

Abstract
The mechanisms underlying the lack of disease progression in natural simian immunodeficiency virus (SIV) hosts are still poorly understood. To test the hypothesis that SIV-infected African green monkeys (AGMs) avoid AIDS due to virus replication occurring in long-lived infected cells, we infected six animals with SIVagm and treated them with potent antiretroviral therapy [ART; 9-R-(2-phosphonomethoxypropyl) adenine (tenofovir) and beta-2,3-dideoxy-3-thia-5-fluorocytidine (emtricitabine)]. All AGMs showed a rapid decay of plasma viremia that became undetectable 36 h after ART initiation. A significant decrease of viral load was observed in peripheral blood mononuclear cells and intestine. Mathematical modeling of viremia decay post-ART indicates a half-life of productively infected cells ranging from 4 to 9.5 h, i.e., faster than previously reported for human immunodeficiency virus and SIV. ART induced a slight but significant increase in peripheral CD4+ T-cell counts but no significant changes in CD4+ T-cell levels in lymph nodes and intestine. Similarly, ART did not significantly change the levels of cell proliferation, activation, and apoptosis, already low in AGMs chronically infected with SIVagm. Collectively, these results indicate that, in SIVagm-infected AGMs, the bulk of virus replication is sustained by short-lived cells; therefore, differences in disease outcome between SIVmac infection of macaques and SIVagm infection of AGMs are unlikely due to intrinsic differences in the in vivo cytopathicities between the two viruses.
One of the most intriguing pathogenic features of simian immunodeficiency virus (SIV) infection is that African nonhuman primate (NHP) natural hosts, such as African green monkeys (AGMs), sooty mangabeys (SMs), mandrills, and chimpanzees, naturally or experimentally infected with their species-specific SIV generally do not progress to AIDS (3, 5, 16, 43, 50, 59, 65, 66). This feature is in striking contrast to pathogenic lentiviral infections of humans and macaques, for which the outcome of infection is disease progression (32). It is now widely acknowledged that a better understanding of the mechanisms of the lack of disease progression in natural SIV infections may be needed for understanding the pathogenesis of AIDS in human immunodeficiency virus (HIV)-infected individuals (70, 71).
Both HIV type 1 (HIV-1) and HIV-2 originated from cross-species transmission events of SIVs naturally infecting chimpanzees/gorillas and SMs, respectively (73). SIVsmm/SIVmac strains that are pathogenic in rhesus macaques (RMs) also originated from naturally infected SMs (2). Infection of SMs or RMs and AGMs or pigtailed macaques with the same strains of SIVsmm/SIVmac or SIVagm, respectively, results in different outcomes, with disease progression in macaques and persistent nonprogressive infection in natural African NHP hosts (17, 25, 29, 65). Therefore, the differences in pathogenic potentials do not appear to be virus related. Importantly, some of the immunological consequences of SIV infection shared by pathogenic and natural SIV infections are very similar, most notably the early and massive CD4+ T-cell depletion at the mucosal sites (19, 33, 38, 48). Hence, the current view is that the main reason behind the lack of disease progression in natural African hosts lies in a better adaptation of the host in response to the highly replicating virus rather than it reflecting an infection with less pathogenic viral strains. This improved adaptation of the host immune system does not mean stronger or broader immune responses to viral antigens (10, 24, 76; I. Pandrea, unpublished data). Moreover, studies by us and others have shown that normal levels of immune activation, T-cell proliferation, and apoptosis are characteristic for the chronic phase of SIV infection in natural hosts (7, 29, 43, 44, 46, 50, 65, 66) and, at least in the case of SIVagm infection of AGMs, may be due to an anti-inflammatory response very rapidly established upon SIVagm infection (30). These responses are different from pathogenic infections in humans and macaques, which are characterized by significant increases in immune activation, the levels of which have been reported to be predictive for disease progression (13, 68). Furthermore, in pathogenic infections, immune cell proliferation and apoptosis are severely compromised (42, 56).
One of the hypotheses proposed to explain the lack of disease progression in natural African NHP hosts is that the better preservation of peripheral CD4+ T cells and the partial immune restoration of mucosal CD4+ T cells in the presence of high levels of viral replication may be due to a different in vivo viral cythopathicity (64), the corollary of this being a significantly longer average life span of infected cells. The in vivo life span of infected cells has been previously measured in pathogenic HIV/SIV infections using potent antiretroviral therapy (ART) (27, 37, 54, 55, 77, 79). These studies reported a two-phase decline in plasma viral load (VL) after the administration of ART: an initial rapid decline of viremia, due to loss of short-lived virus-producing cells (activated CD4+ T cells), followed by a slower decline, occurring as a consequence of loss of longer-lived virus-producing cells (resting T cells or macrophages) (53, 55). Mathematical modeling showed that the bulk of HIV replication (93 to 99%) occurs in recently infected cells that die soon after infection, with an average life span calculated on the order of 1 day after the start of viral production, and only 1 to 7% of virus production derives from long-lived cells, which have an average life span on the order of 1 to 4 weeks (53).
In this study, we followed a similar approach and estimated, from the kinetics of viral decline in vivo, the life span of virus-producing cells in SIVagm-infected AGMs receiving ART during chronic infection. We report that the bulk of SIVagm replication in vivo is sustained by short-lived infected cells. The decay of viremia following ART was more rapid in SIVagm.sab-infected AGMs than in HIV-infected individuals and SIV-infected RMs. These data suggest that the lack of disease progression in SIVagm-infected AGMs is unlikely to be related to reduced intrinsic virus cytopathicity, and the data suggest a key role for species-specific host factors in determining the outcome of a primate lentiviral infection.
MATERIALS AND METHODS
Animals.
Six Caribbean AGMs (Chlorocebus sabaeus), originating from St. Kitts Island, were included in this study. The animals were adults (mean age, 7 years). All animals were negative for simian T-cell lymphotropic virus (Vironostika human T-cell lymphotropic virus types I and II enzyme-linked immunosorbent assay; BioMerieux, Durham, NC) and SIV by an in-house SIVagm.sab-specific enzyme-linked immunosorbent assay and were housed at the Tulane National Primate Research Center, an AAALAC International-accredited facility. Housing and handling of animals were in accordance with the Guide for the Care and Use of Laboratory Animals (40) and the Animal Welfare Act. All protocols and procedures for the animal studies were reviewed and approved by the Tulane University Institutional Animal Care and Use Committee.
SIVagm.sab infection.
To avoid selection of viral variants in vitro, inocula used in this study consisted of plasma obtained from an experimentally SIVagm.sab92018-infected AGM. Plasma titers were determined on SupT1 as described elsewhere (9), and all six AGMs were inoculated with plasma containing the equivalent of 300 50% tissue culture infective doses of SIVagm.sab92018.
Antiretroviral therapy.
All AGMs included in this study were treated with nucleotide reverse transcriptase inhibitors (NRTIs) 9-R-(2-phosphonomethoxypropyl) adenine (PMPA; tenofovir) and beta-2,3-dideoxy-3-thia-5-fluorocytidine (FTC; emtricitabine) for 21 days. Antiretroviral (ARV) drugs were administered starting from day 254 post-SIV inoculation, when levels of plasma viremia had reached the set point and were remarkably stable. Subcutaneous injections of both drugs were given at doses of 30 mg/kg of body weight/day. Prior to treatment, monkeys were trained for subcutaneous injections, which allowed drugs to be administered without repeatedly undergoing anesthesia. PMPA and FTC were kindly provided by Gilead (Foster City, CA).
Blood and tissue collection.
In order to model the dynamics of virus replication, thorough sampling schedules during the primary SIVagm.sab infection and antiretroviral treatment were designed. Blood (4.9 ml, EDTA anticoagulated) was collected from the femoral vein at days −7 and 0, 3, 6, 8, 10, 13, 15, 18, 20, 28, 30, 42, 56, 72, 100, 132, 185 post-SIVagm.sab inoculation. Moreover, every 7 days between day 100 and day 132, 4.9 ml of blood was collected (with EDTA) to determine the weekly variation of VLs in SIVagm.sab-infected AGMs. Then, the animals were not sampled for 4 months to minimize stress prior to treatment.
