

Abstract
In flaviviruses it has been proposed that there is a coupling between genome replication and virion assembly and that nonstructural proteins are involved in this process. It was previously reported that mutations in yellow fever virus (YFV) nonstructural protein NS2A blocked production of infectious virus and that this block could be released by a suppressor mutation in NS3. Here, based on studies using a YFV replicon-based trans-packaging system as well as full-length YFV cDNA, we report that mutation of a conserved tryptophan at position 349 in the helicase domain of NS3 blocks production of infectious virus particles, revealing an as-yet-unknown role for NS3 in virus assembly. Mutation of tryptophan 349 to alanine (W349A) had no effect on viral replication, as demonstrated by wild-type levels of viral RNA amplification and protein expression in W349A-transfected cells. Although release of infectious virus was not detected, release of capsidless subviral particles was not blocked. The assembly defect in W349A could be trans-complemented inefficiently using BHK-REP cells (a cell line containing persistently replicating YFV replicon RNA). trans-complementation was also demonstrated by supplying wild-type NS2B-3 or NS3 protein alone as well as by supplying inactive NS2B-3 protein, indicating that this function of NS3 in virus assembly was independent of its known enzymatic functions.
The family Flaviviridae consists of three genera—Pestivirus, which includes agriculturally important viruses like bovine viral diarrhea virus; Hepacivirus, the sole member being hepatitis C virus (HCV); and Flavivirus, the largest of the three genera, comprising more than 80 members (16). Many of these flaviviruses, including pathogens such as yellow fever virus (YFV), dengue virus (DEN), West Nile virus (WNV), Japanese encephalitis virus, and tick-borne encephalitis virus, cause severe human disease with diverse and complex pathologies and have a significant social and economic impact worldwide. Flaviviruses are small, spherical, enveloped viruses containing an approximately 11,000-nucleotide, positive-sense RNA genome. The genomic RNA has a type I cap at its 5′ end to allow translation similar to that seen with cellular messenger RNAs but lacks a polyadenine tract at the 3′ end (16). The viral RNA contains a single long open reading frame, which when translated gives rise to a long polyprotein that is co- and posttranslationally processed by viral and cellular proteases into three structural proteins (capsid [C], premembrane [prM; the precursor form of M], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The viral protease (NS2B-3) is formed by a complex made up of NS3 and its cofactor NS2B, and it cleaves the polyprotein on the cytoplasmic side of the endoplasmic reticulum (ER) membrane between C/prM, NS2A/NS2B, NS2B/NS3, NS3/NS4A, and NS4B/NS5 proteins. Additional cleavages mediated by NS2B-3 have been observed within NS2A, NS3, and NS4A proteins (15). The nonstructural proteins are responsible for various enzymatic activities, including RNA-dependent RNA polymerase (NS5 [RdRp]), helicase (NS3), and protease, and together form the replicase complex (RC) that primarily functions to carry out viral genome replication. The structural proteins, along with the host-derived lipid bilayer and a single copy of the RNA genome, assemble to form the flavivirus virion (15).
The flavivirus virion is ∼50 nm in diameter, with a relatively smooth outer surface that is constructed from 180 copies of both the major E protein and the small M protein. The E and M proteins are anchored to the underlying viral membrane via C-terminal transmembrane domains. This outer glycoprotein and lipid shell encloses a nucleocapsid core (NC), which is composed of the C protein in complex with the viral genomic RNA (13, 23, 35). Our knowledge of the different steps involved in the assembly of flavivirus particles is rudimentary. Presumably, one of the first steps is the association of the C protein with the genomic RNA to form the NC. The NC is then thought to bud into the rough ER in conjunction with the envelope glycoproteins within the ER lumen. However, apart from one observation in the Sarafend strain of WNV, NC or NC-like structures have not been observed in flavivirus-infected cells, suggesting that the formation of NC occurs in parallel with subsequent steps in virus assembly (26). Interestingly, cryoelectron microscopy reconstructions of both DEN2 (DEN serotype 2) and WNV reveal a nucleocapsid core that apparently lacks icosahedral organization or any connectivity with the surrounding viral membrane, indicating that few if any interactions exist between the C protein and the outer E and M proteins (13, 23). Additionally, the E and prM proteins, by themselves, are able to assemble into noninfectious virus-like particles. Virus-like particles are small T=1 particles that are normally produced during flavivirus infections (6, 15). Similar, if not identical, particles have also been produced by expression of the E and prM proteins in several heterologous expression systems, indicating that the structural determinants of enveloped virus assembly (i.e., the ability to form a closed spherical shell) can function independently of the C protein (1, 2, 8, 12, 30).
Taken together, these observations indicate that assembly of infectious virus particles containing NC involves mechanisms other than direct interactions between C and prM-E proteins. Studies of Kunjin virus (KUN) suggest a coupling between flavivirus genome replication and assembly (11). It was shown that infectious virus is produced only in the presence of functionally active RC. Using KUN replicons, it was shown that replication-incompetent RNA fails to be packaged into virus particles despite accumulation of a large amount of positive-strand RNA as well as viral proteins. In contrast, the same RNA could be packaged when its replication was restored by supplying wild-type (WT) replication proteins in trans by use of a helper replicon. These data suggest a functional coupling between replication and packaging and hint at the possibility of the involvement of nonstructural proteins in genome packaging and virus assembly (11). Immunofluorescence and cryoimmunoelectron microscopy of KUN-infected cells revealed the presence of structural proteins within virus-induced membranous structures, termed convoluted membranes (CM), that are presumed to be the sites of virus polyprotein processing and the initial steps in virion assembly. CM are closely associated with membrane-bound structures called vesicle packets which are the sites of flavivirus RNA replication (20). This is not surprising, since the structural proteins would then be able to interact with and package the newly synthesized genomic RNA. This also suggested that there were microenvironments in which the structural and nonstructural proteins could interact in a specific manner.
It was previously reported that mutation of isoleucine 59 to asparagine (I59N) in NS2A of KUN had no effect on genome replication but blocked production of infectious virus, implying the involvement of NS2A in production of infectious KUN particles (17). Previously, studies of an internal cleavage site within NS2A had revealed an unexpected role for NS2A in YFV particle assembly (14). In YFV-infected cells, two forms of NS2A were detected—full-length NS2A (22 kDa) and a truncated form, NS2Aα (20 kDa). It was shown that NS2Aα is generated as a result of a cleavage in the C-terminal region of NS2A by the NS2B-3 protease, specifically after lysine 190 (QK↓T) (24). Replacement of the lysine residue with serine (K190S), which resulted in inhibition of cleavage, also blocked production of infectious virus (14). Surprisingly, RNA and protein levels in cells transfected with in vitro-transcribed YFV RNA containing the K190S mutation were similar to WT levels, indicating that genome replication was not affected by the mutation but that some stage in the life cycle post-RNA synthesis was being blocked. Additionally, second-site revertants that rescued this assembly defect were isolated and mapped to the helicase domain of NS3. These nucleotide changes led to the replacement of aspartate at position 343 in NS3 with valine (D343V), alanine (D343A), or glycine (D343G). These revertants suggested that NS2A and NS3 were together involved in one or more steps in virion morphogenesis and/or release (14). It was also previously shown by Pijlman et al. that the NS3 gene product in KUN was required for efficient RNA packaging (29).
Here we report mutations within the helicase domain of NS3 in YFV that block production of infectious virus. Mutation of a conserved tryptophan at position 349 in NS3 to alanine (W349A) had no effect on the expression of viral proteins or amplification of RNA within transfected cells; however, no infectious virus was detected. This assembly defect could be trans-complemented by supplying the entire array of YFV replicase proteins (by use of BHK-REP cells) or only NS3. Furthermore, it was also demonstrated that protease- or helicase-inactive NS2B-3 protein could trans-complement the assembly defect in W349A, indicating that this as-yet-unknown role for NS3 in virus assembly was independent of its known enzymatic functions.
