

Abstract
The human cytomegalovirus (HCMV) protein US6 inhibits the transporter associated with antigen processing (TAP). Since TAP transports antigenic peptides into the endoplasmic reticulum for binding to major histocompatibility class I molecules, inhibition of the transporter by HCMV US6 impairs the presentation of viral antigens to cytotoxic T lymphocytes. HCMV US6 inhibits ATP binding by TAP, hence depriving TAP of the energy source it requires for peptide translocation, yet the molecular basis for the interaction between US6 and TAP is poorly understood. In this study we demonstrate that residues 89 to 108 of the HCMV US6 luminal domain are required for TAP inhibition, whereas sequences that flank this region stabilize the binding of the viral protein to TAP. In parallel, we demonstrate that chimpanzee cytomegalovirus (CCMV) US6 binds, but does not inhibit, human TAP. The sequence of CCMV US6 differs from that of HCMV US6 in the region corresponding to residues 89 to 108 of the HCMV protein. The substitution of this region of CCMV US6 with the corresponding residues from HCMV US6 generates a chimeric protein that inhibits human TAP and provides further evidence for the pivotal role of residues 89 to 108 of HCMV US6 in the inhibition of TAP. On the basis of these observations, we propose that there is a hierarchy of interactions between HCMV US6 and TAP, in which residues 89 to 108 of HCMV US6 interact with and inhibit TAP, whereas other parts of the viral protein also bind to TAP and stabilize this inhibitory interaction.
The major histocompatibility (MHC) class I antigen presentation pathway plays a central role in the immune response to viral infection. MHC class I molecules are expressed on the plasma membranes of all nucleated cells and present peptides derived from intracellular proteins to cytotoxic T lymphocytes (CTLs). CTLs monitor these MHC class I molecules for peptides derived from viral proteins and kill the infected cells. The presentation of antigenic peptides to CTLs represents the end point of a complex multistep pathway. The vast majority of peptides presented by MHC class I molecules are generated by the degradation of proteins in the cytosol by the proteasome complex (38). These peptides are then translocated into the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) (1). MHC class I molecules are assembled in the ER by a process that involves ER-resident chaperones (5). Peptide binding represents the final step of MHC class I molecule folding and is facilitated by the TAP-associated molecule tapasin, which links the nascent MHC class I molecules to TAP to form the “peptide-loading complex” (5). Only once they have bound peptides can MHC class I molecules dissociate from this complex and exit the ER for transport to the plasma membrane.
TAP is a member of the ATP binding cassette (ABC) family of transporters, whose members couple ATP hydrolysis to the translocation of a broad spectrum of different substrates across cellular membranes (1, 29, 42). All ABC transporters have two nucleotide-binding domains (NBDs) and two polytopic membrane domains (29). The highly conserved NBDs of ABC transporters bind and hydrolyze ATP energizing transport, whereas the membrane domains translocate the substrate across the membrane. TAP is a heterodimer, composed of the TAP1 and TAP2 subunits (19), both of which have an N-terminal polytopic membrane domain and a cytosolic C-terminal NBD. The TAP1 membrane domain spans the membrane 10 times, with both the N and C termini in the cytosol (14, 39). TAP2 is predicted to span the membrane nine times, with the N terminus of the membrane domain in the ER lumen and the C terminus in the cytosol (14, 39). The C-terminal six transmembrane domains of TAP1 and TAP2 combine to form the core unit of the transporter that is responsible for peptide binding and translocation, whereas the N-terminal transmembrane domains of TAP1 and TAP2 interact with tapasin (21, 22, 28, 36).
Since the vast majority of peptides presented to CTLs require translocation into the ER by TAP, it is not surprisingly that many viruses interfere with this transport step (3, 8, 12, 17, 18, 23-25, 30, 35, 37, 41). Human cytomegalovirus (HCMV) encodes the protein US6, which is an ER-localized type I integral membrane glycoprotein that inhibits peptide translocation by TAP (2, 15, 27). By inhibiting TAP, HCMV US6 decreases cell surface expression of MHC class I molecules and hence reduces CTL killing (2, 15, 27). HCMV US6 does not block peptide binding by TAP but inhibits ATP binding and hydrolysis by the transporter (2, 15, 16, 26, 27). In effect, HCMV US6 starves TAP of the energy source that it requires for peptide translocation. The inhibition of ATP binding is an indirect conformational effect, as only the ER luminal domain of HCMV US6 is required to inhibit ATP binding by the cytosolic TAP1 NBD (16, 26).
How HCMV US6 binds to TAP and inhibits ATP binding and peptide translocation by the transporter is poorly understood. Therefore, in order to investigate the molecular basis for the interaction between HCMV US6 and TAP, we performed a systematic analysis of the HCMV US6 sequence. We establish that residues 89 to 108 of the HCMV US6 luminal domain are required for TAP inhibition, whereas other HCMV US6 sequences stabilize the interaction with TAP. In addition, we characterize the chimpanzee cytomegalovirus (CCMV) US6 protein, which we show in contrast to its HCMV orthologue, can bind but cannot inhibit human TAP. We demonstrate that this is because CCMV US6 differs in its sequence within the region that corresponds to residues 89 to 108 of HCMV US6 and that substitution of these residues with those from HCMV US6 generates a chimeric US6 protein that inhibits human TAP. Together, these data demonstrate that distinct regions of HCMV US6 are required for TAP inhibition and for stabilization of the interaction between the viral protein and transporter.
MATERIALS AND METHODS
Materials.
Unless otherwise stated, all reagents were supplied by Sigma.
DNA constructs. (i) HLA-B2705 and control constructs.
pCR3-HLA-B2705 and pCR3-HLA-B2705-V5, which encode HLA-B2705 and a V5 epitope-tagged HLA-B2705 (4), respectively, were kind gifts from A. Antoniou (University College London); pcDNA3.1(−)Pac (16) was used as the control vector in transfections.
(ii) Full-length HCMV US6 constructs.
pcDNA3.1(−)Pac-US6 encodes HCMV US6, with a FLAG epitope tag fused to the C terminus of US6 (16). pEGFP-N1-HCMV US6 encodes a green fluorescent protein (GFP)-tagged HCMV US6. To generate this construct, HCMV US6 was amplified with the primers HUS6-5′ and HUS6-3′ no stop using pcDNA3.1(−)Pac-US6 as a template; the resultant PCR product was cloned as a XbaI-EcoRI fragment into the NheI-EcoRI sites of pEGFP-N1 (Clontech).
(iii) N-terminal HCMV US6 deletions.
The constructs pcDNA3.1(−)Pac-US6ΔN20 and pcDNA3.1(−)Pac-US6ΔN40 encode FLAG-tagged N-terminal HCMV US6 deletions that lack the N-terminal 20 (US6ΔN20) and 40 residues (US6ΔN40), respectively (16). Further N-terminal deletion constructs were generated with a C-terminal FLAG epitope tag, and these constructs were cloned into pcDNA3.1(−)Pac-hGHSS (16) downstream of the human growth hormone signal sequence (hGHSS) to replace the HCMV US6 signal sequence that was removed in the construction of these deletions. HCMV US6ΔN80 was amplified with the primers HUS6ΔN80-5′ and HUS6-3′; HCMV US6ΔN90 was amplified with the primers HUS6ΔN90-5′ and HUS6-3′. The PCR products were cloned as EcoRI-KpnI fragments into the same sites of pcDNA3.1(−)Pac-hGHSS to generate pcDNA3.1(−)Pac-US6ΔN80 and pcDNA3.1(−)Pac-US6ΔN90.
(iv) C-terminal HCMV US6 deletions.
The constructs pcDNA3.1(−)PacUS6ΔC39 and pcDNA3.1(−)PacUS6ΔC59 encode FLAG-tagged N-terminal deletions that lack the C-terminal 39 (US6ΔC39) and 59 residues (US6ΔC59) of HCMV US6, respectively (16). Further C-terminal deletion constructs were generated in which the C terminus of the deletion was fused to a FLAG epitope tag. HCMV US6ΔC49 was amplified by PCR with the primers HUS6-5′ and HUS6ΔC49-3′; HCMV US6ΔC79 was amplified with the primers HUS6-5′ and HUS6ΔC79-3′. The PCR products were cloned as XbaI-KpnI fragments into the NheI-KpnI sites in pcDNA3.1(−)Pac to generate pcDNA3.1(−)Pac-US6ΔC49 and pcDNA3.1(−)Pac-US6ΔC79.
