

Abstract
Clinical and basic science data support an integral role of calcitonin gene-related peptide (CGRP) in migraine pathology. Following trigeminal nerve activation, afferent release of CGRP causes vasodilation while efferent release leads to pain. Although CGRP can also be secreted from cell bodies of trigeminal neurons located within the ganglion, the function of CGRP released in the ganglion is poorly understood. Initially, we showed that SNAP-25, a protein required for CGRP release, was localized in cell bodies of trigeminal ganglia neurons. We also found that satellite glial cells in the ganglia express the CGRP1 receptor protein RAMP1. To determine whether CGRP could directly activate glial cells, primary cultures of rat trigeminal ganglia were utilized to study the effects of CGRP on glial nitric oxide (NO) synthesis and release. Under our culture conditions, >95% of the cells expressed glial fibrillary acidic protein and RAMP1. While weak iNOS staining was observed in glia under basal conditions, CGRP treatment greatly increased glial iNOS expression and NO release. This stimulatory effect was blocked by the CGRP1 receptor antagonist, CGRP8–37 peptide. Treatment of glial cultures with forskolin or cAMP also increased iNOS expression and stimulated NO release to levels similar to CGRP. To our knowledge, this is the first evidence that activation of CGRP1 receptors regulates glial iNOS and NO release. We propose that following trigeminal nerve activation, CGRP secretion from neuronal cell bodies activates satellite glial cells that release NO and initiate inflammatory events in the ganglia that contribute to peripheral sensitization in migraine.
Keywords: CGRP, NO, iNOS, Glia, Trigeminal, RAMP1
1. Introduction
Calcitonin gene-related peptide (CGRP) is a 37-amino acid regulatory neuropeptide that is generated by alternative processing of the calcitonin gene (Amara et al., 1982; Rosenfeld et al., 1983). It is one of the most abundant peptides in nerve tissue and is widely expressed in both peripheral and central neurons (van Rossum et al., 1997; Wimalawansa, 1996). CGRP levels in serum, cerebrospinal fluid, and saliva have been reported to be elevated during migraine (Bellamy et al., 2006b; Goadsby and Edvinsson, 1993, 1994), a painful neurovascular disease that afflicts 12% of adults in the United States (Ferrari, 1998; Lipton et al., 2001). Further evidence that supports an important role of CGRP in migraine comes from clinical studies where CGRP infusion was shown to cause migraine in susceptible individuals (Lassen et al., 2002), a CGRP receptor antagonist was reported effective in aborting migraine attacks (Olesen et al., 2004), and treatment with the anti-migraine drugs known as triptans, lower CGRP levels to near normal levels coincident with relief of migraine symptoms (Edvinsson and Goadsby, 1994; Goadsby and Edvinsson, 1991). CGRP is thought to play an important role in the underlying pathology of migraine due to its ability to regulate cerebral blood flow, mediate vasodilation of meningeal vessels, cause dural mast cell degranulation, and relay nociceptive information to second order neurons within the CNS (Pietrobon, 2005).
The effects of CGRP in mediating vasodilation, hyperalgesia, and central sensitization are thought to be mediated by CGRP1 receptors. In general, CGRP receptors are formed by a complex of seven membrane domains and includes a calcitonin receptor-like receptor (CLR), a receptor activity-modifying protein (RAMP), and a receptor component protein (RCP) that defines the G protein to which the receptor couples (Poyner et al., 2002). The associated RAMP is critical to receptor function for it defines the relative potency of ligands for the receptor (Mallee et al., 2002). The CGRP1 receptor, which is composed of the proteins CLR, RAMP1, and RCP, binds CGRP and the truncated peptide CGRP8–37 with highest affinity. Activation of CGRP1 receptors has been reported to couple to stimulation of adenylate cyclase and increases in intracellular cAMP levels in many cell types (Poyner et al., 2002; Wimalawansa, 1996). CGRP1 receptors are found on the major cerebral blood vessels and meningeal vessels, dural mast cells, and second order neurons located in the trigeminal nucleus caudalis and other brainstem nuclei (van Rossum et al., 1997). More recently, the presence of functional CGRP receptors has been reported on cultured trigeminal ganglia neurons as well as glial cells (Thalakoti et al., 2007; Zhang et al., 2007).
Most studies on the role of trigeminal nerve activation in migraine have focused on the afferent or peripheral release of CGRP that mediates vasodilation and efferent or centrally released CGRP, which promotes nociception and central sensitization. However, it is known that neuropeptides can be released from the cell bodies of trigeminal neurons that are located in the ganglia in association with glial cells (Matsuka et al., 2001; Neubert et al., 2000). While the function of CGRP release from neuronal cell bodies within the ganglion is not well understood, it is likely that CGRP could function as a paracrine signaling molecule to cause activation of glial cells. There are two types of glial cells present in adult trigeminal ganglia: satellite glial cells that surround neuronal cell bodies, and Schwann cells that associate with nerve fibers and are responsible for myelin production (Lazarov, 2002; Pannese, 2002). Recently, studies of CNS diseases have demonstrated the importance of glial cells in the pathogenesis of inflammation and pain (DeLeo and Yezierski, 2001; Meller et al., 1994; Watkins et al., 2001a,b). Activation of glial cells associated with the CNS causes the release of a number of cytokines and chemokines as well as nitric oxide (NO) (Murphy, 2000).
The bioactive free radical NO is an important messenger involved in the development and maintenance of inflammation and pain (Yun et al., 1996). NO is generated by the activity of NO synthases (NOS) during the enzymatic conversion of l-arginine to l-citruliline. The primary NO-producing enzymes in mammalian tissues are neuronal or nNOS, endothelial or eNOS, and inducible or iNOS (Faraci and Heistad, 1998). The constitutive NOS isoforms, eNOS and nNOS, rapidly produce small amounts of NO that exert direct, short-acting effects on target cells. In contrast, iNOS produces large quantities of NO for hours to days, sustaining high levels that can cause tissue damage and cell death. Glial cells in the CNS are known to express inducible iNOS (Fiebich et al., 2002). Increased iNOS expression in glial cells is implicated in the etiology of CNS diseases by causing inflammation and cytotoxicity (Murphy, 2000; Paakkari and Lindsberg, 1995). However, the pathophysiological consequences of increased iNOS expression and NO release from sensory ganglion glial cells are not known.
