

Abstract
Regardless of the type of arterial reconstruction, luminal narrowing (stenosis or restenosis) develops in approximately one third of the vessels. In the past, the focus of research has been on the mechanisms of stenosis (intimal hyperplasia, pathologic remodeling) and pharmacologic approaches to prevention. An alternative approach is to induce intimal atrophy after luminal narrowing has developed, thus limiting treatment to only those patients that develop a problem. This approach to treat established disease by reducing wall mass through induction of cell death and extracellular matrix removal would be particularly useful for treating stenosis in synthetic bypass grafts or stented vessels, in which intimal hyperplasia is the primary mechanism of stenosis. This approach may be applicable as well to other vascular proliferative disorders, such as pulmonary hypertension and chronic transplant arteriopathy. Proof of principle has been shown in experiments with antibodies to PDGF receptors that cause neointimal regression in baboon PTFE grafts and with ACE inhibitors that induce medial atrophy in hypertensive arteries. Possible molecular targets could include PDGF receptors, A20, and BMP4. Further studies are needed to determine the utility of such a therapeutic approach to vascular disease.
INTRODUCTION
Arterial occlusive diseases are treated by various open and endovascular approaches including bypass graft, endarterectomy, atherectomy, balloon angioplasty and stent angioplasty. Regardless of the type of intervention, stenosis or restenosis develops in a significant number of patients, often leading to limb loss or death1,2.
Research on restenosis has focused on the biological mechanisms of vascular hyperplasia caused by vascular injury and on pharmacological strategies to prevent hyperplasia. Drug eluting stents are a successful application of these strategies, although late stent thrombosis may be a result of inhibiting endothelial cell healing as well as smooth muscle cell (SMC) proliferation3. An alternative approach might be to induce intimal atrophy after restenosis has developed. This approach would be particularly useful for treating stenotic and restenotic disease in synthetic bypass grafts or stented vessels, since restenosis in these rigid vessels only involves intimal hyperplasia and not vessel remodeling. Furthermore, it would permit the surgeons to treat only the subset of patients with restenosis. Since patients with restenosis often have symptoms (worsening claudication or angina), they are easily identified. This strategy might also be applied to pulmonary arterial hypertension and chronic transplant arteriopathy, which share the pathophysiologic features of increased arterial wall mass and luminal narrowing. In this review, we will summarize the evidence that vascular hyperplasia is reversible and that a strategy to reduce wall mass through inhibition of proliferation and induction of cell death and extracellular matrix loss could be applied to many vascular disorders. As background we will first briefly review mechanisms of vascular hyperplasia and hypertrophy.
Mechanisms of Vascular Hyperplasia and Hypertrophy
The vascular response to injury has been studied in animal models for four decades and the cellular and molecular mechanisms of intimal hyperplasia and medial hypertrophy are understood in some detail4,5. For example, mechanical or hemodynamic endothelial injury (e.g. after angioplasty and stent or vein graft placement) may expose the subendothelial matrix and induce platelet adhesion, aggregation and activation. Activated platelets release various cytokines, chemokines and growth factors, which initiate SMC proliferation, leukocyte recruitment and activation of the coagulation cascade. Substances released or activated after injury include platelet-derived growth factor (PDGF), transforming growth factor (TGF)-β interleukin (IL)-1, IL-6, IL-8, thrombin, adenosine diphosphate, and thromboxane A2. The maximal intimal response requires medial damage, as well as endothelial cell injury6. Indeed, SMCs around the area of injury begin to undergo apoptosis within 1 hour of injury, and while blockade of apoptosis after endothelial and medial injury inhibits intimal hyperplasia7, SMC apoptosis without endothelial injury does not lead to intimal hyperplasia8.
Rat medial SMCs begin to proliferate within 48 hours9. Fibroblast growth factor (FGF)-2 plays a major role in this initial proliferative event. It is released from damaged endothelial and SMCs, and, while antibody blockade prevents the proliferation of SMC in the rat carotid media by ~80%10, it has no effect on subsequent cell proliferation in the intima11. In contrast to the injured rat carotid in which PDGF plays a minor role in medial SMC proliferation12, a blocking antibody to the PDGF receptor β inhibits medial SMC proliferation >90% in the baboon saphenous artery13. Other factors such as insulin-like growth factor (IGF)-114,15, thrombin16, TGF- β17 together with cytokines IL-1β18 and IL-6 all contribute to SMC proliferation. A host of inhibitory factors also moderate the proliferative response to injury, such as adiponectin19, heparan sulfate proteoglycans (e.g. perlecan20; syndecan-121), interleukin 1022, adrenomedullin23 and somatostatin24. In addition, high blood flow inhibits intimal hyperplasia after arterial injury25,26.
After 4 days, medial SMC proliferation reaches a peak in both rodent and primate models of injury and SMCs begin to migrate to the intima. PDGF clearly plays a major stimulatory role for SMC migration in both rodents27 and primates.28 It is released from platelets, and is also upregulated in the vessel wall29 in endothelial cells, SMCs, and macrophages. Insulin30, tissue factor31 and FGF232 also contribute to SMC migration in vivo.
Intimal SMCs are derived primarily from the media, but they may also be derived from adventitial myofibroblasts, pericytes associated with infiltrating microvessels, and circulating progenitor cells33,34. Intimal SMCs proliferate for up to 2 weeks and begin to express major extracellular matrix (ECM) genes, such as elastin and collagen I, by approximately 7 days after injury 35. The intima grows as elastin, collagen, glycoproteins and proteoglycans are synthesized and secreted. Between 1 and 3 months, a steady state is reached at which time the intima is ~20% cells and ~80% ECM35.
The response of previously injured vessels to reinjury has also been investigated. Koyama and Reidy36,37 observed intimal and medial SMC proliferation in response to a second balloon injury to the rat carotid, but found that the intimal thickening was entirely from increased ECM synthesis not SMCs. Hanke et al38 performed balloon injury to the rabbit carotid previously injured by electrical stimulation. They found that proliferation continued at low but significant rates up to three weeks and that the number of intimal SMCs nearly doubled.
Vascular atrophy in normal development
Vessel regression is an essential aspect of the development of the vascular system, which includes the processes of vasculogenesis (de novo formation of blood vessels) and angiogenesis (budding of new vessels from preexisting vessels)39,40. Regression occurs during development after formation of a primary plexus of capillary-like vessels. In addition, after birth there is regression of the infrarenal aorta where blood flow decreases dramatically, leading to apoptosis and reduction of vessel diameter41,42. In addition, there is a postnatal loss of a network of vessels in the vitreous and around the lens of the eye. This latter vessel loss depends on macrophage-induced endothelial cell death43. These processes are orchestrated by physical forces, such as blood flow, as well as various stimulators and inhibitors whose expression is tightly regulated both in a temporal and spatial manner44 and which are reinitiated in vascular disease in adults45.
Regression is an inherent component of any angiogenic program40. For new vessels to bud off, stable quiescent vessels must first be destabilized. This process involves the loss of surrounding pericytes. At this stage the vessel can either form a new vessel or regress, a process controlled by relative activities of PI3 kinase and PLCγ46. The presence or absence of growth factors (e.g. VEGF) and other angiomodulators (e.g. endostatin), which can be controlled by proteinases like cathepsins47, dictate the fate of the destabilized vessel.
While induction of microvessel regression may be a therapeutic strategy in many disorders associated with abnormal or excessive angiogenesis, such as cancer, psoriasis, arthritis, retinopathy, obesity and atherosclerosis48, this anti-angiogenic strategy may not be applicable to larger vessels. Nevertheless, it may be possible to induce atrophy in large diseased arteries by various means described in the next section of this review.