(v) HCMV US6 tetra-alanine mutants.
Tetra-alanine mutations were introduced into HCMV US6, spanning residues 81 to 115. Mutagenesis was performed using the Stratagene QuikChange site-directed mutagenesis kit per the manufacturer's instructions using pcDNA3.1(−)Pac-US6 as a template. To mutate residues 81 to 84, 85 to 88, 89 to 92, 93 to 96, 97 to 100, 101 to 104, 105 to 108, 109 to 112, and 112 to 115 of HCMV US6, the sense oligonucleotides used were HUS6 81-84, HUS6 85-88, HUS6 89-92, HUS6 93-96, HUS6 97-100, HUS6 101-104, HUS6 105-108, HUS6 109-112, and HUS6 112-115, respectively. In each instance, the antisense oligonucleotide was the exact complement.
(vi) CCMV US6 constructs.
The open reading frame of CCMV US6 was amplified from CCMV genomic DNA (10) (a kind gift from A. Davison, University of Glasgow) by PCR with primers CUS6-5′ and CUS6-3′. The resultant product was cloned into pcDNA3.1(−)Pac as an EcoRI-BamHI fragment to generate pcDNA3.1(−)Pac-CCMV-US6 encoding the CCMV protein with a C-terminal FLAG epitope tag. The chimeric US6 construct was generated such that the DNA encoding amino acid residues 97 to 101 of CCMV US6 was replaced with that of residues 92 to 96 of HCMV US6. The CCMV US6 S99A glycosylation mutant was generated by mutating serine 99 to an alanine, hence abolishing the second predicted glycosylation site. The CCMV US6 N97D/S99D mutant was generated by substituting CCMV US6 asparagine 97 and serine 99 to aspartate residues. Mutagenesis was performed with the Stratagene QuikChange kit using pcDNA3.1(−)Pac-CCMV-US6 as a template. The sense oligonucleotides used for the US6 chimera, CCMV US6 glycosylation mutant, and CCMV US6 N97D/S99D mutant were US6-chimera, CUS6-S99A, and CUS6-N97D/S99D, respectively; the antisense oligonucleotides were the exact complement.
(vii) Oligonucleotides.
The oligonucleotides used were as follows: HUS6-5′, TATATCTAGAATGGATCTCTTGATTCGTCT; HUS6-3′, ATATGGTACCTTATTTATCATCATCATCTTTGTAATCGGAGCCACAACGTCGAATCCC; HUS6-3′ no stop, CGGAATTCCGGAGCCACAACGTCGAATCCC; HUS6ΔN80-5′, ACACGAATTCACCTGTCAACGAGTCCGTCGG; HUS6ΔN90-5′, GACCTCAGAATTCTGTGATTTGGATATTCATCCTAGCC; HUS6ΔC49-3′, ACACGAATTCTTATTTATCATCATCATCTTTGTAATCCCAAACGGCCCCGTCACAGAC; HUS6ΔC79-3′, ATATGGTACCTTATTTATCATCATCATCTTTGTAATCGACTCGTTGACAGGTCAGCCT; HUS6 81-84, AACCTTGAGGGTAGGCTGGCCGCTGCAGCAGTCCGTCGGCTACTGCC; HUS6 85-88, AGGCTGACCTGTCAACGAGCCGCTGCGGCACTGCCCTGTGATTTGGAT; HUS6 89-92, CAACGAGTCCGTCGGCTAGCGGCCGCTGCTTTGGATATTCATCCTAGC; HUS6 93-96, CGGCTACTGCCCTGTGATGCGGCTGCTGCTCCTAGCCACCGGTTGTTA; HUS6 97-100, TGTGATTTGGATATTCATGCTGCCGCCGCGTTGTTAACGCT-TATGAAT; HUS6 101-104, TATTCATCCTAGCCACCGGGCGGCAGCGGCTATGAATAACTGCGTCTGTG; HUS6 105-108, CACCGGTTGTTAACGCTTGCGGCTGCCGCCGTCTGTGACGGGGCCGTTTG; HUS6 109-112, ACGCTTATGAATAACTGCGCCGCTGCCGCGGCCGTTTGGAACGCGTTTCG; HUS6 112-115, GAATAACTGCGTCTGTGACGCGGCCGCTGCGAACGCGTTTCGCTTG ATAG; CUS6-5′, ACACGAATTCACCATGCGGTACCTAGGACTTTTG; CUS6-3′, ATATGGATCCTTATTTATCATCATCATCTTTGTAATCGGAGCCGCAACGTCGGGAACT; US6-chimera, TCAGCCGTCTCCTGCCCTGCGATCTGGATATCCACCCCAGCCACCGACTGCTGTT; CUS6-S99A, GTCTCCTGCCCTGCAACGCCGCCGGCCTGCGGCCCAGCCACCG; and CUS6-N97D/S99D, AGCCGTCTCCTGCCCTGCGACGCCGACGGCCTGCGGCCCAGCCAC.
Antibodies.
The following antibodies were used: ME1 (11), a monoclonal antibody (MAb) that recognizes HLA-B2705 (a kind gift from S. Powis, University of St. Andrews); R.RING4C, a rabbit antipeptide antibody raised against the C terminus of TAP1 (34); SV5-Pk1, a MAb that recognizes the V5 epitope tag (Serotec); M2, a MAb that recognizes the FLAG epitope tag (Sigma); and a rabbit anticalreticulin antibody (Calbiochem). Secondary antibodies were from Jackson ImmunoResearch.
Cell culture and transfections.
The cervical carcinoma HeLa-M cell line and chimpanzee fibroblast cell line WES (American Type Culture Collection) were grown in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum. For transfection, cells were plated into six-well dishes in 1.5 ml of medium per well; the next day, 10 μl of 1 μg/ml polyethylenimine (Polysciences Inc.) in 20 mM HEPES (pH 7.4) and 2 μg of DNA were added to 1 ml of serum-free medium, mixed by vortexing, incubated for 15 min, and added to the cells. In cotransfection experiments, 0.5 μg of pCR3-HLA-B2705 was mixed with 1.5 μg of either the control vector or US6 expression construct. Two milliliters of fresh medium containing serum was added 24 h posttransfection, and the cells were analyzed 48 h posttransfection. Stable transfectants of HeLa-M cells were selected with 1.2 μg/ml puromycin and subcloned into 96-well plates. Clones expressing the FLAG-tagged constructs were screened by immunoblotting with the anti-FLAG epitope tag MAb M2.
Immunofluorescence microscopy.
Prior to transfection, HeLa-M cells were seeded onto glass coverslips in six-well dishes. Two days posttransfection, the cells were washed twice with phosphate-buffered saline (PBS), fixed in PBS containing 4% paraformaldehyde for 20 min, washed with PBS, and permeabilized in PBS containing 0.1% Triton X-100 for 10 min. After a further three washes with PBS, the coverslips were blocked with PBS containing 10% FCS for 1 h, then incubated with primary antibodies diluted into PBS for 1 h, washed with PBS, and incubated with fluorophore-conjugated secondary antibodies for 1 h. Cells were washed twice with PBS and rinsed in distilled water, and coverslips mounted onto glass slides. Antibody fluorescence was visualized with a Zeiss Axioplan 2 microscope.
Endo H digestion of cell lysates.
Cells were harvested by centrifugation at 400 × g in a benchtop centrifuge, washed once with PBS, and lysed at a density of 2 × 107/ml into Tris-buffered saline (TBS), pH 7.4, 1% Triton X-100, 5 mM iodoacetimide, and 1 mM phenylmethylsulfonyl fluoride. After 30 min on ice, the lysate was spun in a microcentrifuge at 13,000 × g and 4°C for 10 min, and the supernatant was retained. The supernatant was divided into two, and endoglycosidase H (Endo H) (New England BioLabs) was added to one of the samples (according to the manufacturer's instructions), and the other sample was mock digested, before analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Immunoprecipitation of cell lysates.