ART was initiated at 254 days post-SIV inoculation. During the ART, the sampling schedule was every 2 hours during the first 6 hours, every 6 hours during the first 2 days, and then every 2 days during the first 2 weeks and every 3 days during the third week. After the treatment interruption, samples were collected every day for 4 days, then every 3 days for 2 weeks, and weekly for another 2 weeks. A final sample was collected 7 weeks after treatment ceased.
Whole blood was used for flow cytometry within 1 h of collection. Plasma was separated within 2 h of collection and stored in aliquots at −80°C until used for VL quantification.
Endoscopically guided pinch biopsies from the proximal jejunum, intestinal resections of the small intestine, and excisional biopsies of axillary and inguinal lymph nodes (LNs) were collected from all the animals as previously described (46, 48). LNs and intestine were sampled before infection (day −14) and at different time points during the acute (days 10, 21, and 28 postinfection [p.i.]) and chronic (days 42, 72, 100, 132, 200, and 230 p.i.) phases of SIVagm infection. Intestinal biopsies and LN biopsies were also performed at the initiation, at the end, and at 72 days post-antiretroviral treatment.
Isolation of lymphocytes.
Mononuclear cells were separated from the blood through Ficoll density gradient centrifugation. Lymphocytes from the intestine and LNs were isolated and stained for flow cytometry as previously described (46, 48). Briefly, lymphocytes were isolated from intestinal biopsies using EDTA followed by collagenase digestion and Percoll density gradient centrifugation (48). Lymphocytes were isolated from the axillary LNs by gently mincing and pressing tissues through nylon mesh screens.
Antibody detection.
Anti-SIVagm.sab92018 antibody dynamics were monitored by using a SIVagm.sab-specific enzyme immunoassay (PIV-EIA) based on peptides mapping the conserved Gp41 immunodominant region and the highly variable V3 loop of SIVagm.sab2 (67). Serological reactivity was confirmed by Western blotting for all the monkeys included in this study (Zeptometrix Corp., Buffalo, NY).
Viral RNA quantification.
Viral RNA was extracted from 540 μl of plasma using the QIAamp viral RNA extraction kit (Qiagen, Valencia, CA). During the ART, in order to improve the efficacy of testing, viral RNA was extracted from the maximum amount of plasma available (840 to 1,000 μl). RNA was also extracted from 3 × 106 mononuclear cells isolated from blood, LNs, and intestinal biopsies using an RNeasy kit (Qiagen, Valencia, CA). A DNase digestion step was applied to the tissue extractions. Viral RNA was extracted from peripheral blood mononuclear cells (PBMCs), LNs, and intestine prior to, at the end of, and at 72 days post-antiretroviral treatment.
VL quantification was done by real-time PCR as previously described (30, 49). Briefly, total RNA was retrotranscribed into cDNA using the TaqMan Gold reverse transcription-PCR kit and random hexamers (PE, Applied Biosystems, Foster City, CA). PCRs were carried out in a spectrofluorometric thermal cycler (ABI Prism 7700; PE). Quantification was based on the amplification of 180 bp located in the long terminal repeat (LTR) region. This region is very conserved within different SIVagm strains. The SIVagm.sab primers and probe were the following: J15S (5′-CTG GGT GTT CTC TGG TAA G-3′), 5′ J15S (5′-CAA GAC TTT ATT GAG GCA AT-3′), and J15P (6-carboxyfluorescein-CGA ACA CCC AGG CTC AAG CTG G-6-carboxytetramethylrhodamine) as previously described (9). SIVagm.sab cDNA was added to the universal master mix (PE, Applied Biosystems) containing a 10 μM concentration of each primer and 10 μM concentration of the probe. All PCRs were carried out in duplicate in parallel with a negative non-RT control reaction. The PCR cycling conditions were as follows: a first cycle of denaturation (95°C, 10 min), followed by 45 cycles of denaturation (95°C, 10 s), annealing (50°C, 30 s), and extension (72°C, 30 s). Absolute viral RNA copy numbers were deduced by comparing the relative signal strength to corresponding values obtained for seven 10-fold dilutions of standard RNA, which were reverse transcribed and amplified in parallel. The RNA standard consisted of a larger LTR region of SIVagm.sab92018 that was PCR amplified with primers LTR2A (5′-AAC TAA GGC AAG ACT TTA TTG AGG-3′) and LTR4S (5′-ACT GGG CGG TAC TGG GAG TGG CTT-3′). The PCR product was cloned into the pCR 2.1 vector (Invitrogen, Carlsbad, CA). In vitro transcription was then performed using the MEGAscript kit (Ambion, Austin, TX). Known amounts of the SIVagm LTR standard RNA were used to determine the target copy numbers. The detection limit of the SIVagm quantification assays was 100 RNA copies/ml of plasma.
Viral RNA quantification in tissues.
Viral RNA was extracted from 5 × 105 to 106 cells from intestine and LNs with RNeasy (Qiagen, Valencia, CA), and VLs were quantified as described elsewhere (46). Simultaneous quantification of 18S rRNA (rRNA control reagent kit; Perkin-Elmer) was used to normalize the RNA input from cells (21). The assay sensitivity was 100 RNA copies/105 cells.
Flow cytometry.
Mononuclear cells derived from peripheral blood, intestinal biopsies, and LNs were stained for flow cytometric analysis using four-color staining combinations with the following monoclonal antibodies: CD3-fluorescein isothiocyanate (FITC), CD20-phycoerythrin (PE), CD8-peridinin chlorophyll A protein (PerCP), CD4-allophycocyanin (APC), CCR5-PE, HLA-DR-PerCP, CD95-FITC, CD28-APC, CD69-APC or CD69-FITC, CD25-PE, Ki-67-FITC, and Annexin V-PE-7-amino actinomycin D (7AAD; BD Biosciences Pharmingen, San Diego, CA). Cells were incubated with an excess amount of monoclonal antibodies at 4°C for 30 min, followed by a phosphate-buffered saline wash (400 × g; 7 min) and fixation in 2% paraformaldehyde. Whole blood was stained using a whole blood lysis technique previously described (46). Samples were stained for Ki-67 and apoptosis using a Ki-67 R-PE-conjugated mouse anti-human monoclonal antibody set and the Annexin V-PE apoptosis detection kit I (BD Pharmingen) as per the manufacturer's instructions. Apoptotic CD4+ T cells were defined as Annexin V+ 7AAD−, whereas the necrotic CD4+ T cells were defined as Annexin V+ 7AAD+. Stained cells were analyzed with a FACSCalibur flow cytometer (BD Immunocytometry Systems) and analyzed with CellQuest software (BD). CD4+ and CD8+ T-cell percentages were obtained by first gating on lymphocytes and then on CD3+ T cells. Memory, activation, proliferation, and apoptosis markers were determined by gating on lymphocytes, then on CD3+ T cells, and finally on CD4+ CD3+ or CD8+ CD3+ T cells.
IHC staining and ISH.
Immunohistochemical (IHC) staining and in situ hybridization (ISH) were performed on formalin-fixed, paraffin-embedded tissues, as described elsewhere (48). SIVagm was detected in tissues by ISH. Sections were subjected to high-temperature unmasking, treated with 0.2 N HCl, and hybridized overnight at 45°C with either sense or antisense SIVagm digoxigenin-UTP-labeled riboprobe, blocked with normal sheep serum, incubated with sheep antidigoxigenin-alkaline phosphatase, and incubated with the HNPP fluorescence detection set (Roche). The SIVagm-infected cell phenotype was determined after ISH by incubating sections with rabbit anti-human CD3 (DAKO, Carpinteria, CA) or mouse anti-human macrophage (HAM56; DAKO), followed by the appropriate goat anti-mouse or goat anti-rabbit antibodies labeled with Alexa 488 (ABC method; Vectastain Elite ABC kit). Negative controls included an antisense probe with uninfected tissues, a sense probe with infected tissues, an antisense probe with infected tissues, and anti-rabbit or anti-mouse secondary antibodies only.
Mathematical modeling of data.
