

Abstract
Rationale
The dopaminergic system, particularly D2-like dopamine receptors, has been strongly implicated in reward processing. Animal studies have emphasized the role of phasic dopamine (DA) signaling in reward-related learning, but these processes remain largely unexplored in humans.
Objectives
To evaluate the effect of a single, low dose of a D2/D3 agonist—pramipexole—on reinforcement learning in healthy adults. Based on prior evidence indicating that low doses of DA agonists decrease phasic DA release through autoreceptor stimulation, we hypothesized that 0.5 mg of pramipexole would impair reward learning due to presynaptic mechanisms.
Methods
Using a double-blind design, a single 0.5 mg dose of pramipexole or placebo was administered to 32 healthy volunteers, who performed a probabilistic reward task involving a differential reinforcement schedule as well as various control tasks.
Results
As hypothesized, response bias toward the more frequently rewarded stimulus was impaired in the pramipexole group, even after adjusting for transient adverse effects. In addition, the pramipexole group showed reaction time and motor speed slowing and increased negative affect; however, when adverse physical side effects were considered, group differences in motor speed and negative affect disappeared.
Conclusions
These findings show that a single low dose of pramipexole impaired the acquisition of reward-related behavior in healthy participants, and they are consistent with prior evidence suggesting that phasic DA signaling is required to reinforce actions leading to reward. The potential implications of the present findings to psychiatric conditions, including depression and impulse control disorders related to addiction, are discussed.
Keywords: Dopamine, D2 agonists, Reward Processing, Depression, Mesolimbic System, Addiction
INTRODUCTION
The dopamine (DA) system has been strongly implicated in reward-related learning, and abnormality in this system has been shown to play a role in the etiology and pathophysiology of various disorders, including depression, addiction, and Parkinson’s disease (Willner 1995; Dunlop and Nemeroff 2007). Phasic DA signals, in particular, have been associated with reinforcement learning (Montague et al. 1996; Schultz et al. 1997; Schultz, 2007). Electrophysiological studies in non-human primates investigating associative (Pavlovian) learning have shown that midbrain DA neurons code reward-related prediction errors using phasic bursts of dopamine: when an unpredicted reward is delivered, a phasic DA increase (i.e., positive-prediction error) is observed, and learning about the behavior that led to reward occurs (Bayer and Glimcher 2005; Garris et al. 1999; Waelti et al. 2001). Omission of a predicted reward, on the other hand, elicits phasic DA decreases (i.e., negative-prediction error; Fiorillo et al. 2003; Hollerman and Schultz 1998), and the behavior that led to lack of reward is extinguished. These findings have been complemented by reports that have emphasized the role of DA signaling in instrumental learning (e.g., Cheng and Feenstra 2006; Pessiglione et al. 2006; Robinson et al. 2007; Schwabe and Koch 2007; Sokolowski et al. 1998), particularly in reinforcing operant behavior (Reynolds et al. 2001). Collectively, these findings suggest that transient bursts of extracellular DA reinforce actions leading to reward, and thus foster the emergence of reward-related learning.
If dopamine-mediated signals are important for reinforcement learning, disruption of DA signaling should lead to deficient prediction error and thus less efficient learning (Montague et al. 2004; Schultz 2002). Findings from pre-clinical studies as well as studies with Parkinson’s disease (PD) patients are consistent with this assumption. In rodents, DA antagonist administration disrupted the ability to link the value of a reward to the actions necessary to obtain it (Ikemoto and Panksepp 1996). Also, DA depletion in the nucleus accumbens impaired approach behavior in an appetitive Pavlovian paradigm (Parkinson et al. 2002). Disruption of reward-based learning has also been observed in PD, a disease characterized by cell death in the substantia nigra pars compacta leading to depletion of DA in the basal ganglia (Hornykiewicz and Kish 1987). Medication-naïve (Nagy et al. 2007) or unmedicated (Shohamy et al. 2005) PD patients showed impaired performance in a task requiring feedback-based stimulus-response learning. Furthermore, Frank et al. (2004) reported that PD patients off medication were impaired in learning from positive, but not negative, feedback. Interestingly, the behavioral impairments of PD patients off medication were simulated by reduced phasic DA bursts following positive feedback in a computational model of the striatal-cortical system (Frank 2005). Whereas the findings reviewed above strongly implicate phasic DA signaling in the acquisition of reward-related behavior, the role of DA in reinforcement learning in healthy human participants remain largely unexplored. The main goal of the present study was to evaluate the effect of a single, low dose of a D2/D3 agonist—pramipexole—on reinforcement learning in psychiatrically and neurologically healthy adults.
Pramipexole dihydrochloride, a non-ergot DA D2/D3 agonist, was selected because the D2/D3 receptors are distributed densely within mesocorticolimbic pathways implicated in reward processing (e.g., Grace 2002; Sokoloff et al. 2006). Critically, D2 and D3 receptors are located both pre- and post-synaptically (Civille 2000; LeFoll et al. 2005; Sokoloff et al. 2006; Chen et al. 2005), and presynaptic D2/D3 receptors can act as autoreceptors reducing phasic DA release via inhibitory feedback (Grace 1991). Moreover, presynaptic D2 receptors have a higher affinity for DA than postsynaptic receptors (Cooper et al. 2003). Based on this empirical evidence, low doses of DA agonists are expected to decrease phasic DA release through autoreceptor stimulation. Consistent with this hypothesis, in rodents, low doses of D2 agonists have been found to (1) reduce phasic DA release (Baudry et al. 1977; Fuller et al. 1982; Tissari et al. 1983; Sumners et al. 1981; cf. Schmitz et al. 2003); (2) suppress DA cell firing rates in the substantia nigra and ventral tegmental area (VTA) (Piercey et al. 1996); and (3) reduce blood volume in the nucleus accumbens (Chen et al. 2005). In healthy participants, the same dose as used in this study (0.5 mg) lowered alertness, caused pupillary dilatation, increased heart rate, reduced thyroid stimulating hormone, and increased growth hormone, (Samuels et al. 2006a,b); this pattern of findings was interpreted as reflecting a reduction in DA tone due to D2 autoreceptor activation (Samuels et al. 2006a,b; see also Frank and O’Reilly 2006;Rye 2004; Rye and Jankovic 2002).
Based on these findings, we hypothesized that administration of 0.5 mg of pramipexole would reduce phasic DA bursts in response to reward feedback (i.e., reduce positive-prediction errors) and thus impair reward learning in a probabilistic reward task. Alternatively, if a single dose of pramipexole acted to enhance DA neurotransmission, we hypothesized that pramipexole administration would increase reward learning (Pessiglione et al. 2006), decrease reaction time (e.g., Servan-Schreiber et al. 1998; Schluck et al. 2002), and increase positive affect (e.g., de Wit et al. 2002).
MATERIALS AND METHODS
Participants
Participants were recruited from the community using an online advertising forum. Subjects deemed eligible to participate after a phone screen were invited for a diagnostic interview. Exclusion criteria included: current unstable medical illness; known hypersensitivity to pramipexole; lifetime history of any Axis I psychiatric disorder as determined by Structured Clinical Interview for the DSM-IV (SCID; First et al. 2002); neurological disorders; disorders in a first-degree relative that involves dopamine system abnormality (schizophrenia spectrum disorders, psychosis, bipolar disorder, major depressive disorder, substance dependence, Parkinson’s disease, Huntington’s disease, Tourette’s syndrome, ADHD); pregnancy or lactation; use of dopamine antagonists in the last month; use of medications that affect metabolism of pramipexole (e.g., cimetidine, diltiazem, triamterine) in the last 7 days; and use of any CNS depressant (e.g., anti-histamines, alcohol) in the last 24 hours.
All participants were right-handed, as assessed by a standard handedness questionnaire (Chapman and Chapman 1987). Based on reports of potential changes in dopaminergic sensitivity during the menstrual cycle (Myers et al. 2003), female participants performed the experiment during the follicular phase (days 1–14) of their menstrual cycle.
Participants received $10/hour for completion of the SCID, $40 for the experimental session, and $24.60 in task earnings. All participants provided written informed consent to a protocol carried out in accordance with the Declaration of Helsinki and approved by the Harvard University Committee on Use of Human Subjects and the Partners Human Research Committee.
Equipment
Stimuli were presented using E-Prime software (version 1.1; Psychology Software Tools, Inc, Pittsburgh, Pennsylvania) on a 17” PC monitor.