Cells were harvested by centrifugation at 400 × g in a benchtop centrifuge and washed once with PBS. Cells were lysed at a density of 2 × 107/ml in TBS (pH 7.4), 1% digitonin (Wako), 5 mM iodoacetimide, and 1 mM phenylmethylsulfonyl fluoride. After 30 min on ice, the lysate was spun in a microcentrifuge at 13,000 × g at 4°C for 10 min, and the supernatant was retained. M2-conjugated agarose beads were added to the supernatant and mixed by rotation for 2 h. The agarose beads were then washed three times with TBS containing 0.1% digitonin, once in TBS before the addition of SDS-PAGE sample buffer and analysis by SDS-PAGE.
Flow cytometry.
Transfectants were stained on ice with ME1 MAb in PBS and 0.1% bovine serum albumin, and antibody staining was visualized with a goat anti-mouse Cy5-conjugated secondary antibody. Cells were fixed in PBS and 4% formaldehyde prior to flow cytometric analysis. Cell-associated fluorescence was analyzed with a Becton Dickinson FACSCalibur. The cell population was gated to exclude debris, and 10,000 gated events were analyzed. For each sample, the mean fluorescence value (MFV) of the total cell population including both transfected and untransfected cells was determined using Cell Quest software. The data were then normalized for each experiment by calculating the level of HLA-B2705 expression as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only).
ATP-agarose binding assay.
ATP-agarose binding was performed as described previously (16).
Peptide translocation assay.
Peptide translocation with streptolysin O (SLO)-permeabilized cells using a fluorescein-labeled peptide with the sequence CVNKTERAY was performed as described previously (23).
Immunoblotting.
Transfer of SDS-polyacrylamide gels onto polyvinylidene difluoride (PVDF) membranes and incubation with specific antibodies was performed as described previously (27).
RESULTS
HCMV US6 inhibits cell surface expression of the TAP-dependent allele HLA-B2705.
In order to delineate the sequence requirements for the interaction with and inhibition of TAP by HCMV US6, we performed a systematic analysis of the HCMV US6 sequence. To test whether the US6 mutants generated in this study were functional, we developed a novel transient-cotransfection flow cytometric assay to assess the inhibition of TAP. This assay utilized HLA-B2705, a MHC class I allele that is dependent on TAP for peptide loading and cell surface expression (9, 40). HeLa cells do not express HLA-B2705 endogenously (6); correspondingly, only background binding by the ME1 antibody, a conformation-specific antibody that recognizes mature peptide-loaded HLA-B2705 (11), was detected by flow cytometry when HeLa-M cells were transfected with the control vector pcDNA3.1(−)Pac (Fig. 1A). Cotransfection of a construct encoding HLA-B2705 with the control vector resulted in the dramatic increase in the level of binding of ME1 to a substantial proportion of the HeLa-M cells, consistent with the expression of HLA-B2705 on the plasma membranes of cells transfected with the HLA-B2705 cDNA (Fig. 1A). Typically, 40 to 60% of the HeLa-M cells transfected with HLA-B2705 had an increased level of ME1 binding, indicating that the transfection efficiency was correspondingly 40 to 60%. In contrast, when HeLa-M cells were cotransfected with HLA-B2705 and HCMV US6, the cell surface expression of HLA-B2705 was substantially reduced compared to that of cells cotransfected with HLA-B2705 and the control vector (Fig. 1A). Furthermore, when cotransfected with a GFP-tagged HCMV US6, HLA-B2705 colocalized with the ER-resident protein US6, whereas HLA-B2705 was localized to the plasma membrane in cells cotransfected with the control vector (Fig. 1B). This demonstrates that HCMV US6 does not block synthesis of HLA-B2705, but rather by inhibiting TAP, US6 prevents peptide binding by HLA-B2705, which is as a consequence retained in the ER. As such, we conclude that cell surface expression of HLA-B2705 can be used as a convenient and sensitive assay with which to assess TAP inhibition by HCMV US6.
FIG. 1.

HCMV US6 inhibits cell surface expression of HLA-B2705. (A) Analysis of the effect of HCMV US6 on the expression of HLA-B2705 at the plasma membrane. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and HCMV US6. To detect cell surface expression of HLA-B2705, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody. Antibody fluorescence was quantified by flow cytometry. The mean fluorescence value (MFV) for each of the transfected cell populations is indicated. (B) Analysis of the localization of HLA-B2705 in cells cotransfected with HCMV US6. HeLa-M cells were transfected with V5 epitope-tagged HLA-B2705 expression construct in combination with either a control vector or with HCMV US6-GFP. The transfected cells were stained with the anti-V5 epitope tag MAb SV5-Pk1, which detects the V5 epitope-tagged HLA-B2705 and an anti-mouse Texas red antibody. Antibody and GFP fluorescence was detected using a Zeiss Axioplan 2 microscope. The right panels are merged images of the red and green channels where yellow indicates colocalization; in these panels, the nuclei are also visualized with 4′,6′-diamidino-2-phenylindole (DAPI) (blue). Bars, 5 μm.
The N-terminal 80 residues of HCMV US6 are not required for the inhibition of TAP.
Previous studies have shown that the luminal domain of HCMV US6 is sufficient to inhibit TAP and that the N-terminal 40 residues of HCMV US6 can be deleted without impairing function (15, 16). To further delineate the sequence requirements for TAP inhibition by HCMV US6, deletions lacking the N-terminal 20, 40, 80, and 90 residues of HCMV US6 were analyzed (Fig. 2A). By analyzing the deletion constructs in transiently transfected cells, we were able to extend the analysis of the HCMV sequence to include deletions that lack the N-terminal 80 (HCMV US6ΔN80) and 90 (HCMV US6ΔN90) residues, which we had previously been unable to express in stable cell lines (16). It was not practicable to normalize the expression levels of the deletion constructs analyzed in this study. The expression level of HCMV US6ΔN40 was lower than that of the other N-terminal deletions (Fig. 2C), but this deletion was nonetheless able to inhibit the cell surface expression of HLA-B2705 to the same extent as the full-length protein was (Fig. 2B). Furthermore, HCMV US6ΔN90 was unable to reduce cell surface expression of HLA-B2705, whereas HCMV US6ΔN80, which was expressed at a comparable level, inhibited HLA-B2705 cell surface expression (Fig. 2B). This demonstrates that the N-terminal 80 residues are dispensable for TAP inhibition, while further deletion of the HCMV US6 sequence abolishes function. The inability of HCMV US6ΔN90 to inhibit cell surface expression of HLA-B2705 was not due to the mislocalization of this deletion. US6ΔN90 colocalized with the ER-resident chaperone calreticulin (data not shown), although we cannot discriminate between either the retention of HCMV US6ΔN90 in the ER due to misfolding or the possibility that the protein with this deletion was properly folded and retained by the same mechanism as that for the full-length protein.
FIG. 2.

Distinct regions of HCMV US6 are required for TAP binding and inhibition. (A) HCMV US6 deletion constructs. The N-terminal signal sequence of HCMV US6 is shown in italic type. Residue 1 corresponds to the N terminus after signal sequence cleavage. The N-linked glycosylation site is shown in bold type, and the predicted transmembrane domain is underlined. Arrows indicate the positions of the N- and C-terminal truncations. (B) Analysis of the effect of HCMV US6 N-terminal and C-terminal truncations on the cell surface expression of HLA-B2705. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each HCMV US6 truncation. Cell surface expression of HLA-B2705 in the transfectants was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the level (mean ± standard deviation [error bar]) of HLA-B2705 expression calculated from four independent experiments. Statistical analysis was performed using the Mann-Whitney U test. Those samples with a significantly increased level (P ≤ 0.05) of cell surface HLA-B2705 expression compared to that in cells cotransfected with the HCMV US6 and HLA-B2705 vectors are indicated by an asterisk. (C) Analysis of the interaction between HCMV US6 truncations and TAP. HeLa-M cells were transfected with the control vector, full-length HCMV US6, or each HCMV US6 deletion construct. US6 proteins were immunoprecipitated with the anti-FLAG epitope tag antibody M2 and resolved by SDS-PAGE, transferred onto PVDF membranes, and probed with either TAP1-specific or M2 antibodies. The position of a background band present in all samples that may correspond to the light chain of the M2 antibody is indicated by an asterisk.