For the dynamic analysis of our data, we used a simplification of the standard model of viral dynamics (41, 52). The model describes the changes in time of the following populations: target cells (T), infected cells (I), and virus (V). Uninfected cells are produced at the total rate λ and die at the per capita rate d. In addition, they can become infected at a rate proportional to the viral load βV, generating infected cells, which are lost at the rate δ, higher than d. Virions are produced from infected cells at a rate of p per cell and cleared by all mechanisms at the rate c (52). Thus, we obtain the following equations:
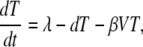 |
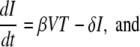 |
(1) |
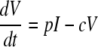 |
When a monkey is first infected, the virus will grow exponentially as V(t) approaches ∼ert, where r is the rate of growth, which can be determined from the slope of the increase of virus in a plot of log Vt versus time. Moreover, it can be shown analytically (60) that this initial growth rate (r) is related to the basic reproduction number of the virus R0, such that
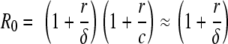 |
(2) |
where the last approximation is valid when c is ≫r. The basic reproduction number is a fundamental quantity, because it expresses the condition for the virus to infect a host: when R0 is >1, the virus can spread and cause chronic infection, but if R0 is <1, the virus cannot cause infection (41).
We assumed that the set point VL (V0) is reached after 4 to 6 weeks of infection. ART was initiated at day 254 p.i. Assuming that this treatment is 100% efficient, as it was assumed before (77), the solution of the system (from equation group 1) is:
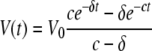 |
(3) |
This equation indicates that the behavior of virus under therapy should follow a double exponential decay pattern. However, the data (see Fig. 1 and Results, below) indicate that the decay follows a single exponential pattern. This result is not surprising, because from other models of infection (such as HIV or SIV in RMs), we know that c is much larger than δ. When this happens, expression (from equation 3) can be approximated by a term containing only the exponential in δ, such that
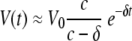 |
(4) |
Again, on a logarithmic scale this represents a simple linear decay, as observed. In addition, the slope of that decay corresponds to the infected cell loss rate, δ.
FIG. 1.

(a) Dynamics of SIVagm replication in AGMs during follow-up. (b) During acute infection, a very active replication, with a pattern that is similar to that of other pathogenic or nonpathogenic infections, was observed. The set point was established between days 28 and 42 postinfection. (c) The administration of antiretroviral treatment induced a rapid reduction in viral loads, followed by a short rebound. This initial effect was followed by a complete control of VLs starting from 42 h. (d to f) Antiretroviral treatment significantly reduced SIVagm RNA VLs in PBMCs (d) and intestine (f) but had a limited impact on RNA VLs in the lymph nodes (e), as illustrated with SIVagm RNA VL quantification before treatment, at the end of treatment, and 7 weeks later.
Statistical analyses.
To estimate both the rate of initial viral expansion upon infection and the infected cell loss rate, we fitted a linear regression to the VL data, using a mixed effects model approach (57). In this method, all the monkeys were fitted simultaneously and the best population estimate for the slopes was obtained. We also estimated rates of virus decline postpeak in acute infection and viral rebound after treatment cessation by using mixed effects models. Results are presented as means ± standard deviations, and P values were considered significant if they were less than 0.05.
RESULTS
Dynamics of SIVagm.sab replication in experimentally infected AGMs.
We measured and modeled quantitatively the dynamics of SIVagm.sab replication in AGMs during the acute phase, the attainment of the viral quasi-steady state (the set point), and while undergoing treatment.
Primary infection.
VLs increased exponentially, reaching a peak around day 8 for all monkeys, with a geometric mean of 3.2 × 107 copies/ml. The average slope of increase was 0.68 ± 0.06 log10 day−1, corresponding to a doubling time in VL of about 11 h. The VL then decreased with a half-life of about 4.5 days, reaching a quasi-steady state of 1.2 × 105 copies/ml by days 28 to 42 p.i. This pattern of viral replication was very similar to that of pathogenic SIVmac infections (26, 38, 48). The steady-state VL was remarkably stable in all animals, with an average coefficient of variation, within individual animals, of only 5% between days 42 and 254, the time of initiation of ART. To confirm that VLs were constant during the steady state, we monitored the weekly variation in VLs between days 100 and 132 p.i. As shown in Fig. 1a, no significant variations in VLs could be observed during this interval.
Combined ISH for SIVagm RNA and IHC for either macrophages or CD3+ T cells confirmed the high viral replication and showed that SIVagm colocalizes with T cells (Fig. 2b) and not macrophages (Fig. 2a), suggesting that SIVagm preferentially infects T cells during acute infection.
FIG. 2.

Combined in situ hybridization for SIV and immunohistochemistry for either macrophage (HAM56) (a) or lymphocyte (CD3) (b) markers demonstrates that during the primary infection, the majority of the SIVagm-infected cells are lymphocytes.
Antiretroviral treatment.
Figure 1 shows the data obtained after treating the six AGMs with daily PMPA plus FTC (see Materials and Methods). After a short delay, possibly due to drug pharmacokinetics, the VL decays very rapidly. Figure 1c shows that there is a clear rebound of virus, starting at about 18 h posttreatment. This rebound is presumably due to the pharmacokinetic elimination of the drugs (20, 75) and is not commonly seen in the setting of HIV/SIV treatment, although the frequency of measurement early posttherapy in our study is higher than other typical macaque studies (18, 41). Nevertheless, starting at 36 h, all monkeys showed a continued decay of virus that eventually fell below the limit of detection (100 copies/ml).
The observed rebounds make it difficult to fit the “standard” model of treatment to the data (equation 3 in Materials and Methods). Nevertheless, the overall decay observed clearly indicates an effect of the drug. Moreover, this decay appears essentially linear on the log scale, once we discount the putative pharmacokinetic rebound. In this case, we can fit an approximation of the standard model, which assumes that the clearance of free virus is much faster than the loss of infected cells, as is well known in both HIV and SIV pathogenic infections (27, 41, 55, 58, 77). From equation 4, we then expect a linear decay, with the slope given by the loss rate of infected cells. We have tried two different strategies to fit the data using simple linear regression: (i) strategy a, using only daily data (i.e., time zero and 1, 2, 3, 4, 5, and 6 days); and (ii) strategy b, fitting only the fastest decay observed on the first day. This analysis was done using a linear mixed effects approach, as described in Materials and Methods.
The average decay rates and their corresponding half-lives were as follows: for strategy a, decay = −0.76 log10/day and t1/2 = 9.5 h; for strategy b, decay = −1.97 log10/day and t1/2 = 3.7 h. These decays are very fast, even if they are minimal estimates, since we assumed 100% efficacious therapy. In fact, these decays are faster than previously reported for HIV in humans (37) and SIV in RMs (41) or cynomolgus macaques (4). They are also faster than those calculated for SIVsmm infection of SMs (18). One contributing factor for the faster virus and cell turnovers in AGMs may be that ART is more effective during SIVagm infection for reasons of bio-distribution or the higher relative dosage.
After the discontinuation of ART, the virus rebounded to levels comparable to those prior to the initiation of treatment. It is interesting that the virus remained undetectable for up to 13 days in some monkeys before showing an exponential rebound, with an average doubling time of 27 h, which is much slower than that observed during primary infection. This slower rebound compared to primary infection might be due to the action of immune responses or to a lower availability of target cells, since during the chronic infection the levels of mucosal CD4+ T cells are never restored to the baseline levels in natural African NHP hosts of SIV, as we have previously reported (19, 48).
SIV RNA quantification in PBMCs, LNs, and intestine indicated that this short-term ART had only a limited impact on viral burden. The reduction in tissue RNA loads was more prominent in PBMCs and intestine (Fig. 1d and f, respectively) than in the LNs (Fig. 1e).
These data indicate that natural SIVagm infection of AGMs is similar to HIV infection of humans in that suppression of de novo infections by ART results in a rapid and profound decline of plasma viremia, thus suggesting that the bulk of virus replication occurs in short-lived infected cells.
Calculation of the basic reproduction number, R0.
The number of infected cells resulting from one infected cell introduced in a totally uninfected population is the basic reproduction number, R0, of the infection. This quantity is important because it has a threshold behavior: when R0 is smaller than 1, the infection will extinguish itself, since it generates less than one infected cell per initial infected cell; if R0 is >1, then the infection will spread. From the data collected in this study, including the initial growth rate of the infection and the loss rate of infected cells, we can estimate R0 for SIVagm in African green monkeys (see Materials and Methods). One unknown, however, is the time interval between cells being infected and starting to produce virus, the intracellular delay. This delay will affect the estimate of R0 (36, 41, 60) when this number is calculated based on the exponential rate of viral growth during early infection: longer delays result in larger R0 values. Thus, we estimated R0 for a fixed intracellular delay of 1 day, as done before (35, 41), and obtained an R0 of 6.3 ± 1.3 (harmonic mean). This relatively small value for R0 is consistent with the fast loss rate of cells estimated above.