Animal models of vascular atrophy
1. High blood flow-induced intimal atrophy
In normal arteries, an increase in blood flow causes acute vasodilation, a process that is dependent on endothelial release of nitric oxide (NO). Chronic adaptation to increased flow is also dependent on the endothelium41. However, because a rigid vessel, such as an artificial graft or calcified artery, cannot dilate, the only way to normalize shear stress is to reduce wall mass. We have investigated this possible mechanism of adaptation to increased blood flow. We have used a polytetraflouroethylene (PTFE) graft model in baboons, in which bilateral PTFE (internodal distance 60um, internal diameter 4mm) aorto-iliac bypass grafts are allowed to heal for 2 months. Unlike the reinforced 30um internodal distance PTFE grafts used clinically, these grafts uniformly heal by the transmural ingrowth of microvessels through the interstices of the PTFE. Complete endothelialization is achieved by 2 weeks, and maximal, evenly distributed neointimal thickening along the graft is complete by 2 months49. When blood flow and shear stress are increased by the creation of a distal femoral arteriovenous fistula, the neointima regresses markedly in the graft, but there is no regression in the normal, adjacent iliac artery50,51. It appears that vessels attempt to maintain shear stress at a constant level between 5 and 25 dynes/cm2 by altering luminal area. The downstream iliac artery, which does not undergo atrophy, increases both overall and lumenal area in response to increased flow without any change in wall mass. The sequence of events leading to neointimal loss is shown in figure 1.
Figure 1.
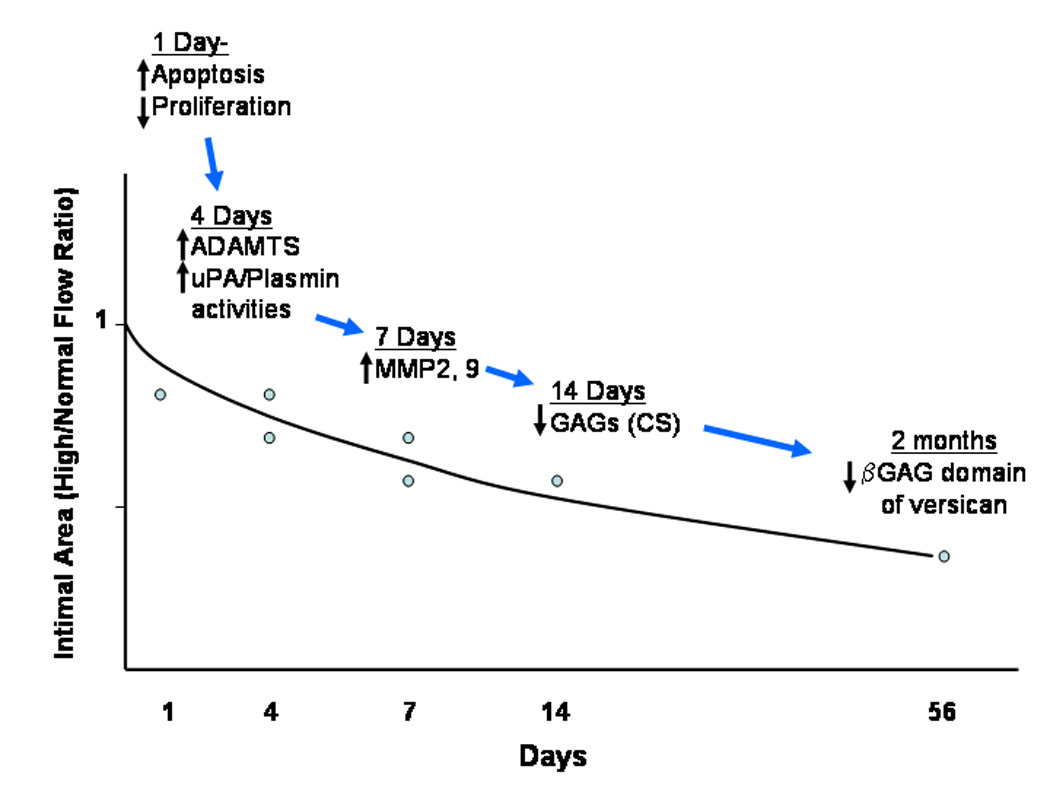
Diagram of temporal changes in the neointima of the baboon PTFE aorto-iliac graft after the switch to high blood flow.
Neointimal SMC death increases and SMC proliferation declines by 1 day after fistula placement52. There is also evidence of ECM degradation beginning at 4 days associated with a proportional loss of cells. There is a particular loss of the ECM proteoglycan versican53,54. This is significant because the sulfate groups of the chondroitin sulfate (CS) glycosaminoglycan chains of of the βGAG domain of versican bind water. Thus early collapse of the neointima could be caused by glycosaminoglycan degradation and loss of water.
The mechanism of flow-mediated regression is not known. An interesting aspect of this baboon model is that, while there is a continuous endothelial layer, it is dividing at a much higher rate than in normal vessels. In baboons as well as humans, SMCs do not accumulate in grafts unless there is an endothelial layer and restenotic lesions usually are endothelialized. The requirement for endothelium in intimal regression has not been tested. However, while increased blood flow clearly inhibits neointimal hyperplasia in the absence of endothelium in rat arterial injury and graft models26,25 and there are unknown endothelium-independent vasodilators (not NO, cyclooxygenase products, cGMP, cAMP, or hypoxia-sensitive) mediating arterial dilation55, regression of established lesions has only been reported for models with endothelium present. For example, Zhuang et al studied a rabbit model utilizing sequential phases of low flow intimal hyperplasia and high flow intimal regression in endothelialized carotid arteries56. In the baboon PTFE graft model, it is an important observation that SMCs only proliferate in the area adjacent to the endothelium51, suggesting endothelial cell regulation of SMC function. Although endothelial cell eNOS is increased50, inhibition of NOS function after the switch to high flow does not inhibit neointimal regression51. Other factors such as hemeoxygenase-1, which catalyses the formation of CO, is induced by high flow after one day (see online data52), and it may inhibit SMC proliferation via CO production57.
2. Tight external wrap-induced medial atrophy
We have previously reported that rabbit vein grafts adapt to arterial pressure by increasing total cross-sectional area of the wall, SMC number, and ECM; this adaptation is suppressed when the grafts are wrapped tightly with PTFE.58. A rigid perivascular polyethylene cuff around the rabbit aorta causes medial, but not intimal, atrophy59. Medial atrophy is also observed in the carotid artery when another carotid artery is used as a cuff, thus proving the foreign body reaction is not required for medial atrophy60. We have recently reproduced these results in baboons; medial, and not intimal, atrophy in the baboon iliac artery can be induced in response to a PTFE wrap. This wrap is designed to limit expansion but not reduce luminal diameter. At 4 days, the tight wrap causes a proportional loss of cells and ECM, while by 28 days there is relatively more loss of ECM61(unpublished data). In both rabbit and baboon the external cuff reduces circumferential strain. Decreased luminal diameter may also contribute to the atrophy process by increasing shear stress.
3. Neointimal and medial regression in an arterialized vein graft
Davies et al reported that wall thickening in vein bypass grafts placed in the arterial circulation of rabbits for 14 days regresses when the grafts are reimplanted into the venous circulation. Both intimal and medial areas decrease and are associated with apoptosis of SMC and a relative increase in collagen. These effects are caused by either a reduction in blood pressure or flow or both62,63.
Regression of in-stent restenosis
Intimal atrophy occurs spontaneously in stented arteries after about 6 months in humans64,65 and pigs66 and after about 2 months in rats67. At these late times in all three species66–68, the neointimal ECM shows a relative loss of versican and a relative gain of collagen compared to earlier times. In addition, the data suggest that collagen is more tightly packed or changes from type III to type I. While human stent neointimas show a loss of SMCs at times greater than 18 months68, these late times have not been studied in animals.
Recently Hong et al conducted an intravascular ultrasound study in humans and at 24 months observed both regression and progression of lesions first observed at 6 months after bare metal stent implantation69. While regression of the intima was observed in 76% of the lesions, progression was demonstrated in 24%. There is no clue as to why lesions regress or progress. However, these results changed the clinical treatment strategy of treatment of asymptomatic in-stent restenosis from deliberate intervention to careful observation and reintervention only in selected cases.