In parallel, we investigated whether these HCMV US6 deletions were able to interact with TAP (Fig. 2C). Cells were transfected with each of the HCMV US6 N-terminal deletions and US6 immunoprecipitated with M2, an antibody that is specific for the C-terminal FLAG epitope tag present in each deletion. TAP coimmunoprecipitated with all of the N-terminal deletions, although substantially less TAP coimmunoprecipitated with HCMV US6ΔN90. Together, these data demonstrate that the N-terminal 80 residues of US6 are not required for either the interaction with, or the inhibition of, TAP. Residues 81 to 90 of HCMV US6 are required for the inhibition of TAP and may also stabilize the interaction between US6 and TAP.
The C-terminal 49 residues of HCMV US6 stabilize the interaction with TAP but are not required for TAP inhibition.
C-terminal deletions were also used to evaluate the sequence requirements for the interaction with and inhibition of TAP by US6 (Fig. 2A). All proteins with deletions were expressed at levels similar to that of the full-length protein with the exception of HCMV US6ΔC79, which was expressed at a lower level (Fig. 2C). Transfection of either HCMV US6ΔC39, which corresponds to the luminal domain of US6, or HCMV US6ΔC49, in which a further 10 residues have been deleted from the C terminus of the protein, reduced cell surface HLA-B2705 expression to the same extent as the full-length protein did. In contrast, in cells transfected with the HCMV US6ΔC59 or US6ΔC79 deletion mutants, the level of cell surface HLA-B2705 was significantly higher than in cells cotransfected with full-length HCMV US6 (Fig. 2B). This demonstrates that the C-terminal 59 residues of HCMV US6 are required for the inhibition of TAP, and this observation is consistent with our previous study (16). The inability of HCMV US6ΔC59 and US6ΔC79 to inhibit TAP was not due to the mislocalization of these proteins, as proteins with all of these C-terminal deletions were localized to the ER (data not shown), although it is possible that the nonfunctional proteins with HCMV US6 deletions may be misfolded and retained by the ER quality control machinery. However, despite inhibiting HLA-B2705 expression, TAP did not efficiently coimmunoprecipitate with HCMV US6ΔC39 and no TAP was detected in HCMV US6ΔC49 immunoprecipitates (Fig. 2C). These results show that while the C-terminal 49 residues of HCMV US6 are dispensable for TAP inhibition, these residues may be required to stabilize the interaction between HCMV US6 and TAP.
Residues 89 to 108 of HCMV US6 are required for TAP inhibition, but not for TAP binding and US6 oligomerization.
To define the minimum sequence requirements for TAP inhibition, a double deletion mutant that lacked the N-terminal 80 and C-terminal 49 residues of HCMV US6 was constructed. This construct encoded residues 81 to 115 of the HCMV US6 sequence, which corresponds to the core region of HCMV US6 required for TAP inhibition, as defined by the deletion analysis. However, we were unable to detect the expression of this protein (data not shown), suggesting that the simultaneous deletion of these HCMV US6 N- and C-terminal sequences results in an unstable protein. An alternative approach was therefore employed to further dissect the sequence requirements for HCMV US6 function. A series of tetra-alanine substitutions were introduced into HCMV US6 spanning residues 81 to 115 (Fig. 3); all of these mutant proteins were expressed at levels comparable to that of the wild-type HCMV US6 (Fig. 4B). However, the introduction of tetra-alanine mutations into residues 89 to 108 of HCMV US6 significantly impaired the ability of the viral protein to inhibit HLA-B2705 expression at the plasma membrane (Fig. 4A).
FIG. 3.

Schematic representation of the HCMV US6 tetra-alanine mutants. The N-terminal signal sequence of US6 is indicated by the hatched box. The arrow indicates the predicted site for signal sequence cleavage. Residue 1 corresponds to the N terminus after signal sequence cleavage. The N-linked glycan (the circled G), predicted transmembrane domain (TMD), and C-terminal epitope tag incorporated into each construct (FLAG) are indicated.
FIG. 4.

Residues 89 to 108 of HCMV US6 are required for TAP inhibition, but not TAP binding. (A) Analysis of the effect of HCMV US6 tetra-alanine mutants on the cell surface expression of HLA-B2705. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each tetra-alanine HCMV US6 mutant. To detect HLA-B2705 cell surface expression, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody; antibody fluorescence was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the mean level of HLA-B2705 cell surface expression calculated from four independent experiments. Error bars show the standard deviations. Statistical analysis was performed using the Mann-Whitney U test. Those samples with a significantly increased level (P ≤ 0.05) of cell surface HLA-B2705 expression compared to that of cells cotransfected with the HCMV US6 and HLA-B2705 vectors are indicated by an asterisk. (C) Analysis of the interaction between HCMV US6 tetra-alanine mutants and TAP. HeLa-M cells were transfected with either the control vector, full-length US6, or US6 tetra-alanine construct. US6 proteins were immunoprecipitated with the anti-FLAG epitope tag antibody M2, resolved by SDS-PAGE, transferred onto PVDF membranes, and probed with either a TAP1-specific or M2 antibody. The position of a background band present in all samples that may correspond to the light chain of the M2 antibody is indicated by an asterisk.
Despite impairing the ability of HCMV US6 to inhibit HLA-B2705 expression, the introduction of tetra-alanine substitutions into residues 89 to 108 did not affect TAP binding (Fig. 4B). As such, TAP binding is in itself insufficient for effective inhibition of the transporter, suggesting that residues 89 to 108 may be required to mediate an interaction either at a distinct site on TAP or with another molecule. One formal possibility was that the mutation of residues 89 to 108 prevents the oligomerization of HCMV US6, which has been proposed to enable HCMV US6 to interact simultaneously with multiple sites on TAP (14). To test whether residues 89 to 108 are required for HCMV US6 oligomerization, wild-type HCMV US6 and the panel of tetra-alanine mutants were analyzed by nonreducing SDS-PAGE (Fig. 4C). Wild-type HCMV US6 was resolved as a ladder of bands, consistent with the previously reported oligomerization of the protein (14). Likewise, all of the tetra-alanine mutants were resolved as a ladder of bands on nonreducing SDS-polyacrylamide gels, hence demonstrating that mutation of residues 89 to 108 does not prevent HCMV US6 oligomerization and by implication suggesting that this region must perform another function that is required for TAP inhibition.
CCMV US6 is localized to the ER but does not inhibit HLA-B2705 expression.