Dynamics of CD4+ T-cell counts during SIVagm infection.
In pathogenic HIV and SIVmac infections of humans and RMs, there is a progressive decline of CD4+ T cells (12). In patients under highly active ART (HAART), the control of viral replication results in increases in CD4+ T-cell counts and significant decreases in the fraction of activated and proliferating T cells, which are associated in untreated patients with a progressive decline of circulating CD4+ T cells (23, 31, 39, 45, 63).
Primary infection.
During acute infection, similar to pathogenic SIVmac infection (38), the initial increase in virus counts was accompanied by a significant loss of CD4+ T cells in the periphery, with counts reaching a nadir between days 6 and 10 and an average depletion of 46% of peripheral CD4+ T cells (an average slope of −15 cells/μl/day between challenge and the nadir; P = 0.002). However, similar to pathogenic infection, the absolute number of CD4+ T cells then showed a partial recovery from day 42 onwards (until the start of treatment on day 254), with the new CD4+ T-cell steady-state level approximately 25% lower than preinfection levels (Fig. 3a). Tissue CD4+ T-cell dynamics during the acute infection showed patterns similar to those previously reported by our group (46, 48).
FIG. 3.

(a) Longitudinal flow cytometric analysis of absolute counts of peripheral CD4+ T cells in SIVagm-infected AGMs showed a transient depletion of CD4+ T cells during acute infection, followed by rebound of CD4+ T cells to near preinfection values and very good preservation during the follow-up. (b) During antiretroviral treatment, a slight but significant increase in CD4+ T-cell counts was observed. After treatment, virus rebound induced a slight but significant depletion of CD4+ T cells.
Antiretroviral treatment.
Over the first 14 days of treatment, the CD4+ T-cell count increased slightly, with an average slope of ∼4 cells/μl/day (P = 0.027) (Fig. 3b). This increase seems to be driven mostly by an increase in naïve CD28+ CD4+ T cells of ∼4.6 cells/μl/day (P = 0.0003) over the first 10 days of treatment. As mentioned above, after treatment discontinuation, the virus remained undetectable for up to 2 weeks. When the virus rebounded, there was an overall decay in CD4+ T-cell numbers in the blood with an average slope of −2.2 cells/μl/day (Fig. 3b). This decay was significant (P = 0.0013) despite the considerable variability of T-cell numbers. Again, naïve T cells contributed to this decay, with an average slope of ∼0.97 cells/μl/day (P = 0.0013) after viral rebound.
During the short-term ART, no significant change in the CD4+ T cell numbers was observed in either LNs (data not shown) or gut-associated lymphoid tissue (data not shown).
Minor fluctuations in the percentages of memory (CD28+ CD95+) and effector (CD28− CD95+) CD4+ T cells were observed during the ART administration (data not shown). The lack of a significant increase in the fraction of these CD4+ T-cell subsets in ART-treated SIVagm-infected AGMs was not entirely unexpected, given that these animals had relatively high baseline levels of CD4+ T cells. In the same way, stopping therapy did not result in a significant decrease in the percentages or numbers for CD4+ T-cell subsets (data not shown), confirming that in natural SIV infection significant CD4+ depletion only occurs in the presence of high levels of viral replication and that maintenance of CD4+ T-cell homeostasis during steady-state viral replication may be related to CD4+ T-cell recovery (48).
Dynamics of T-cell turnover, immune activation, and apoptosis during ART.
In HIV-infected individuals, suppression of virus replication by HAART results in a rapid decrease in T-cell turnover and reduced T-cell activation (22, 23, 34, 39). To investigate the impact of viral replication on the level of T-cell activation, we longitudinally assessed the expression of the immune activation markers DR, CD69, and CD25 on T cells. As previously reported (46, 48), during acute infection, both CD4+ and CD8+ T cells showed transient increases in the levels of immune activation (data not shown) However, during ART administration and posttreatment virus rebound, no significant dynamics of immune activation were observed in SIVagm-infected AGMs, as illustrated by the percentage of DR+ CD4+ T cells which remained more or less constant throughout treatment and virus rebound (Fig. 4a). In contrast, the percentage of DR+ CD8+ T cells increased during the virus rebound (Fig. 4b), but this increase was not statistically significant (P = 0.09). No significant changes in DR+ CD4+ T cells or DR+ CD8+ T cells were observed in the LNs or intestine (data not shown), probably because the treatment was administered only for a short time interval and did not significantly impact the tissue VL dynamics (Fig. 1d to f). The peripheral blood and tissue dynamics of other immune activation markers on the CD4+ and CD8+ T cells (CD25 and CD69) were very similar to those of DR+ CD4+ and DR+ CD8+ T cells (data not shown), showing that short-term suppression of viral replication induces only minor changes in the level of immune activation in AGMs naturally infected with SIVagm.
FIG. 4.

Dynamics of immune activation (DR), cell proliferation (Ki-67), and apoptosis (Annexin V) markers during antiretroviral treatment of SIVagm-infected AGMs. No significant changes in the dynamics of CD4+-DR+ (a) or CD8+-DR+ (b) cells were observed. A significant increase in proliferation for both CD4+ (c) and CD8+ (d) cells was observed that was simultaneous with the posttreatment virus rebound. No impact on the dynamics of apoptotic cells in the intestine was observed (e).
To assess ART-induced changes in T-cell turnover, we measured the expression of the proliferation marker Ki-67. As we previously reported (48), a transient increase in Ki-67 expression by CD4+ T cells occurs during acute SIVagm infection of AGMs (data not shown), followed by a return to baseline levels during steady-state infection. The initiation of ART did not induce any significant reduction in the fraction of proliferating CD4+ T cells of SIVagm-infected AGMs. Interruption of therapy was associated with an increase in the percentage and numbers of proliferating CD4+ T cells that reached levels higher than those observed prior to treatment in five of six animals (Fig. 4c). These data were quite variable for each monkey over time, and we smoothed the data by calculating a moving average. As seen in Fig. 4c, there was a clear increase in the percentage of Ki-67+ CD4+ T cells in all but one (FV76) of the AGMs, starting from the end of treatment for 2 or 3 weeks. This slight increase of ∼0.06% per day was highly significant (P < 0.0001), even when the outlier (FV76) was included in the analysis (P = 0.002). The analysis of Ki-67+ CD4+ T-cell counts showed a trend for increases in these cells from the start of treatment to approximately day 43 post-initiation of treatment, when the virus had rebounded back to pretreatment levels. This increase, with an average across the monkeys of 0.08 cells/μl/day, was significant (P = 0.024) (Fig. 4c). Interestingly, the percentage of CD4+ T cells expressing Ki-67 in the LNs also tended to increase from the end of therapy to day 51, after the viral rebound (P = 0.062) (data not shown).
No significant changes in the levels of proliferating CD8+ T cells were observed, except in one AGM (FV71), during ART or post-treatment interruption (Fig. 4d). A slight transient increase in CD8+ T-cell proliferation occurred at the time of treatment interruption, likely as a response to the increased VLs. Ki-67+ CD8+ T-cell levels returned to pretreatment levels when viral replication reached the preinfection set point (Fig. 4d).
The dynamics of apoptosis of CD4+ T cells in peripheral blood was only measured at selected time points: at the initiation of ART, at the end of treatment, and 28 days post-treatment discontinuation. As shown in Fig. 4e, there was no significant change in the percentage of apoptotic cells during the follow-up, which confirmed our previous data showing no discernible change in the apoptosis of CD4+ T cells during SIVagm infection of AGMs (48).
Dynamics of CCR5+ CD4+ T cells (target cells) during ART.
Our previous results showed that natural hosts of SIV have significantly lower expression levels of CCR5 on their CD4+ T cells (47). Since CCR5+ CD4+ T cells are rapidly depleted during HIV or SIV infection, it has been suggested that the low levels of target cells might be a reason for the nonpathogenicity of infection in natural hosts of SIV (62). However, we recently showed that there is massive mucosal CD4+ T-cell depletion in acute SIV infection of natural hosts (19, 48). Therefore, we analyzed the evolution of the CCR5+ CD4+ T cells during ART. No significant change in the population of CCR5+ CD4+ T cells was observed during the first 10 days of therapy. However, a significant increase of CCR5+ CD4+ T cells with an average slope of 0.11 cell/μl/day (P = 0.0012) (Fig. 5) was observed between days 10 and 43 post-treatment initiation.