While it is clear that the neointima created by stent-mediated injury undergoes spontaneous regression, there is a lack of agreement on whether the intima of a traumatized artery in the absence of a stent undergoes intimal regression at late times70,71. One rat carotid injury study observed regression and one study did not. In the study in which late regression was observed71 there was also a significant decrease in lumenal area before regression began. This constrictive remodeling might increase shear stress that may in turn mediate regression. This idea is supported by the observations of Zhuang et al56, who studied the effects of multiple rounds of increasing and decreasing blood flow through the normal, uninjured rabbit carotid artery by opening and closing downstream arterio-venous fistulas. They observed that increased blood flow could cause neointimal regression in this arterial model as was observed in the baboon PTFE model.
Regression of cardiovascular hypertrophy in hypertension
Cardiovascular hypertrophy and hyperplasia are common features of hypertension and are important components of target organ damage. The regression of cardiovascular hypertrophy is now considered an important therapeutic objective to reduce the complications of hypertension72,73. In addition to lowering blood pressure, some antihypertensive drugs, such as losartan, enalapril and nifedipine, stimulate SMC apoptosis and reduce DNA synthesis, vascular mass and medial cell number in the thoracic aorta of spontaneously hypertensive rats (SHR), but not in normotensive Wistar Kyoto rats (WKY)74. This regression of medial hypertrophy in response to medication is also observed in hypertensive humans75. In balloon-injured arteries, nifedipine induces SMC apoptosis in established neointimas in both SHR and WKY rats and causes lesion regression76. DeBlois and Hamet have suggested that induction of SMC apoptosis might be an important way to reverse the structural changes in the vasculature caused by hypertension77.
Primary pulmonary hypertension is a disorder characterized by hypertensive vasculopathy, vasoconstriction, and vascular wall remodeling. The histologic findings in idiopathic pulmonary hypertension are obliteration of the lumen of small- and medium-sized pulmonary arteries in association with medial hypertrophy, concentric laminar intimal fibrosis, fibrinoid degeneration, and formation of plexiform lesions and in situ thrombosis78. While the pathogenesis and genetic basis of this disease are poorly understood, it is now clear that mutations in a bone morphogenetic protein receptor predispose to both idiopathic and familial forms of the disease (see review by Said79). Current treatment relies on various pulmonary vasodilators.
The balance of cell proliferation and apoptosis in pulmonary artery SMC maintains the thickness and tissue mass of the arterial walls at an optimal level. If this balance is disturbed such that there is more proliferation or less apoptosis, the arterial wall thickens, narrowing the lumen and ultimately leading to the obliteration of the vessel and to an overall increased pulmonary vascular resistance. Prevention or reversal of constrictive vascular remodeling by inhibiting proliferation and promoting apoptosis in pulmonary artery SMC would be an effective future therapeutic modality.
In the monocrotaline rat model of pulmonary hypertension, inhibition of either elastase, matrix metalloproteinases, or the tyrosine kinase activity of the EGF receptor reduces expression of the SMC survival factor tenascin C, increases SMC apoptosis, and causes regression of the hypertrophied vessel wall by a coordinated loss of cells and ECM80–82.
The structural changes associated with pulmonary hypertension can be reversed through reducing hemodynamic stress83. Of particular interest from the clinical perspective are the results of blockade of the tyrosine kinase activity of the PDGF receptors by Gleevec (imatinib mesylate). In both chronic hypoxia and monocrotaline models in rats and mice, Gleevec reverses the symptoms of pulmonary hypertension84. Based on this work, two case reports showed that Gleevec successfully reverses symptoms of idiopathic pulmonary hypertension in patients refractory to conventional treatments85,86 leading to the suggestion by Patterson et al that selective PDGFR blockade may represent a novel therapy to target vascular remodeling in pulmonary arterial hypertension. Because Gleevec also blocks KIT, LCK, and ABL kinases to the same extent and others to a lessor degree87, further work is required to determine the mechanism of action of this drug.
Regression of intimal hyperplasia in transplant arteriopathy
Transplant vasculopathy is characterized by concentric, heterogeneous proliferative thickening of the intima of the allograft vasculature and is initiated and propagated by both immunological and nonimmunological factors88,89. The main pathobiologic manifestations of the disease are the acquisition of an inflammatory endothelial phenotype, increased SMC proliferation, and defective SMC apoptosis. Bach et al reported that a number of cytoprotective genes, including antiapoptotic Bcl family members, Bcl-2 and Bcl-xL, the heat shock protein heme oxygenase-1, and the zinc finger protein A20, are expressed in endothelium and SMCs of long-term surviving cardiac xenografts devoid of transplant arteriosclerosis90. These investigators proposed a cytoprotective recipe that provides the endothelium with potent anti-inflammatory, anticoagulant, and antiapoptotic potential and maintains the contractile medial SMC phenotype91. While there are reports in animal models showing that PDGFR blockade by Gleevec prevents the development of allograft arteriosclerosis in transplanted hearts92–94, the definitive role of PDGF in the development of transplant vasculopathy is still unknown.
It may be possible to induce regression of transplant vasculopathy. One pharmacologic agent that has shown promise is sirolimus, a natural fermentation product produced by Streptomyces hygroscopicus. The cellular action of sirolimus (rapamycin) is mediated by binding to the FK506 binding protein-12 (FKBP-12)95,96. The resulting sirolimus/FKBP-12 complex inhibits the kinase activity of mammalian target of rapamycin (mTOR), subsequently increasing levels of the cyclin-dependent kinase inhibitor p27Kip196-98 and reducing the activity of multiple kinases associated with mitogen-induced cell proliferation (e.g. p70s6k, cyclin E/CDK-2). This leads to cell cycle arrest at the G1/S transition point96,99–101. Ruygrok et al reported a case of angiographic regression of aggressive cardiac allograft vasculopathy after 1 year of sirolimus treatment started 2 years post-transplantation102. Park et al reported regression of transplant coronary artery disease during chronic low-density lipoprotein-apheresis103. They found a 7.9% increase of mean luminal diameter due to either atherosclerotic regression or vessel remodeling.
Strategies to induce vascular atrophy
The main pathologic features of vessel wall atrophy are loss of SMCs and ECM resulting from decreased SMC growth and matrix synthesis, SMC apoptosis, and ECM lysis. There are several molecular pathways that might be utilized to induce vascular atrophy.
1. Platelet-derived growth factors and receptors (PDGF/ PDGFR)
As discussed above, PDGF is a major regulator of SMC proliferation, migration, and ECM protein synthesis. Tanizawa et al 104 reported PDGF B chain and PDGFR β receptor were present by immunostaining and Ueda et al105 found PDGF A and B chain were present by in situ after PCTA in coronaries. Blockade of PDGFR-β by antibodies inhibits intimal hyperplasia in injured arteries and PTFE grafts in baboons28,106. Although by itself it does not block the induction of intimal thickening, PDGFR-α blockade does cause a decrease in SMC density and number106. This latter effect is consistent with the known survival role of PDGF107. Of greater interest, however, is the observation that concomitant antibody blockade of PDGFR-α and -β induces atrophy of established intima in baboon PTFE grafts under normal flow conditions (figure 2)108. Simultaneous inhibition of cell proliferation and stimulation of cell death by the administration of antibodies to both PDGFR α and β is required to induce atrophy. While a fully humanized version of the chimeric antibody to PDGFR-β had no effect on restenosis in patients undergoing coronary stenting109, it may be that simultaneous blockade of both receptors is required for an effect. This goal may be achieved with Gleevec, a PDGFR kinase inhibitor that appears to reverse pulmonary vascular hypertrophy and pulmonary hypertension and might have a similar effect on intima in vascular reconstructions.
Figure 2.