In this study we utilized primary cultures of trigeminal ganglia to study the effect of CGRP on the synthesis and release of NO from glial cells. Our data show that satellite glia cells both in vivo and maintained in vitro express the CGRP1 receptor protein RAMP1. Activation of CGRP1 receptors on glial cells was shown to increase iNOS expression and NO production. Furthermore, we provide evidence that elevated intracellular levels of cAMP increase both iNOS activity and NO release to a level comparable to that seen with CGRP. Based on our findings, we propose that neuronal–glial cell signaling mediated by CGRP release from neuronal cell bodies within trigeminal ganglia is likely to play an important role in the underlying pathology of migraine.
2. Results
2.1. Glial cells in vivo and maintained in vitro express RAMP1
Initially, trigeminal ganglion sections from untreated adult animals were stained with the fluorescent dye DAPI to morphologically identify neuronal and glial cell nuclei. Multiple image alignment was used to obtain images of the entire ganglion so that the distribution of neuronal cell bodies and glia were clearly visible (Fig. 1A). As seen in Fig. 1B, neuronal cell bodies are localized in numerous clusters or bands within the trigeminal ganglion. In adult animals, both types of glia, satellite glial cells and Schwann cells, are present within the ganglion. Neuronal cell bodies are completely surrounded by satellite glial cells, while Schwann cells are associated with neuronal processes projecting both to the periphery and to the central nervous system (Fig. 1B, C). The neuropeptide CGRP was expressed in ∼50% of neuronal cell bodies associated with the ophthalmic (V1), maxillary (V2), and mandibular (V3) regions of the ganglion. While a similar staining pattern was observed for the CGRP1 receptor protein RAMP1 within the ganglion, the percentage of RAMP1 expressing neuronal cells was higher than that observed for CGRP. In fact, almost all the CGRP positive cells also expressed RAMP1 (>95%, data not shown). If viewed at higher magnification, RAMP1 staining was not only observed on neuronal cell bodies but also was detected on satellite glial cells (Fig. 1C). However, in regions of the ganglion where only Schwann cells are present (i.e., posterior region of ganglion devoid of neuronal cell bodies and between neuronal cell clusters), RAMP1 staining was not observed. As a control, no staining was observed if the RAMP1 antibodies were incubated with RAMP1 peptide (Fig. 1D). Similar to CGRP and RAMP1, the vesicle docking protein SNAP-25 was localized the cell bodies of the majority of neurons in all regions of the trigeminal ganglion (Fig. 1A, B), which supports the capability of neuronally-mediated paracrine signaling within the ganglia. Thus, while CGRP and SNAP-25 are localized in neurons, RAMP1 staining was observed on both neuronal cell bodies and satellite glial cells. To our knowledge, this is the first report of RAMP1 being expressed by trigeminal satellite glial cells, and therefore expressing CGRP1 receptors.
Fig. 1.
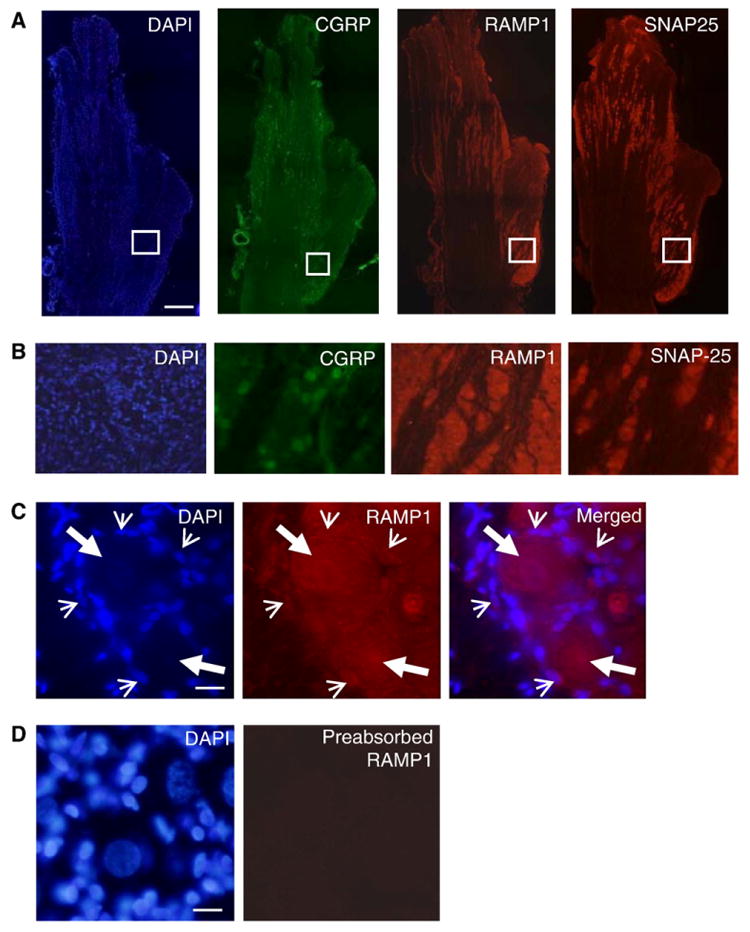
Expression of CGRP, RAMP1, and SNAP-25 in trigeminal ganglia. (A) Rat trigeminal ganglion sections of the whole ganglion stained with the fluorescent dye DAPI to identify neuronal and glial cell nuclei. Multiple image alignment was used to generate a view of the entire ganglia. Other sections were immunostained for the expression of CGRP, RAMP1, and SNAP-25. (B) Enlargement of boxed areas shown in A. (C) The same section was stained with DAPI or immunostained for expression of RAMP1. The panel on the right is a merged image of the DAPI and RAMP1 images. Large arrows identify neuronal cell body nuclei while small arrows identify satellite glial cell nuclei. (D) Ganglion section incubated with RAMP1 antibodies preabsorbed with RAMP1 peptide. Scale bar represents 1000 μm in A and 50 μm in C and D.