MATERIALS AND METHODS
Cell culture.
BHK-15 cells obtained from the American Type Culture Collection were maintained in minimal essential medium (MEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS). BHK-REP cells were maintained in Dulbecco's modification of MEM (DMEM) containing 10% FBS and 0.8 mg/ml G418 (Invitrogen). All cells were grown at 37°C in the presence of 5% CO2.
Virus infection and plaque assays.
YFV stocks were generated using standard procedures from pYF23, a plasmid derived from pACNR/FLYF, a full-length cDNA clone of YFV 17D kindly provided by C. M. Rice. For virus infection, supernatants of transfected cells were adsorbed on a monolayer of BHK cells for 1 h at room temperature with gentle rocking. Cells were then overlaid with the appropriate volume of MEM containing 5% FBS. For plaque assays, 10-fold serial dilutions of culture supernatants were made in phosphate-buffered saline (PBS) containing 1% FBS. BHK cells at ∼90% confluence were inoculated with the dilutions for 1 h at room temperature and then overlaid with MEM containing 5% FBS and 1% agarose. Cells were incubated at 37°C for 3 days and stained with 2% crystal violet, and the plaque phenotypes were analyzed.
Plasmids and cloning procedures.
The previously described YF23 plasmid was derived from pACNR/FLYF, a full-length cDNA clone of YFV 17D. The YF-R.luc2A-RP replicon of YFV was derived from YF23, and details of cloning are described elsewhere (9). Sindbis virus (SINV)-based replicons expressing the YFV structural proteins were constructed from pToto64, a full-length cDNA clone of SINV; construction of the SIN-CprME and SIN-DSCprME helper replicon constructs is described by Jones et al. (9). For cloning SINV helper constructs expressing YFV NS2B-3 or NS3 genes, the SINV structural proteins were deleted in pToto64 and an XhoI restriction site was introduced at the end of the subgenomic promoter by use of standard PCR techniques. NS2B-3 or NS3 coding sequences were amplified using PCR and inserted into the XhoI site. All mutations were engineered in YF23 and YF-R.luc2A-RP by use of mutagenic primers and standard PCR mutagenesis techniques. Mutagenized plasmid DNAs were sequenced to confirm the presence of the mutation.
RNA transcription and transfection.
RNA transcripts of YF23, YF-R.luc2A- RP constructs, and SIN-CprME replicons were generated using in vitro transcription and SP6 RNA polymerase (Amersham Biosciences) from DNA templates linearized by digestion with XhoI (for YFV plasmids) and SacI (for SINV replicon) restriction enzymes and were subsequently purified using GFX columns (Amersham Biosciences). For DEAE-dextran transfection, BHK-15 cells were treated with DEAE-dextran for 1 h at 37°C, following which the DEAE-dextran was aspirated and RNA diluted in PBS was added onto the cells. Cells were kept at room temperature for 30 min and then overlaid with MEM containing 5% FBS and 1% agarose. Cells were incubated at 37°C for 3 days and stained with 2% crystal violet, and plaque phenotypes were analyzed. For electroporation of BHK-15 cells (or BHK-REP cells), subconfluent monolayers of cells grown in T-75 culture flasks (∼1 × 106 cells) were harvested by trypsinization and washed twice with PBS before final resuspension in 400 μl PBS. The resulting cells were combined with ∼10 μg of in vitro-transcribed RNA, placed in a 2-mm-gap cuvette (Bio-Rad), and electroporated (two pulses at settings of 1.5 kV, 25 μF, and 200 Ω) using a GenePulser II apparatus (Bio-Rad). Following 5 min of recovery at room temperature, cells were resuspended in MEM supplemented with 10% FBS and incubated at 37°C in the presence of 5% CO2 (for BHK-REP cells, DMEM supplemented with 10% FBS and 0.8 mg/ml G418 was used).
Generation of PIPs.
BHK-15 cells (or BHK-REP cells) were electroporated with ∼10 μg of in vitro-transcribed YF-R.luc2A-RP replicon RNA, and resuspended cells were plated in a T-75 flask and incubated at 37°C for 24 h. To initiate production of pseudoinfectious particles (PIPs), the electroporated cells were subjected to a second round of electroporation using SIN-CprME transcripts and incubated at 37°C. Cell supernatants from the cotransfected cells were harvested at 12 to 15 h following the second transfection, and 100 μl of supernatant was used to infect naïve BHK cells. Cell extracts were taken from these cells at 24 h postinfection, and luciferase assays were performed.
Luciferase assays.
To determine Renilla luciferase activity, cells were washed with PBS and lysed using Renilla lysis buffer, and lysates were stored at −80°C. Prior to assay, frozen extracts were thawed and then homogenized by brief vortexing. Luciferase activity was initiated by addition of 10 μl of cytoplasmic extracts to 50 μl of Renilla luciferase substrate (Promega). Luciferase activity was detected using a Lumat luminometer system (Berthold) and measured in raw light units (RLU).
RNA extraction and RT-PCR.
Electroporated cells were washed three times with PBS at 24 h postelectroporation before lysis to obtain cytoplasmic extracts. Cytoplasmic RNA was then extracted using an RNeasy Mini kit (Qiagen). Reverse transcriptase PCRs (RT-PCRs) were set up for different dilutions of the isolated RNA transcripts (in water) by use of a cMaster RTplusPCR system (Eppendorf) one-step RT-PCR method according to the manufacturer's recommendations. Nucleotides 5974 to 6579 of the YFV genome were amplified using the following primers: 5′-ATCCCAACAGAGATGGAGACTCAT/+ and 5′-CATTGCCTCAGGCATCATTGA/−.
Western blot analysis of cytoplasmic extracts.
Cytoplasmic extracts of transfected cells were generated using Renilla lysis buffer (Promega) and centrifuged at 3,000 rpm for 5 min to pellet membranous cell debris. The supernatants were separated by electrophoresis on a 13% Bis-Tris acrylamide gel under denaturing conditions. Separated proteins were transferred onto a nitrocellulose membrane, and immunoblotting was performed using standard procedures. Primary polyclonal antibodies against the YFV E protein (obtained from J. Strauss), YFV helicase (obtained from J. Strauss), and α-actin (Chemicon) were diluted 1:100, 1:4,000, and 1:2,000, respectively, in PBS containing 0.1% Tween 20. For the YFV E and NS3 blots, secondary antibody against mouse conjugated with IR680 fluorescent dye was diluted 1:10,000 in PBS containing 0.1% Tween 20. For the actin blot, secondary antibody against rabbit conjugated with IR800 fluorescent dye was diluted 1:10,000 in PBS containing 0.1% Tween 20. The blots were visualized and analyzed using an Odyssey scanner (LI-COR).
IFA.
Immunofluorescence assays (IFAs) were performed using BHK-15 cells grown on plastic coverslips. At 24 h postelectroporation, cells were fixed with methanol at room temperature for 15 min. Following fixation, cells were washed with PBS and incubated with 10 mg/ml bovine serum albumin (BSA) solution in PBS. Cells were then incubated with the primary antibody for 45 min at room temperature with gentle rocking. A monoclonal primary antibody against YFV NS1 (obtained from J. Schlesinger) was diluted 1:500, and monoclonal antibodies against YFV E (Chemicon) and polyclonal YFV helicase were both diluted 1:100 in 10 mg/ml BSA solution. Following primary antibody incubation, cells were washed with PBS and incubated with a 1:200 dilution of Texas red-conjugated monoclonal anti-mouse antibody (Chemicon) in 10 mg/ml BSA solution. Following secondary antibody incubation, cells were washed with PBS and coverslips were mounted on a slide, cell side down, in Fluorosave reagent (Gibco) to be examined by fluorescence microscopy. Fluorescent images were visualized using an epifluorescent microscope equipped with a digital charge-coupled device camera (Nikon). For calculating the number of cells expressing E protein, five random fields were counted and used to calculate the total number of cells using the formula N ↔ (Swell/SIA), where N is the average number of E-expressing cells within an image area determined from four randomly chosen fields, Swell is the surface area of the coverslips (200 mm2), and SIA is the surface area of the image (0.53 mm2).