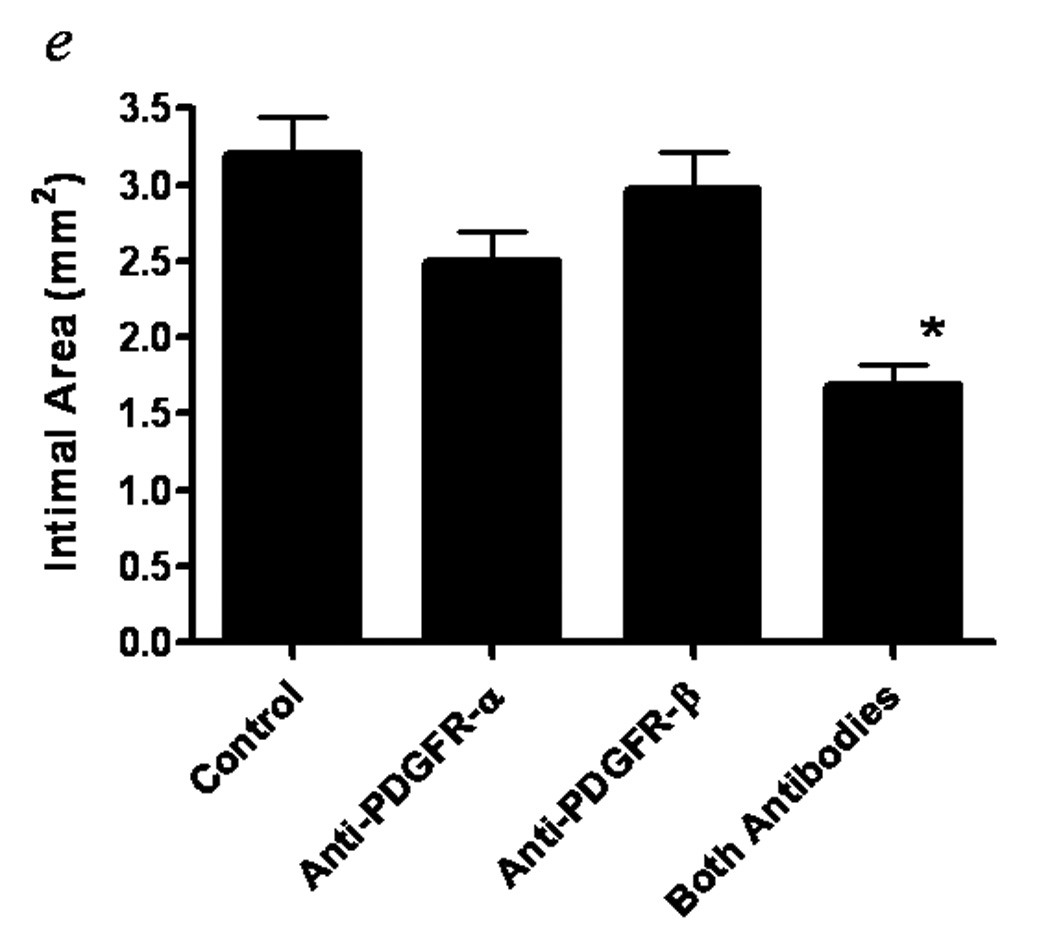
Histologic cross sections of normal flow PTFE grafts at 2 weeks after initiation of treatment with vehicle control (a), blocking antibodies to PDGFR α (b), blocking antibodies to PDGFR β (c), or blocking antibodies to both PDGFR α and β (d). (hemotoxylin-eosin, 16X). Intimal areas of 4–7 animals per group are presented as the mean ± standard error (e). Reproduced from Engelsbe et al93 by permission from Elsevier Publishing.
2. Bone morphogenic protein 4 (BMP4) and its inhibitor noggin
BMPs are members of the transforming growth factor (TGF)-β superfamily that regulate cell proliferation, migration, differentiation, and apoptosis. High shear stress induces BMP4 mRNA and protein expression in the PTFE graft neointima in baboons, while noggin (a BMP inhibitor) is decreased. BMP4 inhibits SMC proliferation in vitro, an effect blocked by noggin. Thus the increase in BMP4 coupled with a decrease noggin may contribute to graft neointimal atrophy by inhibiting SMC proliferation and increasing SMC death52.
3. A20
A20 is a zinc finger protein induced by tumor necrosis factor-α, interleukin-1 or CD-40 cross-linking in a variety of cell types110. A20 prevents intimal hyperplasia when transfected acutely, but induces regression when transfected in established intimal lesions of the injured rat carotid artery111. Expression of A20 in medial SMC prevents neointima formation by shutting down inflammatory and proliferative responses of SMC via inhibition of NF-κB and increased expression of the cell cycle dependent kinase inhibitors p21waf1 and p27kip1. SMCs are also sensitized by A20 to undergo apoptosis in response to cytokines and Fas/FasL via a NO-dependent mechanism. This causes regression of established neointimal lesions. In endothelial cells, A20 is anti-inflammatory by inhibiting NFκB like in SMCs, but unlike SMC it is antiapoptotic via inhibition of the proteolytic activation of caspase 8112. Therefore, A20 has protective effects for endothelial cells and proapoptotic effects for SMCs making it a good candidate for inducing vascular atrophy while minimizing endothelial damage91.
4. Others
Recently we performed microarray analysis of our two vascular atrophy models in baboons, namely the PTFE graft, high flow-induced intimal atrophy model and the PTFE wrap-induced medial atrophy model. Of ~28,000 genes we found several genes commonly up- or down-regulated in both models. Upregulated genes included extracellular matrix degrading factors and possible growth regulatory factors. Downregulated genes included survival factors and matrix genes113. These molecules may have important roles in vascular atrophy and may lead to pharmacological treatments for established lesions.
Conclusion
After vascular reconstruction, luminal narrowing is in part caused by intimal thickening, the consequence of endothelial injury and inflammation, smooth muscle cell hyperplasia, and extracellular matrix accumulation. It may be possible to induce these lesions to shrink (figure 3). This novel approach to the treatment of restenosis is supported by animal experiments and a few clinical observations demonstrating vascular atrophy in response to drugs such as Gleevec. A potential limitation to this approach might be the formation of aneurysms. For example, it is known that venous or arterial aneurysms often form at arterio-venous fistulas and drug-eluting stents may cause aneurysms114. It is clear that a means for targeted delivery of limited duration would need to be developed. Additional studies are needed to determine whether this therapy will be broadly applicable.
Figure 3.
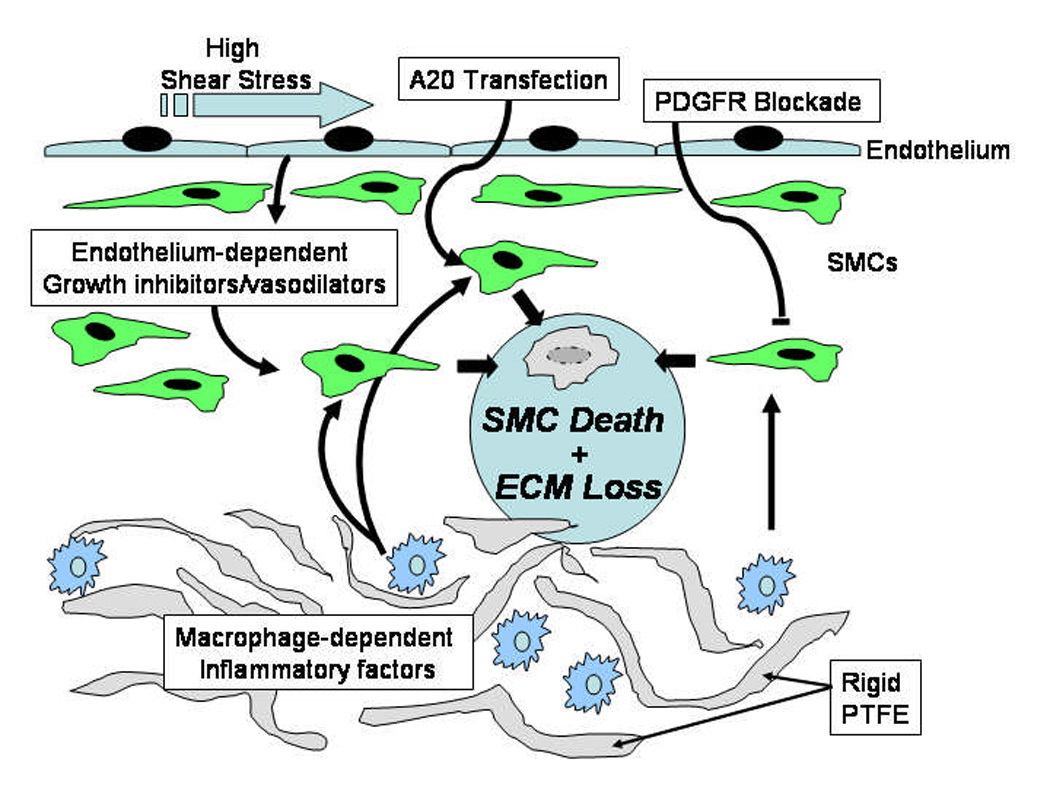
Diagram of possible pathways of vascular regression. Either high shear stress, transfection of A20, or blockade of PDGFR in combination with inflammatory factors could cause regression of neointima.