FIG. 5.

Dynamics of CCR5+ CD4+ T cells under antiretroviral treatment of AGMs chronically infected with SIVagm. A significant increase was observed between day 10 of treatment and day 43 posttreatment.
DISCUSSION
In this study, we performed experimental SIVagm infections of AGMs, treated the animals with potent antiretroviral treatment during the chronic, steady-state SIVagm infection, and measured the turnover of infected cells by applying the same mathematical modeling previously used for pathogenic infections (52, 55, 79). We showed that the bulk of SIVagm replication during acute and chronic infection of AGMs occurs in short-lived infected cells. Therefore, our results demonstrate that the lack of disease progression in SIVagm infection in its natural host is unlikely due to a slower virus and cell turnover compared to pathogenic infections.
SIV infection in African NHPs that are natural hosts of SIVs is characterized by the following: (i) high levels of viral replication during acute and chronic SIV infection in the same range, if not higher, than in pathogenic HIV-1 infection of humans and SIV infection of RMs (1, 43, 44, 46, 49-51, 65, 66); (ii) good preservation of peripheral CD4+ T cells for long periods of time (46, 50, 66, 72); (iii) low levels of target cells (CCR5+ CD4+ T cells) (47). Altogether, these characteristics of SIV infection in natural hosts generated the hypothesis that the lack of disease progression might be due to a more limited impact of SIV replication on the homeostasis of CD4+ T cells (64). This limited impact could result from either (i) a longer life span of the bulk of infected cells (activated effector memory CD4+ T cells) or (ii) virus replication occurring in long-lived cells (most likely macrophages). However, recent data showed that the impact of SIV infection on CD4+ T cells from natural African NHP hosts during acute infection is very similar to that observed during pathogenic infections (19, 48), with early and severe depletion of mucosal CD4+ T cells in both AGMs and SMs (19, 48). This observation suggested that both CD4+ T-cell turnover and SIV cytopathicity are similar between progressive and nonprogressive infections.
With regard to the hypothesis that virus replication mainly occurs in macrophages, there is some evidence against this: (i) the dynamics of virus replication during acute infection is strikingly similar between progressive and nonprogressive SIV infections (51), which likely would not be the case if replication were supported by long-lived infected cells that produce virus slowly; (ii) the massive acute depletion of mucosal CD4+ T cells suggests that these cells are the SIV targets (19, 48); moreover, although this massive mucosal CD4+ T-cell depletion involves all the CD4+ T-cell subsets, the effector memory CD4+ T-cell pool is the most susceptible and shows only minor restoration during chronic infection (19, 48), similar to pathogenic HIV-1 and SIV infections (38, 56); (iii) we and others (15) have provided direct evidence by in situ hybridization that, during acute infection of AGMs, SIVagm replicates in lymphocytes and not in macrophages (Fig. 2) (47). Collectively, these experimental results suggest that the in vivo biology of SIV is comparable between progressive and nonprogressive infections and that the bulk of virus replication also occurs in short-lived infected cells during SIV infections of African NHP hosts. However, the data presented thus far pertain only to acute infection (when the virus can be studied by in situ techniques). Moreover, the significant mucosal CD4+ T-cell restoration during chronic infection in the presence of high viral replication indeed raises the question of whether or not the bulk of viral replication during the chronic phase of infection occurs in macrophages.
To address this question, we performed the current set of experiments, to investigate the source of the virus during chronic SIVagm infection of AGMs. The advantage of using AGMs for these types of studies lies in the fact that, unlike other African NHP species that are currently being used as animal models for natural SIV infection (such as SMs and mandrills), AGMs are not highly endangered and, therefore, more complete studies can be carried out in this species. The frequent testing during acute infection confirmed that the dynamics of SIVagm replication in experimentally infected AGMs is very similar to those of other progressive and nonprogressive SIV infections (46, 48, 49, 51) and that during the chronic infection there are negligible variations in the steady state in SIVagm-infected AGMs.
In this study, chronic SIVagm-infected AGMs were administered a combination of two NRTIs: PMPA and FTC. The choice of drugs was based on previous reports showing excellent antiretroviral activity of these two compounds in monkeys (28, 74). Moreover, other ARV classes, such as nonnucleoside RT inhibitors, are not suitable for studies like these because SIVs are intrinsically resistant to these drugs (78). Similarly, SIVs reportedly have variable susceptibilities to protease inhibitors (14). Conversely, NRTIs are drugs of choice for these types of studies because they are potent enough to induce significant and rapid reduction in viral replication (28, 74) and also because they do not affect preformed virus or the ability of previously infected cells to continue to produce new virions (11), thus allowing a reliable quantification of the in vivo turnover of infected cells and providing indirect information on what cell type(s) supports virus replication in this nonpathogenic model of infection.
Administration of PMPA and FTC resulted in a rapid decline of plasma VLs, which became undetectable in all six animals. Interestingly, an initial rapid decline in plasma VL observed during the first 18 h of treatment was followed by a short viral rebound during the second day of treatment (Fig. 1c), probably the result of reduced drug exposure prior to the establishment of steady-state pharmacokinetics (20). With continuous dosing, this rebound was followed by a sharp decline in VLs, which became undetectable by the fourth day of treatment. Our mathematical modeling of these data clearly indicated that, in AGMs, the bulk (≫90%) of SIVagm replication occurred in cells with an average in vivo life span of 4 to 9.5 h, shorter than estimated in HIV-1-infected humans and SIVmac-infected RMs, when similar methods were used for the calculation (41, 55). This result suggests that SIVagm is cytopathic in vivo for AGM-infected cells, as are HIV-1 for human cells and SIVmac for RM cells. Similar data were reported for another natural host of SIV, the sooty mangabey (18).
Previous studies carried out in HIV-infected humans and SIV-infected macaques using different approaches reported that lentiviral infection is characterized by rapid viral and infected cell turnover. Initial studies, using HAART and modeling the dynamics of viral replication control, estimated the average half-life of virions at approximately 6 h and that for infected cells at 1 day or less (55), and also the fraction of virus replication occurring in short-lived cells (90 to 99%) versus long-lived cells (1 to 10%) (53). More refined analyses, through apheresis, have estimated an even faster viral clearance in humans, with a viral half-life of approximately 20 to 45 min (58), and with more potent therapy, the half-life of virus-producing cells was estimated at 0.7 days (37). In RMs, measurements of viral clearance following bolus injection or continuous infusion showed that clearance was even faster in this species, with a half-life of around 3.3 min (79). We note that in the present study, the observed decay was so fast that we could not reliably estimate the half-life of free virions in SIVagm-infected AGMs.
We also estimated the basic reproduction number (R0) of SIVagm in AGMs at 6.3 ± 1.3, considering that the intracellular delay is ∼1 day. This is similar to estimates for HIV in humans (a harmonic mean of 13.9 in one study with n of 4 [35] and a mean of 5.4 in a study with n of 10 [69]), but probably smaller than SIVsmE660 in pigtailed macaques (harmonic mean, 28.9 [41]). This smaller R0 is likely due to the faster loss of infected cells in the AGM model, which is, for example, ∼6 times faster than in the study on pigtailed macaque infection referred to above.
At the cessation of antiretroviral therapy, the rebound in viral replication was slower than during the acute infection, occurring at 1 to 2 weeks post-treatment interruption to a set point level that was strikingly similar to that observed prior to therapy. This delay in virus rebound compared to the exponential increase observed during acute infection may be due to either the action of immune responses (which are absent in primary infection) or to the lower availability of target cells compared to primary infection. Interestingly, in sooty mangabeys this rebound is faster (18).