Pharmacological Manipulation
Pramipexole dihydrochloride is an FDA-approved treatment for Parkinson’s disease (Parkinson Study Group 2000). The recommended starting dosage is 0.375 mg/day in three divided doses. This is titrated gradually to a therapeutic dose in the range of 1.5–4.5 mg/day across a period of up to seven weeks. Consistent with prior studies (Wright et al. 1997; Samuels et al. 2006a,b), a single 0.5 mg dose of pramipexole or identical placebo was administered orally, with peak brain concentrations expected after two hours (Wright et al. 1997).
Procedure
The study consisted of two sessions. In the first, participants were informed about the study, provided written consent, completed the SCID with a research psychiatrist or masters-level interviewer, and filled out the Beck Depression Inventory-II (BDI-II; Beck et al. 1996) and the trait form of Spielberger State-Trait Anxiety Inventory (STAI; Spielberger et al. 1970) to assess depression and anxiety symptoms, respectively.
The second session involved the pharmacological manipulation and behavioral testing (Figure 1). Participants performed pre-drug assessments of vigilance (Simple Reaction Time Task; Perbal et al. 2003) and motor function (BRAIN Task: Bradykinesia Akinesia Inco-ordination Test; Giovannoni et al. 1999). Subsequently, an adverse effects checklist was administered and baseline assessments of mood were collected using the Visual Analog Mood Scales (VAMS; Bond and Lader 1974).
Figure 1. Schematic summary of the experimental session timeline.
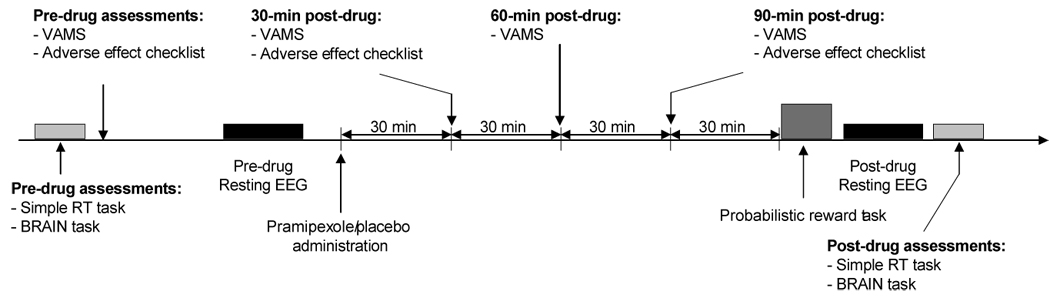
VAMS: Visual Analog Mood Scale (Bond and Lader, 1974). EEG: electroencephalogram.
Next, 6 minutes of resting electroencephalographic (EEG) data were collected (data will be reported separately). Study medication was then administered under the direct supervision of a physician. Using a randomized, double-blind design, participants received either a single dose of pramipexole (0.5 mg) or an identical placebo capsule and then waited two hours to allow medication absorption. During this time, participants watched affectively neutral or slightly positive movies and completed the VAMS at 30, 60, and 90 min and the adverse effect checklist at 30 and 90 min post-administration. At 90 min post-dose, participants were provided with a snack.
Two hours after study medication administration, participants performed a probabilistic reward task followed by another 6-minute resting EEG, the Simple Reaction Time Task, and the BRAIN Task. Participants then completed the adverse effect checklist again. Finally, participants were assessed for orthostatic changes in blood pressure, fully debriefed and paid, and released from the laboratory only after the attending physician confirmed it was safe to do so.
Assessments and Experimental Tasks
Simple Reaction Time (RT) Task
The task was used to assess vigilance. In two separate blocks, participants were instructed to use either the left or right index finger to press a key as quickly as possible when a red square appeared on the computer screen (ISI: 1000–2500 ms). The task consisted of 48 trials (24 right, 24 left) and lasted approximately 2 minutes.
BRAIN Task
The BRAIN task was developed to provide an empirical measure of motor functions (Giovannoni et al. 1999). Participants were instructed to use their right index finger to alternately press two keys (“s” and “;”) on a keyboard as quickly and accurately as possible for 60 seconds.
Probabilistic Reward Task
The task uses an asymmetric reinforcement schedule to produce a response bias towards the more frequently rewarded of two possible stimuli, and provides an objective assessment of the participants’ propensity to modulate behavior as a function of prior reinforcement history (Tripp and Alsop 1999; Pizzagalli et al. 2005). The task consisted of three blocks of 100 trials. Each trial started with a fixation cross, followed 500 ms later, by a mouthless line-drawing of a face. After a 500-ms delay, either a short (11.5 mm) or a long (13 mm) mouth appeared on the screen for 100 ms. The participant’s task was to judge whether a long or short mouth was presented by pressing either the “z” or “/” key (counterbalanced across participants). In each block, a pseudo-random sequence of 50 long and 50 short mouths was presented and 40 correct responses were followed by reward feedback (“Correct!! You won 20 cents!”), which was displayed for 1750 ms. Critically, correct responses for one stimulus (“rich stimulus”) were associated with reward three times more frequently (30:10) than correct responses for the other stimulus (“lean stimulus”). Prior to the task, participants were informed that only a portion of correct responses would be rewarded, but were not informed that correct identification of one of the stimuli would be disproportionally rewarded.
VAMS
At various time points, participants rated their current mood using five 230-mm horizontal lines which represented the following bipolar dimensions: happy—sad, relaxed—tense, friendly—hostile, sociable—withdrawn, and quick-witted—mentally slow.
Adverse Effects
A self-report checklist assessing the severity of 12 different physical symptoms was administered at various time points (headache, cold or chilled, hot or flushed, dizziness, sleepiness, sweating, blurred vision, nausea, fast heartbeat, dry mouth, abdominal pain, diarrhea).
Data Reduction
Simple RT Task
RT data were processed in three steps. First, RT <100 ms or >1000 ms were removed, and a log-transformation was applied to normalize the data. Next, mean and SD values were calculated for each participant at time 1 and 2, and RT > 3SD from the mean were removed. After outlier removal, a mean RT was computed for each participant and time point separately.
BRAIN Task
Kinesia, dysmetria, and incoordination scores were computed (Giovannoni et al. 1999). The kinesia score represents the total number of alternating keystrokes in 60 seconds. The dysmetria score is a weighted index of incorrectly hit keys that assesses movement accuracy (corrected for speed). The incoordination score assesses rhythmicity (the variance of time intervals between keystrokes). Square root transformations were applied for normalization purposes.
Probabilistic Reward Task
The main variables of interest were response bias (log b) and discriminability (log d), which were computed using the following formulas:
Response bias assesses the systematic preference for the response paired with the more frequent reward (rich stimulus). Response bias increases if participants tend to (1) correctly identify the rich stimulus, and/or (2) misclassify the lean stimulus as the rich stimulus. Discriminability assesses the subject’s ability to distinguish between the two stimuli and is an indicator of general task performance. Control analyses also considered accuracy and RT scores.
Adverse Effect Scales
Self-reported adverse effects were summed for each time point. To maximize our ability to detect the influence of potential adverse effects, two difference scores were computed: one between the maximal adverse effect score and the pre-administration score (“△Adverse EffectMAX”) and one between the adverse effect score 90-min post-administration and the pre-administration score (“△Adverse Effect90min”).
VAMS
Scores were calculated as percent negative affect (Harrison et al. 2002). To assess changes independent of individual differences in affect before the drug administration, three difference scores were computed: VAMS30min−VAMSpre-drug, VAMS60min−VAMSpre-drug, and VAMS90min−VAMSpre-drug.
Statistics
Simple RT Task
Mean RT were entered in a mixed ANOVA with Medication Group (placebo, pramipexole), Time (pre-, post-drug), and Hand (left, right) as factors.
BRAIN task
A Medication Group × Time ANOVA was performed separately for the kinesia, dysmetria, and incoordination scores.
Probablistic Reward Task
For response bias and discriminability, a mixed ANOVA with Medication Group and Block (1,2,3) was performed. For accuracy and RT data, Stimulus (Rich, Lean) was added.
VAMS
Mixed Medication Group × Time ANOVAs were conducted for each subscale independently.
For ANOVAs, Greenhouse-Geisser corrections were used when appropriate. In case of significant ANOVA findings, post-hoc Newman-Keuls tests were performed.
Adverse Effects
Two sets of analyses were performed to assess the potential role of adverse effects. In the first, Pearson correlations between the adverse effect scores and variables of interest were performed for the pramipexole group. In the second, hierarchical regression analyses were run to evaluate whether Medication Group explained unique variance of a given outcome variable after adjusting for adverse effects.
