2005. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: a prospective study in men. J. Natl. Cancer Inst. 97265-272. [DOI] [PubMed] [Google Scholar]
- 33.Yuen, M. F., Y. Tanaka, M. Mizokami, J. C. Yuen, D. K. Wong, H. J. Yuan, S. M. Sum, A. O. Chan, B. C. Wong, and C. L. Lai. 2004. Role of hepatitis B virus genotypes Ba and C, core promoter and precore mutations on hepatocellular carcinoma: a case control study. Carcinogenesis 251593-1598. [DOI] [PubMed] [Google Scholar]