ART induced only minor, although statistically significant, changes in the number of peripheral CD4+ T cells in AGMs chronically infected with SIVagm. No significant changes in CD4+ T cells in the LNs and intestine were observed. This difference with data reported in HIV-1-infected patients receiving HAART, in which there is an increase in the pool of CD4+ T cells (61), may be due to several factors: (i) AGMs received only short-term ARV treatment, which might have prevented us from observing a significant CD4+ T-cell restoration under treatment; (ii) in chronically infected AGMs, the levels of peripheral CD4+ T-cell counts are close to normal levels, suggesting that chronic virus replication does not induce peripheral CD4+ T-cell depletion; our previous data showing that mucosal CD4+ T cells can be restored in AGMs chronically infected with SIVagm (48) support these results; (iii) it has been reported that in humans, the initial CD4+ T-cell rebound during HAART is also due to redistribution of CD4+ T cells from lymphoid tissues (6, 8); it is possible that in natural hosts, the CD4+ T-cell redistribution under ART is less significant, as a consequence of less impact of infection on immune parameters, such as T-cell activation and proliferation; (iv) finally, in HIV-1-infected humans, CD4+ T-cell restoration is associated with a significant decrease in the levels of immune activation (8). In SIV natural hosts, immune activation, proliferation, and apoptosis markers are normal during chronic infection (7, 30, 44, 46, 48, 50, 64-66, 73) and, as we have shown here, not significantly influenced by ART, which may also explain the lack of significant immunologic changes in the homeostasis of CD4+ T cells in ARV-treated AGMs. Note that only minor increases in CD4+ T-cell proliferation were observed in our AGMs and that these were correlated with the rebound in viral replication after the cessation of therapy.
In conclusion, our results show that, in chronically infected AGMs, SIVagm replication occurs in short-lived infected cells, thus extending our previous observation of SIVagm replication in lymphocytes and not in macrophages during acute infection (48). Our results corroborate those obtained in another animal model for natural SIV infection, the sooty mangabey (18), and together suggest the bulk of virus replication occurs in activated CD4+ T cells in African natural hosts of SIV. Therefore, the lack of disease progression in natural hosts is likely not due to a prolonged survival of infected cells and/or a reduced intrinsic cytopathicity of SIVs. Since the in vivo dynamics of chronic lentiviral infection is very similar between progressive and nonprogressive infections, the outcome of infection must be dependent on other factors, such as immune activation and/or other unknown factors.
Acknowledgments
We thank Andrew Lackner and Preston Marx for helpful discussions and the veterinary staff of the Tulane National Primate Research Center for their assistance with the animal studies. PMPA and FTC were kindly provided by Gilead Sciences.
This work was supported by NIH grants RO1 AI064066 and R21AI069935 (I.P.), RO1 AI065325 (C.A.), RR06555 and AI28433 (A.S.P.), RR18745 (R.M.R.), and RR-00168 (Tulane National Primate Research Center).
Footnotes
Published ahead of print on 23 January 2008.
REFERENCES
- 1.Apetrei, C., R. Gautam, B. Sumpter, A. C. Carter, T. Gaufin, S. I. Staprans, J. Else, M. Barnes, R. Cao, Jr., S. Garg, J. M. Milush, D. L. Sodora, I. Pandrea, and G. Silvestri. 2007. Virus-subtype specific features of natural SIVsmm infection in sooty mangabeys. J. Virol. 817913-7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apetrei, C., A. Kaur, N. W. Lerche, M. Metzger, I. Pandrea, J. Hardcastle, S. Fakelstein, R. Bohm, J. Kohler, V. Traina-Dorge, T. Williams, S. Staprans, G. Plauche, R. S. Veazey, H. McClure, A. A. Lackner, B. Gormus, D. L. Robertson, and P. A. Marx. 2005. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J. Virol. 798991-9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beer, B., J. Denner, C. R. Brown, S. Norley, J. zur Megede, C. Coulibaly, R. Plesker, S. Holzammer, M. Baier, V. M. Hirsch, and R. Kurth. 1998. Simian immunodeficiency virus of African green monkeys is apathogenic in the newborn natural host. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 18210-220. [DOI] [PubMed] [Google Scholar]
- 4.Brandin, E., R. Thorstensson, S. Bonhoeffer, and J. Albert. 2006. Rapid viral decay in simian immunodeficiency virus-infected macaques receiving quadruple antiretroviral therapy. J. Virol. 809861-9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broussard, S. R., S. I. Staprans, R. White, E. M. Whitehead, M. B. Feinberg, and J. S. Allan. 2001. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J. Virol. 752262-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bucy, R. P., R. D. Hockett, C. A. Derdeyn, M. S. Saag, K. Squires, M. Sillers, R. T. Mitsuyasu, and J. M. Kilby. 1999. Initial increase in blood CD4+ lymphocytes after HIV antiretroviral therapy reflects redistribution from lymphoid tissues. J. Clin. Investig. 1031391-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakrabarti, L. A., S. R. Lewin, L. Zhang, A. Gettie, A. Luckay, L. N. Martin, E. Skulsky, D. D. Ho, C. Cheng-Mayer, and P. A. Marx. 2000. Normal T-cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J. Virol. 741209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark, D. R., R. J. de Boer, K. C. Wolthers, and F. Miedema. 1999. T cell dynamics in HIV-1 infection. Adv. Immunol. 73301-327. [DOI] [PubMed] [Google Scholar]
- 9.Diop, O. M., A. Gueye, M. Dias-Tavares, C. Kornfeld, A. Faye, P. Ave, M. Huerre, S. Corbet, F. Barre-Sinoussi, and M. C. Muller-Trutwin. 2000. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J. Virol. 747538-7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunham, R., P. Pagliardini, S. Gordon, B. Sumpter, J. Engram, A. Moanna, B. Lawson, H. M. McClure, H. Xian-Xu, C. Ibegbu, N. Katz, I. Pandrea, C. Apetrei, D. L. Sodora, M. B. Feinberg, S. I. Staprans, and G. Silvestri. 2006. The AIDS-resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 108209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fung, H. B., E. A. Stone, and F. J. Piacenti. 2002. Tenofovir disoproxil fumarate: a nucleotide reverse transcriptase inhibitor for the treatment of HIV infection. Clin. Ther. 241515-1548. [DOI] [PubMed] [Google Scholar]
- 12.Giorgi, J. V., J. L. Fahey, D. C. Smith, L. E. Hultin, H. L. Cheng, R. T. Mitsuyasu, and R. Detels. 1987. Early effects of HIV on CD4 lymphocytes in vivo. J. Immunol. 1383725-3730. [PubMed] [Google Scholar]
- 13.Giorgi, J. V., L. E. Hultin, J. A. McKeating, T. D. Johnson, B. Owens, L. P. Jacobson, R. Shih, J. Lewis, D. J. Wiley, J. P. Phair, S. M. Wolinsky, and R. Detels. 1999. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 179859-870. [DOI] [PubMed] [Google Scholar]