RESULTS
Thirty-two participants were enrolled in the experiment. Of these, 16 received 0.5 mg pramipexole and 16 received placebo. One participant in the placebo group was excluded from the analysis due to failure to follow directions. The pramipexole (n = 16) and placebo (n = 15) groups did not differ with respect to gender ratio (9 male/7 female vs. 8 male/7 female; Chi-square = 0.027, df = 1, p > 0.50), age (26.0 ± 5.0 years vs. 24.4 ± 3.3; t(29) = 1.05, p > 0.30), baseline depressive symptoms (BDI-II: 0.38 ± 0.74 vs. 1.20 ± 1.75; p > 0.25) or anxiety symptoms (STAI: 28.0 ± 5.5 vs. 31.9 ± 5.7; p > 0.15).
Adverse Effects and Final Sample Sizes
Of the 16 participants receiving pramipexole, six did not complete some part of the experimental session due to adverse effects that ranged in self-rated scores from mild to severe, and most commonly included nausea, dizziness, and sleepiness. These effects were frequently accompanied by a transient decrease in diastolic blood pressure. Similar incidence of adverse effects was described by Samuels et al. (2006a,b) who administered a single 0.5 mg dose of pramipexole to healthy male volunteers.
For the probabilistic reward task, data from an additional three participants were lost because they failed to follow task instructions (final sample: placebo: n = 13; pramipexole: n = 11). For the Simple RT Task, data from 1 additional participant were excluded due to outlier status (placebo: n = 15; pramipexole: n = 11). For the BRAIN Task, data from 15 participants in the placebo group and 12 in the pramipexole group were available.
Probabilistic Reward Task
Response Bias
Replicating prior studies (Pizzagalli et al. 2005), a significant main effect of Block emerged (F(2,44) = 9.71, p < 0.001, partial η² = 0.31), due to significantly higher response bias in both Block 2 and 3 compared to Block 1 (Newman-Keuls p’s < 0.0007). Importantly, the Medication Group × Block interaction was also significant: F(2,44) = 5.06, p < 0.01, partial η² = 0.19 (Figure 2a). Compared to the placebo group, participants receiving pramipexole showed significantly lower response bias in Block 2 and 3 (Newman-Keuls p’s < 0.006). For the placebo but not the pramipexole group, response bias toward the more rewarded stimulus in Blocks 2 and 3 was higher compared to Block 1 (p’s < 0.002). Finally, a direct contrast indicated that the pramipexole group had significantly lower reward learning than the placebo group across the blocks, as assessed by computing a difference score between response bias in block 3 and block 1 (pramipexole 0.03 ± 0.13 vs. placebo 0.25 ± 0.17, t(22) = −3.47, p < 0.002; Cohen’s d = −1.45).
Figure 2. Probabilistic reward task.
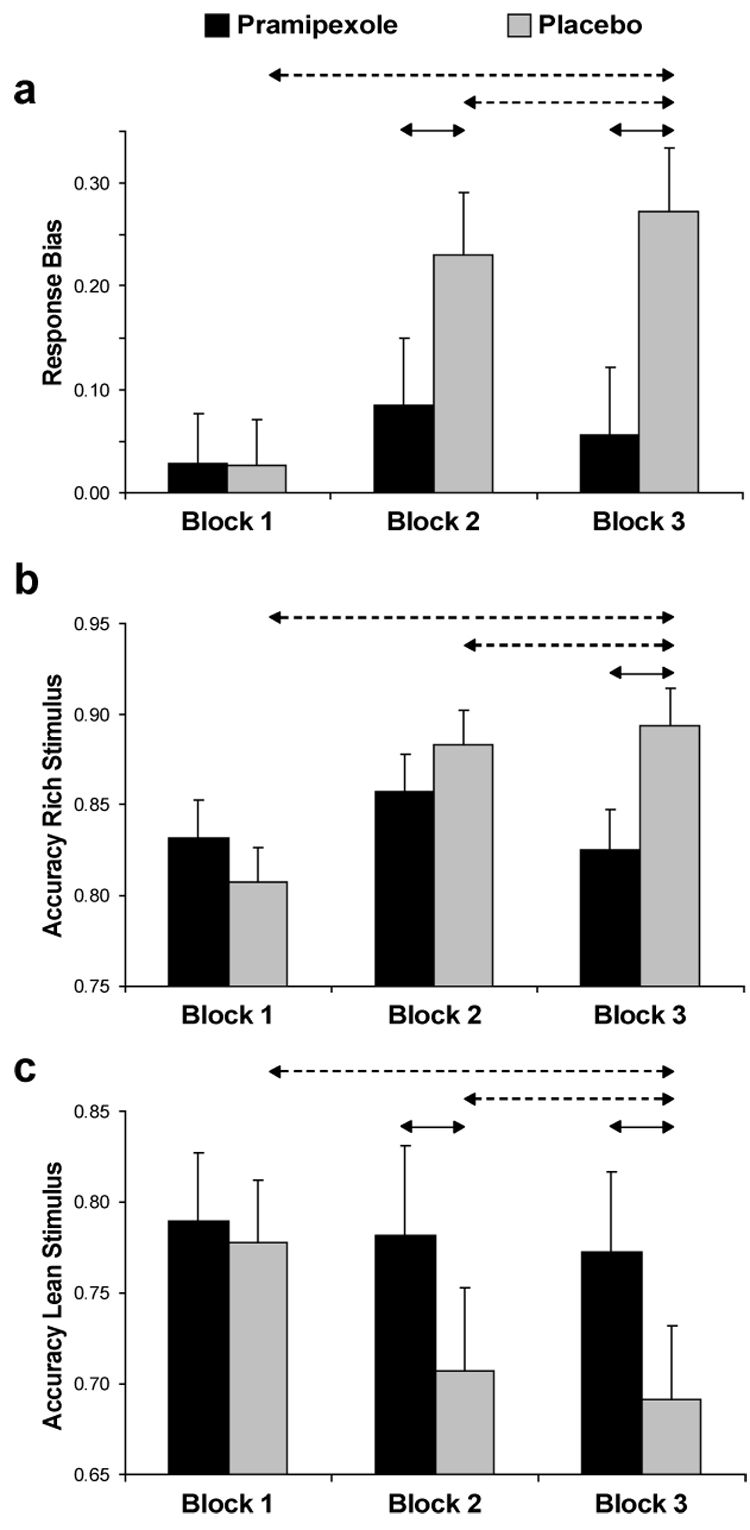
(a) Response bias, (b) Accuracy for the more frequently rewarded stimulus (“rich stimulus”), and (c) Accuracy for the less frequently rewarded stimulus (“lean stimulus”) as a function of Blocks for the pramipexole (n = 11) and placebo group (n = 13). Error bars correspond to S.E. Solid arrows and dashes arrows denote significant between-group and within-group differences, respectively.
Discriminability
No significant effects emerged (all F’s < 0.88, all p’s > 0.35).
Accuracy
The main effect of Condition (rich vs. lean) [F(1,22) = 12.99, p < 0.005, partial η² = 0.37] and the Block x Condition interaction [F(2,44) = 9.50, p < 0.005, partial η² = 0.30] were significant. Replicating independent findings from our laboratory (Bogdan and Pizzagalli 2006; Pizzagalli et al. 2005), accuracy for the rich stimulus was significantly higher than for the lean stimulus, and this difference increased over time [linear contrast of the Block × Condition interaction: F(1,22) = 14.28, p < 0.001]. Of primary relevance for the present study, a significant Block × Medication Group × Stimulus (rich vs. lean) effect also emerged: F(2, 44) = 6.00, p < 0.005, partial η² = 0.21. Follow-up ANOVAs were conducted separately for the two stimulus types. For the rich stimulus, a significant Medication Group × Block interaction was observed, F(2,44) = 5.41, p < 0.01, partial η² = 0.20. Post-hoc tests revealed significantly lower rich accuracy in the pramipexole than placebo group for Block 3 (0.83 ± 0.09 vs. 0.89 ± 0.06; p < 0.015; Figure 2b). The placebo, but not the pramipexole, group was characterized by increases in rich accuracy from Block 1 to both Block 2 and 3 (p’s < 0.005). For the lean stimulus, the Medication Group × Block interaction reached trend level: F(2,44) = 2.873, p = 0.067, partial η² = 0.116. Notably, for both Blocks 2 and 3, participants in the pramipexole group had significantly higher accuracy for the lean stimulus compared to the placebo group (p < 0.005) (Figure 2c). Moreover, whereas the placebo group showed significant lower lean accuracy in Blocks 2 and 3 compared to Block 1 (p’s < 0.01), the pramipexole group show no differences across the blocks.