Purification of viral and subviral particles.
BHK-15 cells were electroporated with YF23 transcripts and incubated at 37°C. Cells were metabolically labeled from 20 h to 40 h postelectroporation in methionine-deficient MEM containing 5% FBS and 50 μCi/ml [35S]methionine (to label viral proteins) and 50 μCi/ml [3H]uridine (to label viral RNA). Cell supernatants were harvested at 40 h, and released particles were pelleted by centrifugation at 100,000 × g for 90 min at 4°C (Beckman Ti-100 rotor) (60,000 rpm). Pellets were resuspended in TNE buffer (100 mM Tris, 2.0 M NaCl, 10 mM EDTA [pH 7.4]) and analyzed by rate zonal gradient centrifugation. Discontinuous sucrose gradients (10%, 35%, and 50% sucrose in TNE buffer containing 0.1% bovine serum albumin) were utilized to achieve an optimal separation of virions from subviral particles (SW 41 rotor [Beckman]) (38,000 rpm, 6 h, 4°C). Gradients were fractionated, and radioactivity in the fractions was measured by scintillation counting.
RESULTS
Mutations of residues within the E338-D354 loop of the YFV NS3 helicase.
As previously reported, a mutation of Asp343 in the helicase domain of NS3 to either valine, alanine, or glycine could rescue the assembly defect of the Lys190Ala mutation of the internal cleavage site within NS2A, suggesting that NS2A and NS3 are involved together in virion assembly (14). However, mutation of Asp343 to valine, alanine, or glycine in the absence of mutation at the Lys 190 within NS2A had no effect on virus growth (14). Asp343 lies on a solvent-accessible loop in domain 2 of the helicase, and this loop, encompassing residues Glu338 to Asp354, points away from the core of the domain (see Fig. 2D). This raised the possibility that residues on this loop other than Asp343 are involved in putative interactions with NS2A (and possibly other proteins) during virus assembly. With the purpose of identifying residues that might be involved in interactions important for virus assembly, alanine-scanning mutagenesis of residues within the Glu338-Asp354 loop was performed.
FIG. 2.

Mutation of Trp349 in YFV NS3 to Ala (W349A) blocks virus production. (A) Plaque phenotype analysis of YF23-W349A. Plaque assays were performed using WT or W349A YF23 transcripts. (B) Replication of YF-R.luc2A-RP-W349A. YF-R.luc2A-RP replicon transcripts, either WT or W349A, were electroporated into BHK-15 cells, and luciferase activity was measured. Data represent the results of three experiments. The error bars indicate standard deviations. (C) Packaging of YF-R.luc2A-RP-W349A by use of the trans-packaging assay. Packaging assay was performed to generate WT or W349A (bar labeled “A”) YF-R.luc2A-RP-containing PIPs. Luciferase activity is denoted as the log of RLU. The dashed line indicates mock levels of packaging. Data represent the results of three experiments. The error bars indicate standard deviations. (D) Ribbon diagram representation of the structure of the helicase domain of YFV NS3. Domain 1 is shown in green, domain 2 is brown, and domain 3 is blue. Trp349 (W349), shown in red, is present on a solvent-accessible loop in domain 2. Asp343 (D343) is also indicated in magenta. Figures were generated using Pymol (DeLano Scientific).
Double or triple alanine substitutions of residues within this loop (including flanking residues) were introduced in YF23, the YFV cDNA, by use of PCR mutagenesis. Full-length transcripts of these substitutions were transfected into BHK-15 cells by use of DEAE-dextran, and the plaque phenotypes were analyzed at 72 h posttransfection. Alanine substitutions of Gln341/Thr342/Asp343 (Q341A/T342A/D343A) produced plaques with a wild-type phenotype, and alanine substitutions of Glu338/Asp339 (E338A/D339A) or Ile344/Pro345/Ser346 (I344A/P345A/S346A) or His353/Asp354 (H353A/D354A) produced small plaques (Fig. 1A). However, no plaques were obtained with substitutions of Glu347/Pro348/Trp349 (E347A/P348A/W349A) or Trp349/Gln350 (W349A/N350A) with alanine, indicating that no infectious virus was produced in those mutants. The D343V mutation was engineered as a control and, as observed by Kümmerer et al. (14), plaques having a wild-type phenotype were obtained.
FIG. 1.

Substitutions in the YFV NS3 helicase E338-D354 loop. (A) Plaque phenotype analysis of mutations of residues within the E338-D354 loop of the YFV NS3 helicase. BHK-15 cells were transfected with either WT or mutated YF23 transcripts. At 3 days posttransfection, cells were stained with crystal violet and plaque phenotypes analyzed. (B) Replication of YF-R.luc2A-RP replicon containing mutations of residues within the E338-D354 loop of the YFV NS3 helicase. YF-R.luc2A-RP replicon transcripts, either WT or containing the alanine substitutions of indicated residues, were electroporated into BHK-15 cells, and luciferase activity was measured at various time points. Luciferase activity is denoted as the log of RLU. Data represent averages of the results of three experiments. The error bars indicate standard deviations. (C) Packaging of YF-R.luc2A-RP containing mutations of residues within the E338-D354 loop of the YFV NS3 helicase by use of the trans-packaging assay. The released PIPs were used to infect naïve BHK-15 cells, and luciferase activity in these cells was measured at 24 h postinfection. Luciferase activity is denoted as the log of RLU. The dashed line indicates mock levels of packaging. Data represent averages of the results of three experiments. The error bars indicate standard deviations.
To determine whether the plaque phenotypes obtained had occurred because of defects in RNA replication or in some subsequent step, the above-mentioned mutations were introduced in the YF-R.luc2A-RP replicon. The YF-R.luc2A-RP replicon expresses the Renilla luciferase gene in a replication-dependent manner, and as demonstrated previously, luciferase activity in transfected cells can be directly correlated to replication of the replicon (9). In vitro-transcribed RNA was electroporated into BHK-15 cells, and cell extracts were taken at 12, 24, 36, and 48 h postelectroporation. Luciferase activity in the cell extracts was measured and compared with the luciferase activity in cells transfected with the wild-type YF-R.luc2A-RP replicon RNA (Fig. 1B). As expected from the plaque phenotype analysis, the Q341A/T342A/D343A mutations had wild-type levels of luciferase activity. However, luciferase levels for alanine substitutions of E338A/D339A and I344A/P345A/S346A were lower than wild-type levels at all time points, suggesting that those mutations have a detrimental effect on RNA replication. At the moment it is unclear how these mutations caused a reduction in RNA replication. In contrast, alanine substitutions of E347A/P348A/W349A, W349A/N350A, and H353A/D354A had no measurable effect on RNA replication, as indicated by the wild-type levels of luciferase activity. This suggested that the defect caused by these mutations is probably at a step subsequent to RNA replication.