Acknowledgements
Supported by grants RO1 HL30946 and RR00166 from the NIH, USPHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Seung-Kee Min, Department of Surgery, Gachon University of Medicine and Science, Incheon, Korea.
Richard D. Kenagy, Department of Surgery, University of Washington, Seattle, Washington, USA.
Alexander W. Clowes, Department of Surgery, University of Washington, Seattle, Washington, USA.
References
- 1.Bauters C, Isner JM. The biology of restenosis. Prog Cardiovasc Dis. 1997;40:107–116. doi: 10.1016/s0033-0620(97)80003-5. [DOI] [PubMed] [Google Scholar]
- 2.Kester M, Waybill C, Kozak M. New strategies to prevent restenosis. Am J Cardiovasc Drugs. 2001;1:77–83. doi: 10.2165/00129784-200101020-00001. [DOI] [PubMed] [Google Scholar]
- 3.Finn AV, Nakazawa G, Joner M, Kolodgie FD, Mont EK, Gold HK, Virmani R. Vascular responses to drug eluting stents: importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27:1500–1510. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- 4.Mitra AK, Gangahar DM, Agrawal DK. Cellular, molecular and immunological mechanisms in the pathophysiology of vein graft intimal hyperplasia. Immunol Cell Biol. 2006;84:115–124. doi: 10.1111/j.1440-1711.2005.01407.x. [DOI] [PubMed] [Google Scholar]
- 5.Allaire E, Clowes AW. Endothelial cell injury in cardiovascular surgery: The intimal hyperplastic response. Ann Thorac Surg. 1997;63:582–591. doi: 10.1016/s0003-4975(96)01045-4. [DOI] [PubMed] [Google Scholar]
- 6.Fingerle J, Au YPT, Clowes AW, Reidy C. Intimal lesion formation in rat carotid arteries after endothelial denudation in absence of medial injury. Arteriosclerosis. 1990;10:1082–1087. doi: 10.1161/01.atv.10.6.1082. [DOI] [PubMed] [Google Scholar]
- 7.Beohar N, Flaherty JD, Davidson CJ, Maynard RC, Robbins JD, Shah AP, Choi JW, MacDonald LA, Jorgensen JP, Pinto JV, Chandra S, Klaus HM, Wang NC, Harris KR, Decker R, Bonow RO. Antirestenotic effects of a locally delivered caspase inhibitor in a balloon injury model. Circulation. 2004;109:108–113. doi: 10.1161/01.CIR.0000105724.30980.CD. [DOI] [PubMed] [Google Scholar]
- 8.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 9.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury.I.Smooth muscle growth in the absence of endothelium. Lab Invest. 1983;49:327–333. [PubMed] [Google Scholar]
- 10.Lindner V, Lappi DA, Baird A, Majack RA, Reidy MA. Role of basic fibroblast growth factor in vascular lesion formation. Circ Res. 1991;68:106–113. doi: 10.1161/01.res.68.1.106. [DOI] [PubMed] [Google Scholar]
- 11.Olson NE, Chao S, Lindner V, Reidy MA. Intimal smooth muscle cell proliferation after balloon catheter injury: The role of basic fibroblast growth factor. Am J Pathol. 1992;140:1017–1023. [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis CD, Olson NE, Raines EW, Reidy MA, Jackson CL. Modulation of smooth muscle proliferation in rat carotid artery by platelet-derived smooth muscle proliferation in rat carotid artery by platelet-derived mediators and fibroblast growth factor-2. Platelets. 2001;12:352–358. doi: 10.1080/09537100120071013. [DOI] [PubMed] [Google Scholar]
- 13.Englesbe MJ, Davies MG, Hawkins SM, Hsieh pch, Daum G, Kenagy RD, Clowes AW. Arterial injury repair in non-human primates - The role of platelet-derived growth factor receptor-β. J Surg Res. 2004;119:80–84. doi: 10.1016/j.jss.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Hayry P, Myllarniemi M, Aavik E, Alatalo S, Aho P, Yilmaz S, Raisanen-Sokolowski A, Cozzone G, Jameson BA, Baserga R. Stabile D-peptide analog of insulin-like growth factor-1 inhibits smooth muscle cell proliferation after carotid ballooning injury in the rat. FASEB J. 1995;9:1336–1344. doi: 10.1096/fasebj.9.13.7557024. [DOI] [PubMed] [Google Scholar]
- 15.Zhu BH, Zhao GS, Witte DP, Hui DY, Fagin JA. Targeted overexpression of IGF-I in smooth muscle cells of transgenic mice enhances neointimal formation through increased proliferation and cell migration after intraarterial injury. Endocrinology. 2001;142:3598–3606. doi: 10.1210/endo.142.8.8331. [DOI] [PubMed] [Google Scholar]
- 16.Gallo R, Padurean A, Toschi V, Bichler J, Fallon JT. Prolonged thrombin inhibition reduces restenosis after balloon angioplasty in porcine coronary arteries. Circulation. 1998;97:588. doi: 10.1161/01.cir.97.6.581. [DOI] [PubMed] [Google Scholar]
- 17.Wolf YG, Rasmussen LM, Ruoslahti E. Antibodies against transforming growth factor-β1 suppress intimal hyperplasia in a rat model. J Clin Invest. 1994;93:1172–1178. doi: 10.1172/JCI117070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rectenwald JE, Moldawer LL, Huber TS, Seeger JM, Ozaki CK. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102:1697–1702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, Kishida K, Komuro R, Ouchi N, Kihara S, Nagai R, Funahashi T, Matsuzawa Y. Role of adiponectin in preventing vascular stenosis. The missing link of adipo-vascular axis. J Biol Chem. 2002;277:37487–37491. doi: 10.1074/jbc.M206083200. [DOI] [PubMed] [Google Scholar]
- 20.Tran PK, Tran-Lundmark K, Soininen R, Tryggvason K, Thyberg J, Hedin U. Increased intimal hyperplasia and smooth muscle cell proliferation in transgenic mice with heparan sulfate-deficient perlecan. Circ Res. 2004;94:550–558. doi: 10.1161/01.RES.0000117772.86853.34. [DOI] [PubMed] [Google Scholar]
- 21.Fukai N, Kenagy RD, Daum G, Li Q, Parks WC, Clowes AW. Syndecan-1 inhibits the development of intimal thickening after vascular injury. FASEB J. 2007;21(6 part II):A746. Ref Type: Abstract. [Google Scholar]
- 22.Feldman LJ, Aguirre L, Ziol M, Bridou JP, Nevo N, Michel JB, Steg PG. Interleukin-10 inhibits intimal hyperplasia after angioplasty or stent implantation in hypercholesterolemic rabbits. Circulation. 2000;101:908–916. doi: 10.1161/01.cir.101.8.908. [DOI] [PubMed] [Google Scholar]
- 23.Rauma-Pinola T, Paakko P, Ilves M, Serpi R, Romppanen H, Vuolteenaho O, Ruskoaho H, Hautala T. Adrenomedullin gene transfer induces neointimal apoptosis and inhibits neointimal hyperplasia in injured rat artery. J Gene Med. 2006 doi: 10.1002/jgm.865. [DOI] [PubMed] [Google Scholar]
- 24.Aavik E, Luoto NM, Petrov L, Aavik S, Patel YC, Hayry P. Elimination of vascular fibrointimal hyperplasia by somatostatin receptor 1.4 selective agonist. FASEB Journal. 2002;16:NIL202–NIL223. doi: 10.1096/fj.01-0272fje. [DOI] [PubMed] [Google Scholar]
- 25.Qin F, Dardik H, Pangilinan A, Robinson J, Chuy J, Wengerter K. Remodeling and suppression of intimal hyperplasia of vascular grafts with a distal arteriovenous fistula in a rat model. J Vasc Surg. 2001;34:701–706. doi: 10.1067/mva.2001.116804. [DOI] [PubMed] [Google Scholar]
