

Abstract
Background
In view of the limited success of available treatment modalities for a wide array of cancer, alternative and complementary therapeutic strategies need to be developed. Virotherapy employing conditionally replicative adenoviruses (CRAds) represents a promising targeted intervention relevant to a wide array of neoplastic diseases. Critical to the realization of an acceptable therapeutic index using virotherapy in clinical trials is the achievement of oncolytic replication in tumor cells, while avoiding non-specific replication in normal tissues. In this report, we exploited cancer-specific control of mRNA translation initiation in order to achieve enhanced replicative specificity of CRAd virotherapy agents. Heretofore, the achievement of replicative specificity of CRAd agents has been accomplished either by viral genome deletions or incorporation of tumor selective promoters. In contrast, control of mRNA translation has not been exploited for the design of tumor specific replicating viruses to date. We show herein, the utility of a novel approach that combines both transcriptional and translational regulation strategies for the key goal of replicative specificity.
Methods
We describe the construction of a CRAd with cancer specific gene transcriptional control using the CXCR4 gene promoter (TSP) and cancer specific mRNA translational control using a 5′ untranslated region (5′-UTR) element from the FGF-2 (Fibroblast Growth Factor-2) mRNA.
Results
Both in vitro and in vivo studies demonstrated that our CRAd agent retains anti-tumor potency. Importantly, assessment of replicative specificity using stringent tumor and non-tumor tissue slice systems demonstrated significant improvement in tumor selectivity.
Conclusions
Our study addresses a conceptually new paradigm: dual targeting of transgene expression to cancer cells using both transcriptional and mRNA translational control. Our novel approach addresses the key issue of replicative specificity and can potentially be generalized to a wide array of tumor types, whereby tumor selective patterns of gene expression and mRNA translational control can be exploited.
Keywords: Virotherapy, Adenovirus, Conditionally replicative, CRAd, E1A, Tumor selective promoter, TSP, Transcription, Transcriptional control, CXCR4, mRNA translation, mRNA translational control 5′-untranslated region, 5′-UTR, Fibroblast growth factor, FGF-2, Eukaryotic initiation factor 4E, eIF4E, Breast cancer, Breast tumor
Introduction
In view of the limited success of available treatment modalities for breast cancer, alternative and complementary strategies need to be developed. Virotherapy marks an innovative approach in the development of new treatment regimes, in which a replicating virus itself is the anticancer agent. In this regard, virotherapy employing conditionally replicative adenoviruses (CRAds) represents a promising targeted intervention relevant to a wide array of neoplastic diseases. Critical to the realization of an acceptable therapeutic index in clinical trials using virotherapy is the achievement of oncolytic replication in tumor cells, while avoiding nonspecific replication in normal tissues [1–4]. In this regard, the elucidation of the critical pathways involved in the adenoviral infectious cycle has provided the basis of multiple levels of potential control to achieve the goal of selective replication. For example, modification of viral tropism via altered cell surface binding provides the practical basis of transductional targeting [5, 6]. Alternatively, controlling expression of critical adenoviral regulatory genes via tumor selective promoters (TSPs) is the basis of transcriptional targeting [7]. These multiple targeting strategies thus provide the potential for a combinatorial approach to achieve an optimized level of selective CRAd replication.
Thus far, transcriptional targeting has been the most frequently applied strategy in the design of CRAd agents. Due to the native adenoviral hepatotropism [4, 8], the ideal TSP should exhibit the widest differential between “tumor on” and “liver off” expression profiles [1]. Whereas candidate promoters have been identified that embody this profile, direct TSP employment in the CRAd context may not necessarily provide the level of cancer specificity required. In this regard, heterologous promoters incorporated into the adenovirus (Ad) genome may exhibit altered induction patterns due to endogenous cis and trans regulatory elements derived from the Ad genome. To address this issue, the addition of insulator elements has been proposed. Such strategies however, may not be generally applicable to all TSPs [9]. These observations demonstrate that additional regulatory strategies may be necessary to achieve an optimized cancer selective replication of CRAds.
To this end, control of gene expression at the level of mRNA translation offers an additional approach relevant to targeting cancer cells. Recently, the control of mRNA stability via tumor-associated proteins has been explored for tumor-specific replicating viruses. This approach utilized sequences from the 3′ untranslated region (3′-UTR) of the PTGS2 (prostaglandin-endoperoxide synthase 2) mRNA to control selective mRNA stability and degradation of the adenovirus E1A gene transcript [10]. In contrast, control of mRNA translation has not been exploited for the design of tumor specific replicating viruses to date. Of note, dysregulation of mRNA translation in cancer is a well-recognized mechanism, which may contribute to neoplastic conversion and progression. In particular, overexpression of the translation eukaryotic initiation factor eIF4E has been documented in cellular transformation and tumorigenesis [11]. The eIF4E factor specifically binds to the cap structure of mRNAs during the first step of mRNA recruitment for translation [12, 13]. It is also a subunit of a pre-initiation complex, which contains a helicase activity that unwinds the secondary structure at the 5′-UTR of mRNA. This latter function is critical during scanning for the translation start site of mRNAs with long 5′-UTRs capable of forming complex stable secondary structures [14, 15].
Recognizing the nearly ubiquitous eIF4E over-expression in solid tumors, we hypothesized that mRNA translation could be exploited as an additional regulatory strategy to control replication of adenoviral vectors for cancer virotherapy. We hypothesized that the addition of a long and highly structured 5′-UTR preceding the adenoviral E1A coding sequence would limit efficient translation exclusively to cells that express high levels of the translation initiation factor eIF4E. We describe herein, the construction of a CRAd with cancer specific gene transcriptional control using the CXCR4 gene promoter as a TSP and cancer specific mRNA translational control utilizing a 5′-UTR element from the FGF-2 (Fibroblast Growth Factor-2) mRNA. Our study thus addresses a conceptually new paradigm: dual targeting of transgene expression to cancer cells using both transcriptional and mRNA translational control. Our novel approach addresses the key issue of replicative specificity and may be very valuable both scientifically and clinically. It can potentially be applied to a wide array of tumor types, whereby tumor selective patterns of gene expression and mRNA translational control can be exploited, resulting in a new class of virotherapeutic agents.
Materials and methods
Cell lines and cell culture
The MDA-MB-231, MDA-MB-361, and MDA-MB-435 breast cancer cell lines were obtained from the American Type Culture Collection (ATCC) and cultured as described. In brief, the cells were maintained in DMEM/F-12 medium (Life Technologies, Inc., Grand Island, NY), containing 10% fetal bovine serum (FBS; Gemini BioProducts, Woodland, Ca), and 1% antibiotic-antimycotic solution (penicillin-streptomycin-fungizone; Sigma Chemicals Co., St. Louis, MO). The cells were maintained in T150 flasks at 37°C and 5% humidified CO2, and were subcultured using 1% trypsin-EDTA (Gibco BRL, Life Technologies). Human mammary epithelial cells (HMEC) were obtained from Clonetics (Walkersville, MD), and were maintained in serum-free mammary epithelial growth media (Clonetics) and passaged as recommended by the vendor.
Primary human cells
Human dermal fibroblasts were derived from neonatal skin by trypsinization as described [16–18]. Human dermal fibroblasts obtained from outgrowth of explant cultures, were grown in DMEM (Dulbecco’s modified Eagle’s medium; Bio Whittaker) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 lg/mL streptomycin and grown as monolayers on plastic tissue culture dishes in a humidified atmosphere of a CO2 incubator at 37°C.
Tissue slices and culture
Approval was obtained from the Institutional Review Board for all studies on human tissue. Human breast cancer samples were obtained (Department of Pathology, University of Alabama at Birmingham) from three patients undergoing mastectomy for primary invasive ductal carcinoma;normal breast tissue samples were obtained from three patients undergoing reduction mammoplasty. Human liver samples were obtained (Department of Surgery, University of Alabama at Birmingham) from three HIV seronegative donor livers prior to transplantation into recipients. All tissue samples were flushed with University of Wisconsin (UW) solution (ViaSpan, Barr Laboratories, Inc. Pomona, NY) before harvesting and kept on ice in UW solution until slicing. Time from harvest to slicing was kept at an absolute minimum (<2 h). The precision tissue cut technique was performed using the Krumdieck Tissue Slicer as previously described [19].
Tissue slices (250 micron) were placed into six-well plates (1 slice per well) containing 2 mL of complete culture media (liver, William’s Medium E with 1% antibiotics, 1% L-glutamine, and 10% FCS; normal breast and breast tumor tissue, RPMI with 1% antibiotics, 1% L-glutamine, and 10% FCS). The plates were then incubated at 37°C and 5% CO2 in a humidified environment under normal oxygen concentrations for up to 48 h. A plate rocker set at 60 rpm was used to agitate slices and ensure adequate oxygenation and viability [20].