To check whether the defect was in genome packaging and virus release, YF-R.luc2A-RP replicons containing the aforementioned loop mutations were packaged into PIPs by use of a trans-packaging protocol established previously that involved supplying the YFV structural proteins in trans by use of a helper SINV construct (SIN-CprME) (9). As expected, the packaging efficiency of Q341A/T342A/D343A was similar to that seen with the wild type (Fig. 1C). In agreement with their plaque phenotypes, the luciferase activity for the E338A/D339A and I344A/P345A/S346A PIPs was significantly reduced and that for the H353A/D354A mutation was moderately reduced compared to wild-type activity. The lower packaging efficiency for E338A/D339A and I344A/P345A/S346A most likely occurred because of the reduced replication of the RNA containing these mutations. Although replication of H353A/D354A, based on luciferase levels, was similar to that seen with the wild type, it is possible that RNA amplification was slightly less than wild-type levels and that this disparity could account for the modest difference in packaging efficiency. The RNA levels in cells transfected with the H353A/D354A mutation need to be accurately determined to confirm this supposition. Interestingly, E347A/P348A/W349A and W349A/N350A PIPs showed mock levels of luciferase activity (i.e., luciferase activity obtained by using SIN-prME helper replicon for packaging YF-R.luc2A-RP replicon) in spite of wild-type levels of RNA replication. Thus, based on plaque phenotype analysis as well as replicon-based replication and packaging assays, it can be concluded that the E347A/P348A/W349A and W349A/N350A mutations had no measurable effect on RNA replication but blocked production of infectious particles.
NS3 W349A mutation blocks production of infectious virus.
Since the E347A/P348A/W349A and W349A/N350A mutations had specific defects in packaging, single amino acid substitutions were engineered in YF23 as well as the YF-R.luc2A-RP replicon to yield constructs containing point mutations E347A, P348A, W349A, and N350A. Full-length transcripts of these point mutations were transfected into BHK-15 cells by use of DEAE-dextran, and plaque phenotypes were analyzed at 72 h posttransfection. Plaques were obtained for E347A, P348A, and N350A (data not shown), but no plaques were observed for the W349A mutation (Fig. 2A). Analysis of mutated YF-R.Iuc2A-RP replicons revealed wild-type levels of luciferase activity, suggesting equivalent levels of replication of wild-type and mutated YF-R.luc2A-RP replicons in transfected cells (Fig. 2B and data not shown). Interestingly, the trans-packaging assay revealed that, except for the YF-R.luc2A-RP replicon containing the W349A mutation, all other mutated replicons were packaged efficiently into PIPs (Fig. 2C and data not shown). Taken together, these data suggest that the W349A mutation had no detectable effect on genome replication but resulted in a block in assembly and/or release of infectious particles. Importantly, W349 is the only residue shared by the E347A/P348A/W349A and W349A/N350A mutations that were shown to have an assembly defect (see previous section).
RNA synthesis and protein expression are not affected in NS3 W349A.
Determination of levels of luciferase activity of the YF-R.luc2A-RP replicon is an indirect method to measure virus protein production and RNA replication. In order to directly compare viral RNA and protein amounts in wild-type or W349A-transfected cells, BHK-15 cells were transfected with in vitro-transcribed YF23 or YF23-W349A RNA. At 24 h posttransfection, viral RNA was isolated from the cell cytoplasm and semiquantitative RT-PCR (20 cycles) was performed using YFV-specific primers to amplify and quantify positive-strand viral RNA. In order to ensure that the RT-PCR was indeed quantitative and that the PCR products obtained were representative of the actual viral RNA amounts, RNA transcripts were diluted 1:2, 1:10, and 1:100 and RT-PCR was performed. As shown in Fig. 3A, the RNA amounts amplified from wild-type and W349A-transfected cells were essentially similar, indicating that RNA replication of W349A was similar to that seen with the wild type. A closer observation reveals that the WT RNA amounts were in fact slightly lower than the W349A RNA amounts, probably as a consequence of virus release from WT-transfected cells but not from W349A-transfected cells.
FIG. 3.

W349A does not impact viral RNA synthesis or protein expression. (A) Analysis of viral RNA amounts by use of RT-PCR. BHK-15 cells transfected with either WT or W349A YF23 transcripts and cytoplasmic viral RNA were isolated. Semiquantitative RT-PCR was performed using different dilutions of the RNA transcripts, and the PCR products obtained were compared. As a control for DNA carryover, parallel reactions without reverse transcriptase (- lanes) were performed (only the undiluted RNA transcripts were used in this reaction). RNA was also extracted from untransfected (un) BHK-15 cells and used for RT-PCR analysis as a negative control (lane M). (B) Analysis of viral protein amounts by use of Western blotting. Viral proteins were detected using anti-YFV E or anti-YFV helicase primary antibody followed by addition of secondary anti-mouse antibody conjugated with IR680 fluorescent dye. Antiactin antibody was used to detect actin (loading control). The blot was visualized and analyzed using an Odyssey scanner (LI-COR). (C) Analysis of viral protein expression using immunofluorescence. At 24 h postelectroporation, immunofluorescence analysis of transfected cells was performed using anti-YFV E, anti-YFV helicases, and anti-YFV NS1 primary antibody. The anti-mouse secondary was conjugated with Texas Red fluorophore. Untransfected BHK-15 cells (mock) served as the negative control. E- or NS3- or NS1-expressing cells are indicated by red fluorescence.
In addition, viral protein amounts in transfected cells were analyzed by Western blotting of cell extracts and immunofluorescence. Cell extracts of transfected cells were examined in a Western blotting assay using anti-E and anti-NS3 antibody. The amounts of E and NS3 in wild-type and W349A-transfected cells were found to be essentially equivalent, confirming that there was no defect in protein expression and stability (Fig. 3B). Immunofluorescence assays were also performed using anti-E, anti-NS1, and antihelicase antibodies to check for expression of E, NS1, and NS3 proteins, respectively. Viral protein expression and distribution results for cells transfected with YF23-W349A RNA were similar to wild-type transfected-cell results (Fig. 3C). Thus, these experiments confirmed that the W349A mutation had no measurable effect on viral genome amplification or protein expression but blocked some step in virus assembly, release, or infection.
NS3 W349A does not block release of subviral particles.
The results thus far indicated that the W349A mutation blocked production of infectious virus despite efficient viral protein expression and RNA amplification. To check whether noninfectious virus particles were released, culture supernatants of transfected cells were analyzed for the presence of viral proteins and RNA. BHK-15 cells were electroporated with either wild-type or W349A transcripts followed by metabolic labeling of the cells using [35S]methionine and [3H]uridine at 20 h postelectroporation. At 36 h postelectroporation, medium on the cells was harvested and pelleted at 38,000 rpm through a 24% sucrose cushion. The pellet was resuspended in TNE buffer, and particles were separated on a discontinuous sucrose gradient by rate-zonal centrifugation at 38,000 rpm for 7 h. A discontinuous gradient of 10%, 35%, and 50% sucrose was employed, since clear separations of subviral and viral particles are obtained at the 35% and 50% boundaries, respectively. Following gradient fractionation, analysis of 35S scintillation counts revealed distinct peaks corresponding to both viral and subviral particles for the wild type (Fig. 4). Interestingly, peaks corresponding to both viral and subviral particles were obtained for W349A as well, but the viral particle peak was much smaller than the peak obtained for wild-type virus particles. Not surprisingly, a 3H peak overlapping with the 35S viral particle peak was obtained for the wild type and for the mutant W349A, indicating that viral particles most likely containing viral RNA genome were released from both wild-type and W349A-transfected cells. To check whether the peaks obtained consisted of virus particles, the viral peak fractions were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis to visualize viral proteins. As expected, bands for E and C were detected for the wild type, but no clear bands were detected for W349A (data not shown). Plaque assays of the peak fractions were performed, and plaques were readily obtained with wild-type peak fractions (titer = ∼1 × 106 PFU/ml) but not with those of W349A, thus confirming that the “virus” peak obtained for W349A was not due to wild-type virus contamination but was perhaps due to the release of limited amounts of noninfectious virus particles. Based on these results we can conclude that, in similarity to the results seen with the K190S mutation in NS2A, the W349A mutation in NS3 blocks production of infectious virus particles but not of subviral particles, although it is unclear whether small amounts of noninfectious virus particles were being released.