- 26.Kohler TR, Jawien A. Flow affects development of intimal hyperplasia after arterial injury in rats. Arterioscler Thromb. 1992;12:963–971. doi: 10.1161/01.atv.12.8.963. [DOI] [PubMed] [Google Scholar]
- 27.Jackson CL, Raines EW, Ross R, Reidy MA. Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler Thromb. 1993;13:1218–1226. doi: 10.1161/01.atv.13.8.1218. [DOI] [PubMed] [Google Scholar]
- 28.Englesbe MJ, Davies MG, Hawkins SM, Hsieh pch, Daum G, Kenagy RD, Clowes AW. Arterial injury repair in nonhuman primates - The role of PDGF receptor-β. J Surg Res. 2004;119:80–84. doi: 10.1016/j.jss.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Majesky M, Reidy M, Benditt EP, Schwartz S. Expression of platelet-derived growth factor (PDGF) A- and B-chain gene in smooth muscle during repair of arterial injury. J Mole Cell Cardiol. 1987;19 suppl.IV:S3. [Google Scholar]
- 30.Indolfi C, Torella D, Cavuto L, Davalli AM, Coppola C, Esposito G, Carriero MV, Rapacciuolo A, Di Lorenzo E, Stabile E, Perrino C, Chieffo A, Pardo F, Chiariello M. Effects of balloon injury on neointimal hyperplasia in streptozotocin-induced diabetes and in hyperinsulinemic nondiabetic pancreatic islet-transplanted rats. Circulation. 2001;103:2980–2986. doi: 10.1161/01.cir.103.24.2980. [DOI] [PubMed] [Google Scholar]
- 31.Roqué M, Reis ED, Fuster V, Padurean A, Fallon JT, Taubman MB, Chesebro JH, Badimon JJ. Inhibition of tissue factor reduces thrombus formation and intimal hyperplasia after porcine coronary angioplasty. J Am Coll Cardiol. 2000;36:2303–2310. doi: 10.1016/s0735-1097(00)01018-4. [DOI] [PubMed] [Google Scholar]
- 32.Jackson CL, Reidy MA. Basic fibroblast growth factor: Its role in the control of smooth muscle cell migration. Am J Pathol. 1993;143:1024–1031. [PMC free article] [PubMed] [Google Scholar]
- 33.Zalewski A, Shi Y, Johnson AG. Diverse origin of intimal cells: smooth muscle cells, myofibroblasts, fibroblasts, and beyond? Circ Res. 2002;91:652–655. doi: 10.1161/01.res.0000038996.97287.9a. [DOI] [PubMed] [Google Scholar]
- 34.Yokote K, Take A, Nakaseko C, Kobayashi K, Fujimoto M, Kawamura H, Maezawa Y, Nishimura M, Mori S, Saito Y. Bone marrow-derived vascular cells in response to injury. J Atheroscler Thromb. 2003;10:205–210. doi: 10.5551/jat.10.205. [DOI] [PubMed] [Google Scholar]
- 35.Nikkari ST, Järveläinen HT, Wight TN, Ferguson M, Clowes AW. Smooth muscle cell expression of extracellular matrix genes after arterial injury. Am J Pathol. 1994;144:1348–1356. [PMC free article] [PubMed] [Google Scholar]
- 36.Koyama H, Reidy MA. Reinjury of arterial lesions induces intimal smooth muscle cell replication that is not controlled by fibroblast growth factor 2. Circulation Research. 1997;80:408–417. [PubMed] [Google Scholar]
- 37.Koyama H, Reidy MA. Expression of extracellular matrix proteins accompanies lesion growth in a model of intimal reinjury. Circulation Research. 1998;82:988–995. doi: 10.1161/01.res.82.9.988. [DOI] [PubMed] [Google Scholar]
- 38.Hanke H, Strohschneider T, Oberhoff M, Betz E, Karsch KR. Time course of smooth muscle cell proliferation in the intima and media of arteries following experimental angioplasty. Circ Res. 1990;67:651–659. doi: 10.1161/01.res.67.3.651. [DOI] [PubMed] [Google Scholar]
- 39.Dimmeler S, Zeiher AM. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res. 2000;87:434–439. doi: 10.1161/01.res.87.6.434. [DOI] [PubMed] [Google Scholar]
- 40.Im E, Kazlauskas A. New insights regarding vessel regression. Cell Cycle. 2006;5:2057–2059. doi: 10.4161/cc.5.18.3210. [DOI] [PubMed] [Google Scholar]
- 41.Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science. 1986;231:405–407. doi: 10.1126/science.3941904. [DOI] [PubMed] [Google Scholar]
- 42.Cho A, Courtman DW, Langille BL. Apoptosis (programmed cell death) in arteries of the neonatal lamb. Circ Res. 1995;76:168–175. doi: 10.1161/01.res.76.2.168. [DOI] [PubMed] [Google Scholar]
- 43.Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, Kurup S, Glass DA, Patel MS, Shu W, Morrisey EE, McMahon AP, Karsenty G, Lang RA. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 45.Chrétien M, Noria SF, Jackson M, Langille BL. Vascular adaptations to altered blood flow. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and thrombosis: Basic principles and clinical practice. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 667–678. [Google Scholar]
- 46.Im E, Kazlauskas A. Regulating angiogenesis at the level of PtdIns-4,5-P2. EMBO J. 2006;25:2075–2082. doi: 10.1038/sj.emboj.7601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Im E, Venkatakrishnan A, Kazlauskas A. Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol Biol Cell. 2005;16:3488–3500. doi: 10.1091/mbc.E04-11-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255:538–561. doi: 10.1111/j.1365-2796.2003.01297.x. [DOI] [PubMed] [Google Scholar]
- 49.Clowes AW, Kirkman TR, Reidy MA. Mechanisms of arterial graft healing. Rapid transmural capillary ingrowth provides a source of intimal endothelium and smooth muscle in porous PTFE prostheses. Am J Pathol. 1986;123:220–230. [PMC free article] [PubMed] [Google Scholar]
- 50.Mattsson EJR, Kohler TR, Vergel SM, Clowes AW. Increased blood flow induces regression of intimal hyperplasia. Arterioscler Thromb Vasc Biol. 1997;17:2245–2249. doi: 10.1161/01.atv.17.10.2245. [DOI] [PubMed] [Google Scholar]
- 51.Berceli SA, Davies MG, Kenagy RD, Clowes AW. Flow-induced neointimal regression in baboon polytetrafluoroethylene grafts is associated with decreased cell proliferation and increased apoptosis. J Vasc Surg. 2002;36:1248–1255. doi: 10.1067/mva.2002.128295. [DOI] [PubMed] [Google Scholar]
- 52.Hsieh pch, Kenagy RD, Mulvihill ER, Jeanette JP, Wang X, Chang CMC, Yao ZH, Ruzzo WL, Justice S, Hudkins KL, Alpers CE, Berceli S, Clowes AW. Bone morphogenetic protein 4: Potential regulator of shear stress-induced graft neointimal atrophy. J Vasc Surg. 2006;43:150–158. doi: 10.1016/j.jvs.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kenagy RD, Fischer JW, Lara S, Sandy JD, Clowes AW, Wight TN. Accumulation and Loss of Extracellular Matrix During Shear Stress-mediated Intimal Growth and Regression in Baboon Vascular Grafts. J Histochem Cytochem. 2005;53:131–140. doi: 10.1369/jhc.4A6493.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenagy RD, Fischer JW, Davies MG, Berceli SA, Hawkins SM, Wight TN, Clowes AW. Increased plasmin and serine proteinase activity during flow-induced intimal atrophy in baboon PTFE grafts. Arterioscler Thromb Vasc Biol. 2002;22:400–404. doi: 10.1161/hq0302.105376. [DOI] [PubMed] [Google Scholar]