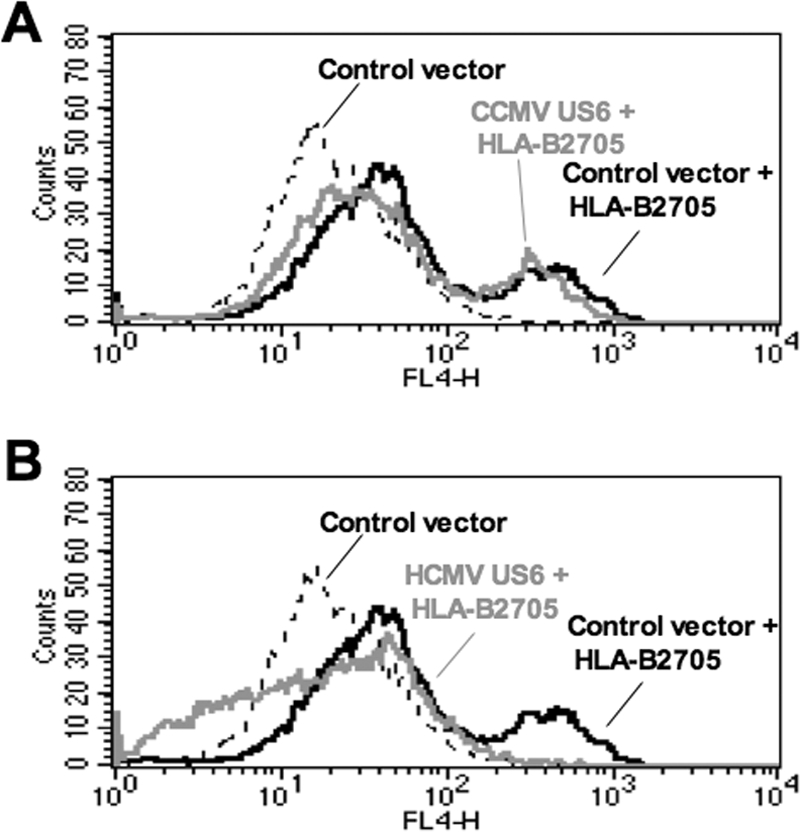
In order to further evaluate the sequence requirements for TAP inhibition and binding by HCMV US6, we characterized the CCMV homologue of US6 (10). CCMV US6 shares 35.2% sequence identity with HCMV US6, and like its HCMV orthologue, it is predicted to be a type I integral membrane protein with an N-terminal signal sequence (Fig. 5). However, of the 20 residues that we have defined as being crucial for TAP inhibition by HCMV US6, only 13 of these amino acids are found in the corresponding region of CCMV US6. The sequence divergence in this region includes the presence of a consensus sequence for a second N-linked glycosylation site (Asn-Ala-Ser) in residues 97 to 99 of CCMV US6, which is absent in the singly glycosylated HCMV US6. When expressed in HeLa-M cells, CCMV US6 colocalized with calreticulin (Fig. 6A), consistent with the localization of this protein in the ER. After signal peptide cleavage, CCMV US6 is predicted to be 6 residues longer than HCMV US6, but when resolved by SDS-PAGE, CCMV US6 was ∼3 kDa larger than HCMV US6 (Fig. 6B). This increase in Mr is consistent with the addition of an N-linked glycan to the second consensus N-linked glycosylation site in the CCMV protein. The glycosylation status of CCMV US6 was investigated with Endo H (Fig. 6B). Endo H can cleave oligosaccharides from N-linked glycoproteins that are resident in the ER, while glycoproteins whose N-linked glycan had been processed in the medial Golgi apparatus are resistant to Endo H digestion. After Endo H digestion, both CCMV US6 and HCMV US6 were reduced to approximately the same size, suggesting that the difference in electrophoretic mobility of the two proteins was primarily due to the presence of additional N-linked glycan in the CCMV protein (Fig. 6B). Furthermore, the cleavage of the N-linked glycans from CCMV US6 by Endo H confirms the localization of the protein in the ER. Since CCMV US6 was localized to the ER, we next tested whether CCMV US6 could inhibit the cell surface expression of the TAP-dependent allele HLA-B2705 in HeLa-M cells. Unlike its HCMV orthologue, CCMV US6 did not affect the cell surface expression of this MHC class I allele (Fig. 6C); from this, we conclude that despite sharing significant sequence identity with HCMV US6 and being localized to the ER, CCMV US6 is unable to inhibit TAP in human cells.
FIG. 5.

Alignment of the CCMV and HCMV US6 protein sequences. The HCMV and CCMV US6 protein sequences were aligned using the Vector NTI Suite software (Invitrogen), and identical residues are highlighted in gray. The transmembrane domains and signal sequences were predicted with PSORT II (32); the predicted signal sequences for both proteins are shown in italic type. The cleavage site is indicated by an arrow. Residue 1 corresponds to the N-terminal residue of the protein after signal peptide cleavage. The predicted transmembrane domains are underlined. Consensus sites for N-linked glycosylation sites are shown in bold type, and residues 89 to 108 in HCMV US6 and the corresponding region in CCMV US6 are boxed.
FIG. 6.

CCMV US6 localizes to the ER but does not inhibit cell surface HLA-B2705 expression. (A) Analysis of the intracellular localization of CCMV US6 by immunofluorescence microscopy. HeLa-M cells were transfected with either a control vector or with CCMV US6 or HCMV US6. The transfected cells were stained with an antibody specific for the ER marker calreticulin and the anti-FLAG epitope tag monoclonal antibody M2, which detects the US6 proteins. Calreticulin staining was detected with an anti-rabbit fluorescein isothiocyanate (FITC) antibody, and FLAG staining was detected with an anti-mouse Texas red antibody. Antibody fluorescence was visualized using a Zeiss Axioplan 2 microscope. The right panels are merged images of the red and green channels where yellow indicates colocalization; in these panels, the nuclei are also visualized with 4′,6′-diamidino-2-phenylindole (DAPI) (blue). Bars, 5 μm. (B) Endo H digestion of CCMV US6. HeLa-M cells were transfected with either a control vector, HCMV US6, or CCMV US6. Cell lysates (L) were immunoprecipitated with the anti-FLAG epitope antibody M2 and either digested with Endo H (+) or mock digested (−). Samples were resolved by SDS-PAGE, proteins transferred onto PVDF membranes and probed with the anti-FLAG epitope tag antibody M2. The position of a background band present in all samples that may correspond to the light chain of the M2 antibody is indicated by the asterisk. (C) Analysis of the effect of CCMV US6 on HLA-B2705 expression. HeLa-M cells were either transfected with the control vector or cotransfected with the control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and the US6 constructs. To detect HLA-B2705 cell surface expression, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody; antibody fluorescence was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the mean level of HLA-B2705 expression calculated from four independent experiments. Error bars show the standard deviations. Statistical analysis was performed using the Mann-Whitney U test. The sample with a significantly increased level (P ≤ 0.05) of cell surface HLA-B2705 expression compared to that of cells cotransfected with the HCMV US6 and HLA-B2705 vectors is indicated with an asterisk.
Mutational analysis of CCMV US6.
The inability of CCMV US6 to inhibit human TAP provided an opportunity to use it as a template for mutagenesis experiments in which to corroborate our findings on the sequence requirements for TAP inhibition by HCMV US6. Specifically, we sought to determine whether the inability of CCMV US6 to inhibit human TAP was due to the differences in the protein sequences within the region that we have identified as important for TAP inhibition by the HCMV US6. Residues 97 to 101 of CCMV US6 were substituted with the equivalent residues from HCMV US6 (residues 92 to 96) to generate a chimeric US6 (Fig. 7A). The resultant chimera, encoded a protein that was identical in the region corresponding to residues 89 to 108 of HCMV US6 in all but 2 of these 20 residues, whereas the remainder of the sequence was that of the CCMV US6. In contrast to the CCMV US6 protein, the chimeric US6 was able to inhibit cell surface expression of the TAP-dependent allele HLA-B2705 (Fig. 7B).
FIG. 7.

A chimeric US6 inhibits cell surface expression of HLA-B2705. (A) Schematic representation of the amino acid substitutions introduced into CCMV US6 to generate the chimeric US6 molecule. The schematic representations are set up as described in the legend to Fig. 3. (B) Analysis of the effect of CCMV US6 and the chimeric US6 on cell surface HLA-B2705 expression. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each US6 construct. To detect HLA-B2705 cell surface expression, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody; antibody fluorescence was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the mean level ± standard deviation (error bar) of HLA-B2705 expression calculated from four independent experiments. Statistical analysis was performed using the Mann-Whitney U test. The sample with a significantly increased level (P ≤ 0.05) of cell surface HLA-B2705 expression compared to that of cells cotransfected with the HCMV US6 and HLA-B2705 vectors is indicated by an asterisk. (C) Analysis of the interaction between chimeric and CCMV US6 with TAP. US6 proteins were immunoprecipitated from stable cell lines that expressed either HCMV US6, CCMV US6, or chimeric US6 constructs with the anti-FLAG epitope tag M2 antibody, resolved by SDS-PAGE, transferred onto PVDF membranes. and probed with TAP1 and M2 antibodies. The position of a background band present in all samples, which may correspond to the light chain of the M2 antibody, is indicated by the asterisk.