FIG. 4.
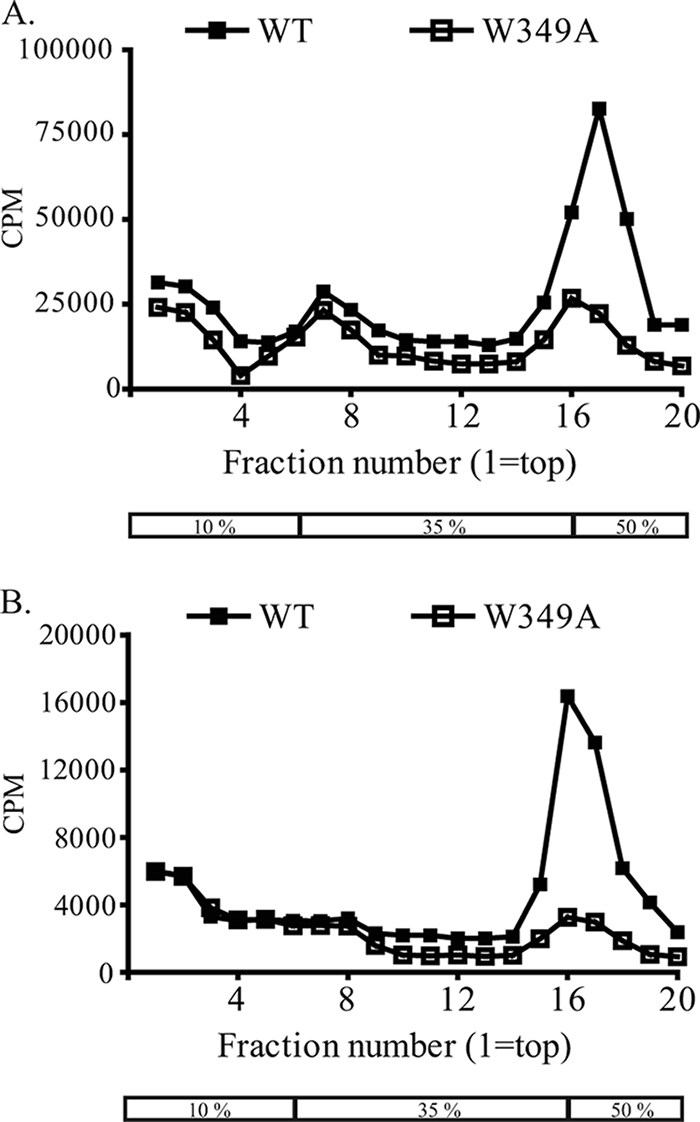
W349A does not impair release of subviral particles. BHK-15 cells were transfected with full-length transcripts of either WT (shown as solid squares) or W349A (shown as open squares) and were metabolically labeled with [35S]methionine (A) and [3H]uridine (B). Supernatants were harvested, pelleted, and sedimented on a discontinuous sucrose gradient; the box indicates the 10%, 35%, and 50% zones. The gradient fractions were analyzed by scintillation counting, and scintillation counts obtained for each fraction were plotted. The scintillation profile for 35S is shown in panel A and for 3H in panel B.
Replacement of W349 with aromatic amino acids is tolerated for virus assembly.
Sequence alignment of flavivirus NS3 shows that W349 is conserved among flaviviruses (Fig. 5A). Alignment of YFV and HCV helicase domain sequences revealed the presence of tyrosine at the position corresponding to W349 in HCV (in genotypes 1a, 1b, and 2a). To check whether an aromatic amino acid can replace W349 in YFV, substitutions of phenylalanine (W349F), tyrosine (W349Y), and histidine (W349H) were made at the 349 position in YF23 and in YF-Rluc2A-RP for plaque phenotype analysis and production of PIPs. Nonaromatic substitutions with glutamine (W349Q), leucine (W349L), and lysine (W349K) were also engineered into YF23 and YF-Rluc2A-RP. YF23 containing W349F produced plaques with a wild-type phenotype, and W349Y produced small plaques, but none of the other substitutions produced detectable plaques, suggesting that these substitutions blocked production of infectious virus (Fig. 5B). Luciferase activity within cells transfected with YF-R.luc2A-RP replicons containing the different substitutions of W349 was essentially similar to wild-type activity, suggesting that genome replication was not affected by these mutations; these were subsequently analyzed for production of PIPs in the trans-packaging assay. In accordance with the plaque assay results, W349F had no defect and W349Y was moderately defective (Fig. 5C). While no plaques were obtained with YF23-W349H, production of PIPs, although significantly reduced, was detected with W349H in the trans-packaging assay. This discrepancy could be attributed to the fact that the luciferase-based trans-packaging assay is more sensitive than a plaque assay, and perhaps the low level of infectious virus produced in the W349H mutant was below the detection levels of a plaque assay. However, YF-R.luc2A-RP replicons containing W349Q, W349L, or W349K mutations were nonfunctional in production of PIPs, in similarity to the results seen with the W349A mutation. These observations point to the requirement of an amino acid with an aromatic ring at position 349 in the helicase domain of YFV NS3.
FIG. 5.

An aromatic amino acid is required at position 349. (A) Multiple sequence alignment of NS3 protein from flaviviruses and HCV, showing the region including the solvent-accessible loop in the helicase domain of NS3. Identical residues are denoted by an asterisk, conserved residues are indicated by double dots, and partially conserved residues are indicated by single dots. JEV, Japanese encephalitis virus; TBE, tick-borne encephalitis virus; HCV, hepatitis C virus genotype 1a H77 strain. (B) Plaque phenotype analysis of YF23 containing W349 substitutions. Plaque assay was performed using either WT YF23 or YF23 containing W349 substitutions as described previously. (C) Replication and packaging of YF-R.luc2A-RP containing W349 substitutions in BHK cells. (D) trans-complementation of YF-R.luc2A-RP containing W349 mutations by use of BHK-REP cells. Luciferase activity of YF-R.luc2A-RP replicon transcripts containing NS3 mutations in BHK or BHK-REP cells was measured at various time points. The gray bars represent luciferase activity at 36 h postelectroporation. Packaging assay was performed as before, and the luciferase activity of PIPs is represented by the white bars. The wild-type YF-R.luc2A-RP replicon is denoted by WT, and W349 mutations are denoted by one-letter codes for the specific amino acid substitutions. Luciferase activity is indicated as the log of RLU. The dashed line indicates mock levels of packaging. Data represent averages of the results of three experiments. The error bars indicate standard deviations.
Assembly defects in NS3 W349 mutations can be trans-complemented.
It was of interest to examine whether the packaging defect in the W349 mutations could be complemented by supplying functional NS3 protein in trans, and for this purpose BHK-REP cells were utilized. BHK-REP is a BHK-15-derived cell line that contains persistently replicating YFV replicon RNA selected using neomycin antibiotic selection (9). Thus, BHK-REP cells function to supply wild-type viral replicase proteins that can be utilized to complement defective viral proteins.
In vitro-transcribed YF-R.luc2A-RP replicon RNAs, either wild type or containing W349 mutations, were electroporated into BHK-REP cells. Cell extracts were taken at 12, 24, 36, and 48 h postelectroporation, and luciferase activity in the cell extracts was measured. As expected, the mutants showed wild-type levels of luciferase activity (Fig. 5D). The different W349 mutations electroporated in the BHK-REP cells were subsequently tested for packaging in the trans-packaging assay. In this case, PIPs were produced by sequential electroporation of either wild-type or mutated YF-R.luc2A-RP replicon RNA followed by SIN-CprME RNA in BHK-REP cells. PIPs produced were then used to infect naïve BHK cells for analysis of the efficiency of virus assembly. As indicated by the luciferase levels within PIP-infected cells, all the mutations were trans-complemented using BHK-REP cells, albeit inefficiently (Fig. 5D). The inefficient trans-complementation also suggests that NS3 might function more effectively in cis.