- 55.Quan A, Ward ME, Kulandavelu S, Adamson SL, Langille BL. Endothelium-independent flow-induced dilation in the mouse carotid artery. J Vasc Res. 2006;43:383–391. doi: 10.1159/000094414. [DOI] [PubMed] [Google Scholar]
- 56.Zhuang Y-J, Singh TM, Zarins CK, Masuda H. Sequential increases and decreases in blood flow stimulates progressive intimal thickening. Eur J Vasc Endovasc Surg. 1998;16:301–310. doi: 10.1016/s1078-5884(98)80049-x. [DOI] [PubMed] [Google Scholar]
- 57.Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H, Schafer AI, Durante W. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood. 2002;99:4443–4448. doi: 10.1182/blood.v99.12.4443. [DOI] [PubMed] [Google Scholar]
- 58.Kohler TR, Kirkman TR, Clowes AW. The effect of rigid external support on vein graft adaptation to the arterial circulation. J Vasc Surg. 1989;9:277–285. [PubMed] [Google Scholar]
- 59.Courtman DW, Cho A, Langille L, Wilson GJ. Eliminating arterial pulsatile strain by external banding induces medial but not neointimal atrophy and apoptosis in the rabbit. Am J Pathol. 1998;153:1723–1729. doi: 10.1016/S0002-9440(10)65687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayer IM, Adamson SL, Langille BL. Atrophic remodeling of the artery-cuffed artery. Arterioscler Thromb Vasc Biol. 1999;19:1499–1505. doi: 10.1161/01.atv.19.6.1499. [DOI] [PubMed] [Google Scholar]
- 61.Kenagy RD, Jeanette JP, Min S-K, Clowes AW. Effects of Wall Tension and Blood Flow on Cuff-Induced Atrophy of the Baboon Iliac Artery. Arterioscler.Thromb.Vasc Biol. 2006;26(e77) [Google Scholar]
- 62.Davies MG, Klyachkin ML, Dalen H, Svendsen E, Hagen P-O. Regression of intimal hyperplasia with restoration of endothelium-dependent relaxing factor-mediated relaxation in experimental vein grafts. Surgery. 1993;114:258–271. [PubMed] [Google Scholar]
- 63.Davies MG, Fulton GJ, Svendsen E, Hagen PO. Time course of the regression of intimal hyperplasia in experimental vein grafts. Cardiovasc Pathol. 1999;8:161–168. doi: 10.1016/s1054-8807(98)00029-5. [DOI] [PubMed] [Google Scholar]
- 64.Kuroda N, Kobayashi Y, Nameki M, Kuriyama N, Kinoshita T, Okuno T, Yamamoto Y, Komiyama N, Masuda Y. Intimal hyperplasia regression from 6 to 12 months after stenting. Am J Cardiol. 2002;89:869–872. doi: 10.1016/s0002-9149(02)02205-1. [DOI] [PubMed] [Google Scholar]
- 65.Asakura M, Ueda Y, Nanto S, Hirayama A, Adachi T, Kitakaze M, Hori M, Kodama K. Remodeling of in-stent neointima, which became thinner and transparent over 3 years - Serial angiographic and angioscopic follow-up. Circulation. 1998;97:2003–2006. doi: 10.1161/01.cir.97.20.2003. [DOI] [PubMed] [Google Scholar]
- 66.Kim WH, Hong MK, Virmani R, Kornowski R, Jones R, Leon MB. Histopathologic analysis of in-stent neointimal regression in a porcine coronary model. Coron Artery Dis. 2000;11:273–277. doi: 10.1097/00019501-200005000-00011. [DOI] [PubMed] [Google Scholar]
- 67.Finn AV, Gold HK, Tang A, Weber DK, Wight TN, Clermont A, Virmani R, Kolodgie FD. A novel rat model of carotid artery stenting for the understanding of restenosis in metabolic diseases. J Vasc Res. 2002;39:414–425. doi: 10.1159/000064518. [DOI] [PubMed] [Google Scholar]
- 68.Farb A, Kolodgie FD, Hwang JY, Burke AP, Tefera K, Weber DK, Wight TN, Virmani R. Extracellular matrix changes in stented human coronary arteries. Circulation. 2004;110:940–947. doi: 10.1161/01.CIR.0000139337.56084.30. [DOI] [PubMed] [Google Scholar]
- 69.Hong MK, Lee CW, Kim YH, Lee BK, Kim MK, Yang TH, Song JM, Han KH, Kang DH, Song JK, Kim JJ, Park SW, Park SJ. Two-year follow-up intravascular ultrasound analysis after bare metal stent implantation in 120 lesions. Catheter Cardiovasc Interv. 2005;65:247–253. doi: 10.1002/ccd.20358. [DOI] [PubMed] [Google Scholar]
- 70.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 71.Nuthakki VK, Fleser PS, Malinzak LE, Seymour ML, Callahan RE, Bendick PJ, Zelenock GB, Shanley CJ. Lysyl oxidase expression in a rat model of arterial balloon injury. J Vasc Surg. 2004;40:123–129. doi: 10.1016/j.jvs.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 72.Fleischmann EH, Schmieder RE. Are all antihypertensive drug classes equal in reducing left ventricular hypertrophy? Curr Cardiol Rep. 2002;4:474–478. doi: 10.1007/s11886-002-0109-2. [DOI] [PubMed] [Google Scholar]
- 73.DeBlois D, Tea BS, Beaudry D, Hamet P. Regulation of therapeutic apoptosis: a potential target in controlling hypertensive organ damage. Can J Physiol Pharmacol. 2005;83:29–41. doi: 10.1139/y05-001. [DOI] [PubMed] [Google Scholar]
- 74.DeBlois D, Tea BS, Than VD, Tremblay J, Hamet P. Smooth muscle apoptosis during vascular regression in spontaneously hypertensive rats. Hypertension. 1997;29:340–349. doi: 10.1161/01.hyp.29.1.340. [DOI] [PubMed] [Google Scholar]
- 75.Schiffrin EL, Deng LY. Structure and function of resistance arteries of hypertensive patients treated with a beta-blocker or a calcium channel antagonist. J Hypertens. 1996;14:1247–1255. doi: 10.1097/00004872-199610000-00014. [DOI] [PubMed] [Google Scholar]
- 76.Lemay J, Tea BS, Hamet P, DeBlois D. Regression of neointimal lesionsin the carotid artery of nifedipine-treated SHR and WKY rats: Possible role of apoptosis. J Vasc Res. 2001;38:462–470. doi: 10.1159/000051079. [DOI] [PubMed] [Google Scholar]
- 77.DeBlois D, Orlov SN, Hamet P. Apoptosis in cardiovascular remodeling--effect of medication. Cardiovasc Drugs Ther. 2001;15:539–545. doi: 10.1023/a:1013723922582. [DOI] [PubMed] [Google Scholar]
- 78.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Said SI. Mediators and modulators of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;291:L547–L558. doi: 10.1152/ajplung.00546.2005. [DOI] [PubMed] [Google Scholar]
- 80.Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105:21–34. doi: 10.1172/JCI6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 82.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 83.O'Blenes SB, Fischer S, McIntyre B, Keshavjee S, Rabinovitch M. Hemodynamic unloading leads to regression of pulmonary vascular disease in rats. J Thorac Cardiovasc Surg. 2001;121:279–289. doi: 10.1067/mtc.2001.111657. [DOI] [PubMed] [Google Scholar]
- 84.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 86.Patterson KC, Weissmann A, Ahmadi T, Farber HW. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann Intern Med. 2006;145:152–153. doi: 10.7326/0003-4819-145-2-200607180-00020. [DOI] [PubMed] [Google Scholar]
- 87.Fabian MA, Biggs WH, III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 88.Mehra MR. Contemporary concepts in prevention and treatment of cardiac allograft vasculopathy. Am J Transplant. 2006;6:1248–1256. doi: 10.1111/j.1600-6143.2006.01314.x. [DOI] [PubMed] [Google Scholar]