To determine whether RAMP1 would be expressed by glial cells maintained in vitro, primary trigeminal cultures enriched for glial cells were established using neonatal rats based on a modification of our previously published methods for studying neuronal cell function (Bellamy et al., 2006a; Bowen et al., 2006). Under our culture conditions, >95% of the cells were identified as glial cells based on cellular and nuclear morphology (Fig. 2A) and expression of the glial cell marker protein GFAP (1194±6.6 GFAP positive cells/1250±6.8 total cells, p<0.01, n=3 independent experiments done in duplicate). While in adult rat trigeminal ganglia two glial cell types are observed, based on the size and morphology of the cells it appears that the majority (>90%) of trigeminal ganglia glial cells in our cultures are satellite glial cells rather than Schwann cells, which are reported to differentiate later in development (Lazarov, 2002). Similar to the GFAP results, most of the glial cells expressed RAMP1 (Fig. 2B, 1202±7.0 RAMP1 positive cells/1245±7.5 total cells, p<0.01, n=3). This finding is in agreement with our in vivo results that demonstrated that RAMP1 is expressed by satellite glial cells. No glial cell staining was observed if cells were incubated with RAMP1 antibodies preabsorbed with RAMP1 peptide (Fig. 2C) or incubated with secondary antibodies only (Fig. 2D).
Fig. 2.
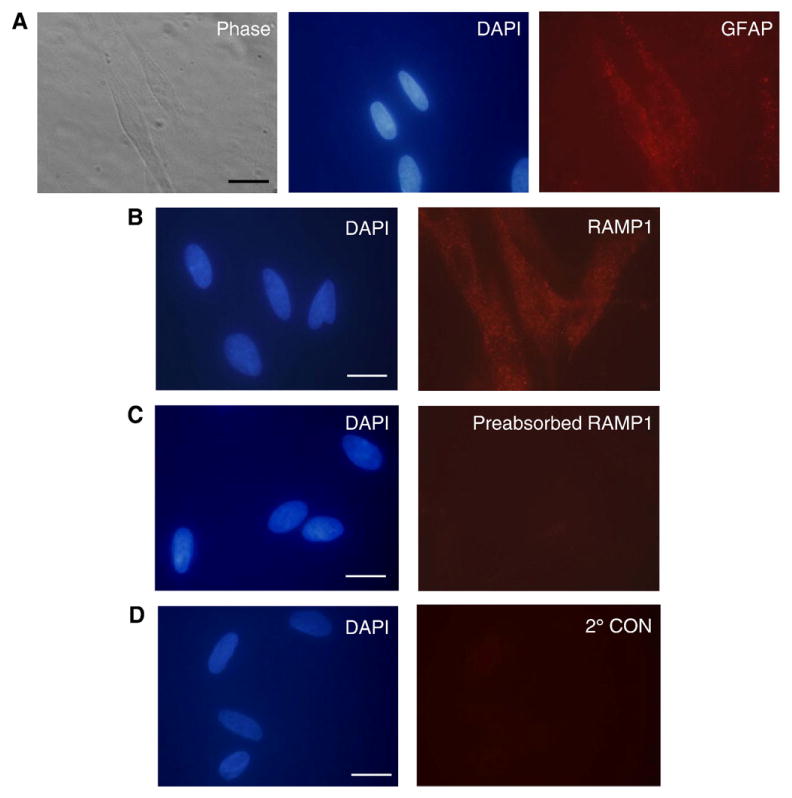
Cultured trigeminal ganglia glial cells express RAMP1. (A) GFAP is expressed in the majority of cultured cells that exhibit glial morphology by phase microscopy. The same cultured cells were costained with DAPI to identify nuclei. (B) Almost all trigeminal ganglion glial cells express the CGRP receptor subunit RAMP1. The same culture was costained with DAPI. (C) Cultured cells incubated with RAMP1 antibodies preabsorbed with RAMP1 peptide and costained with DAPI are shown. (D) Cultured cells incubated with secondary antibodies only and costained with DAPI. Scale bar represents 20 μm.
2.2. CGRP stimulates iNOS and NO release from cultured glial cells
Having shown that glial cells express RAMP1, we next wanted to determine whether CGRP could stimulate iNOS expression and NO release from cultured glial cells. The basal level of iNOS was barely detectable in cultured glial cells (Fig. 3A, B). In contrast, trigeminal ganglion glia satellite cells treated with 1 μM CGRP for 24 h showed increased expression of iNOS compared to control levels (Fig. 3C, D). Pretreatment of cultures with the CGRP receptor antagonist, CGRP8–37 (10 μM), blocked CGRP increased iNOS expression (Fig. 3E, F). CGRP also stimulated NO release from cultured trigeminal glial satellite cells as measured by the Griess reaction (Fig. 4). NO values were normalized to the total amount of protein in each sample to control for small differences in the number of plated cells. The basal NO level measured in media collected from untreated, control cultures maintained for 48 h was around 5 μM. Even at a concentration of 0.2 μM, the amount of NO released into the culture medium 48 h after CGRP treatment was significantly elevated (∼8.5 μM, p<0.05) when compared to control levels. The amount of NO released was only slightly increased (to ∼10 μM) following treatment at higher CGRP concentrations (1 or 2 μM). However, even at the higher CGRP concentrations, the amount of NO released into the media after 24 h was not significantly different than control levels (data not shown). This finding is in agreement with results from a previously published study on iNOS and NO release from glial cells that showed increased iNOS expression at 24 h but changes in NO release were only seen at 48 h (Platten et al., 2001). No significant loss of viability was noted after 48 h for the control or CGRP-treated cultures (>90% of the control value at each CGRP concentration) as measured by the CellTiter 96 viability assay (n=3 independent experiments performed in duplicate). Similar to the iNOS data, the stimulatory effect of 1 μM CGRP was suppressed to near control levels in response to pretreatment with the CGRP8–37 peptide (Fig. 4). The concentrations of CGRP (1 μM) and CGRP8–37 (10 μM) used in our study are similar to values used in other studies on the functional role of CGRP (Guo et al., 2007; Zhang et al., 2007). To demonstrate that NO production was being mediated primarily by iNOS activity and not nNOS, cultures were pretreated with the selective and potent nNOS inhibitor Nω-propyl-l-arginine 60 min prior to CGRP addition and the levels of NO in the medium determined. Pretreatment with 60 nM and 600 nM Nω-propyl-l-arginine did not cause a significant change in the basal or CGRP-stimulated release of NO (data not shown, n=3 independent experiments performed in duplicate). These results provide evidence that CGRP stimulation of NO production and release from glial cells involves RAMP1 and increased iNOS activity.