Construction and production of adenoviruses
The plasmid pBSKCAT/CXCR4, which contains a 279 bp sequence from the human CXCR4 promoter (−191 to +88), was a kind gift of Dr. Nelson L. Michael [21]. The CXCR4 promoter sequence (NCBI Accession Number AY728138, from 1780 to 2059 bp) was cloned by PCR into the SpeI/PmeI site of pAdenoVator-CMV5 (Qbiogene; Irvine, CA) replacing the CMV promoter to generate pAdenoVator-CXCR4. This construct contained the 279 bp CXCR4 promoter and the simian virus 40 (SV40) polyadenylation (poly A) signal. The adenoviral type 5 E1A gene (NCBI Accession Number AY728138, from 560 to 1545 bp) was then cloned by PCR into the PmeI/BamHI site of pAdenoVator-CXCR4 to generate the pAdenoVator-CXCR4-E1A plasmid. The 5′ upstream-untranslated region sequence of the rat FGF-2 cDNA (NCBI Accession Number NM_019305, from 1 to 532 bp) was inserted in the PmeI site immediately upstream of the E1A sequence to produce the pAdenoVator-CXCR4-UTR-E1A plasmid. After cleavage with FseI, the pAdenoVator-CXCR4-E1A and the pAdenoVator-CXCR4-UTR-E1A plasmids were used for homologous recombination in BJ5183 bacteria with the pAdEasy-1 genome (Stratagene; La Jolla, CA). The resulting pAdEasy-CXCR4-E1A and pAdEasy-CXCR4-UTR-E1A recombinant plasmids were identified by PCR screening.
The recombinant plasmids were linearized with PacI and transfected into 293 cells using Superfect reagent (Qiagen; Valencia, CA) to generate the Ad5-CXCR4-E1A and Ad5-CXCR4-UTR-E1A adenoviruses. The adenoviruses were propagated in the FaDu cell line (a human head and neck cancer cell line in which CXCR4 is over expressed), and purified by double CsCl density gradient centrifugation, followed by dialysis against phosphate buffered saline (PBS) containing 10% glycerol. The concentration of total viral particle numbers (PN) were determined by measuring absorption at 260 nm. Infectious PN (IFN) were determined by measuring the concentration of viral hexon protein-positive 293 cells after a 48 h infection period, using an Adeno-X Rapid Titer Kit (Clontech; Mountain View, CA). Wild type Ad5 (Ad-wt-dE3) and Ad-CXCR4-GL3 [22] were used for replication positive and negative control, respectively, in the CRAd agent analysis.
Western blot analysis
Levels of endogenous human eIF4E, CXCR4, and b-actin expressed in uninfected cells, were determined by Western immunoblot analysis, from 20 lg of total cellular protein. Cells were solubilized in lysis buffer (150 mM NaCl, 1.0% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris pH 8.0) and the protein concentration was determined using the Bradford protein assay (Bio-Rad Laboratories, Richmond, CA, USA). For eIF4E expression, cells were harvested and solubilized in lysis buffer. Samples were electrophoresed on a 10% SDS/polyacrylamide gel and subsequently transferred to nitrocellulose in a semidry electrophoretic blotting system (NovaBlot; Pharmacia, Piscataway, NJ, USA). The nitrocellulose membranes were incubated for 1 h in a blocking buffer of 5% bovine serum albumin (BSA) in TTBS buffer (500 mM NaCl, 100 mM Tris, 0.1% Tween 20), followed by incubation with buffer containing 1% BSA and either (a) a mouse anti-human eIF4E monoclonal antibody (BD Transduction Labs), (b) a mouse anti-human CXCR4 monoclonal antibody (BD Transduction Labs), or (c) a mouse anti-human b-actin monoclonal antibody (Pharmingen). Subsequently, membranes were washed in TTBS buffer and incubated with a horseradish peroxidase (HRP) conjugated secondary anti-mouse or anti-rabbit antibody (Santa Cruz Biotechnology; Santa Cruz, CA). Specific protein bands were detected using the ECL Western blotting system (Amersham Biosciences; Piscataway, NJ). Finally, the membranes were washed and incubated with ECL reagent and analyzed by autoradiography. Densitometry analysis was performed using a VersaDoc 3000 imaging system (Bio-Rad).
Levels of adenoviral E1A protein were determined by Western immunoblot analysis. Cells infected Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3, or Ad-wtdE3 at a multiplicity of infection (MOI) of 100:1 were harvested at 48 h. Cells were harvested and solubilized in lysis buffer. Samples were subjected to electrophoresis on a 10% SDS/polyacrylamide gel and subsequently transferred to nitrocellulose. The nitrocellulose membranes were incubated for 1 h in a blocking buffer of 5% BSA in TTBS buffer, followed by incubation with TTBS buffer containing 1% BSA and a mouse anti-Adenovirus type 5 E1A monoclonal antibody (Lab Vision; Fremont, CA). Subsequently, the membranes were washed in TTBS buffer and incubated with a HRP conjugated goat anti-mouse IgG secondary antibody (Santa Cruz Biotechnology). Specific protein bands were detected using the ECL reagent and analyzed by autoradiography.
Quantifying viral transcription
For quantification of E1A mRNA expression 1.5 × 105 cells were seeded per well in a six-well plate. The next day, cells were infected with the indicated viruses at 100 MOI, or mock infected. After 48 h, purification of total RNA and quantitative real-time PCR for E1A were performed as previously described [23]. Viral infection of the tissue slices was performed as previously described [19]. After infection and frozen storage, tissue slices were thawed and placed in RLT buffer (RNEasy RNA extraction kit, Qiagen, Valencia, CA) with β-mercaptoethanol (Sigma, St. Louis, MO). Slices were homogenized immediately with an ultrasonic sonicator (Fisher Scientific Model 100) at a setting of 15 watts for 10 sec. The homogenate was centrifuged, and the supernatant was removed to separate Eppendorf tubes for subsequent RNA purification with the RNEasy RNA purification kit according to the manufacturer’s directions. Comparison of transcription rates of different treatment groups were performed with a Student’s t-test.
Quantifying virus replication
Purification of the DNA and quantitative real-time PCR for E4 was performed as previously described [24]. 1.5 × 105 cells were seeded per well in a six-well plate. The next day cells were infected with the indicated viruses at 100:1 MOI or mock infected. Growth medium was collected at the indicated time points, and assessment of viral copy number in 200 lL aliquots was performed as previously described [19]. Negative controls without template were performed for each reaction series, and an internal control (human GAPDH) was used to normalize the copy number for the E4 genes. Comparison of replication rates of different treatment groups were performed with a Student’s t-test.
In vitro cytotoxicity assay
For determination of virus-mediated cytotoxicity, 1.5 · 104 cells were seeded in 24 well tissue culture plates and were infected with adenoviruses at indicated titers or were mock-infected [25]. To visualize cell killing, cells were fixed and stained with 1% crystal violet in 70% ethanol for 20 min, followed by washing with tap water to remove excess dye. The plates were dried and images were captured with a Kodak DC260 digital camera (Eastman Kodak, Rochester, NY, USA).
In vivo oncolysis
All experimental protocols were approved by the Institutional Animal Care and Use Committees of LSU Health Sciences Center and University of Alabama at Birmingham. To establish subcutaneous tumors, 4–5 week old BALB/c nude mice were inoculated in both flanks with 5 · 106 MDA-MB-361 cells (n = 5 animals/group; 2 tumors/animal). Before injection, the tumor cells were verified to have >95% viability by trypan blue exclusion. When the tumor reached 5 mm in widest diameter, 1 · 108 IFN of Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 or Ad-wt-dE3 was injected intratumorally, in a 0.05 mL volume. Control tumors were injected with an equal volume of PBS only. Animals were examined every other day and killed if tumor size reached 1.0 · 1.0 cm. The tumor volume was calculated by the formula: 1/2 xy2, where x is the longest distance of the tumor and y is the distance perpendicular to x.
Results
A model for using differential expression of eIF4E
We constructed a dual-level targeted CRAd with the Ad E1A gene that is essential for viral replication, with transcriptional control using the CXCR4 gene promoter and mRNA translational control using the FGF-2 5′-UTR sequence placed upstream of the coding sequence (Fig. 1). This CRAd construct (Ad-CXCR4-UTR-E1A) was compared to a single-level targeted CRAd (Ad-CXCR4-E1A) with transcriptional control using the CXCR4 gene promoter alone. Experiments were performed to compare regulation of E1A expression in cells in infected with Ad-CXCR4-E1A (using gene transcriptional control alone) with regulation of E1A expression in cells infected with Ad-CXCR4-UTR-E1A (using mRNA translational and gene transcriptional control). E1A expression was compared in infected breast cancer cell lines (MDA-MB-231, MDA-MB-361, and MDA-MB-435) and in infected normal breast epithelial cells (HMEC) and normal human fibroblasts.