- 14.Giuffre, A. C., J. Higgins, R. W. Buckheit, Jr., and T. W. North. 2003. Susceptibilities of simian immunodeficiency virus to protease inhibitors. Antimicrob. Agents Chemother. 471756-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein, S., C. R. Brown, I. Ourmanov, I. Pandrea, A. Buckler-White, C. Erb, J. S. Nandi, G. J. Foster, P. Autissier, J. E. Schmitz, and V. M. Hirsch. 2006. Comparison of simian immunodeficiency virus SIVagmVer replication and CD4+ T-cell dynamics in vervet and sabaeus African green monkeys. J. Virol. 804868-4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein, S., I. Ourmanov, C. R. Brown, B. E. Beer, W. R. Elkins, R. Plishka, A. Buckler-White, and V. M. Hirsch. 2000. Wide range of viral load in healthy African green monkeys naturally infected with simian immunodeficiency virus. J. Virol. 7411744-11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein, S., I. Ourmanov, C. R. Brown, R. Plishka, A. Buckler-White, R. Byrum, and V. M. Hirsch. 2005. Plateau levels of viremia correlate with the degree of CD4+-T-cell loss in simian immunodeficiency virus SIVagm-infected pigtailed macaques: variable pathogenicity of natural SIVagm isolates. J. Virol. 795153-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon, S., R. M. Dunham, J. C. Engram, J. Estes, Z. Wang, N. R. Klatt, M. Paiardini, I. Pandrea, C. Apetrei, D. L. Sodora, H. Y. Lee, A. T. Haase, M. Miller, A. Kaur, S. I. Staprans, A. S. Perelson, M. B. Feinberg, and G. Silvestri. 2008. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. J. Virol. 823725-3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon, S., N. R. Klatt, J. M. Milush, J. Engram, R. M. Dunham, M. Paiardini, E. A. Strobert, C. Apetrei, I. Pandrea, S. Staprans, D. L. Sodora, and G. Silvestri. 2007. Severe depletion of mucosal CD4+ T cells in AIDS-free SIV-infected sooty mangabeys. J. Immunol. 1793026-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grim, S. A., and F. Romanelli. 2003. Tenofovir disoproxil fumarate. Ann. Pharmacother. 37849-859. [DOI] [PubMed] [Google Scholar]
- 21.Gueye, A., O. M. Diop, M. J. Ploquin, C. Kornfeld, A. Faye, M. C. Cumont, B. Hurtrel, F. Barre-Sinoussi, and M. C. Muller-Trutwin. 2004. Viral load in tissues during the early and chronic phase of non-pathogenic SIVagm infection. J. Med. Primatol. 3383-97. [DOI] [PubMed] [Google Scholar]
- 22.Hazenberg, M. D., J. W. Stuart, S. A. Otto, J. C. Borleffs, C. A. Boucher, R. J. de Boer, F. Miedema, and D. Hamann. 2000. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95249-255. [PubMed] [Google Scholar]
- 23.Hellerstein, M. K., R. A. Hoh, M. B. Hanley, D. Cesar, D. Lee, R. A. Neese, and J. M. McCune. 2003. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J. Clin. Investig. 112956-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirsch, V. M. 2004. What can natural infection of African monkeys with simian immunodeficiency virus tell us about the pathogenesis of AIDS? AIDS Rev. 640-53. [PubMed] [Google Scholar]
- 25.Hirsch, V. M., G. Dapolito, P. R. Johnson, W. R. Elkins, W. T. London, R. J. Montali, S. Goldstein, and C. Brown. 1995. Induction of AIDS by simian immunodeficiency virus from an African green monkey: species-specific variation in pathogenicity correlates with the extent of in vivo replication. J. Virol. 69955-967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirsch, V. M., T. R. Fuerst, G. Sutter, M. W. Carroll, L. C. Yang, S. Goldstein, M. Piatak, Jr., W. R. Elkins, W. G. Alvord, D. C. Montefiori, B. Moss, and J. D. Lifson. 1996. Patterns of viral replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara. J. Virol. 703741-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373123-126. [DOI] [PubMed] [Google Scholar]
- 28.Hurwitz, S. J., M. J. Otto, and R. F. Schinazi. 2005. Comparative pharmacokinetics of Racivir, (+/−)-beta-2′,3′-dideoxy-5-fluoro-3′-thiacytidine in rats, rabbits, dogs, monkeys and HIV-infected humans. Antivir. Chem. Chemother. 16117-127. [DOI] [PubMed] [Google Scholar]
- 29.Kaur, A., R. M. Grant, R. E. Means, H. McClure, M. Feinberg, and R. P. Johnson. 1998. Diverse host responses and outcomes following simian immunodeficiency virus SIVmac239 infection in sooty mangabeys and rhesus macaques. J. Virol. 729597-9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kornfeld, C., M. J. Ploquin, I. Pandrea, A. Faye, R. Onanga, C. Apetrei, V. Poaty-Mavoungou, P. Rouquet, J. Estaquier, L. Mortara, J. F. Desoutter, C. Butor, R. Le Grand, P. Roques, F. Simon, F. Barre-Sinoussi, O. M. Diop, and M. C. Muller-Trutwin. 2005. Anti-inflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J. Clin. Investig. 1151082-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovacs, J. A., R. A. Lempicki, I. A. Sidorov, J. W. Adelsberger, B. Herpin, J. A. Metcalf, I. Sereti, M. A. Polis, R. T. Davey, J. Tavel, J. Falloon, R. Stevens, L. Lambert, R. Dewar, D. J. Schwartzentruber, M. R. Anver, M. W. Baseler, H. Masur, D. S. Dimitrov, and H. C. Lane. 2001. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J. Exp. Med. 1941731-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lackner, A. A., and R. S. Veazey. 2007. Current concepts in AIDS pathogenesis: insights from the SIV/macaque model. Annu. Rev. Med. 58461-476. [DOI] [PubMed] [Google Scholar]
- 33.Li, Q., L. Duan, J. D. Estes, Z. M. Ma, T. Rourke, Y. Wang, C. Reilly, J. Carlis, C. J. Miller, and A. T. Haase. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 4341148-1152. [DOI] [PubMed] [Google Scholar]
- 34.Li, T. S., R. Tubiana, C. Katlama, V. Calvez, H. Ait Mohand, and B. Autran. 1998. Long-lasting recovery in CD4 T-cell function and viral-load reduction after highly active antiretroviral therapy in advanced HIV-1 disease. Lancet 3511682-1686. [DOI] [PubMed] [Google Scholar]
- 35.Little, S. J., A. R. McLean, C. A. Spina, D. D. Richman, and D. V. Havlir. 1999. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd, A. L. 2001. Destabilization of epidemic models with the inclusion of realistic distributions of infectious periods. Proc. Biol. Sci. 268985-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio, A. S. Perelson, and D. D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 775037-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattapallil, J. J., D. C. Douek, B. Hill, Y. Nishimura, M. Martin, and M. Roederer. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 4341093-1097. [DOI] [PubMed] [Google Scholar]
- 39.Mohri, H., A. S. Perelson, K. Tung, R. M. Ribeiro, B. Ramratnam, M. Markowitz, R. Kost, A. Hurley, L. Weinberger, D. Cesar, M. K. Hellerstein, and D. D. Ho. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 1941277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC.
- 41.Nowak, M. A., A. L. Lloyd, G. M. Vasquez, T. A. Wiltrout, L. M. Wahl, N. Bischofberger, J. Williams, A. Kinter, A. S. Fauci, V. M. Hirsch, and J. D. Lifson. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 717518-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okoye, A., M. Meier-Schellersheim, J. M. Brenchley, S. I. Hagen, J. M. Walker, M. Rohankhedkar, R. Lum, J. B. Edgar, S. L. Planer, A. Legasse, A. W. Sylwester, M. Piatak, Jr., J. D. Lifson, V. C. Maino, D. L. Sodora, D. C. Douek, M. K. Axthelm, Z. Grossman, and L. J. Picker. 2007. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J. Exp. Med. 2042171-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onanga, R., C. Kornfeld, I. Pandrea, J. Estaquier, S. Souquiere, P. Rouquet, V. P. Mavoungou, O. Bourry, S. M'Boup, F. Barre-Sinoussi, F. Simon, C. Apetrei, P. Roques, and M. C. Muller-Trutwin. 2002. High levels of viral replication contrast with only transient changes in CD4+ and CD8+ cell numbers during the early phase of experimental infection with simian immunodeficiency virus SIVmnd-1 in Mandrillus sphinx. J. Virol. 7610256-10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onanga, R., S. Souquiere, M. Makuwa, A. Mouinga-Ondeme, F. Simon, C. Apetrei, and P. Roques. 2006. Primary simian immunodeficiency virus SIVmnd-2 infection in mandrills (Mandrillus sphinx). J. Virol. 803303-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orendi, J. M., A. C. Bloem, J. C. Borleffs, F. J. Wijnholds, N. M. de Vos, H. S. Nottet, M. R. Visser, H. Snippe, J. Verhoef, and C. A. Boucher. 1998. Activation and cell cycle antigens in CD4+ and CD8+ T cells correlate with plasma human immunodeficiency virus (HIV-1) RNA level in HIV-1 infection. J. Infect. Dis. 1781279-1287. [DOI] [PubMed] [Google Scholar]