If unpredicted rewards elicit phasic DA bursts, and this positive-prediction error supports learning about the consequences of the behavior leading to reward (Schultz 2007), we reasoned that a single dose of pramipexole reducing phasic DA response would negatively impact the participants’ probability of choosing a specific action after having received reward for that action in the preceding trial. To test this hypothesis, we calculated the probability that participants correctly identified a rich stimulus after a preceding rewarded rich stimulus trial (“win-stay” strategy). As hypothesized, participants receiving pramipexole had significantly lower probabilities than placebo participants of making the subsequent correct identification (80.50 ± 8.88% vs. 87.43 ± 7.41%, t(25) = 2.23, p < 0.03; Figure 3). In other words, the pramipexole group had a 19.50% probability of missing the more frequently rewarded stimulus after this stimulus had been rewarded in the trial before, compared to a 12.57% miss probability in the placebo group. To test whether these differences were driven by the delivery of reward, we calculated the probability that the participants correctly identified a rich stimulus after they had correctly identified a rich stimulus in the preceding trial, but there had been no scheduled reward feedback. In this scenario, no significant differences emerged between the pramipexole and placebo groups (79.07% ± 12.84 vs. 84.42% ± 9.47, t(25) = 1.27, p > 0.20). Although these findings suggest that group differences were larger in trials immediately following a rewarded rich trial, it is important to emphasize that a Medication Group × Probability ANOVA considering the two types of probabilities revealed a main effect of Medication Group, F(1,25) = 4.35, p < 0.050, partial η² = 0.15, whereas the interaction was not significant [F(1,25) = 0.08, p > 0.78]. Thus, from a statistical perspective, group differences were not specific to trials immediately following a rewarded identification of the rich stimulus.
Figure 3. Probabilistic reward task.
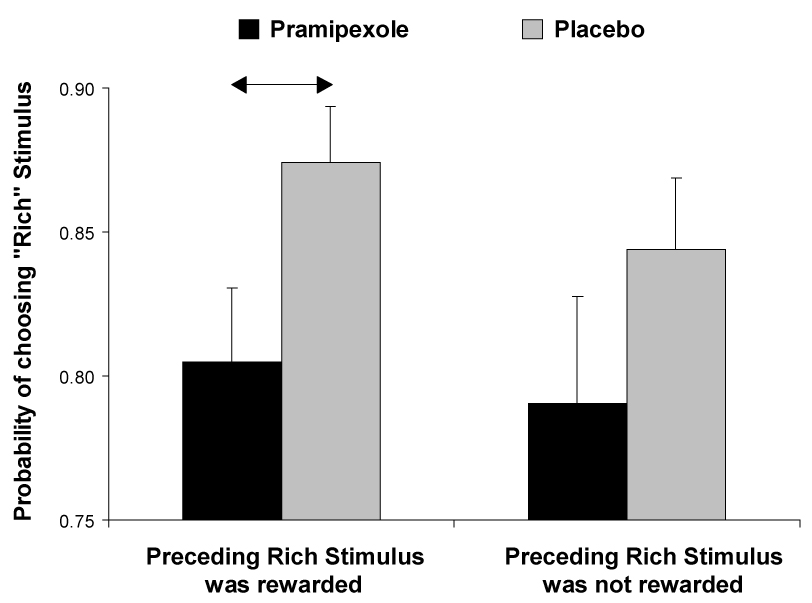
Probability of choosing the more frequently rewarded stimulus (“rich stimulus”) as a function of whether the immediately preceding rich stimulus had been rewarded (left side) or not (right side) for the pramipexole (n = 11) and placebo group (n = 13). See Figure 2 for more detail.
Reaction Time
A Medication Group × Block × Condition ANOVA revealed no significant effects involving Medication Group.
Control Analyses
For the pramipexole group, Pearson correlation analyses revealed no significant correlations between (1) △Adverse EffectMAX or △Adverse Effect90min and (2) response bias in Block 1, Block 2, or Block 3, or the reward learning (Block 3 - Block 1) (all |r| < 0.47, all p’s > 0.14). Importantly, hierarchical regression analyses revealed that Medication Group (coded as a dummy variable; entered in the third step) was a significant predictor of Response Bias in Block 3, even after adjusting for Response Bias in Block 1 (entered in the first step) and △Adverse EffectMAX (entered in the second step), △R² = 0.169, △F(1,20) = 8.81, p < 0.009. Similar findings emerged when considering △Adverse Effect90min, △R² = 0.134, △F(1,20) = 6.88, p < 0.017.
Additional control analyses showed that the pramipexole and placebo group did not differ in the number of rewards received for the rich stimulus (88.27 ± 1.95 vs. 88.77 ± 1.83; t(22) = −0.64, p > 0.52) or the lean stimulus (29.27 ± 0.79 vs. 29.08 ± 1.12; t(22) = 0.49, p > 0.63), indicating that the two groups were exposed to similar reinforcement schedules.
Simple RT Task
The only effect emerging from the ANOVA was a significant Medication Group × Time interaction, F(1,24) = 7.70, p < 0.015, partial η² = 0.24). Post-hoc Newman-Keuls tests indicated that participants receiving pramipexole showed a significant RT slowing after the drug administration (p < 0.005); post-drug RTs for the pramipexole group were significantly slower than the RTs for the placebo group (p < 0.050; Figure 4).
Figure 4. Simple RT task.
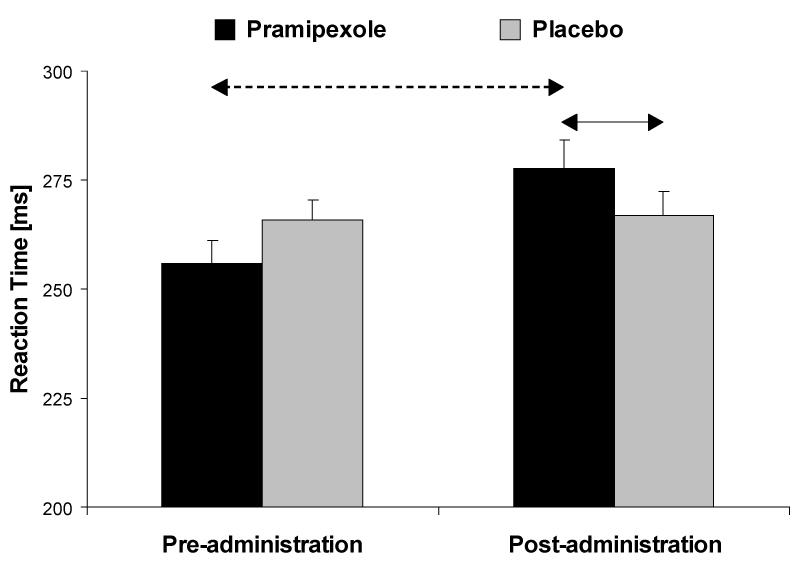
Reaction time before and after administration of study medication for the pramipexole (n = 11) and placebo group (n = 15). See Figure 2 for more detail.
Control Analyses
To assess whether the RT findings were associated with adverse effects, difference scores were computed (post-drug - pre-drug), and entered in correlation analyses with the Adverse effect scores. For the pramipexole group, Pearson correlation analyses revealed a trend for a positive correlation between mean RT changes (averaged across hands) and △Adverse Effect90min (r = 0.54, p = 0.090). To further explore this finding, hierarchical regression analyses were performed, in which pre-drug mean RT, Adverse effect scores, and Medication Group were entered sequentially to predict post-drug mean RT. For both △Adverse EffectMAX and △Adverse Effect90min, the model was not significant, indicating that Group did not account for a significant amount of variance after adjusting for baseline RT difference and, more importantly, for adverse effects, both △R² < 0.041, △F(1,22) < 1.73, p’s>0.20).
BRAIN Task
For the dysmetria scores, no significant effects emerged (all F(1,25) < 3.21, all p’s > 0.85). For the kinesia score, a significant Medication Group × Time interaction was found, F(1,25) = 14.49, p <0.001, partial η² = 0.37. This interaction was due to a significant decrease in kinesia score from pre- to post-drug administration in the pramipexole group (p <0.010), and an opposite pattern in the placebo group (p <0.015). Compared to the placebo group, participants receiving pramipexole showed significantly lower kinesia scores post-drug (Figure 5).
Figure 5. BRAIN task.