Fig. 1.
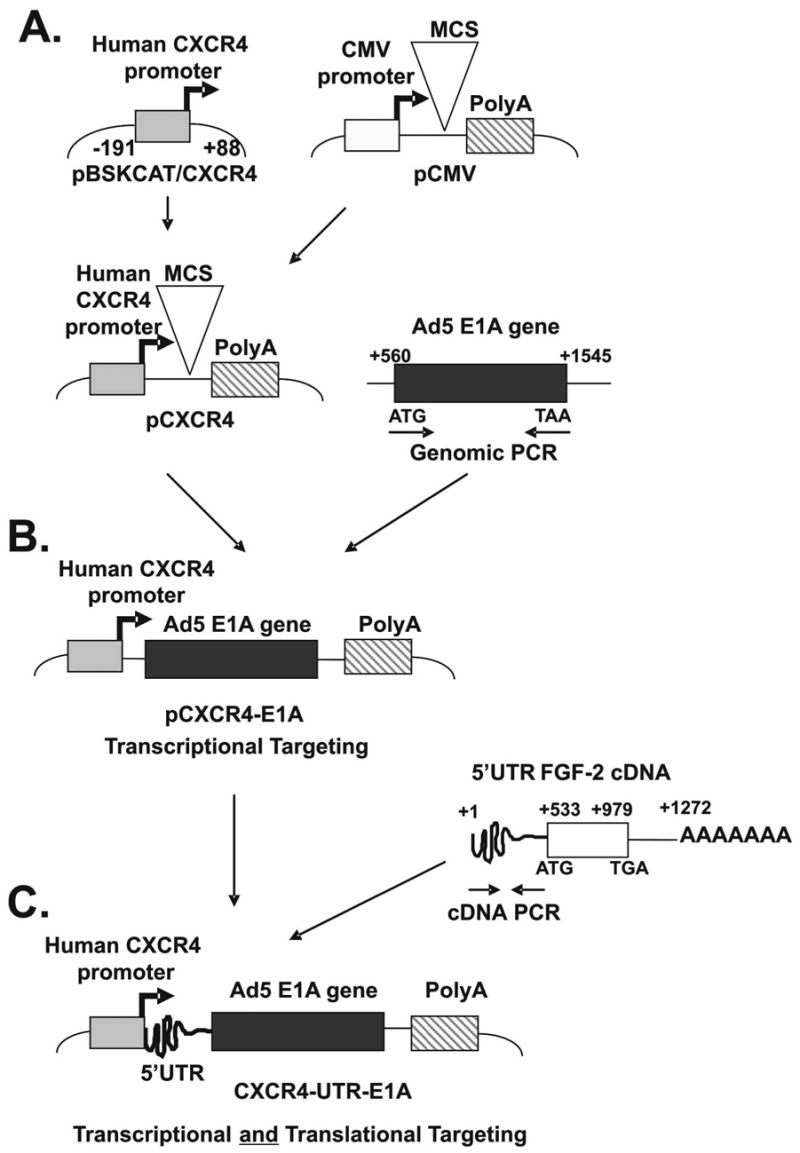
Strategy to exploit translational targeting as an adjunct to transcriptional targeting. (A) Construction of pAdenoVator-CXCR4. This construct contains the 279 bp CXCR4 promoter and the simian virus 40 (SV40) polyadenylation (poly A) signal. (B) The adenoviral type 5 E1AcDNA was cloned by PCR into pAdenoVator-CXCR4 to generate the pAdenoVator-CXCR4-E1A plasmid. (C) The 5′ upstream-untranslated region sequence of the rat FGF-2 cDNA was inserted immediately upstream of the E1A sequence to produce the pAdenoVator-CXCR4-UTR-E1A plasmid.
First, to correlate the differential expression of endogenous expression of eIF4E and CXCR4 between normal and cancer cell lines, Western blot analysis was performed, and the amounts of each protein were quantified by densitometry and normalized using the relative expression of b-actin. As shown in Fig. 2, normal HMEC and fibroblasts showed low levels of eIF4E expression. However, the breast cancer cell lines MDA-MB-231, MDA-MB-361, and MDA-MB-435 showed between 37- and 41-fold higher levels of eIF4E compared to normal HMEC, and between 23- and 25-fold higher levels compared to normal fibroblasts. In contrast, to the differential expression of eIF4E between normal and breast cancer cell lines, CXCR4 showed high levels of expression in normal HMEC and fibroblasts as well as in breast cancer cell lines. These results suggest that using gene transcriptional control alone may be insufficient to target cancer cells, and that the addition of mRNA translational control may be necessary to achieve tumor specificity.
Fig. 2.
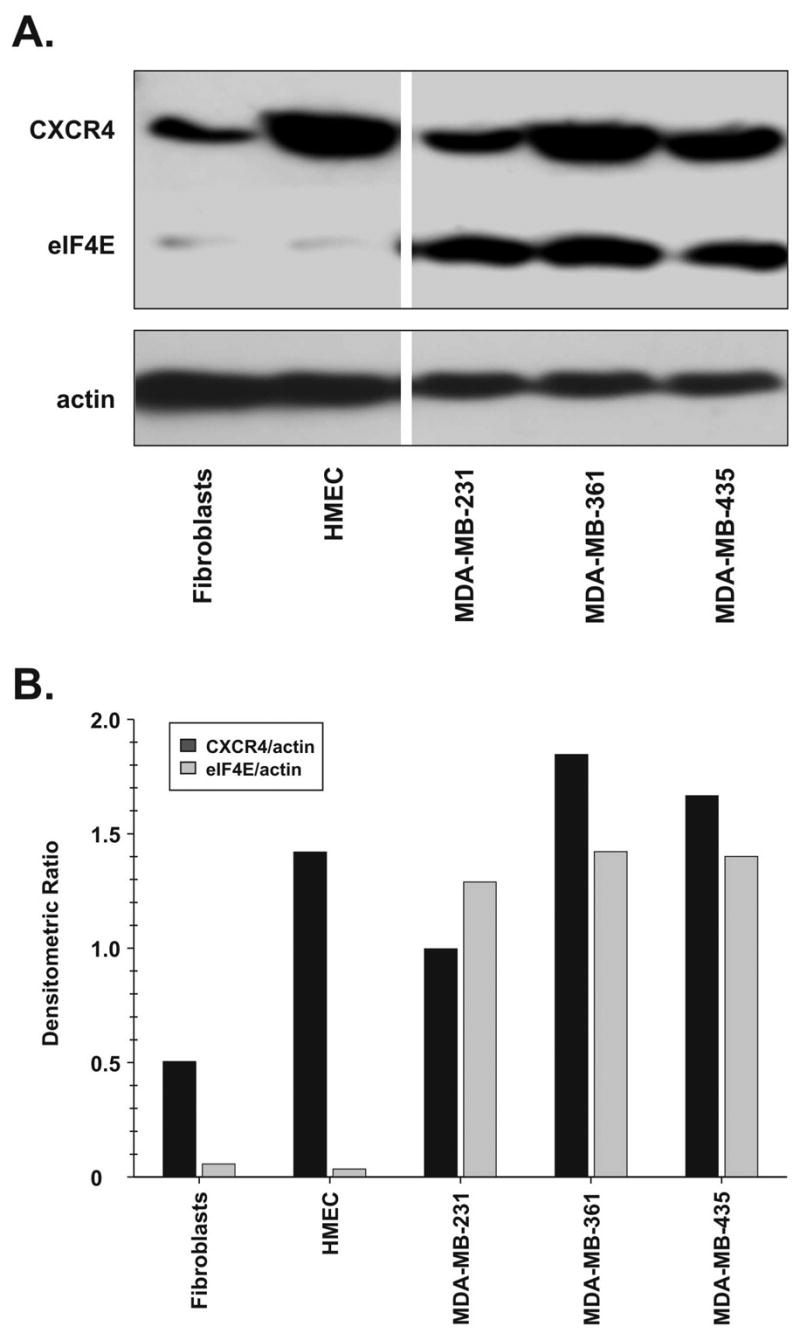
Western blot analysis of eIF4E protein levels in normal breast epithelial and stromal cells and breast cancer cell lines. An analysis was performed for each cell line used (fibroblasts, HMEC, MDA-MB-231, MDA-MB-361, and MDA-MB-435) for expression of CXCR4, eIF4E and b-actin. (A) A representative of three independent Western blot experiments. (B) Densitometric ratios of CXCR4 and eIF4E levels normalized to b-actin.