- 46.Pandrea, I., C. Apetrei, J. Dufour, N. Dillon, J. Barbercheck, M. Metzger, B. Jacquelin, R. Bohm, P. A. Marx, F. Barre-Sinoussi, V. M. Hirsch, M. C. Muller-Trutwin, A. A. Lackner, and R. Veazey. 2006. Simian immunodeficiency virus SIVagm.sab infection of Caribbean African green monkeys: a new model for the study of SIV pathogenesis in natural hosts J. Virol. 804858-4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandrea, I., C. Apetrei, S. Gordon, J. Barbercheck, J. Dufour, R. Bohm, B. Sumpter, P. Roques, P. A. Marx, V. M. Hirsch, A. Kaur, A. A. Lackner, R. S. Veazey, and G. Silvestri. 2007. Paucity of CD4+ CCR5+ T cells is a typical feature of natural SIV hosts. Blood 1091069-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandrea, I., R. Gautam, R. Ribeiro, J. M. Brenchley, I. F. Butler, M. Pattison, T. Rasmussen, P. A. Marx, G. Silvestri, A. A. Lackner, A. S. Perelson, D. C. Douek, R. S. Veazey, and C. Apetrei. 2007. Acute loss of intestinal CD4+ T cells is not predictive of SIV virulence. J. Immunol. 1793035-3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandrea, I., C. Kornfeld, M. J.-I. Ploquin, C. Apetrei, A. Faye, P. Rouquet, P. Roques, F. Simon, F. Barré-Sinoussi, M. C. Müller-Trutwin, and O. M. Diop. 2005. Impact of viral factors on very early in vivo replication profiles in SIVagm-infected African green monkeys. J. Virol. 796249-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandrea, I., R. Onanga, C. Kornfeld, P. Rouquet, O. Bourry, S. Clifford, P. T. Telfer, K. Abernethy, L. T. White, P. Ngari, M. Muller-Trutwin, P. Roques, P. A. Marx, F. Simon, and C. Apetrei. 2003. High levels of SIVmnd-1 replication in chronically infected Mandrillus sphinx. Virology 317119-127. [DOI] [PubMed] [Google Scholar]
- 51.Pandrea, I., G. Silvestri, R. Onanga, R. S. Veazey, P. A. Marx, V. M. Hirsch, and C. Apetrei. 2006. Simian immunodeficiency viruses replication dynamics in African non-human primate hosts: common patterns and species-specific differences. J. Med. Primatol. 35194-201. [DOI] [PubMed] [Google Scholar]
- 52.Perelson, A. S. 2002. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 228-36. [DOI] [PubMed] [Google Scholar]
- 53.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387188-191. [DOI] [PubMed] [Google Scholar]
- 54.Perelson, A. S., P. Essunger, and D. D. Ho. 1997. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS 11(Suppl. A)S17-S24. [PubMed] [Google Scholar]
- 55.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 2711582-1586. [DOI] [PubMed] [Google Scholar]
- 56.Picker, L. J., S. I. Hagen, R. Lum, E. F. Reed-Inderbitzin, L. M. Daly, A. W. Sylwester, J. M. Walker, D. C. Siess, M. Piatak, Jr., C. Wang, D. B. Allison, V. C. Maino, J. D. Lifson, T. Kodama, and M. K. Axthelm. 2004. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J. Exp. Med. 2001299-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinheiro, J. C., and D. M. Bates. 2002. Mixed-effects models in S and S-Plus. Springer-Verlag, New York, NY.
- 58.Ramratnam, B., S. Bonhoeffer, J. Binley, A. Hurley, L. Zhang, J. E. Mittler, M. Markowitz, J. P. Moore, A. S. Perelson, and D. D. Ho. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 3541782-1785. [DOI] [PubMed] [Google Scholar]
- 59.Rey-Cuille, M. A., J. L. Berthier, M. C. Bomsel-Demontoy, Y. Chaduc, L. Montagnier, A. G. Hovanessian, and L. A. Chakrabarti. 1998. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J. Virol. 723872-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ribeiro, R. M., N. M. Dixit, and A. S. Perelson. 2006. Modeling the in vivo growth rate of HIV: implications for vaccination, p. 231-246. In R. Paton and L. A. McNamara (ed.), Multidisciplinary approaches to theory in medicine. Elsevier, Amsterdam, The Netherlands.
- 61.Richman, D. D. 2001. HIV chemotherapy. Nature 410995-1001. [DOI] [PubMed] [Google Scholar]
- 62.Roederer, M., and J. Mattapallil. 2007. CCR5 and HIV: the less, the better. Blood 109854. [Google Scholar]
- 63.Sachsenberg, N., A. S. Perelson, S. Yerly, G. A. Schockmel, D. Leduc, B. Hirschel, and L. Perrin. 1998. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 1871295-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silvestri, G. 2005. Naturally SIV-infected sooty mangabeys: are we closer to understanding why they do not develop AIDS? J. Med. Primatol. 34243-252. [DOI] [PubMed] [Google Scholar]
- 65.Silvestri, G., A. Fedanov, S. Germon, N. Kozyr, W. J. Kaiser, D. A. Garber, H. McClure, M. B. Feinberg, and S. I. Staprans. 2005. Divergent host responses during primary simian immunodeficiency virus SIVsm infection of natural sooty mangabey and nonnatural rhesus macaque hosts. J. Virol. 794043-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silvestri, G., D. L. Sodora, R. A. Koup, M. Paiardini, S. P. O'Neil, H. M. McClure, S. I. Staprans, and M. B. Feinberg. 2003. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18441-452. [DOI] [PubMed] [Google Scholar]
- 67.Simon, F., S. Souquiere, F. Damond, A. Kfutwah, M. Makuwa, E. Leroy, P. Rouquet, J. L. Berthier, J. Rigoulet, A. Lecu, P. T. Telfer, I. Pandrea, J. C. Plantier, F. Barre-Sinoussi, P. Roques, M. C. Muller-Trutwin, and C. Apetrei. 2001. Synthetic peptide strategy for the detection of and discrimination among highly divergent primate lentiviruses. AIDS Res. Hum. Retrovir. 17937-952. [DOI] [PubMed] [Google Scholar]
- 68.Sousa, A. E., J. Carneiro, M. Meier-Schellersheim, Z. Grossman, and R. M. Victorino. 2002. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 1693400-3406. [DOI] [PubMed] [Google Scholar]
- 69.Stafford, M. A., L. Corey, Y. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203285-301. [DOI] [PubMed] [Google Scholar]
- 70.Stebbing, J., B. Gazzard, and D. C. Douek. 2004. Where does HIV live? N. Engl. J. Med. 3501872-1880. [DOI] [PubMed] [Google Scholar]
- 71.Stevenson, M. 2003. HIV-1 pathogenesis. Nat. Med. 9853-860. [DOI] [PubMed] [Google Scholar]
- 72.Sumpter, B., R. Dunham, S. Gordon, J. Engram, M. Hennessy, A. Kinter, M. Paiardini, B. Cervasi, N. Klatt, H. McClure, J. M. Milush, S. Staprans, D. L. Sodora, and G. Silvestri. 2007. Correlates of preserved CD4+ T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J. Immunol. 1781680-1691. [DOI] [PubMed] [Google Scholar]
- 73.VandeWoude, S., and C. Apetrei. 2006. Going wild: lessons from T-lymphotropic naturally occurring lentiviruses. Clin. Microbiol. Rev. 19728-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Rompay, K. K., M. L. Marthas, J. D. Lifson, C. J. Berardi, G. M. Vasquez, E. Agatep, Z. A. Dehqanzada, K. C. Cundy, N. Bischofberger, and N. C. Pedersen. 1998. Administration of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) for prevention of perinatal simian immunodeficiency virus infection in rhesus macaques. AIDS Res. Hum. Retrovir. 14761-773. [DOI] [PubMed] [Google Scholar]
- 75.Verhoeven, D., S. Sankaran, and S. Dandekar. 2007. Simian immunodeficiency virus infection induces severe loss of intestinal central memory T cells which impairs CD4+ T-cell restoration during antiretroviral therapy. J. Med. Primatol. 36219-227. [DOI] [PubMed] [Google Scholar]
- 76.Wang, Z., B. Metcalf, R. M. Ribeiro, H. McClure, and A. Kaur. 2006. Th-1-type cytotoxic CD8+ T-lymphocyte responses to simian immunodeficiency virus (SIV) are a consistent feature of natural SIV infection in sooty mangabeys. J. Virol. 802771-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373117-122. [DOI] [PubMed] [Google Scholar]
- 78.Witvrouw, M., C. Pannecouque, K. Van Laethem, J. Desmyter, E. De Clercq, and A. M. Vandamme. 1999. Activity of non-nucleoside reverse transcriptase inhibitors against HIV-2 and SIV. AIDS 131477-1483. [DOI] [PubMed] [Google Scholar]
- 79.Zhang, L., P. J. Dailey, T. He, A. Gettie, S. Bonhoeffer, A. S. Perelson, and D. D. Ho. 1999. Rapid clearance of simian immunodeficiency virus particles from plasma of rhesus macaques. J. Virol. 73855-860. [DOI] [PMC free article] [PubMed] [Google Scholar]