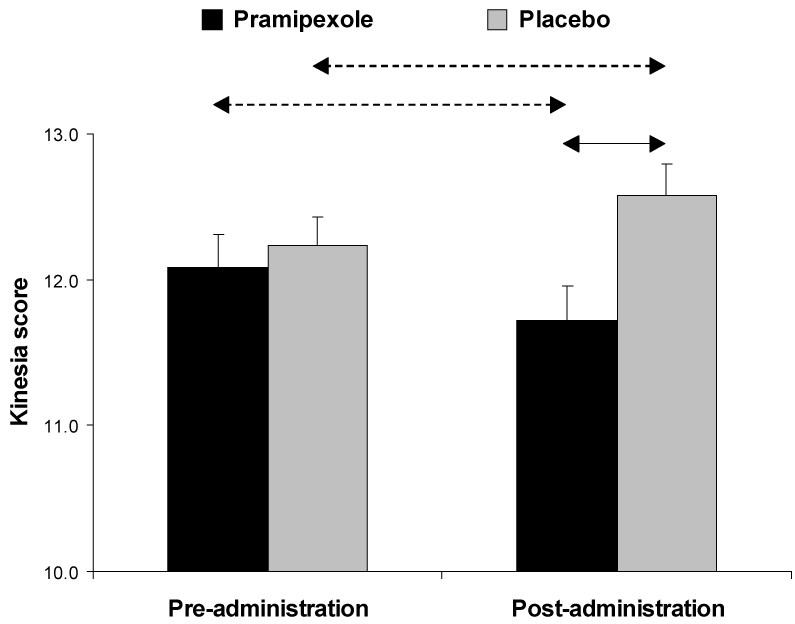
Square-root transformed kinesia score before and after administration of study medication for the pramipexole (n = 12) and placebo group (n = 15). See Figure 2 for more detail.
For the incoordination score, the Medication Group × Time interaction approached significance, F(1,25) = 3.97, p = 0.057, partial η² = 0.14. No significant differences emerged in post-hoc tests, however.
Control Analyses
For both the kinesia and dysmetria variables, a difference score was computed (post-drug - pre-drug), and entered in correlation analyses with the Adverse effect scores. For the pramipexole group, both difference scores were negatively correlated with △Adverse Effect90min (both r’s = -0.81, n = 12, p’s <0.001), indicating that increasing Adverse effects were associated with decreasing kinesia and dysmetria scores. Hierarchical regression analyses further suggested that Medication Group did not explain a significant amount of variance in the post-drug dysmetria scores after adjusting for the effects of pre-drug dysmetria scores and, more importantly, Adverse effects scores (△Adverse EffectMAX: △R² = 0.002, △F(1,23) = 0.09, p’s > 0.70; △Adverse Effect90min: △R² = 0.002, △F(1,23) = 0.13, p > 0.70). For the kinesia score, however, Medication Group remained a significant predictor of post-drug scores, even after adjusting for baseline score and adverse effects (both △R² > 0.05, both △F(1,23) = 5.33, both p’s < 0.030).
VAMS
For the “sadness” and “hostile” subscale, no effects involving Medication Group emerged. For the “withdrawn” subscale, a main effect of Medication Group emerged, F(1,28) = 4.15, p = 0.05, whereas for the “mentally slow” subscale, the Medication Group × Time interaction was significant, F(2,56) = 3.95, p < 0.025. Finally, trends for the “tense” subscale also emerged (Medication Group: F(1,28) = 3.26, p = 0.08; Medication Group × Time: F(2,56) = 2.75, p = 0.07). Compared to the placebo group, pramipexole participants were (1) more withdrawn across all time points; (2) more mentally slow 90 min post-drug administration; and (3) more tense 90 min post-drug administration (Figure 6).
Figure 6. VAMS.
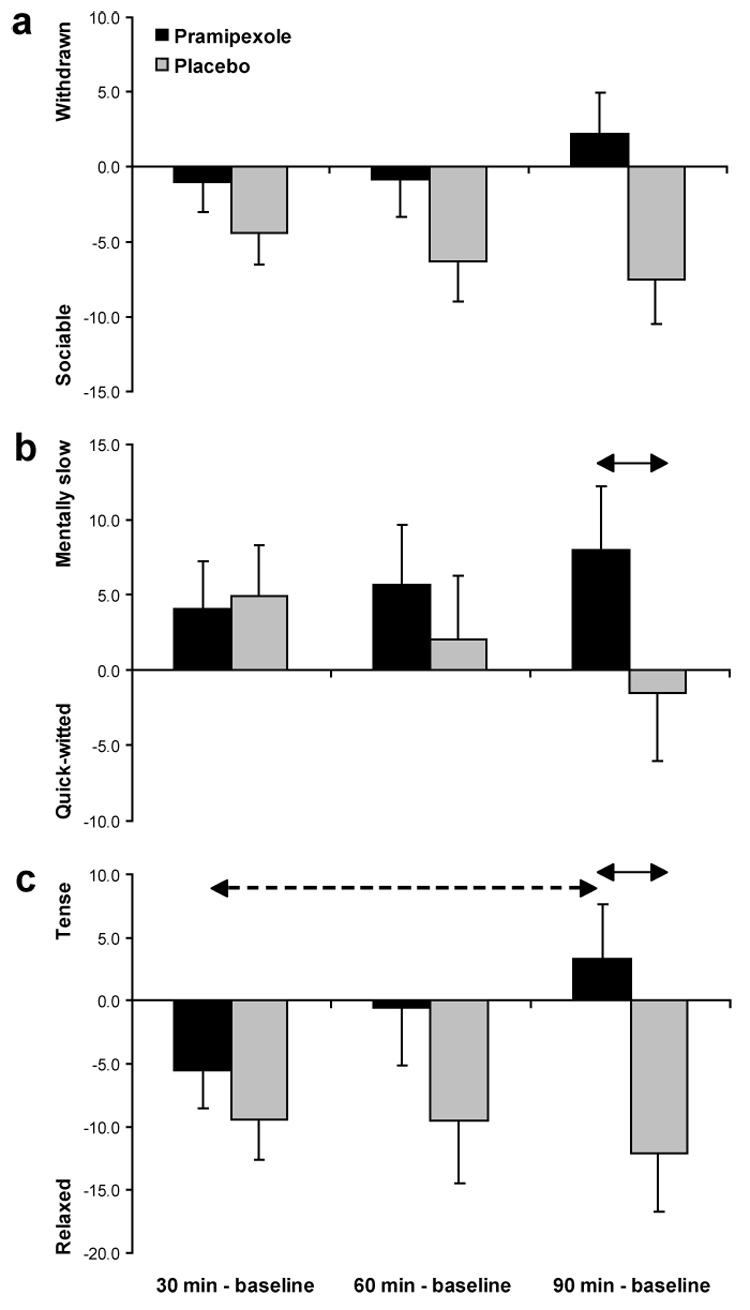
Baseline-corrected VAMS scores at several time points during the experimental session for the pramipexole (n = 16) and placebo group (n = 14). See Figure 2 for more detail.
Control Analyses
For each VAMS subscale, Pearson correlations were computed between the difference score at 90 min (VAMS90min - VAMSpre-drug) and the adverse effect scores. All correlations were positive, indicating that increases in adverse effects were associated with more negative affect. For the withdrawn (r = 0.59, n = 16, p < 0.017) and hostile (r = 0.43, n = 16, p = 0.098) subscale, the correlation with △Adverse Effect90min was significant or nearly significant. For each VAMS subscale, hierarchical regression analyses sequentially entering pre-drug VAMS scores, △Adverse Effect90min, and Medication Group suggested that Group was not a significant predictor of VAMS score at 90 min after adjusting for the effects of baseline VAMS scores and, more importantly, adverse effects (all △R² < 0.048, all △F(1,26) < 2.10, all ps > 0.16).
DISCUSSION
Animal studies have emphasized the role of phasic DA signaling in reward-related learning (Bayer and Glimcher 2005; Garris et al. 1999; Schultz 2007). The main goal of the current study was to evaluate the effect of a single dose of a D2/D3 agonist—pramipexole—on reinforcement learning in psychiatrically and neurologically healthy adults. Based on prior animal (Baudry et al. 1977; Fuller et al. 1982; Tissari et al. 1983; Sumners et al. 1981; cf. Schmitz et al. 2003) and limited human (Frank and O’Reilly 2006; Samuels et al. 2006a,b) evidence, we hypothesized that administration of 0.5 mg of pramipexole would blunt phasic DA bursts to reward feedback via presynaptic autoreceptor mechanisms and thus impair reward learning. This hypothesis was confirmed. Compared to the placebo group, participants receiving pramipexole showed significantly (1) reduced response bias and overall reward learning; (2) reduced accuracy for the stimulus associated with more frequent reward (“rich stimulus”) in block 3; and (3) reduced probability of using a “win-stay” strategy (choosing “rich” when a current rich stimulus followed a rewarded rich stimulus). Highlighting the specificity of these findings, no group differences emerged for RT and discriminability scores in the probabilistic reward task; furthermore, the pramipexole group showed significantly higher accuracy for the stimulus associated with less frequent reward (“lean stimulus”) in Block 2 and 3. Thus, blunted response bias was not due to generally poor performance or global effects of D2 agonist administration. Critically, control analyses indicated that group differences in response bias were independent from adverse effects or were not due to exposure to a different reinforcement schedule. In addition to impacting reward learning, pramipexole led to (1) RT slowing in a vigilance task (Simple Reaction Time Task); (2) reduced motor speed (kinesia score in the BRAIN Task); and (3) elevated self-report of withdraw, mental slowness, and tension. Whereas group differences in reward learning and motor speed remained after adjusting for adverse effects, RT slowing and increases in negative affect did not, suggesting that the latter findings were associated with the adverse effects experienced by the pramipexole group.