We next sought to investigate whether NS3 alone could trans-complement the assembly defect in W349A. A SINV helper replicon, in which the genes coding for the SINV structural proteins were replaced with coding sequences for either YFV NS2B-3 (SIN-NS2B3) or NS3 (SIN-NS3), was utilized for supplying functional NS2B-3 or NS3. BHK-15 cells were coelectroporated with transcripts of W349A and SIN-NS2B3 or SIN-NS3 or were mock electroporated. Wild-type YF23 transcripts were also transfected in parallel as a positive control. Electroporated cells were then diluted fourfold with naïve BHK-15 cells, divided into aliquots, added to a 24-well plate, and incubated at 37°C. Immunofluorescence using anti-E antibody was performed at 20 h and 40 h postelectroporation. At 20 h, equivalent numbers (approximately 5% of plated cells) of E-positive cells were observed for all transfections, indicating that similar amounts of cells were initially transfected. At 40 h for the wild-type transfection, almost all the cells in the observed well were expressing E protein, indicating that there was virus release from the transfected cells and reinfection of the untransfected BHK cells. A 16-fold increase in the number of cells expressing E protein was observed at 40 h postelectroporation. As expected, no increase in the number of cells expressing E protein was observed for the W349A-transfected cells, suggesting that no infectious virus was released from the cells transfected with W349A (Fig. 6A). There was a modest increase in the number of E-positive cells in wells with cotransfections of W349A plus SIN-NS2B3 and of W349A plus SIN-NS3, suggesting trans-complementation of the assembly defect in W349A by NS2B-3 and by NS3, respectively. The approximate numbers of E-expressing cells at 40 h postelectroporation in wells with transfections of W349A plus SIN-NS2B3 and of W349A plus SIN-NS3 increased by approximately 2.4- and 2.3-fold, respectively. Additionally, naïve BHK cells were infected with the culture media of electroporated cells and immunostaining using anti-E antibody was performed on these cells. E-expressing cells were observed on infection with culture supernatants of W349A plus SIN-NS2B3 or of W349A plus SIN-NS3 but not with W349A-transfected cells, suggesting that infectious virus was released into the media (data not shown). To eliminate the possibility that the observed trans-complementation was a consequence of genetic recombination, plaque assays were performed with supernatants of coelectroporated cells. No plaques were detected, suggesting that the virus particles released contained the W349A genome, thus confirming that rescue of the assembly defect was a result of true trans-complementation using a nonstructural replication protein.
FIG. 6.

Assembly defects in W349A can be complemented by supplying NS3 protein in trans. (A) trans-complementation of W349A assembly defect by supplying NS3 or NS2B-3 protein alone. BHK-15 cells were electroporated with YF23-W349A transcripts, either alone or together with SIN-helper constructs expressing either NS2B-3 (+SIN-NS2B3) or NS3 (+SIN-NS3) proteins. (B) trans-complementation of W349A assembly defect by supplying enzymatically inactive NS3 protein. BHK-15 cells were electroporated with YF23-W349A transcripts, either alone or together with SIN helper constructs expressing either protease-inactive [+SIN-NS2B3(S138A)] or helicase-inactive [+SIN-NS2B3(R461Q)] NS2B-3 proteins. For both panels A and B, immunofluorescence assays were performed at 20 h (top panel) and 40 h (bottom panel) postelectroporation to detect E protein. The changes in the numbers of positive cells at 40 h compared to the numbers of positive cells at 20 h postelectroporation are reported as severalfold increases.
Enzymatic functions of NS3 are not required for trans-complementation of W349A.
As mentioned previously, NS3 functions as the viral protease and helicase. To determine whether the enzymatic functions of NS3 that are vital for virus replication are also important for virus assembly, mutations of Ser138Ala and of Arg461Gln were engineered into the SIN-NS2B3 construct to obtain SIN-NS2B3(S138A) and SIN-NS2B3(R461Q), respectively. Ser138, along with His51 and Asp76, is part of the catalytic triad that is conserved among all serine proteinases, and the S138A mutation has been shown previously to render the protease inactive (3). Arg461 lies within the conserved motif VI of the helicase, which is involved in ATP binding; thus, the R461Q mutation results in inactivation of the ATPase and helicase functions of NS3 (33) (A. Bera, unpublished data). BHK-15 cells were coelectroporated with transcripts of W349A and SIN-NS2B3(S138A) or SIN-NS2B3(R461Q), and immunofluorescence was performed at 20 h and 40 h postelectroporation. As can be seen in Fig. 6B, the number of cells expressing E protein for the W349A plus SIN-NS2B3(S138A) electroporation increased by 2.7-fold at 40 h postelectroporation compared to the results seen with 20 h postelectroporation, indicating release of infectious virus by trans-complementation of the assembly defect in W349A followed by reinfection of neighboring untransfected cells. Similarly, the number of E-positive cells for the W349A plus SIN-NS2B3(R461Q) electroporation increased approximately threefold at 40 h postelectroporation. Thus, the enzymatic activities of NS3 were not required for trans-complementation of the assembly defect in W349A.
DISCUSSION
Coupling between RNA replication and packaging has been observed or suggested to occur in several positive-strand RNA viruses (11, 27). Such coupling imposes a requirement that the RNAs to be packaged must be generated from active replication complexes, which minimizes the amplification and transmission of defective viral RNA that arise because of the high copy-error rate seen with the viral RdRp. NS3 is an approximately 70-kDa (YFV NS3 contains 623 amino acids), highly conserved protein and performs several enzymatic functions that are critical for virus replication. The N-terminal third of NS3 (the first 180 amino acids) is a serine protease; however, the viral protease becomes active only after forming a stable complex with NS2B (3). The C-terminal two-thirds of NS3 contain regions with homology to the DEAD family of RNA helicases and to an RNA-stimulated nucleoside triphosphatase (NTPase) (32). Atomic structures for the complex of NS2B with the protease domain of NS3 (NS3pro) of DEN2 and WNV and of the helicase domain of NS3, with bound nucleotide as well as free enzyme, have been solved for DEN2 and YFV, and the structure of the entire NS3 protein has recently been reported (5, 19, 33, 34). Observations suggest a role for NS3 in virus assembly as well. In KUN, expression of full-length NS3 protein was required in cis for the production of infectious virus (18, 29). However, it was demonstrated that YFV assembly did not require NS3 in cis, suggesting that these distantly related viruses may carry out the process of assembly by different mechanisms (9). In any case, these observations point to the involvement of nonstructural proteins such as NS3 in virus assembly.
This idea was strongly supported by observations that second-site mutations in YFV NS3 (at an Asp343 that lies on a solvent-accessible loop in domain 2 of YFV helicase) could compensate for mutations in NS2A (Lys190) that blocked production of infectious virus (14). This led to the hypothesis that NS2A and NS3 are involved together in virus assembly, although the mechanism of virus assembly is obscure. Interestingly, mutations of NS3 Asp343 in the absence of mutations in NS2A had no effect on infectious virus production. Based on these results, we speculated that residues adjacent to Asp343, perhaps within this loop of the helicase domain of NS3, were involved in interactions with other viral (possibly NS2A) or host proteins that mediate infectious virus assembly and/or release. Indeed, as shown here, analyses of alanine by scanning mutations of residues in the loop revealed a role for tryptophan 349 in the helicase domain of YFV NS3 in virion assembly. Substitution of Trp349 with Ala (W349A) had no measurable effect on viral RNA replication or protein expression but blocked some step in the assembly and/or release of infectious virus particles. Furthermore, virus was produced with mutation of W349 to Phe (W349F) or Tyr (W349Y) or His (W349H), suggesting the requirement of an aromatic amino acid at position 349 in NS3 for infectious virus production.