To determine why the chimeric US6, but not CCMV US6, was able to inhibit HLA-B2705 expression, we analyzed the interaction of these proteins with TAP. Stable HeLa-M cell lines expressing either the CCMV US6 or chimeric US6 protein were generated and compared to cells transfected with HCMV US6. TAP was coimmunoprecipitated with each US6 protein, hence demonstrating that like the HCMV protein, both the CCMV and chimeric US6 proteins interact with human TAP (Fig. 7C). Next TAP-dependent peptide translocation was assayed in the stable cell lines (Fig. 8A). In these experiments, peptide translocation was assayed in SLO-permeabilized cells using a fluorescein-labeled peptide that incorporated an N-linked glycosylation consensus sequence (23). Peptides that are translocated into the ER can be recovered on agarose containing concanavalin A, and peptide translocation was quantified by measuring fluorescein fluorescence. As predicted, the HCMV and chimeric US6 proteins inhibited peptide translocation into the ER, whereas the level of peptide translocation in cells expressing CCMV US6 was comparable to that in untransfected HeLa-M cells. ATP binding and hydrolysis are essential for peptide translocation by TAP and is inhibited by HCMV US6 (16, 20, 26, 31, 33). We therefore analyzed whether CCMV US6 was able to inhibit ATP binding by TAP. The ATP binding capacity of TAP was assayed with ATP-agarose beads (20) (Fig. 8B). TAP binding to the ATP-agarose beads was inhibited by HCMV US6, but not by CCMV US6. Hence, despite interacting with human TAP, CCMV US6 cannot block ATP binding by the transporter, and correspondingly, this explains why CCMV US6 is unable to inhibit peptide translocation by TAP and hence the cell surface expression of the TAP-dependent allele HLA-B2705. In contrast, the chimeric US6 protein was able to inhibit ATP-agarose binding by TAP. Since the chimeric US6 is identical in all but two of the amino acids corresponding to HCMV US6 residues 89 to 108 but otherwise has the same sequence as CCMV US6, these data provide further evidence for the crucial role of this region of the HCMV protein in the inhibition of TAP.
FIG. 8.

A chimeric US6 inhibits peptide translocation and ATP binding by TAP. (A) Analysis of the effect of CCMV and chimeric US6 on peptide translocation by TAP. SLO-permeabilized cells were incubated with the fluorescein-labeled peptide at 37°C for 10 min in the absence (−) and presence (+) of ATP. The reaction was stopped by the addition of ice cold 1% Triton X-100. Translocation into the ER was quantified by measuring the recovery of glycosylated fluorescein-labeled peptides on agarose containing concanavalin A. AFU, arbitrary fluorescence units. (B) Analysis of the effect of the CCMV US6 and chimeric US6 on ATP binding by TAP. Cell lysates were incubated with ATP-agarose, and bound proteins were eluted and resolved by SDS-PAGE. Proteins were transferred onto PVDF membranes and probed with a TAP1-specific antibody.
One potential reason for the inability of CCMV US6 to inhibit human TAP was the presence of an additional N-linked glycan, which may interfere with the interaction between CCMV US6 and TAP. This glycosylation site was therefore abolished by substitution of serine 99 with alanine (Fig. 9A). The mutant protein had a reduced Mr compared to that of the wild-type CCMV protein (data not shown), consistent with the loss of the glycosylation site; however, elimination of this glycosylation site did not enable the CCMV protein to inhibit cell surface expression of HLA-B2705 (Fig. 9B). This demonstrates that the inability of CCMV US6 to inhibit TAP is not simply due to steric interference caused by the N-linked glycan at this position and that instead the amino acid sequence of this region is critical for TAP inhibition.
FIG. 9.

Mutation of the C-terminal N-linked glycosylation site does not enable CCMV US6 to inhibit cell surface expression of HLA-B2705. (A) Schematic representation of the amino acid amino acid substitution introduced into CCMV US6 to generate the CCMV US6 S99A glycosylation mutant. The schematic representations are set up as described in the legend to Fig. 3. (B) Analysis of the effect of CCMV US6 and the chimeric US6 on cell surface HLA-B2705 expression. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each US6 construct. To detect HLA-B2705 cell surface expression, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody; antibody fluorescence was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the mean level of HLA-B2705 expression calculated from four independent experiments. Error bars show the standard deviations. Statistical analysis was performed using the Mann-Whitney U test. Those samples with a significantly increased level (P ≤ 0.05) of cell surface HLA-B2705 expression compared to that of cells cotransfected with the HCMV US6 and HLA-B2705 vectors are indicated by an asterisk.
To further evaluate the sequence requirements for TAP inhibition, additional mutational analysis of CCMV US6 was performed. There are multiple arginine residues within the predicted ER exposed loops of TAP1 and TAP2 (39). Residues 92 and 94 of HCMV US6 correspond to aspartate residues but are absent in CCMV US6, where the corresponding residues are asparagine 97 and serine 99. These two HCMV US6 aspartate residues may form salt bridges with the positively charged arginine residues of TAP. To test whether these aspartate residues are critical for TAP inhibition, we used CCMV US6 as a template for mutagenesis and substituted residues 97 and 99 with aspartate-generating CCMV US6 N97D/S99D (Fig. 10A). While the CCMV US6 N97D/S99D mutant was able to reduce cell surface expression of HLA-B2705, it was nonetheless functionally impaired in comparison to the HCMV US6 protein (Fig. 10B). We therefore conclude that while aspartate residues 92 and 94 contribute to the inhibition of TAP by HCMV US6, other residues are also important.
FIG. 10.

The substitution of residues 97 and 99 of CCMV US6 with aspartate residues enhances the ability of CCMV US6 to inhibit cell surface expression of HLA-B2705. (A) Schematic representation of the amino acid amino acid substitutions introduced into CCMV US6 to generate the CCMV US6 N97D/S99D mutant. The schematic representations are set up as described in the legend to Fig. 3. (B) Analysis of the effect of the CCMV US6 N97D/S99D mutant on the cell surface expression of HLA-B2705. HeLa-M cells were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each US6 construct. To detect HLA-B2705 expression, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody; antibody fluorescence was quantified by flow cytometry. The level of HLA-B2705 expression is expressed as a percentage of the MFV of the positive control (cells transfected with HLA-B2705 and control vector) after subtracting the MFV of the negative control (cells transfected with control vector only). Each bar represents the mean level of HLA-B2705 expression calculated from four independent experiments. Error bars show the standard deviations. Statistical analysis was performed using the Mann-Whitney U test.
CCMV US6 does not inhibit the cell surface expression of HLA-B2705 in the chimpanzee cell line WES.
CCMV US6 does not inhibit human TAP and as a consequence cannot inhibit expression of HLA-B2705 in the human cell line HeLa-M. We therefore wondered whether CCMV US6 could function as a TAP inhibitor in chimpanzee cells. HLA-B2705 was cotransfected with either HCMV or CCMV US6 into the chimpanzee fibroblast cell line WES (Fig. 11). Transfection of the control vector and HLA-B2705 resulted in an increase in cell surface staining by the HLA-B2705-specific monoclonal ME1 antibody, consistent with the expression of HLA-B2705 on the cell surface of the transfected cells. Cell surface expression of HLA-B2705 was reduced when WES cells were cotransfected with HCMV US6, but not by CCMV US6. Therefore, these data suggest that while HCMV US6 can inhibit chimpanzee TAP, CCMV US6 cannot, and as such, these data do not support a role for CCMV US6 as a TAP inhibitor in chimpanzee cells.
FIG. 11.
CCMV US6 does not inhibit cell surface expression of HLA-B2705 in a chimpanzee cell line. Analysis of the effect of CCMV US6 (A) and HCMV US6 (B) on the expression of HLA-B2705 at the plasma membrane. WES chimpanzee fibroblasts were either transfected with a control vector or cotransfected with a control vector and the HLA-B2705 expression construct or cotransfected with HLA-B2705 and each US6 construct. To detect cell surface expression of HLA-B2705, the transfectants were stained with the ME1 MAb and a Cy5-conjugated secondary antibody. Antibody fluorescence was quantified by flow cytometry.