The present findings of blunted reward responsiveness and reward learning in participants receiving a single dose of a D2/D3 agonist extend prior observations of dopamine-dependent modulation of reward processing. In a recent study, Sevy et al. (2006) observed that a transient reduction of DA levels worsened performance in the IOWA gambling task, specifically by diminishing the influence of past outcomes on decision making. Along similar lines, Leyton et al. (2007) found that an acute DA precursor depletion impaired the ability to preferentially respond to stimuli predicting reward in a GO/NOGO task in healthy participants; this effect was reversed by L-DOPA administration. Finally, and of particular relevance to the present study, Frank and O’Reilly (2006) administered low doses of D2 agents—cabergoline and haloperidol—to healthy adults performing a probabilistic selection task involving reward and punishment feedback. Based on animal findings indicating that low doses of DA (D2) agonists (e.g., cabergoline) and antagonists (e.g., haloperidol) preferentially activate and inhibit DA presynaptic autoreceptors respectively, the authors hypothesized that cabergoline would impair and haloperidol would increase learning from positive reinforcement. These hypotheses were confirmed.
Notably, in Frank and O’Reilly (2006), the effects of cabergoline and haloperidol on reinforcement learning were predicted by opposite effects of these DA agents on presynaptic DA bursts in a computational model of striatal-cortical function (Frank 2005). This model, which is grounded on a variety of anatomical, physiological, and pharmacological findings concerning DA-dependent cortico-striatal mechanisms, postulates that phasic DA bursts during positive feedback lead to a strengthening of the selected response (Go signal), and thus foster learning of the action that leads to reward. Negative feedback, on the other hand, is assumed to induce DA dips, which support NoGo signals to avoid the bad choice in the future. In the computational model of DA-driven learning, the effects of cabergoline were mimicked by impaired Go learning due to reduced phasic presynaptic DA bursts. Conversely, the effects of haloperidol were explained by enhanced Go learning from positive feedback due to inhibition of presynaptic DA autoreceptors.
In a companion paper focusing on the event-related potentials collected during the probabilistic reward task, we recently applied Frank’s computational model of striatal-cortical function (Frank 2005) to evaluate whether the blunted reward learning described here could be explained by reduction of presynaptic DA bursts (Santesso, Evins, Frank, Schetter, and Pizzagalli, in preparation). Indeed, consistent with our hypotheses and replicating prior findings with cabergoline (Frank and O’Reilly 2006), we found that a neural network simulating presynaptic effects of pramipexole showed the observed effects on reward processing. Specifically, weaker Go pathways due to blunted DA bursts after reward delivery produced lower response bias and lower accuracy toward the more frequently rewarded stimulus but higher accuracy for the lean stimulus (Santesso et al. in preparation). Note that a corollary of the computational model is that reduced phasic DA responses in the pramipexole group would lead to a lower ability to resolve Go/NoGo conflict and thus lower the ability to differentiate between two competing responses associated with different reinforcement histories. In line with this prediction, in the present study, participants receiving pramipexole, as compared to those receiving placebo, were characterized by smaller accuracy differentiation between the rich and lean stimuli.
Self-report assessments revealed increased negative affect (withdrawal, tension) and sedation in the pramipexole group, although these effects were associated with drug-related adverse effects. These findings are, again, consistent with pre-synaptic mechanisms. With respect to alertness, in the animal literature, a biphasic dose-response curve has been hypothesized for DA receptor agonists: whereas low doses act pre-synaptically and lead to sedation, higher doses are assumed to act post-synaptically, leading to an increase of alertness (Monti et al. 1988; Rye 2004; Keating and Rye 2003). Consistent with these animal data, low doses of D2 agonists have been found to cause sedation in humans (Ferreira et al. 2002; Samuels et al. 2006a,b), a finding that has been attributed to activation of D2-like inhibitory autoreceptors on the cell bodies of VTA neurons (Rye 2004).
Although the present findings provide evidence that disruption of DA signaling can impair reinforcement learning in humans, three important limitations of the current study should be emphasized. First, although the current data concur with prior animal and human findings that low single doses of DA agonists might reduce DA signaling (e.g., Baudry et al. 1977; Ferreira et al. 2002; Piercey et al. 1996; Rye and Jankovic 2002; Samuels et al. 2006a,b), interpretations about the potential pharmacological mechanisms responsible for the present findings remain tentative. To further elucidate underlying mechanisms, studies using a dose-response curve will be required to determine the effects of progressively increasing doses of pramipexole on reward learning.
Second, the probabilistic reward task used in the current study involved only positive feedback. Although the present behavioral findings and computational modeling of these data (Santesso et al. in preparation) suggest that blunted reward learning can be produced by reduced DA bursts that disrupt the ability to learn from positive feedback (Go pathway), prior findings have clearly highlighted the role of phasic DA suppression after negative feedback in reinforcement learning (e.g., Frank et al. 2004; Holroyd and Coles 2002).
Third, the 0.5 mg pramipexole dose used in the present study elicited transient adverse effects in a substantial portion of the participants. Although a similar profile and extent of adverse effects have been observed in recent studies using an identical dose of pramipexole (Samuels et al. 2006a,b), and the main findings of impaired reward learning remained after adjusting for the effects of the adverse events, several participants were unable to complete portions of the experimental session. Future studies aimed at evaluating the effects of pramipexole on the healthy dopaminergic system should consider using a titration approach, in which a low starting dose of pramipexole is gradually increased over a 1–2 week period to a level used in treatment of Parkinson’s disease. A titration approach would not only minimize drug-related adverse effects but also allow to test the hypothesis that higher doses of pramipexole would increase, rather than decrease, reward responsiveness due to a preferential post-synaptic DA action. This speculation is consistent with (1) findings in rodents showing that pramipexole is effective in reversing anhedonia produced by the chronic mild stress paradigm (Willner et al. 1994) and has antidepressant properties (Maj and Rogoz 1999; (Lehr 2002); and (2) clinical findings showing that higher doses of pramipexole ameliorate depressive symptoms in PD patients (Lemke et al. 2006) and treatment-resistant depression (Corrigan et al. 2000; Cassano et al. 2004). Collectively, these preclinical and clinical findings raise the possibility that sustained pramipexole administration may alleviate depressive—particularly anhedonic—symptoms through its action on D3 receptors concentrated in the nucleus accumbens.
Finally, a possible alternative explanation for the present findings is that pramipexole led to tonically increased DA levels, which in turn reduced the dynamic range of phasic DA bursts necessary for reinforcement learning. According to this account, tonically increased DA may have impaired the ability of neurons to detect transient fluctuations in DA levels. Recent studies support this interpretation since medicated Parkinson’s disease patients demonstrate impaired learning in response to punishment (Frank et al. 2004) as well as impaired performance in reversal learning tasks (Cools et al. 2006,(2007). Furthermore, these patients sometimes exhibit impulse control disorders (e.g., compulsive gambling), failing to inhibit behaviors with negative consequences (Weintraub et al. 2006). These impairments have been attributed to “over-dosing” of unaffected brain regions with the DA agonist treatment for PD (Cools et al. 2006,2007; Frank et al. 2004). Although the present behavioral findings cannot rule out either of these competing hypotheses, we believe that presynaptic autoreceptor mechanisms provide the most parsimonious explanation for the overall pattern of findings, including impaired reinforcement learning, motor slowing, and increased negative affect and sedation.
Acknowledgments
This work was supported by grants from NIMH (R01 MH68376; DAP) and Harvard College Research Program (ECS). Dr. Evins and Ms. Culhane were supported by a grant from the National Institute on Drug Abuse (K23 DA00510-01; AEE). Dr. Frank was supported by a grant from the National Institute on Drug Abuse (DA022630). The authors would like to thank Dr. Catherine Fullerton, Kyle Ratner, Elena Goetz, and Jeffrey Birk for their assistance with the project, Dr. David Standaert for his helpful review of the results, and three anonymous reviewers for their constructive criticisms.