Transcription of the dual-level targeted CRAd is Independent of eIF4E protein level
A real-time RT-PCR was used to quantify transcription levels of E1A in breast epithelial and stromal cell lines expressing low levels of eIF4E (HMEC and fibroblasts) and in breast cancer cell lines having high levels of eIF4E (MDA-MB-231, MDA-MB-361, and MDA-MB-435), at 48 h after infection with either Ad-CXCR4-E1A or with Ad-CXCR4-UTR-E1A. Transcription levels of E1A were also quantified in cells infected with an untargeted replicative Ad-wt-dE3 as a positive control and in cells infected with a non-replicative Ad-CXCR4-GL3 as a negative control. As shown in Fig. 3, there were no statistically significant differences in the levels of E1A mRNA observed for any of the cell lines, whether the cells were infected with Ad-CXCR4-E1A or with Ad-CXCR4-UTRE1A. Importantly, these levels were similar to levels of E1A in cells infected with Ad-wt-dE3. These results serve to confirm our hypothesis that the transcription of E1A in these systems is independent of eIF4E protein levels and is unaffected by the FGF-2 5′-UTR sequence.
Fig. 3.
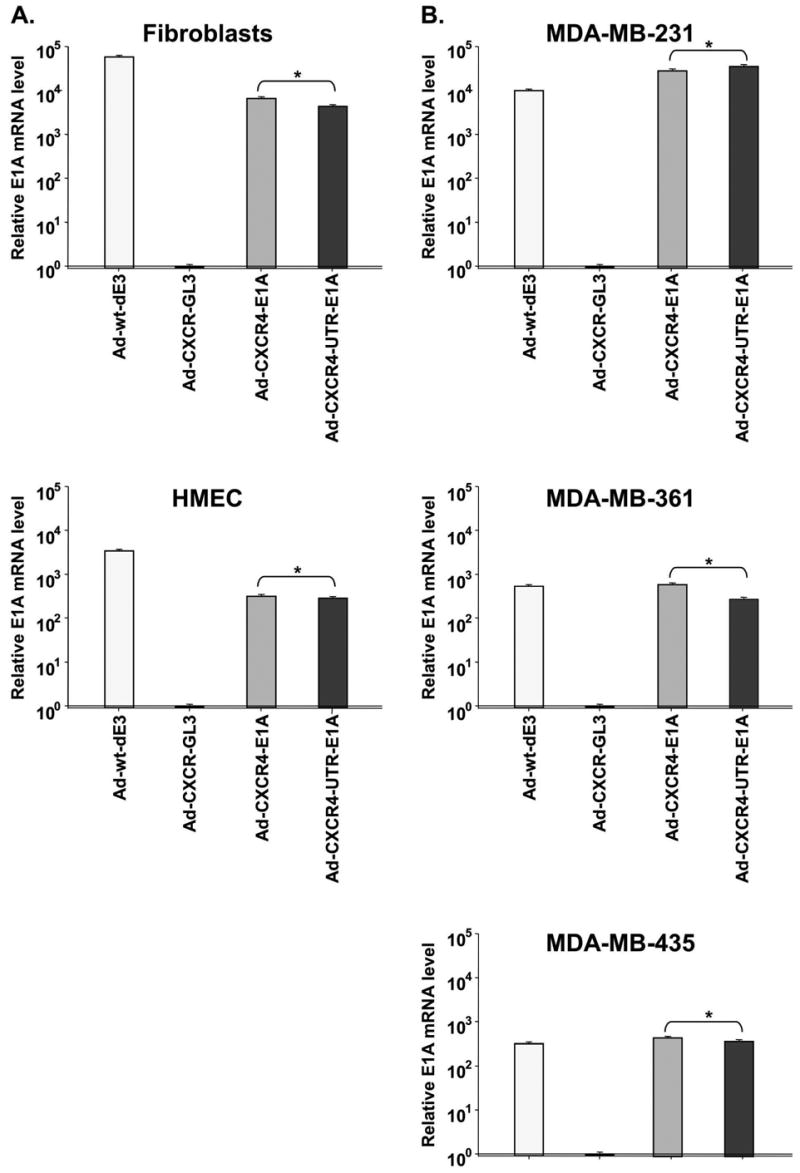
Evaluation of transcription of the E1A gene of the single and dual-level targeted CRAd byuantitative Real-time PCR. (A) Quantification of E1A mRNA expression after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control) in normal cells expressing relatively low levels of eIF4E. (B) Quantification of E1A mRNA expression after infection with AdCXCR4-GL3, Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A and Ad-wt-dE3 in breast cancer cell lines expressing relatively high levels of eIF4E. The mRNA copy numbers are normalized by the GAPDH copy number. Each bar represents the mean of three experiments ± SD. *P = ns.
Dual-Level targeting allows specific 5′-UTR-E1A
mRNA translation during CRAd infection In the following experiments we tested the feasibility of translational regulation in the context of CRAd, based on eIF4E dependent translational control. The breast cancer cell lines (MDA-MB-231, MDA-MB-361, and MDA-MB-435) and normal cell lines (HMEC and fibroblasts) were infected with either Ad-CXCR4-E1A or Ad-CXCR4-UTRE1A, and Western blot analysis of E1A protein levels was performed (Fig. 4). Western blot analysis was also performed on cells infected with an untargeted replicative Adwt-dE3 as a positive control and on cells infected with a non-replicative Ad-CXCR4-GL3 as a negative control. In breast cell lines that express relatively high levels of eIF4E, the E1A protein expression levels in cells infected with Ad-CXCR4-UTR-E1A were similar to cells infected with Ad-CXCR4-E1A or with Ad-wt-dE3. Importantly, in breast epithelial and stromal cell lines that express relatively low levels of eIF4E, the E1A expression level was significantly lower (in fact, lower than the detection limit) after infection with Ad-CXCR4-UTR-E1A compared to infection with Ad-CXCR4-E1A or Ad-wt-dE3. Thus, these results confirm our hypothesis that the translation of the 5′-UTR-E1A mRNA in the dual-level targeted CRAd is strongly dependent on the presence of elevated levels of eIF4E.
Fig. 4.
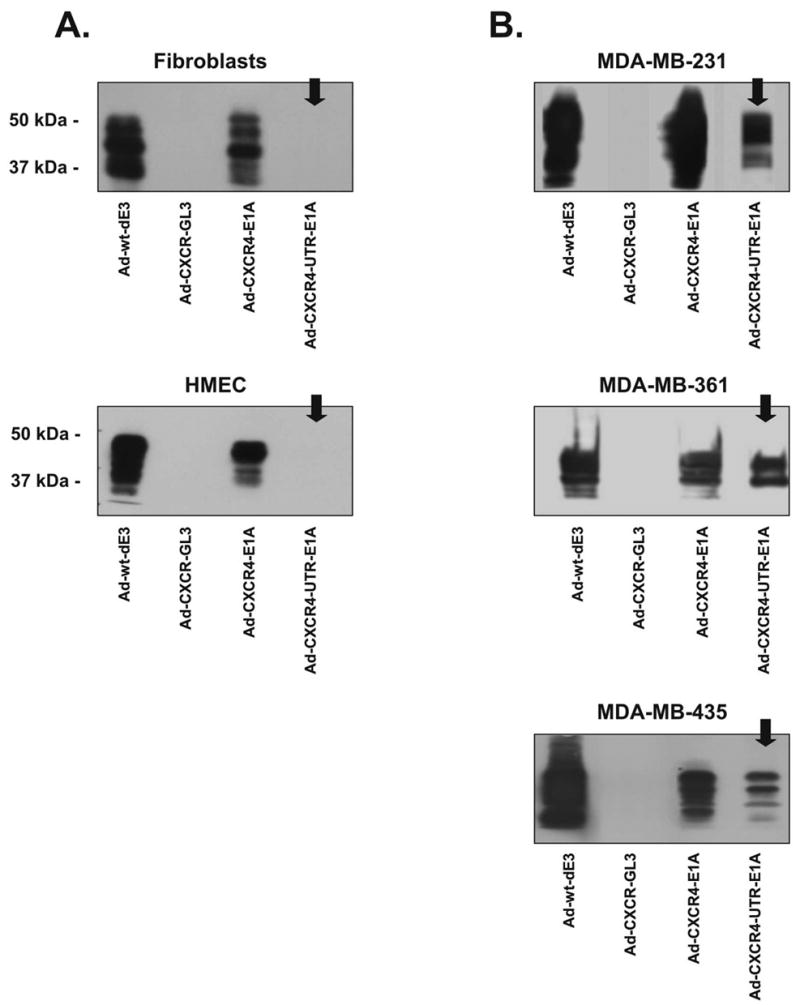
Dual-level targeting allows specific 5′-UTR-E1A mRNA translation during CRAd infection. Western blot analysis was performed using lysates from normal cells expressing relatively low levels of eIF4E (A) and breast cancer cell lines expressing relatively high levels of eIF4E (B) after infection with after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control).