Fig. 3.
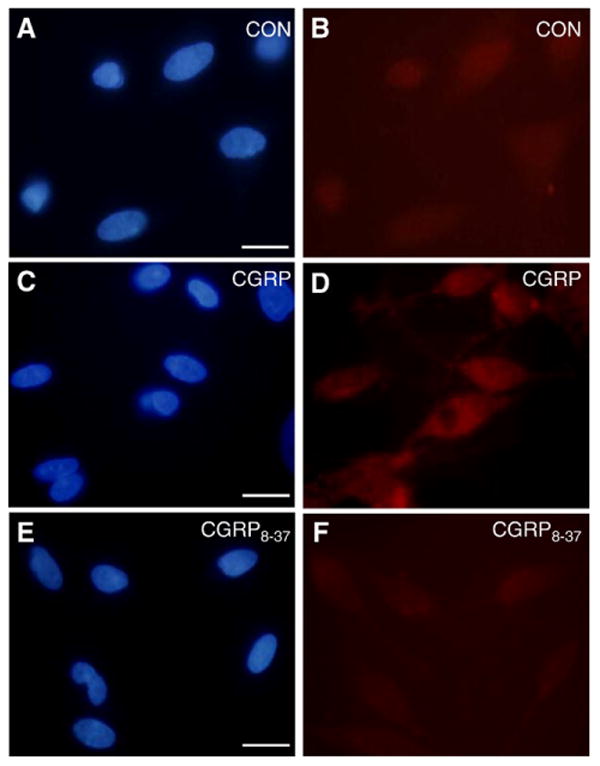
CGRP stimulation of iNOS involves RAMP1. Untreated (control; CON) trigeminal ganglion glial cells were costained with DAPI and antibodies directed against iNOS (A, B) or after 24 h stimulation with 1 μm CGRP (C,D), or treatment with 10 μm CGRP8–37 prior to CGRP stimulation (E, F). Scale bar represents 20 μm.
Fig. 4.
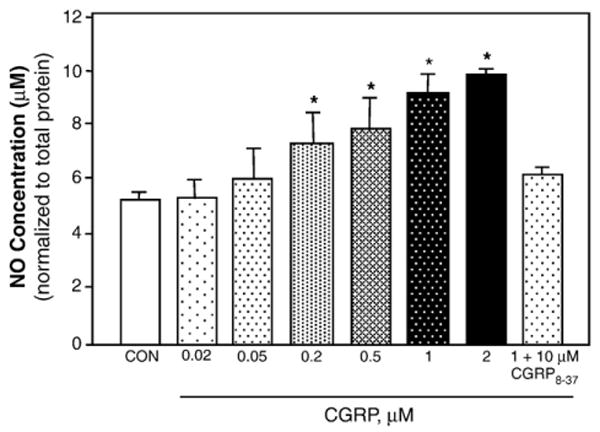
CGRP-stimulated NO release from trigeminal ganglion glial cells. NO levels were determined by the Griess reaction in media collected from untreated control cultures (CON) or 48 h after treatment with 0.02, 0.05, 0.2, 0.5, 1, or 2 μM CGRP or cotreatment with 1 μM CGRP and 10 μM CGRP8–37. *p<0.05.
2.3. Forskolin and cAMP stimulate iNOS and NO release from cultured glial cells
The cellular effects of CGRP mediated by activation of CGRP receptors are reported to involve activation of adenylate cyclase and increased intracellular cAMP levels in many cell types (van Rossum et al., 1997). As expected, CGRP treatment (200 nM and 1 μM) caused a marked increase in the intracellular cAMP levels in cultured trigeminal ganglia glia cells (>20-fold) when compared to control levels (data not shown). To determine the effects of adenylate cyclase activation and cAMP on iNOS induction and NO production, glial enriched cultures were treated with forskolin or a cAMP analogue. As shown in Fig. 5, treatment of glial cells with 1 μM forskolin, which selectively stimulates adenylate cyclase activity, or with 1 μM dibutyryl cAMP resulted in increased iNOS expression in almost all glial cells (728±8.4 iNOS positive cells/742±8.8 total cells, p<0.01, n=3). As shown before, iNOS levels in untreated control cultures were barely detectable. Treatment of glia cells with either forskolin or cAMP also caused a dose dependent increase in the amount of NO released into the culture media 48 h after addition of stimulatory agents (Fig. 6). The NO concentration in the media increased from around 5 μM for the control to almost 13 μM in response to 10 μM forskolin. A similar pattern of NO stimulation was observed in response to increasing cAMP concentrations. While the NO level for the control cultures was less than 3.5 μM, the NO concentration in the media increased significantly after treatment with 1, 10, or 100 μM cAMP. The concentration of NO was significantly elevated by all concentrations of forskolin and cAMP tested.
Fig. 5.
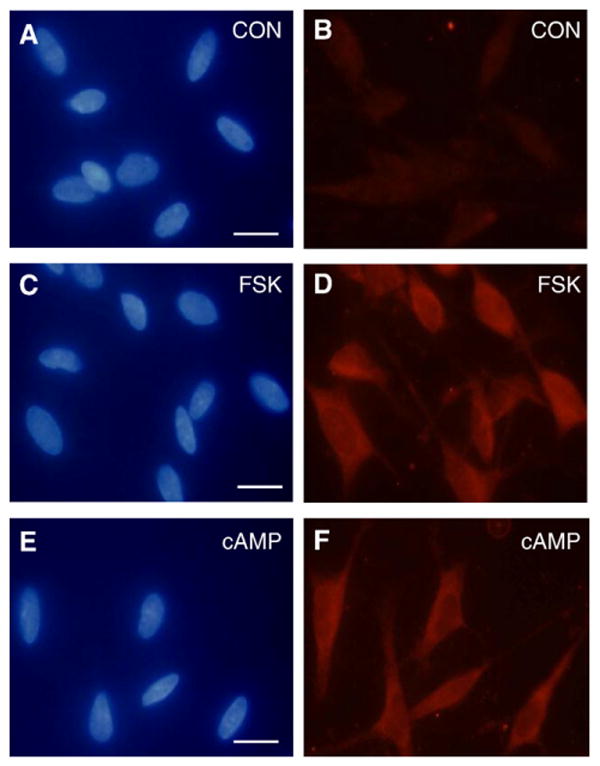
Forskolin and cAMP stimulate iNOS expression. Trigeminal ganglion glial cells that were left untreated (control; CON) or treated for 24 h with 1 μM forskolin (FSK) or 1 μM cAMP were costained with DAPI (left panels) and antibodies directed against iNOS (right panels). Scale bar represents 20 μm.