DISCUSSION
In this study we have identified regions of HCMV US6 that are required for the interaction with and inhibition of TAP. We show that residues 89 to 108 of HCMV US6 are necessary for efficient inhibition of TAP. However, mutation of these residues does not impair TAP binding nor does it affect oligomerization of HCMV US6. This demonstrates that TAP binding and the oligomerization of HCMV US6 are by themselves insufficient for the inhibition of the transporter. This conclusion is supported by the observation that CCMV US6 can also bind to human TAP and form oligomers (data not shown) but does not inhibit the transporter. Residues 89 to 108 of HCMV US6 may therefore mediate an interaction with TAP that is critical for inhibition of the transporter, whereas other parts of HCMV US6 may stabilize this inhibitory interaction (Fig. 12). Indeed, deletion of the C-terminal 49 residues of HCMV US6 results in a protein that can inhibit TAP but that does not coimmunoprecipitate with TAP, suggesting that the C terminus of HCMV US6 functions to stabilize the interaction between the viral protein and TAP. Although the C-terminal 49 residues were dispensable in these experiments, in which HCMV US6 was overexpressed, the C terminus of HCMV US6 may play an important role in the context of natural infection. Likewise, deletion of the N-terminal 90 residues also reduces the amount of TAP that coimmunoprecipitates with HCMV US6, suggesting that this region also stabilizes the interaction between HCMV US6 and TAP.
FIG. 12.

Schematic representation of the interaction between HCMV US6 and TAP. The relative positions of the functional HCMV US6ΔN80 and ΔC49 truncations are indicated by arrows. The nucleotide binding domains of TAP (NBD), transmembrane domain of HCMV US6 (TMD), and N-linked glycan of HCMV US6 (circled G) are indicated. Residues 89 to 108 of HCMV US6 are required for the inhibition of TAP, whereas residues 116 to 125 are not required for TAP inhibition but stabilize the interaction between US6 and TAP. In addition, residues 81 to 90 and the C-terminal 39 residues of HCMV US6 may also contribute to the stabilization of the interaction between US6 and TAP.
Although the three-dimensional structure of HCMV US6 is unknown, by comparison with HCMV US2, whose structure has been solved, US6 has been proposed to have an immunoglobulin fold (13). Immunoglobulin domains often mediate protein-protein interactions, and it is conceivable that residues 89 to 108 may form a TAP binding surface analogous to the MHC class I binding surface formed by US2. However, when expressed in Escherichia coli, HCMV US6 has significant α-helical content, which would be inconsistent with an immunoglobulin fold (26). Nonetheless, irrespective of the precise fold assumed by HCMV US6, residues 89 to 108 are likely to form a surface-exposed region that binds to TAP. The other sequences that stabilize the interaction of HCMV US6 with TAP may either be required for the proper assembly of this TAP binding site or alternatively they may also interact with TAP. While our data cannot discriminate between these two possibilities, a recent study has identified up to four binding sites for HCMV US6 on the TAP heterodimer (14). As such, the different regions of HCMV US6 may bind TAP at distinct sites (Fig. 12). HCMV US6 can bind the N-terminal part of the TAP1 and TAP2 membrane domains, but these interactions do not result in the inhibition of peptide translocation (14), which is analogous to HCMV US6 proteins that have mutations in residues 89 to 108. The interaction of HCMV US6 with the C-terminal parts of the TAP1 and TAP2 subunit membrane domains is necessary for the inhibition of peptide translocation (14). Intriguingly, the interaction of HCMV US6 with this region of TAP2 cannot be detected by immunoprecipitation (14), a result that is analogous to the effect of deleting the C-terminal 49 residues of HCMV US6. As such, there are clear parallels between our observations and those of Halenius et al. (14), with both studies suggesting that TAP inhibition by US6 involves a hierarchy of interactions. We therefore speculate that residues 89 to 108 of the HCMV US6 luminal domain inhibit TAP by interacting with the C termini of the membrane domains of TAP1 and/or TAP2 that are within the core transporter domain of TAP, which is inhibited by HCMV US6 (21). The C terminus of HCMV US6, and potentially residues 81 to 90, may interact with the N-terminal parts of the TAP1 and/or TAP2 subunits. Interaction at these sites thus would be predicted to stabilize the interaction between the HCMV protein and TAP but would not result in the inhibition of the transporter.
In contrast to its HCMV orthologue, CCMV US6 was unable to inhibit human TAP. While CCMV US6 binds to human TAP, it may not interact with the C-terminal regions of the TAP1 and TAP2 membrane domains that constitute the core transporter domain (21) and as a consequence cannot inhibit peptide translocation. CCMV US6 could instead be a highly species-specific inhibitor of chimpanzee TAP. Indeed, HCMV US6 displays species specificity, in that it can inhibit human, African green monkey, and rabbit TAP, but not rat and mouse TAP (14). However, HCMV US6, but not CCMV US6, blocks cell surface expression of cotransfected HLA-B2705 in the chimpanzee cell line WES. Hence, our data do not support a role for CCMV US6 as a TAP inhibitor in either human or chimpanzee cells. This contrasts with the rhesus monkey cytomegalovirus protein Rh185, which despite having only 28.8% sequence identity with HCMV US6 can inhibit peptide translocation by human TAP (35). If CCMV US6 is not a TAP inhibitor, what is its function? Since CCMV US6 can bind to TAP, it may recruit another viral protein that inhibits peptide translocation. Alternatively, CCMV US6 may interfere with other components of the peptide-loading complex, such as tapasin; for example, the adenovirus protein E19 binds to TAP and inhibits tapasin (7). Clearly, further work is required to elucidate the function of CCMV US6. Nonetheless, the comparison of this CCMV protein with its HCMV orthologue has provided valuable information about the sequence requirements for the binding and inhibition of TAP by HCMV US6. In summary, we propose that distinct regions of HCMV US6 mediate a hierarchy of interactions with TAP, in which residues 89 to 108 form an inhibitory interaction with TAP that is stabilized by other parts of the viral protein.
Acknowledgments
Gillian Dugan is a BBSRC-funded Ph.D. student.
We thank Andrew Davison, Antony Antoniou, and Simon Powis for reagents; Graham Bottley for advice on flow cytometry; Emmanuel Wiertz for advice on the peptide translocation assay; and Mark Harris for helpful discussions.
Footnotes
Published ahead of print on 16 January 2008.
REFERENCES
- 1.Abele, R., and R. Tampe. 2004. The ABCs of immunology: structure and function of TAP, the transporter associated with antigen processing. Physiology 19216-224. [DOI] [PubMed] [Google Scholar]
- 2.Ahn, K., A. Gruhler, B. Galocha, T. R. Jones, E. J. Wiertz, H. L. Ploegh, P. A. Peterson, Y. Yang, and K. Fruh. 1997. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6613-621. [DOI] [PubMed] [Google Scholar]
- 3.Ambagala, A. P., S. Hinkley, and S. Srikumaran. 2000. An early pseudorabies virus protein down-regulates porcine MHC class I expression by inhibition of transporter associated with antigen processing (TAP). J. Immunol. 16493-99. [DOI] [PubMed] [Google Scholar]
- 4.Antoniou, A. N., S. Ford, J. D. Taurog, G. W. Butcher, and S. J. Powis. 2004. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J. Biol. Chem. 2798895-8902. [DOI] [PubMed] [Google Scholar]
- 5.Antoniou, A. N., S. J. Powis, and T. Elliott. 2003. Assembly and export of MHC class I peptide ligands. Curr. Opin. Immunol. 1575-81. [DOI] [PubMed] [Google Scholar]
- 6.Benham, A. M., M. Gromme, and J. Neefjes. 1998. Allelic differences in the relationship between proteasome activity and MHC class I peptide loading. J. Immunol. 16183-89. [PubMed] [Google Scholar]