eIF4E selective replication of dual-level targeted cRAds
To assay and compare eIF4E selective replication of the single-level targeted CRAd (Ad-CXCR4-E1A) and the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) after infection of cells with different levels of eIF4E, viral replication was determined by viral copy number. In this assay, the presence of the E4 gene was determined using real-time PCR from DNA samples extracted from medium after 1, 3, and 9 days post-infection [26, 27]. Viral replication was also determined in cells infected with an untargeted replicative Ad-wt-dE3 as a positive control and in cells infected with a non-replicative Ad-CXCR4-GL3 as a negative control. Infection of breast cancer cell lines (MDA-MB-231, MDA-MB-361, and MDA-MB-435) expressing relatively high levels of eIF4E with either the single-level targeted CRAd or the dual-level targeted CRAd generated comparably high DNA copy numbers, which were similar to the untargeted replicative Ad-wt-dE3 control (Fig. 5). In contrast, in normal cells (HMEC and fibroblasts) expressing low levels of eIF4E the dual-level targeted CRAd displayed significantly (P < 0.05) lower viral copy number in comparison to the single-level targeted CRAd. Thus, the dual-level targeted CRAd demonstrated specific eIF4E selective replication.
Fig. 5.
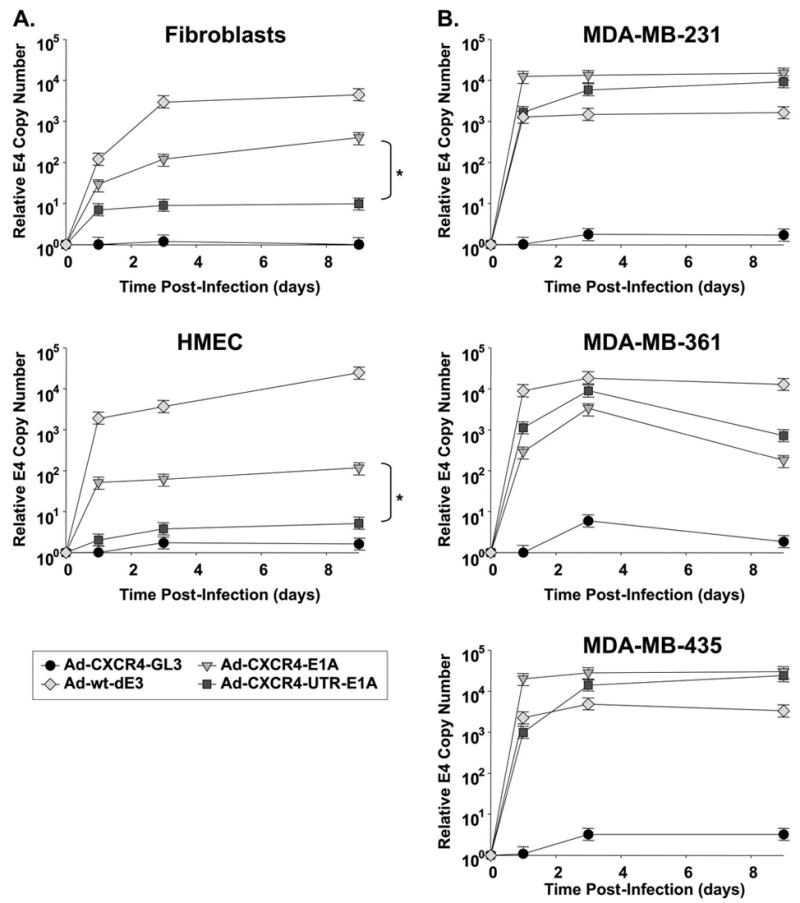
eIF4E selective replication of dual-level targeted CRAds. Viral copy number as an indicator of viral replication in normal cells expressing relatively low levels of eIF4E (A) and breast cancer cell lines expressing relatively high levels of eIF4E (B). Growth medium was collected at day 1, 3 and 9 after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control). Each bar represents the mean of three experiments ±SD. *P < 0.05 Ad-CXCR4-E1A versus Ad-CXCR4-UTR-E1A
Dual-Level targeting exploiting both gene transcriptional and mRNA translational control displays oncolytic specificity in cells over expressing eIF4E
We next compared the oncolytic profile of the single-level targeted and the dual-level targeted CRAd in cells expressing different levels of eIF4E (Fig. 6). Cell survival was determined by crystal violet staining. In all the breast cancer cell lines tested, the single and dual-level targeted CRAds displayed oncolytic potency comparable to Ad-wt-dE3. In contrast, in the normal HMEC and fibroblasts expressing low levels of eIF4E, the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) showed a cytotoxicity that was severely attenuated relative to the wild type adenovirus control (Adwt-dE3). Importantly, Ad-CXCR4-UTR-E1A displayed an oncolytic activity that was one order of magnitude lower compared to Ad-CXCR4-E1A in fibroblasts and HMEC. In the aggregate, these data clearly demonstrate that differential E1A protein expression and replication by the dual-level targeted CRAd Ad-CXCR4-UTR-E1A in cells expressing low and high levels of eIF4E results in eIF4E specific oncolytic activity.
Fig. 6.
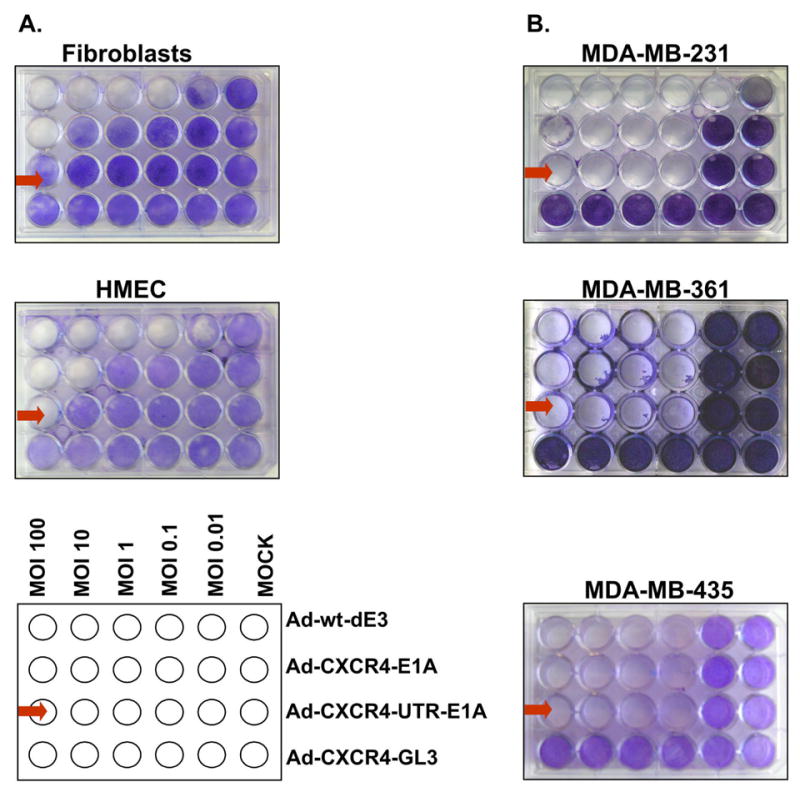
Dual-level targeting exploiting both gene transcriptional and mRNA translational control displays cytotoxic specificity in cells over expressing eIF4E. Oncolysis was evaluated by crystal violet staining in normal cells expressing relatively low levels of eIF4E (A) and breast cancer cell lines expressing relatively high levels of eIF4E (B) after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control) as described in Material and methods.
Dual-Level targeted CRAd maintains its cancer specificity in a human tissue slice model
To more closely mimic the clinical situation, we aimed to determine eIF4E selective replication of the dual-level targeted CRAd in the most stringent available preclinical model, precision cut tissue slices of normal breast and breast cancer samples. To measure viral copy number, the presence of the E4 gene was determined using real-time PCR from DNA samples extracted from medium at 1, 3, and 9 days post-infection [26, 27]. Infection of breast cancer tissue samples expressing relatively high levels of eIF4E with either the single-level targeted CRAd or the dual-level targeted CRAd generated comparably high DNA copy numbers, (Fig. 7), which were similar to the untargeted replicative Ad-wt-dE3 control. However, in normal breast tissue samples expressing low levels of eIF4E the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) showed a significantly lower (P < 0.05) viral copy number in comparison to the single-level targeted CRAd (Ad-CXCR4-E1A). Importantly, the results determined in normal human breast tissues and primary human breast cancer samples correspond to the results obtained in cell lines expressing low and high levels of eIF4E. Thus, exploiting translational targeting as an adjunct to transcriptional targeting in a CRAd allows distinctive cancer-selective replication.