Fig. 6.
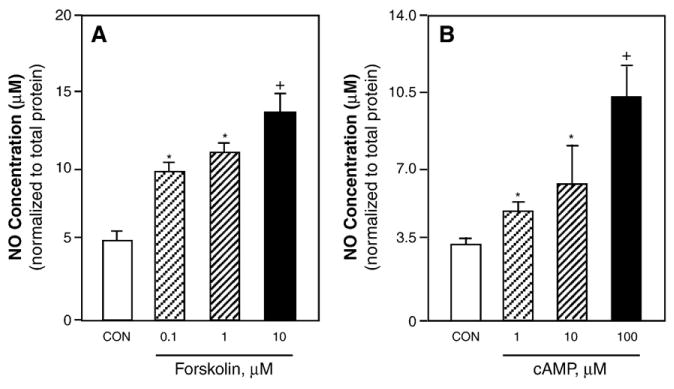
Stimulated NO release from trigeminal ganglion glial cells. NO levels were determined by the Griess reaction in media collected from untreated control cultures (CON) or 48 h after treatment with 0.1, 1, or 10 μM forskolin (A) or 1, 10, or 100 μM cAMP (B). *p<0.05; +p<0.01.
3. Discussion
We found that the membrane protein RAMP1, which is required to make a functional CGRP1 receptor, was expressed in most neuronal and satellite glial cells throughout all regions of the trigeminal ganglion. The trigeminal ganglion, which provides somatosensory innervation of distinct regions of most structures of the face, head, and oral cavity is divided into three main branches that include the ophthalmic (V1), maxillary (V2), and mandibular (V3) (Shankland, 2000). Trigeminal neurons are classified as pseudounipolar neurons since a single axon, which divides into a peripheral and central branch, extends from the cell body or soma that is located in the ganglion (Lazarov, 2002). While there are two reported glial cell types in trigeminal ganglion, we detected RAMP1 staining only in association with satellite glial cells, but not Schwann cells. Satellite glial cells, which surround neuronal cell bodies, are reported to modulate the excitability state of ganglion neurons and play an important role in peripheral sensitization (Dublin and Hanani, 2007; Hanani, 2005). Furthermore, it is now thought that activation of satellite glial cells in the trigeminal ganglion may trigger enhancement of neuron excitability in response to nerve injury and inflammation associated with hyperalgesia (Takeda et al., 2007).
It is well established that chemical or electrical activation of trigeminal nerves leads to afferent and efferent release of neuropeptides that facilitate peripheral inflammatory responses and cause activation of second order neurons involved in pain transmission (Buzzi, 2001; Pietrobon, 2005). However, it has previously been reported that neuropeptides can be released from neuronal cell bodies within the ganglia following nerve activation (Matsuka et al., 2001; Neubert et al., 2000). In this study, we provide evidence that the vesicle docking protein SNAP-25 is expressed in many neuronal cell bodies located throughout the ganglia. This finding is in agreement with our previously published results that showed SNAP-25 staining of trigeminal neuron cell bodies and processes (Thalakoti et al., 2007). SNAP-25 is a member of the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) family of proteins that facilitate binding and fusion of vesicles (Leabu, 2006; Snyder et al., 2006) and is a molecular target of botulinum neurotoxin type A (Dolly and Aoki, 2006). Previously, we had demonstrated the importance of SNAP-25 in facilitating CGRP release by showing that botulinum toxin type A greatly reduced neuronal SNAP-25 levels and dose and time-dependently blocked stimulated CGRP release from trigeminal neurons (Durham et al., 2004). Based on our findings and others, it is likely that activation of trigeminal nerves will lead to release of CGRP within the ganglion and bind to CGRP receptors present on neuronal cells as well as satellite glial cells. In this way, CGRP would be expected to exert both autocrine and paracrine effects in the ganglia. Towards this end, Zhang et al. (2007) recently demonstrated that CGRP can function in an autocrine manner to stimulate CGRP promoter activity and increase mRNA levels in cultured trigeminal neurons. Similarly, we have recently provided evidence that CGRP can function in a paracrine manner to regulate the release of inflammatory cytokines from cultured trigeminal ganglia glial cells (Thalakoti et al., 2007).
In this study, we utilized primary cultures of trigeminal ganglia enriched for glial cells to provide evidence that CGRP can also stimulate iNOS activity and NO release from glial cells. Initially, we demonstrated that under our culture conditions RAMP1 is expressed by almost all glial cells, as identified by staining for glial fibrillary associated protein or GFAP. The stimulatory effects of CGRP observed in our study are likely mediated via activation of CGRP1 receptors since pretreatment of cultures with the peptide CGRP8–37 blocked both iNOS induction and NO release. This peptide has been shown to selectively bind RAMP1 and inhibit physiological and cellular functions of CGRP in a number of cell types (Poyner et al., 2002; van Rossum et al., 1997; Wimalawansa, 1996). To our knowledge, our findings provide the first evidence that CGRP activation of RAMP1 can couple to increased iNOS activity and NO release from trigeminal glial cells. While the physiological effects of elevated NO levels in the trigeminal ganglia are not known, the importance of increased iNOS activity and NO release from glial cells in CNS diseases is well established (Saha and Pahan, 2006). Within the CNS, the glial cell types, astrocytes and microglial, do not appear to express iNOS under normal conditions but are known to express iNOS in response to a wide variety of stimuli released following ischemic, traumatic, neurotoxic, or inflammatory damage. Thus, NO generated by iNOS is thought to contribute to the pathology of Alzheimer's disease, Parkinson's disease, Huntington's disease, multiple sclerosis, brain ischemia, and trauma (Liu et al., 2002). In addition, it is now thought that activation of glial cells within the CNS leads to alterations in neuronal excitability that help maintain the pain sensation even after the original injury or inflammatory event has subsided (Wieseler-Frank et al., 2004). Based on the importance of glial cells and iNOS expression in CNS diseases, it is likely that CGRP stimulation of iNOS activity and increased NO release from satellite glial cells within the trigeminal ganglia will play a main role in the underlying pathology of migraine as well as other diseases that involve trigeminal ganglia nerves.