Analysis of culture supernatants of transfected cells revealed that, although the W349A mutation blocked production of RNA and C containing viral particles, in similarity to the results seen with the cleavage site mutations in NS2A, the release of M and E containing subviral particles from transfected cells was detected. Analysis of gradient fractions of W349A supernatants revealed a small peak that corresponds with the peak obtained for wild-type viral particles, suggesting that there might be viral particles released from W349A-transfected cells. Although it could not be conclusively demonstrated, it is likely that this peak contained larger particles, as observed previously in culture supernatants of cells expressing recombinant tick-borne encephalitis virus prM and E proteins (2). However, although unlikely, it is also possible that noninfectious virus particles were released from W349A-transfected cells or that mature viral particles were released but not in amounts required for initiating a productive infectious cycle. Unfortunately, the correlation between the total amount of viral particles and the number of infectious particles released is not well understood for YFV, and so the possibility that in addition to subviral particles, small amounts of viral particles (either infectious or noninfectious) are being released cannot be completely eliminated.
As mentioned previously, YFV replicons harboring deletions within NS3 could be packaged into PIPs when trans-complemented using BHK-REP cells, suggesting that NS3 was not required in cis for YFV assembly (9). Not surprisingly, the assembly defect in W349A could be complemented using BHK-REP cells, albeit inefficiently, suggesting that the activity of NS3 in virus assembly is more efficient in cis than in trans. trans-complementation was also demonstrated by supplying NS2B-3 or NS3 proteins, although it was even less efficient. NS3 has been shown to be involved in interactions with the viral RNA, and with NS2B and NS5 proteins, and is an integral part of the viral RC. trans-complementation of defective NS3 would presumably involve its dissociation from multiple interactions and insertion of functional NS3 into the RC. Not surprisingly, it has been previously shown that trans-complementation of replication of YFV and KUN replicons harboring large deletions within NS3 by use of appropriate BHK-REP cells was inefficient (9, 18). The W349A mutation had no effect on viral protein expression and genome replication, suggesting that the protease and helicase enzymatic functions of NS3 that are vital for viral genome replication were independent of the function of NS3 in virus assembly. Not unexpectedly, the assembly defect in W349A could be trans-complemented by supplying enzymatically deficient NS3 protein harboring either Ser138Ala or Arg461Gln mutations.
Perhaps the simplest explanation for the defect in the W349A mutation is the possibility that the nucleotide changes in the W349A RNA template alter an encapsidation signal that is required for specific genomic RNA packaging into infectious particles. Although no encapsidation sequences have been recognized for flaviviruses, such signals have been identified on the genomes for other RNA viruses, including alphaviruses (7). Mutations of these encapsidation signals result in virus production defects. However, the fact that W349F, W349Y, and W349H mutations are viable strongly suggests that it is the amino acids encoded that are important and not the template sequence. Moreover, the W349A replicon could be trans-packaged into infectious particles by supplying wild-type NS3 protein, implying that the RNA sequences in the vicinity of the mutations in NS3 do not represent part(s) of the packaging signal.
The NS2B-3 protease cleaves the signal sequence at the C terminus of the C protein on the cytoplasmic side of the ER to release the mature C protein. It was shown that this cleavage is required for production of infectious virus particles, and it was proposed that this cleavage by NS2B-3 is the critical step that initiates virion assembly (31). It is appealing to suggest that the W349A mutation hinders the NS2B-3-mediated C-prM cleavage, leading to a block in virus production. In order to check whether the block in virus assembly in W349A occurred because of anomalous cleavage at the C-prM site, a SIN helper construct (termed SIN-DSCprME) in which the YFV C and prM-E proteins were expressed from two separate subgenomic promoters was utilized to package the W349A replicon in the trans-packaging assay. Since the C and prM proteins were expressed separately, there was no need for NS2B-3-mediated cleavage. However no PIPs were obtained, suggesting that the defect in W349A was not dependent on NS2B-3-mediated C-prM cleavage (data not shown). W349 is located in domain 2 of the helicase domain of NS3 and has not been shown to be involved in the protease activity of NS3. Indeed, the W349A mutation has no measurable effect on RNA replication, suggesting that NS2B-3-mediated cleavages of the polyprotein occurred normally.
Although direct interactions have not yet been demonstrated, NS3 and NS2A have been proposed to interact with each other as well as with the viral RNA during formation of the RC as well as during transportation of the RNA template to virus-induced vesicle packets that are the sites of viral genome replication (10, 21). Following RNA synthesis and translation, the nascent viral RNA is transported to the rough ER where assembly of the NC and immature virion is thought to occur (20). One possible scenario is that NS3 and NS2A together are involved in mediating the transport of nascent RNA to the rough ER and that the W349A mutation precludes such an interaction with NS2A and/or RNA to form a functional complex that is competent for RNA transport. Amino acids with aromatic ring structures have been shown to be involved in base-stacking interactions with nucleic acids, thus suggesting a possible function for the aromatic side chain at position 349.
As mentioned above, although it is unlikely that the assembly defect in the W349A mutation is a consequence of inefficient NS2B-3-mediated cleavage of the C protein, the possibility of direct interactions between the C and a NS2A-NS3 complex (or NS3 alone) during assembly cannot be ruled out. HCV core protein has been shown to interact with host cell protein DDX3, a putative RNA helicase and a member of the highly conserved superfamily of DEAD/H-box proteins that includes the flavivirus helicase (28). It is unlikely that NS2A-NS3 (or NS3) interacts with the prM or E proteins during particle assembly, since subviral particles can be produced in the absence of nonstructural proteins by expression of prM and E proteins (1). Not surprisingly, there was no defect in the production of subviral particles from cells transfected with W349A.
Another possibility is that the NS2A-NS3 complex might be incorporated into the virus particle during assembly. Additionally, this complex could also mediate the incorporation of other nonstructural proteins into the virus particle and this inclusion of nonstructural proteins could be important for specific RNA encapsidation. In fact, detectable amounts of RdRp have been isolated from foot-and-mouth disease virions (25). Although radiolabeling of mature flavivirus particles has revealed only the three structural proteins, a more sensitive method such as mass spectroscopy of purified flavivirus particles could be utilized to check for the presence of minute amounts of nonstructural proteins (B. Kim and R. J. Kuhn, unpublished data). One other possible scenario could be that NS3 interacts with host proteins and that these interactions facilitate genome packaging and virion assembly/release. Colocalization and coimmunoprecipitation assays using Japanese encephalitis virus-infected cells revealed interaction of NS3 with tumor susceptibility protein 101 (TSG101) (4). TSG101 is a host cell protein localized in the Golgi apparatus that has been shown to interact with the late domain of Gag protein from human immunodeficiency virus type 1 and Ebola virus to facilitate virus budding (22). TSG101 together with NS2A-NS3 could serve a similar function in assembly and/or release of flavivirus particles. The W349A mutation could interfere with one or more of these scenarios, leading to a block in virus assembly.