Fig. 7.
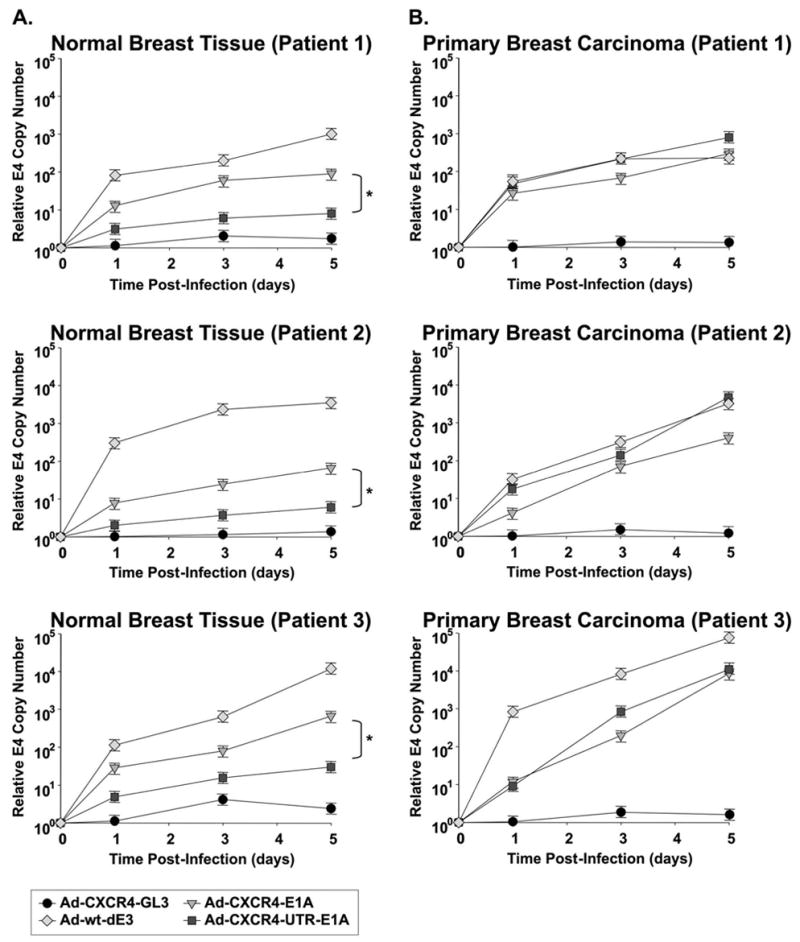
Evaluation of dual-level targeted CRAds in primary normal breast and breast cancer cells (4 patients). Viral copy number as an indicator of viral replication in precision cut tissue slices of normal breast (A) and breast cancer samples (B) obtained from three patients. Growth medium was collected at day 1, 3, 5 after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control). Each bar represents the mean of three experiments ±SD. *P < 0.05 Ad-CXCR4-E1A versus Ad-CXCR4- UTR -E1A.
Dual-Level targeted CRAd is oncolytic in vivo in a murine model of breast cancer
Next, the anti-tumor effects of the single-level targeted CRAd (Ad-CXCR4-E1A) and the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) were analyzed in vivo using MDA-MB-361 breast cancer cells injected subcutaneously in an athymic mouse xenograft model. As shown in Fig. 8, the decrease of the relative tumor volume in Ad-CXCR4-UTR-E1A treated mice was comparable to the Ad-CXCR4-E1A injected mice and the untargeted replicative Ad-wt-dE3 control. Taken together, these in vivo results are consistent with the in vitro data demonstrating a comparable oncolytic effect between the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) and the single-level targeted CRAd (Ad-CXCR4-E1A). Interestingly, treatment with a negative control (non-replicative) Ad-CXCR4-GL3 virus resulted in a partial therapeutic effect. The effect of transient tumor inhibition by a non-therapeutic adenovirus has previously been observed [28–31], and has been attributed to the recruitment and activation of the innate immune system (e.g., natural killer cells and macrophages) [32–35]. These findings may explain the nonspecific tumor inhibition we observed.
Fig. 8.
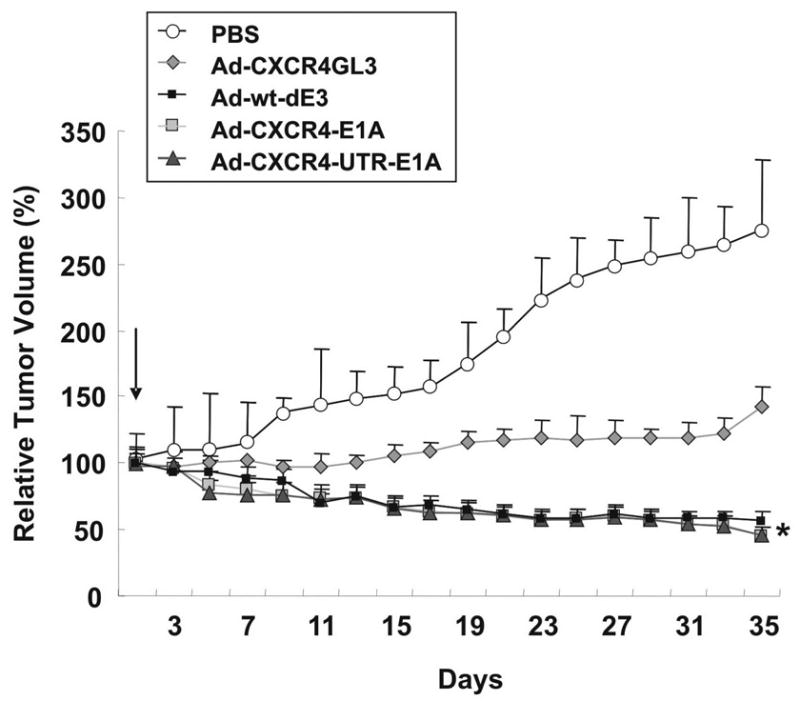
Oncolytic activity of dual-level targeted controlled CRAds is maintained in an orthotopic murine model of breast cancer. Antitumor effect of CRAd agents were determined using MDA-MB-361 mouse xenograft model. When the tumor reached 5 mm in widest diameter, 1 · 108 IFN of Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control) were injected intratumorally in a 0.05 ml volume. Control tumors were injected with an equal volume of PBS only. The tumor volume was calculated by the formula: 1/2 xy2, where x is the longest distance of the tumor. The relative tumor volume = tumor volume/tumor volume at day 1 · 100%. Each bar represents the mean of 10 tumors ±SD.
Dual-Level targeted CRAd displays reduced replication in a human liver tissue slice model
To preclinically evaluate the liver toxicity of human CRAds is a challenge. It is known that human adenoviruses replicate only poorly in mice. Therefore, we evaluated adenoviral replication in the most relevant preclinically available model, the human liver tissue slice model. To measure viral copy number, the presence of the E4 gene was determined using real-time PCR from DNA samples extracted from medium 2 and 4 days postinfection [26, 27]. As shown in Fig. 9, the dual-level targeted CRAd (Ad-CXCR4-UTR-E1A) showed the lowest copy number among the replication competent viruses tested, which was significantly lower compared to the single-level targeted CRAd (Ad-CXCR4-E1A) or Ad-wt-dE3. Thus, exploiting translational targeting as an adjunct to transcriptional targeting in a CRAd allows a significant reduction of replication in human liver tissue.
Fig. 9.