The expression of iNOS in glial cells of the CNS has been shown to be increased in response to bacterial lipopolysaccharide, viral products including the human immunodeficiency virus, cytokines, and neurotoxins associated with several neurodegenerative diseases (Saha and Pahan, 2006). Similar to other inducible genes, iNOS is thought to be regulated primarily at the transcriptional level and involve multiple intracellular signaling pathways. A number of extracellular signaling molecules are known to couple via receptor activation to increases in intracellular cAMP, a second messenger reported to regulate iNOS induction (Saha and Pahan, 2006). Importantly, CGRP receptor activation induced by CGRP binding to RAMP1 has been reported to stimulate adenylate cyclase and cause large increases in intracellular cAMP levels in a number of different cell types (van Rossum et al., 1997). Data from our study demonstrate that iNOS activity and NO production are increased in trigeminal glial cells in response to exogenously added cAMP or treatment with forskolin, which causes elevated intracellular cAMP levels via stimulation of adenylate cyclase activity. The observed increase in NO levels following forskolin or cAMP treatment was similar in magnitude to that seen in response to CGRP treatment. Our finding that increases in intracellular cAMP cause induction of iNOS in glial cells is in agreement with results from some studies but is in contrast to the inhibitory effects reported in other studies (Kozuka et al., 2007; Markovic et al., 2003; Won et al., 2004; Zidek, 2001). Based on numerous studies, cAMP regulation of iNOS activity in glial cells is reported to be quite complex since the effect of cAMP has been shown to be dependent on the cell type and the presence of other stimulatory factors (Saha and Pahan, 2006).
CGRP release from the cell body of activated trigeminal nerves and subsequent stimulation of iNOS activity and NO release from satellite glial cells within the ganglia may have important implications for understanding migraine pathology and current abortive treatments. Migraine is a recurring, episodic neurovascular disorder characterized by painful headache, as well as neurological, gastrointestinal, and other somatic symptoms (Ferrari, 1998). Sensitization and activation of trigeminal ganglion nerves is thought to play a central role in the underlying pathology of migraine. Current theories propose that migraine-specific triggers promote primary brain dysfunction, which evokes changes in meningeal blood vessel dilation and a reflex-like response of perivascular trigeminal nerves (Pietrobon, 2005). Based on animal studies, it has been proposed that activation of peripheral trigeminovascular afferents of V1 in the meninges releases CGRP that mediates neurogenic vasodilation, while central efferent release of CGRP contributes to pain, central sensitization, and allodynia associated with migraine. The hyperalgesia and allodynia associated with migraine are thought to involve both peripheral and central sensitization (Dodick and Silberstein, 2006). Peripheral sensitization, which is the result of increased activity of trigeminal nociceptors, is thought to initiate a migraine attack, while central sensitization, which involves enhanced excitability of second order neurons, leads to pain. Peripheral sensitization is characterized by increased neuronal excitability and a lowering of the threshold for activation. Glia cells, which were thought to serve only a supportive role, are now known to directly modulate neuronal function and activity (Hanani, 2005; Watkins and Maier, 2002). While the majority of studies to date have focused on understanding the role of CGRP release from peripheral or central terminals, there is evidence that sensory ganglion cell bodies are transiently depolarized and become more excitable by repetitive action potential activity in neighboring axons in the same ganglion (Amir and Devor, 1996, 2000). Based on other studies that have reported that neuropeptides can be released from trigeminal ganglia cell bodies (Matsuka et al., 2001; Neubert et al., 2000) and our results, it is likely that CGRP released from the cell body of stimulated neurons can cause excitation of other neuronal cells as well as satellite glial cells. Thus, CGRP release from neuronal cell bodies would be expected to function as an autocrine signal by modulating neuronal cell activities and also function as a paracrine factor by regulating cellular activities of trigeminal glial cells. The stimulatory effects of CGRP would be mediated via activation of CGRP1 receptors expressed by neuronal and glial cells located within the trigeminal ganglia. Based on data from this study, we propose that CGRP release in the ganglia would lead to activation of adjacent glial cells and increased NO production, and increased release of other inflammatory cytokines from cultured trigeminal glial cells as reported previously (Thalakoti et al., 2007). It is possible that the glial-derived NO would stimulate trigeminal neurons to increase the synthesis and release of CGRP. In support of this notion, we have previously shown that NO could stimulate CGRP promoter activity and release from cultured trigeminal ganglia neurons (Bellamy et al., 2006a). Thus, a neuron–glia inflammatory loop within the trigeminal ganglia would be established. In addition, CGRP is likely to function in an autocrine manner within the ganglia to stimulate the further synthesis of CGRP as recently demonstrated in trigeminal neurons (Zhang et al., 2007). Taken together, these events would be expected to contribute to peripheral sensitization of trigeminal neurons.
Interestingly, data is accumulating to advocate treatment of migraine attacks while head pain is mild, presumably during peripheral sensitization but before central sensitization, since early intervention has been shown to be the most effective acute treatment strategy (Dowson et al., 2006). Based on animal studies, triptan drugs are thought to function to block the pain and associated symptoms of migraine by preventing the peripheral release of CGRP that is involved in a neurogenic inflammatory response in the dura, and blocking its release at the level of second order neurons to prevent pain transmission. However, results from our study and the study by Zhang et al. (2007) provide evidence to support the notion that triptans may also function within the trigeminal ganglia to block CGRP activation of neurons and glia cells that are involved in peripheral sensitization. In addition, BIBN4096BS, a member of the new class of migraine drugs designed to competitively block CGRP receptor activation (Olesen et al., 2004), would be predicted to block neuronal and glial cell activation by CGRP within the trigeminal ganglia. Inhibition of neuronal–glial cell signaling within the trigeminal ganglia is likely to be clinically important since neuronal–glial signaling within the dorsal root ganglia has been reported to contribute to chronic inflammatory pain (Dublin and Hanani, 2007).