- 89.Rahmani M, Cruz RP, Granville DJ, McManus BM. Allograft vasculopathy versus atherosclerosis. Circ Res. 2006;99:801–815. doi: 10.1161/01.RES.0000246086.93555.f3. [DOI] [PubMed] [Google Scholar]
- 90.Bach FH, Ferran C, Hechenleitner P, Mark W, Koyamada N, Miyatake T, Winkler H, Badrichani A, Candinas D, Hancock WW. Accommodation of vascularized xenografts: expression of "protective genes" by donor endothelial cells in a host Th2 cytokine environment. Nat Med. 1997;3:196–204. doi: 10.1038/nm0297-196. [DOI] [PubMed] [Google Scholar]
- 91.Ferran C. Protective genes in the vessel wall: Modulators of graft survival and function. Transplantation. 2006;82:S36–S40. doi: 10.1097/01.tp.0000231445.62162.d5. [DOI] [PubMed] [Google Scholar]
- 92.Sihvola RK, Tikkanen JM, Krebs R, Aaltola EM, Buchdunger E, Laitinen O, Koskinen PK, Lemström KB. Platelet-derived growth factor receptor inhibition reduces allograft arteriosclerosis of heart and aorta in cholesterol-fed rabbits. Transplantation. 2003;75:334–339. doi: 10.1097/01.TP.0000045056.82561.0F. [DOI] [PubMed] [Google Scholar]
- 93.Nykanen AI, Krebs R, Tikkanen JM, Raisky O, Sihvola R, Wood J, Koskinen PK, Lemstrom KB. Combined vascular endothelial growth factor and platelet-derived growth factor inhibition in rat cardiac allografts: beneficial effects on inflammation and smooth muscle cell proliferation. Transplantation. 2005;79:182–189. doi: 10.1097/01.tp.0000147199.60464.f9. [DOI] [PubMed] [Google Scholar]
- 94.Savikko J, Taskinen E, von Willebrand E. Chronic allograft nephropathy is prevented by inhibition of platelet-derived growth factor receptor: tyrosine kinase inhibitors as a potential therapy. Transplantation. 2003;75:1147–1153. doi: 10.1097/01.TP.0000062836.93496.CE. [DOI] [PubMed] [Google Scholar]
- 95.Koltin Y, Faucette L, Bergsma DJ, Levy MA, Cafferkey R, Koser PL, Johnson RK, Livi GP. Rapamycin sensitivity in Saccharomyces cerevisiae is mediated by a peptidyl-prolyl cis-trans isomerase related to human FK506-binding protein. Mol Cell Biol. 1991;11:1718–1723. doi: 10.1128/mcb.11.3.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marx SO, Jayaraman T, Go LO, Marks AR. Rapamycin-FKBP inhibits cell cycle regulators of proliferation in vascular smooth muscle cells. Circ Res. 1995;76:412–417. doi: 10.1161/01.res.76.3.412. [DOI] [PubMed] [Google Scholar]
- 97.Luo Y, Marx SO, Kiyokawa H, Koff A, Massague J, Marks AR. Rapamycin resistance tied to defective regulation of p27Kip1. Mol Cell Biol. 1996;16:6744–6751. doi: 10.1128/mcb.16.12.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, Massague J, Crabtree GR, Roberts JM. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 99.Marks AR. Sirolimus for the prevention of in-stent restenosis in a coronary artery. New England Journal of Medicine. 2003;349:1307–1309. doi: 10.1056/NEJMp038141. [DOI] [PubMed] [Google Scholar]
- 100.Parry TJ, Brosius R, Thyagarajan R, Carter D, Argentieri D, Falotico R, Siekierka J. Drug-eluting stents: Sirolimus and paclitaxel differentially affect cultured cells and injured arteries. Eur J Pharmacol. 2005;524:19–29. doi: 10.1016/j.ejphar.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 101.Gustafsson F, Ross HJ. Proliferation signal inhibitors in cardiac transplantation. Curr Opin Cardiol. 2007;22:111–116. doi: 10.1097/HCO.0b013e328012545e. [DOI] [PubMed] [Google Scholar]
- 102.Ruygrok PN, Webber B, Faddy S, Muller DW, Keogh A. Angiographic regression of cardiac allograft vasculopathy after introducing sirolimus immunosuppression. J Heart Lung Transplant. 2003;22:1276–1279. doi: 10.1016/s1053-2498(02)01239-1. [DOI] [PubMed] [Google Scholar]
- 103.Park JW, Merz M, Braun P. Regression of transplant coronary artery disease during chronic low-density lipoprotein-apheresis. J Heart Lung Transplant. 1997;16:290–297. [PubMed] [Google Scholar]
- 104.Tanizawa S, Ueda M, Van der Loos CM, Van Der Wal AC, Becker AE. Expression of platelet derived growth factor B chain and β receptor in human coronary arteries after percutaneous transluminal coronary angioplasty: An immunohistochemical study. Br Heart J. 1996;75:549–556. doi: 10.1136/hrt.75.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ueda M, Becker AE, Kasayuki N, Kojima A, Morita Y, Tanaka S. In situ detection of platelet-derived growth factor-A and -B chain mRNA inhuman coronary arteries after percutaneous transluminal coronary angioplasty. Am J Pathol. 1996;149:831–843. [PMC free article] [PubMed] [Google Scholar]
- 106.Davies MG, Owens EL, Mason DP, Lea H, Tran PK, Vergel S, Hawkins SA, Hart CE, Clowes AW. Effect of platelet-derived growth factor receptor-α and -β blockade on flow-induced neointimal formation in endothelialized baboon vascular grafts. Circulation Research. 2000;86:779–786. doi: 10.1161/01.res.86.7.779. [DOI] [PubMed] [Google Scholar]
- 107.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Englesbe MJ, Hawkins S, Hsieh pch, Davies MG, Daum G, Kenagy RD, Clowes AW. Concomitant Blockade of PDGF Receptors –α and –β Induces Intimal Atrophy in Baboon PTFE Grafts. J Vasc Surg. 2004;39:440–446. doi: 10.1016/j.jvs.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 109.Serruys PW, Heyndrickx GR, Patel J, Cummins PA, Kleijne JA, Clowes AW. Effect of an anti-PDGF-beta-receptor-blocking antibody on restenosis in patients undergoing elective stent placement. Int J Cardiovasc Intervent. 2003;5:214–222. doi: 10.1080/14628840310017177. [DOI] [PubMed] [Google Scholar]
- 110.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60:1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 111.Patel VI, Daniel S, Longo CR, Shrikhande GV, Scali ST, Czismadia E, Groft CM, Shukri T, Motley-Dore C, Ramsey HE, Fisher MD, Grey ST, Arvelo MB, Ferran C. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. FASEB J. 2006;20:1418–1430. doi: 10.1096/fj.05-4981com. [DOI] [PubMed] [Google Scholar]
- 112.Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, Mahiou J, Sun DW, Mottley C, Grey ST, Ferran C. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104:2376–2384. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 113.Min S-K, Defawe O, Kenagy RD, Mulvihill ER, Clowes AW. ADAMTS4, tissue plasminogen activator, and hyaluronidase 2 are upregulated while GAS6 is downregulated in two baboon vascular atrophy models. FASEB J. 2007;21(6 part II):A1128. [Google Scholar]
- 114.Bavry AA, Chiu JH, Jefferson BK, Karha J, Bhatt DL, Ellis SG, Whitlow PL. Development of coronary aneurysm after drug-eluting stent implantation. Ann Intern Med. 2007;146:230–232. doi: 10.7326/0003-4819-146-3-200702060-00146. [DOI] [PubMed] [Google Scholar]