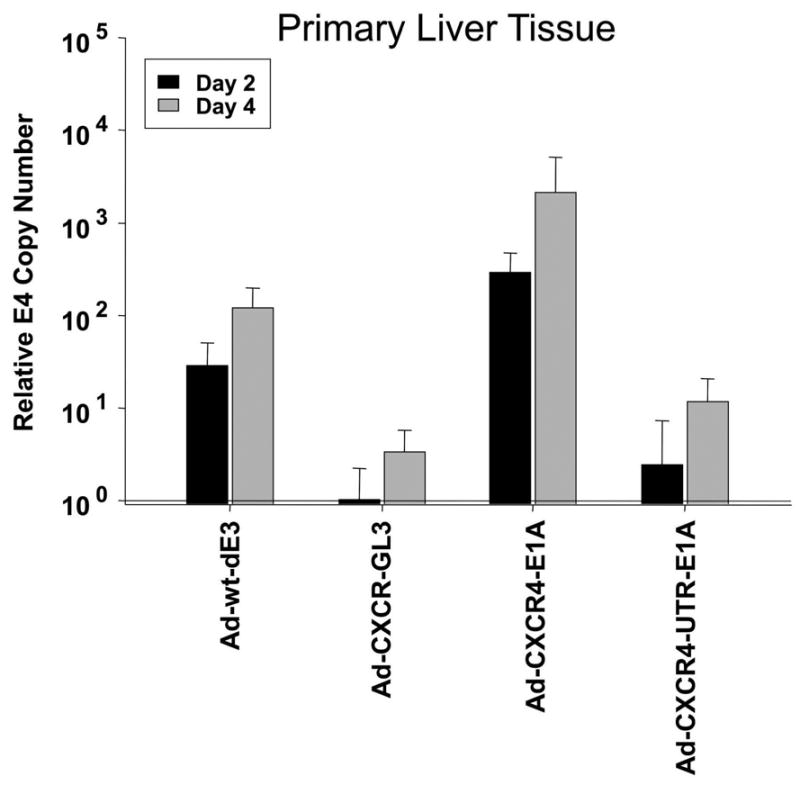
Evaluation of liver toxicity of the single-level and dual-level controlled CRAd in a human liver tissue slice model. Viral copy number as an indicator of viral replication in precision cut tissue slices of normal human liver obtained from three individual donors. Growth medium was collected at day 2 and 4 after infection with Ad-CXCR4-E1A, Ad-CXCR4-UTR-E1A, Ad-CXCR4-GL3 (negative control) or Ad-wt-dE3 (positive control). Each bar represents the mean of three experiments ±SD. *P < 0.05 Ad-CXCR4-UTR-E1A versus Ad-CXCR4-E1A and Ad-wt-dE3.
Discussion
In this report, we exploited cancer-specific control of mRNA translation initiation in order to achieve enhanced replicative specificity of CRAd virotherapy agents. Both in vitro and in vivo studies demonstrated that the CRAd agent, which utilized a heterologous mRNA translational control element, retained anti-tumor potency. Importantly, assessment of replicative specificity using stringent tumor and non-tumor tissue slice systems demonstrated significant improvement in tumor selectivity. Heretofore, the achievement of replicative specificity of CRAd agents has been accomplished solely with gene transcriptional control elements based upon incorporation of tumor selective promoters. Our report herein demonstrates the utility of a novel approach that combines both transcriptional and translational regulation strategies for the key goal of replicative specificity.
In cancer cells, translational regulation has become a well-recognized mechanism contributing to the neoplastic phenotype. Processes linked to aberrant translation regulation include cell cycle alterations, motility and invasiveness, angiogenesis, metastasis, and resistance to apoptosis. Therefore, translational regulation represents a very attractive point of attack to for therapy of cancer, because of the multitude of downstream effectors that can be modulated at one time. Clearly, eIF4E is a central player in translational regulation and is critical to the genesis and/or progression of cancer cells as indicated by the fact that it is over-expressed in the majority of solid tumors studied to date [11, 36, 37]. In addition to the importance of eIF4E in early changes in the primary tumor, eIF4E has been identified as one of eight genes that when elevated constitutes a molecular signature of metastasis potential [38]. In this regard, elevated eIF4E expression results in drastically increased synthesis of basic fibroblast growth factor (FGF-2) [39] and vascular endothelial growth factor (VEGF) [40], both of which are encoded by mRNAs with a long and complex 5′-UTR. Both of them powerful mitogens for vascular endothelia and are essential for tumor vascularization [41].
In this study, use of an mRNA translational control element in combination with the use of a gene transcriptional control element maintained oncolytic CRAd activity, both in vitro in tumor cells and in vivo in a xenograft breast cancer mouse model. Importantly, our CRAd agent demonstrated selective cytotoxicity in vitro, which was dependent upon eIF4E activity. Murine models are not an appropriate in vivo model to test non-specific adenoviral replication and spread, because normal murine tissues are poorly permissive for Ad replication [42, 43]. Thus we utilized the human tissue slice model, the most stringent preclinical substrate system currently available [44, 45]. In this model, we showed using normal human liver and breast tissue that the dual-level targeted CRAd significantly reduced non-specific adenoviral replication compared to a transcriptional targeted CRAd.
Our study exploited the 5′-UTR sequence from the FGF-2 mRNA as an mRNA translational control element. In our construct, protein expression of the modified Ad5 E1A transcript, which is essential for CRAd replication function, was restricted to cancer cells expressing high levels of elF-4E. Recently, another study exploited mRNA stabilization in the context of virotherapy for tumor specific replication [10]. In this design, the 3′-UTR sequence from the PTGS2 (prostaglandin-endoperoxide synthase 2) mRNA was exploited as a targeting element that allowed activated RAS/P-MAPK dependant mRNA stability of a modified Ad5 E1A transcript in cancer cells. Importantly, however, this design only tested a single targeting approach, with the use of a non-specific CMV promoter for E1A expression. In our approach, we extended CRAd targeting using mRNA translational control by placing the E1A gene under a second targeting element, using gene transcriptional control from a candidate TSP, the CXCR4 gene promoter [22].
To our knowledge, the conditionality of adenoviral replication and oncolysis through a post-transcriptional level of control of E1A expression using an mRNA translational control element has not been previously demonstrated. At this point, it is clear that reliance on a single targeting control of adenovirus replication could allow toxicity in non-target tissues. Thus, the combination of CRAd replication control at the level of both transcription and translation should provide greater cancer selectivity in vivo than either control element alone.
Our study has highlighted specificity gains that accrue from the employment of methods combining transcriptional and translational targeting. In this regard, the parent vector of our CRAd construct is based on adenovirus serotype 5. Genetic capsid modifications have recently been shown to allow for cell-specific targeted infection. This transductional targeting represents an additional strategy for the achievement of specificity, which can be employed with the methods we describe in this study. In conclusion, our future efforts are aimed toward the development of multiple-component targeted CRAds that will be the most effective at generating truly tumor selective vectors. Based on our studies, we propose that mRNA translation may be considered as one of the targeting component of such multiple targeted CRAds.
Augmentation of the specificity of adenoviruses for cancer targets is urgently required for deriving their full benefit and safety profile with regard to clinical trials. We presently describe an innovative dual cancer-specific targeting approach resulting in a new class of CRAds. We envisage/believe that the mRNA translational control element will play an important role in the vector design of virotherapeutic CRAds in future virotherapeutic clinical trials.
Acknowledgments
This work was supported by Grant of the Deutsche Forschungsgemeinschaft Sto 647/1-1 (to M. A. Stoff-Khalili), by grants from the NIH P01CA104177, R01CA083821, and R01CA113454 (to D. T. Curiel), R01CA93796, R01CA98543 and AR46031 (to G. Siegal) and Department of Defense W81XWH-05-1-035 (to D. T. Curiel) and grants from the Department of Defense W81XWH-05-1-5500, NIH R41CA114921, and the Louisiana Gene Therapy Research Consortium, Inc. (to J. M. Mathis).