In summary, data from this study provide evidence that CGRP can cause activation of cultured trigeminal ganglia glial cells that leads to increased iNOS activity and NO release. In addition, we have recently shown that CGRP can differentially modulate the release of cytokines from trigeminal glial cells (Thalakoti et al., 2007), while Zhang et al. (2007) have shown that CGRP can increase its own synthesis in trigeminal neurons. Based on these findings, we propose that neuronal–glial cell interactions within the trigeminal ganglion are likely to play an important role in the underlying pathology of migraine as well as other diseases that involve trigeminal nerve activation.
4. Experimental procedures
4.1. Animals
All animal care and procedures were conducted in accordance with institutional and National Institutes of Health guidelines. Adult male and female Sprague–Dawley rats (Charles River, Wilmington, MA) were housed in clean plastic cages on a 12-h light/dark cycle with unrestricted access to food and water.
4.2. Immunohistochemistry of trigeminal ganglia
For immunohistochemical studies, ganglia were collected from untreated male animals and immediately placed in Neg 50 mounting medium (Richard-Allan Scientific, Kalamazoo, MI) at −25 °C. Sections of the entire ganglion (20 μm or 50 μm) were made using a cryostat (Microm HM525, Richard-Allan Scientific) set at −25 °C. Two sections were placed on each Superfrost plus microscope slide (Fischer Scientific, Pittsburgh, PA). Sections were fixed using 4% paraformaldehyde for 30 min, permeabilized with 0.3% Triton X-100 for 30 min, and then incubated in 5% donkey serum for another 30 min. Sections were then incubated overnight at 4 °C with rabbit primary antibodies against either CGRP (Neuromics, Edina, MN), receptor activity-modifying protein type 1 (RAMP1; Alpha Diagnostics, Inc. San Antonio, TX), or synaptosomal-associated protein 25 (SNAP-25; Sigma). As a control in some experiments, the RAMP1 antibody was incubated with 20 μg of RAMP1 peptide (Alpha Diagnostics, Inc.) for 2 h at room temperature and spun for 10 min at 13,000 ×g before being used for immunostaining. Immunoreactive proteins were visualized following incubation for 1 h at room temperature with FITC-conjugated donkey anti-rabbit IgG (for CGRP) or Rhodamine Red X-conjugated donkey anti-rabbit IgG (for RAMP1 and SNAP-25) secondary antibodies (Jackson Immuno Research Laboratories, West Grove, PA; diluted 1:100 in PBS). In some experiments, samples were only incubated with the secondary antibody. Following the staining procedure, sections were mounted in Vectashield medium (H-1200, Vector Laboratories, Burlingame, CA) containing 4′,6 diamidino-2-phenylindole (DAPI) to allow for identification of neuronal and glial cell nuclei and images (at 40× or 400×) collected using an Olympus DP70 camera mounted on an Olympus BX41 fluorescent microscope and image analysis performed using Olympus MicroSuite Five image processing software (Olympus, Center Valley, PA). Multiple image alignment was utilized to view the entire ganglion in a single image at 40× magnification. Briefly, a total of 12 images were collected and aligned to produce a much larger view of the tissue.
4.3. Trigeminal ganglia cultures
Primary cultures of trigeminal ganglia enriched in glial cells were established based on our previously published protocol (Bowen et al., 2006; Durham and Russo, 1999, 2003). Briefly, ganglia were isolated from 20 to 24 two to three day-old neonatal rat pups and incubated in 10 mL L15 media (Leibovitz, Sigma, St. Louis, MO) containing 10 mg/mL Dispase II (Invitrogen Corp., Carlsbad, CA), and 1 U/μL RQ1 DNase (Promega, Madison WI) for 30 min at 37 °C. Following centrifugation at 250 ×g for 1 min, pellets were resuspended and dissociated in plating medium by vigorous trituration and then spun at 250 ×g for 3 min to pellet neuronal cells, and the resultant supernatant respun at 500 ×g for 5 min to concentrate glial cells. The resulting glial cell pellet was resuspended in L-15 medium containing 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 50 mM glucose, 250 μM ascorbic acid, 8 μM glutathione, 2 mM glutamine, and 10 ng/mL mouse 2.5 S nerve growth factor (Alomone Laboratories, Jerusalem, Israel). Penicillin (100 U/mL), streptomycin (100 μg/mL), and amphotericin B (2.5 μg/mL, Sigma) were also added to the supplemented L15 media, which will be referred to as L15 complete medium. For NO studies, dissociated cells were plated on 24-well tissue culture plates (Becton Dickinson Transduction Laboratories, Franklin Lakes, NJ) at a density equivalent of two ganglia per well. For the immunocytochemistry studies, glial cells were plated at a density of half a ganglion on 11 mm glass or plastic coverslips coated with poly-d-lysine (relative MW 30,000–70,000; Sigma). Cultures were incubated at 37 °C at ambient CO2. The culture medium was changed after 24 h and every other day thereafter.
4.4. Immunocytochemistry of primary trigeminal ganglia cultures
Initially, cultures maintained for 24 h were rinsed briefly with PBS and treated with 4% paraformaldehyde for 30 min at room temperature and with 0.2% Triton X-100 in PBS for an additional 15 min to fix and permeabilize the cells. Cultures were incubated overnight at 4 °C with goat anti-glia fibrillary acidic protein (GFAP) antibodies (1:500 in PBS; Chemicon International, Inc., Temecula, CA), RAMP1 antibodies (1:100 in PBS; RAMP1; Alpha Diagnostics, Inc.), or RAMP1 antibody preabsorbed with RAMP1 peptide as described for tissue staining. Immunoreactive proteins were detected following incubation with Rhodamine Red X-conjugated donkey anti-goat (Jackson Immuno Research Laboratories, Inc., for GFAP) or Rhodamine Red X-conjugated donkey anti-rabbit antibodies (Jackson Immuno Research Laboratories, Inc., for RAMP1) for 1 h at room temperature. Prior to viewing, cells were mounted using Vectashield mounting media (Vector Laboratories) containing DAPI to identify nuclei. In addition, cells maintained for 24–48 h were left untreated (control), treated for 24 h with CGRP (1 μM; American Peptide, Inc., Sunnyvale, CA), or pretreated for 60 min with 10 μM CGRP8–37 peptide (Sigma) prior to addition of CGRP were incubated overnight at 4 °C with mouse antibody directed against iNOS (Becton Dickinson Transduction Laboratories). Cultures were incubated for 1 h with Rhodamine-X conjugated donkey anti-mouse IgG antibodies (1:100 in PBS, Jackson Immuno Research Laboratories, Inc.) and immunoreactive proteins visualized by fluorescent microscopy. The number of immunopositive cells was determined from analysis of >10 random fields of cells from several coverslips. The reported numbers are the average±SEM of the counts obtained by two laboratory members that were blinded to the experimental design. Statistical analysis was performed using the nonparametric Mann–Whitney U-test. Differences were considered statistically significant at p<0.05. All statistical tests were performed using Minitab Statistical Software, Release 14.