Footnotes
Disclosure/Conflict of Interest Dr. Pizzagalli has received research support from GlaxoSmithKline and Merck & Co., Inc., manufactures of antidepressants. Dr. Evins has received research grant support from Janssen Pharmaceutica, Sanofi-Aventis, Astra Zeneca; research materials from GSK and Pfizer, and honoraria from Primedia, Inc. Moreover, Dr. Evins is an investigator in a NIDA-funded collaborative study with GSK. Dr. Frank, Ms. Schetter, Ms. Culhane, and Ms. Pajtas report no competing interests.
Previous presentation The data in this paper were presented in preliminary form at the 44th annual meeting of the American College of Neuropsychopharmacology, Waikoloa Village, HI, December 11–15, 2005.
REFERENCES
- Baudry M, Martres MP, Schwartz JC. In vivo binding of 3H-pimozide in mouse striatum: effects of dopamine agonists and antagonists. Life Sci. 1977;21:1163–1170. doi: 10.1016/0024-3205(77)90116-3. [DOI] [PubMed] [Google Scholar]
- Bayer HM, Glimcher PW. Midbrain dopamine neurons encode a quantitative reward prediction error signal. Neuron. 2005;47:129–141. doi: 10.1016/j.neuron.2005.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AT, Steer RA, Brown GK. Beck Depression Inventory Manual. 2nd. San Antonio, TX: The Psychological Corporation; 1996. [Google Scholar]
- Bogdan R, Pizzagalli DA. Acute stress reduces reward responsiveness: implications for depression. Biol Psychiatry. 2006;60:1147–1154. doi: 10.1016/j.biopsych.2006.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211–218. [Google Scholar]
- Cassano P, Lattanzi L, Soldani F, Navari S, Mattistini G, Gemignani A, Cassano G. Pramipexole in treatment-resistant depression: an extended follow-up. Depress Anxiety. 2004;20:131–138. doi: 10.1002/da.20038. [DOI] [PubMed] [Google Scholar]
- Chapman LJ, Chapman JP. The measurement of handedness. Brain Cognition. 1987;6:175–183. doi: 10.1016/0278-2626(87)90118-7. [DOI] [PubMed] [Google Scholar]
- Chen YC, Choi JK, Andersen SL, Rosen BR, Jenkins BG. Mapping dopamine D2/D3 receptor function using pharmacological magnetic resonance imaging. Psychopharmacology (Berl) 2005;180:705–715. doi: 10.1007/s00213-004-2034-0. [DOI] [PubMed] [Google Scholar]
- Cheng J, Feenstra MG. Individual differences in dopamine efflux in nucleus accumbens shell and core during instrumental learning. Learn Mem. 2006;13:168–177. doi: 10.1101/lm.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O. Molecular biology of dopamine receptor subtypes. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 2000. [Google Scholar]
- Cools R, Altamirano L, D’Esposito M. Reversal learning in Parkinson's disease depends on medication status and outcome valence. Neuropsychologia. 2006;44:1663–1673. doi: 10.1016/j.neuropsychologia.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Cools R, Lewis SJ, Clark L, Barker RA, Robbins TW. L-DOPA disrupts activity in the nucleus accumbens during reversal learning in Parkinson's disease. Neuropsychopharmacology. 2007;32:180–189. doi: 10.1038/sj.npp.1301153. [DOI] [PubMed] [Google Scholar]
- Cooper JR, Bloom FE, Roth RH. The biochemical basis of neuropharmacology. 8th. Oxford: Oxford University Press; 2003. [Google Scholar]
- Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety. 2000;11:58–65. doi: 10.1002/(sici)1520-6394(2000)11:2<58::aid-da2>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- de Wit H, Enggasser JL, Richards JB. Acute administration of d-amphetamine decreases impulsivity in healthy volunteers. Neuropsychopharmacology. 2002;27:813–825. doi: 10.1016/S0893-133X(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64:327–337. doi: 10.1001/archpsyc.64.3.327. [DOI] [PubMed] [Google Scholar]
- Ferreira JJ, Galitzky M, Thalamas C, Tiberge M, Montastruc JL, Sampaio C, Rascol O. Effect of ropinirole on sleep onset: a randomized, placebo-controlled study in healthy volunteers. Neurology. 2002;58:460–462. doi: 10.1212/wnl.58.3.460. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W. Discrete coding of reward probability and uncertainty by dopamine neurons. Science. 2003;299:1898–1902. doi: 10.1126/science.1077349. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBM. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition(SCID-I/P) New York: Biometrics Research, New York State Psychiatric Institute; 2002. [Google Scholar]
- Frank MJ. Dynamic dopamine modulation in the basal ganglia: a neurocomputational account of cognitive deficits in medicated and nonmedicated Parkinsonism. J Cogn Neurosci. 2005;17:51–72. doi: 10.1162/0898929052880093. [DOI] [PubMed] [Google Scholar]
- Frank MJ, O’Reilly RC. A mechanistic account of striatal dopamine function in human cognition: psychopharmacological studies with cabergoline and haloperidol. Behav Neurosci. 2006;120:497–517. doi: 10.1037/0735-7044.120.3.497. [DOI] [PubMed] [Google Scholar]
- Frank MJ, Seeberger LC, O'Reilly RC. By carrot or by stick: cognitive reinforcement learning in parkinsonism. Science. 2004;306:1940–1943. doi: 10.1126/science.1102941. [DOI] [PubMed] [Google Scholar]
- Fuller RW, Clemens JA, Hynes MD., 3rd Degree of selectivity of pergolide as an agonist at presynaptic versus postsynaptic dopamine receptors: implications for prevention or treatment of tardive dyskinesia. J Clin Psychopharmacol. 1982;2:371–375. [PubMed] [Google Scholar]
- Garris PA, Kilpatrick M, Bunin MA, Michael D, Walker QD, Wightman RM. Dissociation of dopamine release in the nucleus accumbens from intracranial self-stimulation. Nature. 1999;398:67–69. doi: 10.1038/18019. [DOI] [PubMed] [Google Scholar]
- Giovannoni G, van Schalkwyk J, Fritz VU, Lees AJ. Bradykinesia akinesia inco-ordination test (BRAIN TEST): an objective computerised assessment of upper limb motor function. J Neurol Neurosurg Psychiatry. 1999;67:624–629. doi: 10.1136/jnnp.67.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Grace AA. Dopamine. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 119–132. [Google Scholar]
- Harrison BJ, Olver JS, Norman TR, Nathan PJ. Effects of serotonin and catecholamine depletion on interleukin-6 activation and mood in human volunteers. Hum Psychopharmacol. 2002;17:293–297. doi: 10.1002/hup.414. [DOI] [PubMed] [Google Scholar]
- Hollerman JR, Schultz W. Dopamine neurons report an error in the temporal prediction of reward during learning. Nat Neurosci. 1998;1:304–309. doi: 10.1038/1124. [DOI] [PubMed] [Google Scholar]
- Holroyd CB, Coles MGH. The neural basis of human error processing: Reinforcement learning, dopamine, and the error-related negativity. Psychol Rev. 2002;109:679–709. doi: 10.1037/0033-295X.109.4.679. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. Adv Neurol. 1987;45:19–34. [PubMed] [Google Scholar]
- Ikemoto S, Panksepp J. Dissociations between appetitive and consummatory responses by pharmacological manipulations of reward-relevant brain regions. Behav Neurosci. 1996;110:331–345. doi: 10.1037//0735-7044.110.2.331. [DOI] [PubMed] [Google Scholar]
- Keating GL, Rye DB. Where you least expect it ' dopamine in the pons and modulation of sleep and REM-sleep. Sleep. 2003;26:788–789. [PubMed] [Google Scholar]
- Lefoll B, Diaz J, Sokoloff P. Neuroadaptations to hyperdopaminergia in dopamine D3-receptor deficient mice. Life Sci. 2005;76:1281–1296. doi: 10.1016/j.lfs.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Lehr E. Potential antidepressant properties of pramipexole detected in locomotor and operant behavioral investigations in mice. Psychopharmacology. 2002;163:495–500. doi: 10.1007/s00213-002-1199-7. [DOI] [PubMed] [Google Scholar]
- Leyton M, aan het Rot M, Booij L, Baker GB, Young SN, Benkelfat C. Mood-elevating effects of d-amphetamine and incentive salience: the effect of acute dopamine precursor depletion. J Psychiatry Neurosci. 2007;32:129–136. [PMC free article] [PubMed] [Google Scholar]
- Lemke MR, Brecht HM, Koester J, Reichmann H. Effects of the dopamine agonist pramipexole on depression, anhedonia and motor functioning in Parkinson's disease. J Neurol Sci. 2006;248:266–270. doi: 10.1016/j.jns.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Maj J, Rogoz Z. Synsergic effect of pramipexole and sertraline in the forced swimming test. Pol J Pharmacol. 1999;51:471–475. [PubMed] [Google Scholar]