Based on the limited information obtained thus far, it is difficult to propose a mechanistic model for the flavivirus virion assembly process. However, it is clear that flavivirus virion assembly involves interactions of NS3 with other nonstructural proteins (presumably at least NS2A), and probably host proteins as well, to form an assembly-competent RC that mediates specific RNA encapsidation and, ultimately, virion assembly. Preliminary results obtained with mutations of the conserved W349 residue in this study represent the first time that the flavivirus NS3 protein has been directly shown to be involved in virus assembly and/or release. Comparison of the structures of domains 1 and 2 of the YFV helicase with the corresponding domains in HCV helicase shows a high degree of similarity, and our sequence alignment reveals the presence of a conserved aromatic residue (tyrosine at position 350) in HCV at the position corresponding to 349 in YFV (33). Although a direct role for NS3 in HCV virus production has not been demonstrated, these data suggest that the results obtained with YFV might be applicable to HCV as well and could serve as a guide for the development of safe and effective vaccine strains of flaviviruses.
Acknowledgments
We thank James Strauss for providing the anti-E and antihelicase antibodies and J. Schlesinger for anti-NS1 antibody. We are grateful to Aloke K. Bera, Yu-hsuan Chang, and Ranjan Sengupta for helpful discussions and Anita Robinson and Jennifer Yoder for assistance.
This work was supported by Public Health Service Program Project grant A155672 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print on 16 January 2008.
REFERENCES
- 1.Allison, S. L., K. Stadler, C. W. Mandl, C. Kunz, and F. X. Heinz. 1995. Synthesis and secretion of recombinant tick-borne encephalitis virus protein E in soluble and particulate form. J. Virol. 695816-5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, S. L., Y. J. Tao, G. O'Riordain, C. W. Mandl, S. C. Harrison, and F. X. Heinz. 2003. Two distinct size classes of immature and mature subviral particles from tick-borne encephalitis virus. J. Virol. 7711357-11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers, T. J., R. C. Weir, A. Grakoui, D. W. McCourt, J. F. Bazan, R. J. Fletterick, and C. M. Rice. 1990. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. USA 878898-8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiou, C. T., C. Hu, P. H. Chen, C. L. Liao, Y. L. Lin, and J. J. Wang. 2003. Association of Japanese encephalitis virus NS3 protein with microtubules and tumor susceptibility gene 101 (TSG101) protein. J. Gen. Virol. 842795-2805. [DOI] [PubMed] [Google Scholar]
- 5.Erbel, P., N. Schiering, A. D'Arcy, M. Renatus, M. Kroemer, S. Lim, Z. Yin, T. Keller, S. Vasudevan, and U. Hommel. 2006. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 13372-373. [DOI] [PubMed] [Google Scholar]
- 6.Ferlenghi, I., M. Clarke, T. Ruttan, S. L. Allison, J. Schalich, F. X. Heinz, S. C. Harrison, F. A. Rey, and S. D. Fuller. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis. Mol. Cell 7593-602. [DOI] [PubMed] [Google Scholar]
- 7.Frolova, E., I. Frolov, and S. Schlesinger. 1997. Packaging signals in alphaviruses. J. Virol. 71248-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunt, A. R., C. B. Cropp, and G. J. Chang. 2001. A recombinant particulate antigen of Japanese encephalitis virus produced in stably-transformed cells is an effective noninfectious antigen and subunit immunogen. J. Virol. Methods 97133-149. [DOI] [PubMed] [Google Scholar]
- 9.Jones, C. T., C. G. Patkar, and R. J. Kuhn. 2005. Construction and applications of yellow fever virus replicons. Virology 331247-259. [DOI] [PubMed] [Google Scholar]
- 10.Khromykh, A. A., P. L. Sedlak, and E. G. Westaway. 1999. trans-complementation analysis of the flavivirus Kunjin ns5 gene reveals an essential role for translation of its N-terminal half in RNA replication. J. Virol. 739247-9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khromykh, A. A., A. N. Varnavski, P. L. Sedlak, and E. G. Westaway. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J. Virol. 754633-4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konishi, E., and A. Fujii. 2002. Dengue type 2 virus subviral extracellular particles produced by a stably transfected mammalian cell line and their evaluation for a subunit vaccine. Vaccine 201058-1067. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn, R. J., W. Zhang, M. G. Rossmann, S. V. Pletnev, J. Corver, E. Lenches, C. T. Jones, S. Mukhopadhyay, P. R. Chipman, E. G. Strauss, T. S. Baker, and J. H. Strauss. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kümmerer, B. M., and C. M. Rice. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 764773-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindenbach, B. D., and C. M. Rice. 2003. Molecular biology of flaviviruses. Adv. Viral Res. 5923-61. [DOI] [PubMed] [Google Scholar]
- 16.Lindenbach, B. D., and C. M. Rice. 2001. Flaviviridae, p. 991-1041. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 17.Liu, W. J., H. B. Chen, and A. A. Khromykh. 2003. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 777804-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu, W. J., P. L. Sedlak, N. Kondratieva, and A. A. Khromykh. 2002. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J. Virol. 7610766-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo, D., T. Xu, C. Hunke, G. Grüber, S. Vasudevan, and J. Lescar. 2008. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 82:173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie, J. M., and E. G. Westaway. 2001. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 7510787-10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie, J. M., A. A. Khromykh, M. K. Jones, and E. G. Westaway. 1998. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 245203-215. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 71278-1280. [DOI] [PubMed] [Google Scholar]
- 23.Mukhopadhyay, S., B. S. Kim, P. R. Chipman, M. G. Rossmann, and R. J. Kuhn. 2003. Structure of West Nile virus. Science 303248. [DOI] [PubMed] [Google Scholar]
- 24.Nestorowicz, A., T. J. Chambers, and C. M. Rice. 1994. Mutagenesis of the yellow fever virus NS2A/2B cleavage site: effects on proteolytic processing, viral replication, and evidence for alternative processing of the NS2A protein. Virology 199114-123. [DOI] [PubMed] [Google Scholar]
- 25.Newman, J. F. E., P. G. Piatti, B. M. Gorman, T. G. Burrage, M. D. Ryan, M. Flint, and F. Brown. 1994. Foot-and-mouth disease virus particles contain replicase protein 3D. Proc. Natl. Acad. Sci. USA 91733-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng, M. L., S. H. Tan, and J. J. H. Chu. 2001. Transport and budding at two distinct sites of visible nucleocapsids of West Nile (Sarafend) virus. J. Med. Virol. 65758-764. [DOI] [PubMed] [Google Scholar]
- 27.Nugent, C. I., K. L. Johnson, P. Sarnow, and K. Kirkegaard. 1999. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 73427-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owsianka, A. M., and A. H. Patel. 1999. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257330-340. [DOI] [PubMed] [Google Scholar]
- 29.Pijlman, G., N. Kondratieva, and A. Khromykh. 2006. Translation of the flavivirus Kunjin NS3 gene in cis but not its RNA sequence or secondary structure is essential for efficient RNA packaging. J. Virol. 8011255-11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pincus, S., P. W. Mason, E. Konishi, B. A. Fonseca, R. E. Shope, C. M. Rice, and E. Paoletti. 1992. Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology 187290-297. [DOI] [PubMed] [Google Scholar]
- 31.Stocks, C. E., and M. Lobigs. 1998. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 722141-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wengler, G., and G. Wengler. 1993. The NS3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 197265-273. [DOI] [PubMed] [Google Scholar]
- 33.Wu, J., A. K. Bera, R. J. Kuhn, and J. L. Smith. 2005. Structure of the flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 7910268-10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu, T., A. Sampath, A. Chao, D. Wen, M. Nanao, P. Chene, S. Vasudevan, and J. Lescar. 2005. Structure of the dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 Å. J. Virol. 7910278-10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, W., P. R. Chipman, J. Corver, P. R. Johnson, Y. Zhang, S. Mukhopadhyay, T. S. Baker, J. H. Strauss, M. G. Rossmann, and R. J. Kuhn. 2003. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 10907-912. [DOI] [PMC free article] [PubMed] [Google Scholar]