- 7.Bennett, E. M., J. R. Bennink, J. W. Yewdell, and F. M. Brodsky. 1999. Adenovirus E19 has two mechanisms for affecting class I MHC expression. J. Immunol. 1625049-5052. [PubMed] [Google Scholar]
- 8.Boname, J. M., B. D. de Lima, P. J. Lehner, and P. G. Stevenson. 2004. Viral degradation of the MHC class I peptide loading complex. Immunity 20305-317. [DOI] [PubMed] [Google Scholar]
- 9.Daniel, S., V. Brusic, S. Caillat-Zucman, N. Petrovsky, L. Harrison, D. Riganelli, F. Sinigaglia, F. Gallazzi, J. Hammer, and P. M. van Endert. 1998. Relationship between peptide selectivities of human transporters associated with antigen processing and HLA class I molecules. J. Immunol. 161617-624. [PubMed] [Google Scholar]
- 10.Davison, A. J., A. Dolan, P. Akter, C. Addison, D. J. Dargan, D. J. Alcendor, D. J. McGeoch, and G. S. Hayward. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 8417-28. [DOI] [PubMed] [Google Scholar]
- 11.Ellis, S. A., C. Taylor, and A. McMichael. 1982. Recognition of HLA-B27 and related antigen by a monoclonal antibody. Hum. Immunol. 549-59. [DOI] [PubMed] [Google Scholar]
- 12.Fruh, K., K. Ahn, H. Djaballah, P. Sempe, P. M. van Endert, R. Tampe, P. A. Peterson, and Y. Yang. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375415-418. [DOI] [PubMed] [Google Scholar]
- 13.Gewurz, B. E., R. Gaudet, D. Tortorella, E. W. Wang, H. L. Ploegh, and D. C. Wiley. 2001. Antigen presentation subverted: structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc. Natl. Acad. Sci. USA 986794-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halenius, A., F. Momburg, H. Reinhard, D. Bauer, M. Lobigs, and H. Hengel. 2006. Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J. Biol. Chem. 2815383-5390. [DOI] [PubMed] [Google Scholar]
- 15.Hengel, H., J. O. Koopmann, T. Flohr, W. Muranyi, E. Goulmy, G. J. Hammerling, U. H. Koszinowski, and F. Momburg. 1997. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 6623-632. [DOI] [PubMed] [Google Scholar]
- 16.Hewitt, E. W., S. Sen Gupta, and P. J. Lehner. 2001. The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J. 20387-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill, A., P. Jugovic, I. York, G. Russ, J. Bennink, J. Yewdell, H. Ploegh, and D. Johnson. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375411-415. [DOI] [PubMed] [Google Scholar]
- 18.Hislop, A. D., M. E. Ressing, D. van Leeuwen, V. A. Pudney, D. Horst, D. Koppers-Lalic, N. P. Croft, J. J. Neefjes, A. B. Rickinson, and E. J. Wiertz. 2007. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 2041863-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly, A., S. H. Powis, L. A. Kerr, I. Mockridge, T. Elliott, J. Bastin, B. Uchanska-Ziegler, A. Ziegler, J. Trowsdale, and A. Townsend. 1992. Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature 355641-644. [DOI] [PubMed] [Google Scholar]
- 20.Knittler, M. R., P. Alberts, E. V. Deverson, and J. C. Howard. 1999. Nucleotide binding by TAP mediates association with peptide and release of assembled MHC class I molecules. Curr. Biol. 9999-1008. [DOI] [PubMed] [Google Scholar]
- 21.Koch, J., R. Guntrum, S. Heintke, C. Kyritsis, and R. Tampe. 2004. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J. Biol. Chem. 27910142-10147. [DOI] [PubMed] [Google Scholar]
- 22.Koch, J., R. Guntrum, and R. Tampe. 2006. The first N-terminal transmembrane helix of each subunit of the antigenic peptide transporter TAP is essential for independent tapasin binding. FEBS Lett. 5804091-4096. [DOI] [PubMed] [Google Scholar]
- 23.Koppers-Lalic, D., E. A. Reits, M. E. Ressing, A. D. Lipinska, R. Abele, J. Koch, M. Marcondes Rezende, P. Admiraal, D. van Leeuwen, K. Bienkowska-Szewczyk, T. C. Mettenleiter, F. A. Rijsewijk, R. Tampe, J. Neefjes, and E. J. Wiertz. 2005. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 1025144-5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koppers-Lalic, D., M. Rychlowski, D. van Leeuwen, F. A. Rijsewijk, M. E. Ressing, J. J. Neefjes, K. Bienkowska-Szewczyk, and E. J. Wiertz. 2003. Bovine herpesvirus 1 interferes with TAP-dependent peptide transport and intracellular trafficking of MHC class I molecules in human cells. Arch. Virol. 1482023-2037. [DOI] [PubMed] [Google Scholar]
- 25.Kutsch, O., T. Vey, T. Kerkau, T. Hunig, and A. Schimpl. 2002. HIV type 1 abrogates TAP-mediated transport of antigenic peptides presented by MHC class I. Transporter associated with antigen presentation. AIDS Res. Hum. Retrovir. 181319-1325. [DOI] [PubMed] [Google Scholar]
- 26.Kyritsis, C., S. Gorbulev, S. Hutschenreiter, K. Pawlitschko, R. Abele, and R. Tampe. 2001. Molecular mechanism and structural aspects of transporter associated with antigen processing inhibition by the cytomegalovirus protein US6. J. Biol. Chem. 27648031-48039. [DOI] [PubMed] [Google Scholar]
- 27.Lehner, P. J., J. T. Karttunen, G. W. Wilkinson, and P. Cresswell. 1997. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA 946904-6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leonhardt, R. M., K. Keusekotten, C. Bekpen, and M. R. Knittler. 2005. Critical role for the tapasin-docking site of TAP2 in the functional integrity of the MHC class I-peptide-loading complex. J. Immunol. 1755104-5114. [DOI] [PubMed] [Google Scholar]
- 29.Linton, K. J. 2007. Structure and function of ABC transporters. Physiology 22122-130. [DOI] [PubMed] [Google Scholar]
- 30.Lipińska, A. D., D. Koppers-Lalic, M. Rychłlowski, P. Admiraal, F. A. M. Rijsewijk, K. Bieńkowska-Szewczyk, and E. J. H. J. Wiertz. 2006. Bovine herpesvirus 1 UL49.5 protein inhibits the transporter associated with antigen processing despite complex formation with glycoprotein M. J. Virol. 805822-5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer, T. H., P. M. van Endert, S. Uebel, B. Ehring, and R. Tampe. 1994. Functional expression and purification of the ABC transporter complex associated with antigen processing (TAP) in insect cells. FEBS Lett. 351443-447. [DOI] [PubMed] [Google Scholar]
- 32.Nakai, K., and P. Horton. 1999. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 2434-36. [DOI] [PubMed] [Google Scholar]
- 33.Neefjes, J. J., F. Momburg, and G. J. Hammerling. 1993. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science 261769-771. [DOI] [PubMed] [Google Scholar]
- 34.Ortmann, B., M. J. Androlewicz, and P. Cresswell. 1994. MHC class I/beta 2-microglobulin complexes associate with TAP transporters before peptide binding. Nature 368864-867. [DOI] [PubMed] [Google Scholar]
- 35.Pande, N. T., C. Powers, K. Ahn, and K. Fruh. 2005. Rhesus cytomegalovirus contains functional homologues of US2, US3, US6, and US11. J. Virol. 795786-5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Procko, E., G. Raghuraman, D. C. Wiley, M. Raghavan, and R. Gaudet. 2005. Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol. Cell Biol. 83475-482. [DOI] [PubMed] [Google Scholar]
- 37.Ressing, M. E., S. E. Keating, D. van Leeuwen, D. Koppers-Lalic, I. Y. Pappworth, E. J. Wiertz, and M. Rowe. 2005. Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J. Immunol. 1746829-6838. [DOI] [PubMed] [Google Scholar]
- 38.Rock, K. L., and A. L. Goldberg. 1999. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 17739-779. [DOI] [PubMed] [Google Scholar]
- 39.Schrodt, S., J. Koch, and R. Tampe. 2006. Membrane topology of the transporter associated with antigen processing (TAP1) within an assembled functional peptide-loading complex. J. Biol. Chem. 2816455-6462. [DOI] [PubMed] [Google Scholar]
- 40.Smith, K. D., and C. T. Lutz. 1996. Peptide-dependent expression of HLA-B7 on antigen processing-deficient T2 cells. J. Immunol. 1563755-3764. [PubMed] [Google Scholar]
- 41.Vambutas, A., J. DeVoti, W. Pinn, B. M. Steinberg, and V. R. Bonagura. 2001. Interaction of human papillomavirus type 11 E7 protein with TAP-1 results in the reduction of ATP-dependent peptide transport. Clin. Immunol. 10194-99. [DOI] [PubMed] [Google Scholar]
- 42.van Endert, P. M., L. Saveanu, E. W. Hewitt, and P. Lehner. 2002. Powering the peptide pump: TAP crosstalk with energetic nucleotides. Trends Biochem. Sci. 27454-461. [DOI] [PubMed] [Google Scholar]