- Montague PR, Dayan P, Sejnowski TJ. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J Neurosci. 1996;16:1936–1347. doi: 10.1523/JNEUROSCI.16-05-01936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague PR, Hyman SE, Cohen JD. Computational roles for dopamine in behavioural control. Nature. 2004;431:760–767. doi: 10.1038/nature03015. [DOI] [PubMed] [Google Scholar]
- Monti JM, Hawkins M, Jantos H, D'Angelo L, Fernandez M. Biphasic effects of dopamine D-2 receptor agonists on sleep and wakefulness in the rat. Psychopharmacology. 1988;95:395–400. doi: 10.1007/BF00181955. [DOI] [PubMed] [Google Scholar]
- Myers RE, Anderson LI, Dluzen DE. Estrogen, but not testosterone, attenuates methamphetamine-evoked dopamine output from superfused striatal tissue of female and male mice. Neuropharmacology. 2003;44:624–632. doi: 10.1016/s0028-3908(03)00043-1. [DOI] [PubMed] [Google Scholar]
- Nagy H, Keri S, Myers CE, Benedek G, Shohamy D, Gluck MA. Cognitive sequence learning in Parkinson's disease and amnestic mild cognitive impairment: Dissociation between sequential and non-sequential learning of associations. Neuropsychologia. 2007;45:1386–1392. doi: 10.1016/j.neuropsychologia.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Parkinson JA, Dalley JW, Cardinal RN, Bamford A, Fehnert B, Lachenal G, Rudarakanchana N, Halkerston KM, Robbins TW, Everitt BJ. Nucleus accumbens dopamine depletion impairs both acquisition and performance of appetitive Pavlovian approach behaviour: implications for mesoaccumbens dopamine function. Behav Brain Res. 2002;137:149–163. doi: 10.1016/s0166-4328(02)00291-7. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. Parkinson Study Group. JAMA. 2000;284:1931–1938. doi: 10.1001/jama.284.15.1931. [DOI] [PubMed] [Google Scholar]
- Perbal S, Couillet J, Azouvi P, Pouthas V. Relationships between time estimation, memory, attention, and processing speed in patients with severe traumatic brain injury. Neuropsychologia. 2003;41:1599–1610. doi: 10.1016/s0028-3932(03)00110-6. [DOI] [PubMed] [Google Scholar]
- Pessiglione M, Seymour B, Flandin G, Dolan RJ, Frith CD. Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature. 2006;442:1042–1045. doi: 10.1038/nature05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piercey MF, Hoffmann WE, Smith MW, Hyslop DK. Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: comparison to other dopamine receptor agonists. Eur J Pharmacol. 1996;312:35–44. doi: 10.1016/0014-2999(96)00454-2. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA, Jahn AL, O'Shea JP. Toward an objective characterization of an anhedonic phenotype: a signal-detection approach. Biol Psychiatry. 2005;57:319–327. doi: 10.1016/j.biopsych.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JN, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67–70. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- Robinson S, Rainwater AJ, Hnasko TS, Palmiter RD. Viral restoration of dopamine signaling to the dorsal striatum restores instrumental conditioning to dopamine-deficient mice. Psychopharmacology (Berl) 2007;191:567–578. doi: 10.1007/s00213-006-0579-9. [DOI] [PubMed] [Google Scholar]
- Rye DB. The two faces of Eve: dopamine's modulation of wakefulness and sleep. Neurology. 2004;63 8 suppl 3:S2–S7. doi: 10.1212/wnl.63.8_suppl_3.s2. [DOI] [PubMed] [Google Scholar]
- Rye DB, Jankovic J. Emerging views of dopamine in modulating sleep/wake state from an unlikely source: PD. Neurology. 2002;58:341–346. doi: 10.1212/wnl.58.3.341. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CW. Comparison of pramipexole and amisulpride on alertness, autonomic and endocrine functions in healthy volunteers. Psychopharmacology (Berl) 2006;187:498–510. doi: 10.1007/s00213-006-0443-y. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM. Comparison of pramipexole and modafinil on arousal, autonomic, and endocrine functions in healthy volunteers. J Psychopharmacol. 2006;20:756–770. doi: 10.1177/0269881106060770. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Benoit-Marand M, Gonon F, Sulzer D. Presynaptic regulation of dopaminergic neurotransmission. J Neurochem. 2003;87:273–289. doi: 10.1046/j.1471-4159.2003.02050.x. [DOI] [PubMed] [Google Scholar]
- Schuck S, Bentue-Ferrer D, Kleinermans D, Reymann JM, Polard E, Gandon JM, Allain H. Psychomotor and cognitive effects of piribedil, a dopamine agonist, in young healthy volunteers. Fundam Clin Pharmacol. 2002;16:57–65. doi: 10.1046/j.1472-8206.2002.00070.x. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Servan-Schreiber D, Carter CS, Bruno RM, Cohen JD. Dopamine and the mechanisms of cognition: Part II. D-amphetamine effects in human subjects performing a selective attention task. Biol Psychiatry. 1998;43:723–729. doi: 10.1016/s0006-3223(97)00449-6. [DOI] [PubMed] [Google Scholar]
- Sevy S, Hassoun Y, Bechara A, Yechiam E, Napolitano B, Burdick K, Delman H, Malhotra A. Emotion-based decision-making in healthy subjects: short-term effects of reducing dopamine levels. Psychopharmacology (Berl) 2006;188:228–235. doi: 10.1007/s00213-006-0450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Grossman S, Sage J, Gluck MA. The role of dopamine in cognitive sequence learning: evidence from Parkinson's disease. Behav Brain Res. 2005;156:191–199. doi: 10.1016/j.bbr.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Diaz J, Le Foll B, Guillin O, Leriche L, Bezard E, Gross C. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord Drug Targets. 2006;5:25–43. doi: 10.2174/187152706784111551. [DOI] [PubMed] [Google Scholar]
- Sokolowski JD, Conlan AN, Salamone JD. A microdialysis study of nucleus accumbens core and shell dopamine during operant responding in the rat. Neuroscience. 1998;86:1001–1009. doi: 10.1016/s0306-4522(98)00066-9. [DOI] [PubMed] [Google Scholar]
- Spielberger CD, Gorsuch RL, Lushere RE. Manual of the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1970. [Google Scholar]
- Schwabe K, Koch M. Effects of aripiprazole on operant responding for a natural reward after psychostimulant withdrawal in rats. Psychopharmacology (Berl) 2007;191:759–765. doi: 10.1007/s00213-006-0520-2. [DOI] [PubMed] [Google Scholar]
- Sumners C, de Vries JB, Horn AS. Behavioural and neurochemical studies on apomorphine-induced hypomotility in mice. Neuropharmacology. 1981;20:1203–1208. doi: 10.1016/0028-3908(81)90065-4. [DOI] [PubMed] [Google Scholar]
- Tissari AH, Rossetti ZL, Meloni M, Frau MI, Gessa GL. Autoreceptors mediate the inhibition of dopamine synthesis by bromocriptine and lisuride in rats. Eur J Pharmacol. 1983;91:463–468. doi: 10.1016/0014-2999(83)90171-1. [DOI] [PubMed] [Google Scholar]
- Tripp G, Alsop B. Sensitivity to reward frequency in boys with attention deficit hyperactivity disorder. J Clin Child Psychol. 1999;28:366–375. doi: 10.1207/S15374424jccp280309. [DOI] [PubMed] [Google Scholar]
- Waelti P, Dickinson A, Schultz W. Dopamine responses comply with basic assumptions of formal learning theory. Nature. 2001;412:43–48. doi: 10.1038/35083500. [DOI] [PubMed] [Google Scholar]
- Weintraub D, Siderowf AD, Potenza MN, Goveas J, Morales KH, Duda JE, Moberg PJ, Stern MB. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol. 2006;63:969–973. doi: 10.1001/archneur.63.7.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P. Dopaminergic mechanisms in depression and mania. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995. pp. 921–931. [Google Scholar]
- Willner P, Lappas S, Cheeta S, Muscat R. Reversal of stress-induced anhedonia by the dopamine receptor agonist, pramipexole. Psychopharmacology. 1994;115:454–462. doi: 10.1007/BF02245568. [DOI] [PubMed] [Google Scholar]
- Wright CE, Sisson TL, Ichhpurani AK, Peters GR. Steady-state pharmacokinetic properties of pramipexole in healthy volunteers. J Clin Pharmacol. 1997;37:520–525. doi: 10.1002/j.1552-4604.1997.tb04330.x. [DOI] [PubMed] [Google Scholar]