References
- 1.Mathis JM, Stoff-Khalili MA, Curiel DT. Oncolytic adenoviruses - selective retargeting to tumor cells. Oncogene. 2005;24(52):7775–7791. doi: 10.1038/sj.onc.1209044. [DOI] [PubMed] [Google Scholar]
- 2.Biederer C, et al. Replication-selective viruses for cancer therapy. J Mol Med. 2002;80(3):163–175. doi: 10.1007/s00109-001-0295-1. [DOI] [PubMed] [Google Scholar]
- 3.Vile RG, Russell SJ, Lemoine NR. Cancer gene therapy: hard lessons and new courses. Gene Ther. 2000;7(1):2–8. doi: 10.1038/sj.gt.3301084. [DOI] [PubMed] [Google Scholar]
- 4.Alemany R, Suzuki K, Curiel DT. Blood clearance rates of adenovirus type 5 in mice. J Gen Virol. 2000;81(Pt 11):2605–2609. doi: 10.1099/0022-1317-81-11-2605. [DOI] [PubMed] [Google Scholar]
- 5.Krasnykh V, et al. Genetic targeting of an adenovirus vector via replacement of the fiber protein with the phage T4 fibritin. J Virol. 2001;75(9):4176–4183. doi: 10.1128/JVI.75.9.4176-4183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18(7):723–727. doi: 10.1038/77283. [DOI] [PubMed] [Google Scholar]
- 7.Siders WM, Halloran PJ, Fenton RG. Transcriptional targeting of recombinant adenoviruses to human and murine melanoma cells. Cancer Res. 1996;56(24):5638–5646. [PubMed] [Google Scholar]
- 8.Bernt KM, et al. The effect of sequestration by nontarget tissues on anti-tumor efficacy of systemically applied, conditionally replicating adenovirus vectors. Mol Ther. 2003;8(5):746–755. doi: 10.1016/j.ymthe.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Ring CJ, et al. Suicide gene expression induced in tumour cells transduced with recombinant adenoviral, retroviral and plasmid vectors containing the ERBB2 promoter. Gene Ther. 1996;3(12):1094–1103. [PubMed] [Google Scholar]
- 10.Ahmed A, et al. A conditionally replicating adenovirus targeted to tumor cells through activated RAS/P-MAPK-selective mRNA stabilization. Nat Biotechnol. 2003;21(7):771–777. doi: 10.1038/nbt835. [DOI] [PubMed] [Google Scholar]
- 11.DeFatta RJ, et al. Elevated expression of eIF4E in confined early breast cancer lesions: possible role of hypoxia. Int J Cancer. 1999;80(4):516–522. doi: 10.1002/(sici)1097-0215(19990209)80:4<516::aid-ijc6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 12.De Benedetti A, et al. Expression of antisense RNA against initiation factor eIF-4E mRNA in HeLa cells results in lengthened cell division times, diminished translation rates, and reduced levels of both eIF-4E and the p220 component of eIF-4F. Mol Cell Biol. 1991;11(11):5435–5445. doi: 10.1128/mcb.11.11.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhoads RE. Cap recognition and the entry of mRNA into the protein synthesis initiation cycle. Trends Biochem Sci. 1988;13(2):52–56. doi: 10.1016/0968-0004(88)90028-x. [DOI] [PubMed] [Google Scholar]
- 14.Kozak M. An analysis of vertebrate mRNA sequences: intimations of translational control. J Cell Biol. 1991;115(4):887–903. doi: 10.1083/jcb.115.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelletier J, Sonenberg N. The involvement of mRNA secondary structure in protein synthesis. Biochem Cell Biol. 1987;65(6):576–581. doi: 10.1139/o87-074. [DOI] [PubMed] [Google Scholar]
- 16.Smola H, Thiekotter G, Fusenig NE. Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. J Cell Biol. 1993;122(2):417–429. doi: 10.1083/jcb.122.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stark HJ, et al. Organotypic keratinocyte cocultures in defined medium with regular epidermal morphogenesis and differentiation. J Invest Dermatol. 1999;112(5):681–691. doi: 10.1046/j.1523-1747.1999.00573.x. [DOI] [PubMed] [Google Scholar]
- 18.Satish L, et al. Keloid fibroblast responsiveness to epidermal growth factor and activation of downstream intracellular signaling pathways. Wound Repair Regen. 2004;12(2):183–192. doi: 10.1111/j.1067-1927.2004.012111.x. [DOI] [PubMed] [Google Scholar]
- 19.Kirby TO, et al. A novel ex vivo model system for evaluation of conditionally replicative adenoviruses therapeutic efficacy and toxicity. Clin Cancer Res. 2004;10(24):8697–8703. doi: 10.1158/1078-0432.CCR-04-1166. [DOI] [PubMed] [Google Scholar]
- 20.Olinga P, et al. Comparison of five incubation systems for rat liver slices using functional and viability parameters. J Pharmacol Toxicol Methods. 1997;38(2):59–69. doi: 10.1016/s1056-8719(97)00060-9. [DOI] [PubMed] [Google Scholar]
- 21.Wegner SA, et al. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J Biol Chem. 1998;273(8):4754–4760. doi: 10.1074/jbc.273.8.4754. [DOI] [PubMed] [Google Scholar]
- 22.Zhu ZB, et al. Transcriptional targeting of adenoviral vector through the CXCR4 tumor-specific promoter. Gene Ther. 2004;11(7):645–648. doi: 10.1038/sj.gt.3302089. [DOI] [PubMed] [Google Scholar]
- 23.Zhu ZB, et al. Transcriptional targeting of tumors with a novel tumor-specific survivin promoter. Cancer Gene Ther. 2004;11(4):256–262. doi: 10.1038/sj.cgt.7700679. [DOI] [PubMed] [Google Scholar]
- 24.Kanerva A, et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol Ther. 2003;8(3):449–458. doi: 10.1016/s1525-0016(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 25.Rivera AA, et al. Combining high selectivity of replication with fiber chimerism for effective adenoviral oncolysis of CAR-negative melanoma cells. Gene Ther. 2004;11(23):1694–1702. doi: 10.1038/sj.gt.3302346. [DOI] [PubMed] [Google Scholar]
- 26.Zhu ZB, et al. Incorporating the surviving promoter in an infectivity enhanced CRAd-analysis of oncolysis and anti-tumor effects in vitro and in vivo. Int J Oncol. 2005;27(1):237–246. [PubMed] [Google Scholar]
- 27.Yamamoto M, et al. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology. 2003;125(4):1203–1218. doi: 10.1016/s0016-5085(03)01196-x. [DOI] [PubMed] [Google Scholar]
- 28.Carroll JL, et al. The role of natural killer cells in adenovirus-mediated p53 gene therapy. Mol Cancer Ther. 2001;1(1):49–60. [PubMed] [Google Scholar]
- 29.Kianmanesh A, et al. Intratumoral administration of low doses of an adenovirus vector encoding tumor necrosis factor alpha together with naive dendritic cells elicits significant suppression of tumor growth without toxicity. Hum Gene Ther. 2001;12(17):2035–2049. doi: 10.1089/10430340152677395. [DOI] [PubMed] [Google Scholar]
- 30.Hall SJ, et al. A novel bystander effect involving tumor cell-derived Fas and FasL interactions following Ad.HSV-tk and Ad.mIL-12 gene therapies in experimental prostate cancer. Gene Ther. 2002;9(8):511–517. doi: 10.1038/sj.gt.3301669. [DOI] [PubMed] [Google Scholar]
- 31.Ruzek MC, et al. Adenoviral vectors stimulate murine natural killer cell responses and demonstrate antitumor activities in the absence of transgene expression. Mol Ther. 2002;5(2):115–124. doi: 10.1006/mthe.2002.0529. [DOI] [PubMed] [Google Scholar]
- 32.Bessis N, GarciaCozar FJ, Boissier MC. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11(Suppl 1):S10–S17. doi: 10.1038/sj.gt.3302364. [DOI] [PubMed] [Google Scholar]
- 33.Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15(12):1157–1166. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Muruve DA. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003;10(11):935–940. doi: 10.1038/sj.gt.3302036. [DOI] [PubMed] [Google Scholar]
- 35.Perricone MA, et al. Enhanced efficacy of melanoma vaccines in the absence of B lymphocytes. J Immunother. 2004;27(4):273–281. doi: 10.1097/00002371-200407000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Rosenwald IB, et al. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene. 1999;18(15):2507–2517. doi: 10.1038/sj.onc.1202563. [DOI] [PubMed] [Google Scholar]
- 37.Miyagi Y, et al. Elevated levels of eukaryotic translation initiation factor eIF-4E, mRNA in a broad spectrum of transformed cell lines. Cancer Lett. 1995;91(2):247–252. doi: 10.1016/0304-3835(95)03737-h. [DOI] [PubMed] [Google Scholar]
- 38.Ramaswamy S, et al. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 39.Kevil C, et al. Translational enhancement of FGF-2 by eIF-4 factors, and alternate utilization of CUG and AUG codons for translation initiation. Oncogene. 1995;1(11):2339–2348. [PubMed] [Google Scholar]
- 40.Kevil CG, et al. Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int J Cancer. 1996;65(6):785–790. doi: 10.1002/(SICI)1097-0215(19960315)65:6<785::AID-IJC14>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Goto F, et al. Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest. 1993;69(5):508–517. [PubMed] [Google Scholar]
- 42.Ginsberg HS, et al. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc Natl Acad Sci USA. 1991;88(5):1651–1655. doi: 10.1073/pnas.88.5.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hjorth RN, et al. A new hamster model for adenoviral vaccination. Arch Virol. 1988;100(3–4):279–283. doi: 10.1007/BF01487691. [DOI] [PubMed] [Google Scholar]
- 44.Stoff-Khalili MA, et al. Employment of liver tissue slice analysis to assay hepatotoxicity linked to replicative and nonreplicative adenoviral agents. Cancer Gene Ther. 2006;13(6):606–618. doi: 10.1038/sj.cgt.7700934. [DOI] [PubMed] [Google Scholar]
- 45.Stoff-Khalili MA, et al. Preclinical evaluation of transcriptional targeting strategies for carcinoma of the breast in a tissue slice model system. Breast Cancer Res. 2005;7(6):R1141–R1152. doi: 10.1186/bcr1353. [DOI] [PMC free article] [PubMed] [Google Scholar]