4.5. Nitric oxide measurement
The amount of NO released into the culture medium from glia-enriched trigeminal ganglion cultures was determined by the Griess reaction using sodium nitrate as the standard (Promega, Madison, WI). Cultures maintained for 1 d were untreated or treated for 24 or 48 h with CGRP (0.02, 0.05, 0.2, 0.5, 1, or 2 μM diluted in water; Sigma), forskolin (0.1, 1, or 10 μM diluted in DMSO, 1% final concentration; Sigma), dibutyryl cAMP (1, 10, or 100 μM diluted in water; Sigma) or pretreated for 30 min with CGRP8–37 peptide (10 μM diluted in water) or the selective and potent nNOS inhibitor Nω-propyl-l-arginine (60 or 600 nM diluted in water; ki=57 nM; Tocris Bioscience, Ellisville, MO) 1 h prior to addition of CGRP. Both the CGRP8–37 peptide and nNOS inhibitor were present during the CGRP treatment. NO concentration for each well was normalized to the total protein level determined by the Bio-Rad Protein Assay (Hercules, CA). Each condition was performed in triplicate in a minimum of three independent experiments. Data are reported as mean±SEM. Cell viability was determined at the conclusion of the 48 h treatment for the unstimulated and CGRP-stimulated cultures using the CellTiter 96 AQ One Solution Cell Proliferation Assay according to manufacturer's instructions (Promega, Madison, WI). Statistical analysis was performed using the nonparametric Mann–Whitney U-test. Differences were considered statistically significant at p<0.05.
Acknowledgments
We would like to thank Marcie Abbey for her technical assistance. This work was supported by NIH grant DE017805.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit:
References
- Amara S, Jonas V, Rosenfeld M, Ong E, Evans R. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 1982;298:240–244. doi: 10.1038/298240a0. [DOI] [PubMed] [Google Scholar]
- Amir R, Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. J Neurosci. 1996;16:4733–4741. doi: 10.1523/JNEUROSCI.16-15-04733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95:189–195. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- Bellamy J, Bowen E, Russo A, Durham P. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006a;23:2057–2066. doi: 10.1111/j.1460-9568.2006.04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy J, Cady R, Durham P. Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache. 2006b;46:24–33. doi: 10.1111/j.1526-4610.2006.00294.x. [DOI] [PubMed] [Google Scholar]
- Bowen E, Schmidt T, Firm C, Russo A, Durham P. Tumor necrosis factor—α stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem. 2006;96:65–77. doi: 10.1111/j.1471-4159.2005.03524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzi M. Trigeminal pain pathway: peripheral and central activation as experimental models of migraine. Funct Neurol. 2001;16:S77–S81. [PubMed] [Google Scholar]
- DeLeo J, Yezierski R. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Dodick D, Silberstein S. Central sensitization theory of migraine: clinical implications. Headache. 2006;46:S182–S191. doi: 10.1111/j.1526-4610.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- Dolly J, Aoki K. The structure and mode of action of different botulinum toxins. Eur J Neurol. 2006;13 4:1–9. doi: 10.1111/j.1468-1331.2006.01648.x. [DOI] [PubMed] [Google Scholar]
- Dowson A, Mathew N, Pascual J. Review of clinical trials using early acute intervention with oral triptans for migraine management. Int J Clin Pract. 2006;60:698–706. doi: 10.1111/j.1742-1241.2006.00981.x. [DOI] [PubMed] [Google Scholar]
- Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007 doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Durham P, Russo A. Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J Neurosci. 1999;19:3423–3429. doi: 10.1523/JNEUROSCI.19-09-03423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham P, Russo A. Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J Neurosci. 2003;23:807–815. doi: 10.1523/JNEUROSCI.23-03-00807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham P, Cady R, Cady R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: implications for migraine therapy. Headache. 2004;44:35–42. doi: 10.1111/j.1526-4610.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Goadsby P. Neuropeptides in migraine and cluster headache. Cephalalgia. 1994;14:320–327. doi: 10.1046/j.1468-2982.1994.1405320.x. [DOI] [PubMed] [Google Scholar]
- Faraci F, Heistad D. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Ferrari MD. Migraine. Lancet. 1998;351:1043–1051. doi: 10.1016/S0140-6736(97)11370-8. [DOI] [PubMed] [Google Scholar]
- Fiebich B, Lieb K, Engels S, Heinrich M. Inhibition of LPS-induced p42/44 MAP kinase activation and iNOS/NO synthesis by parthenolide in rat primary microglial cells. J Neuroimmunol. 2002;132:18–24. doi: 10.1016/s0165-5728(02)00279-5. [DOI] [PubMed] [Google Scholar]
- Goadsby P, Edvinsson L. Sumatriptan reverses the changes in calcitonin gene-related peptide seen in the headache phase of migraine. Cephalalgia. 1991;11:3–4. [Google Scholar]
- Goadsby P, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48–56. doi: 10.1002/ana.410330109. [DOI] [PubMed] [Google Scholar]
- Goadsby P, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies Brain. 1994;117:427–434. doi: 10.1093/brain/117.3.427. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize S, Wei F, Dubner R, Ren K. Glial–cytokine–neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Kozuka N, Kudo Y, Morita M. Multiple inhibitory pathways for lipopolysaccharide- and pro-inflammatory cytokine-induced nitric oxide production in cultured astrocytes. Neuroscience. 2007;144:911–919. doi: 10.1016/j.neuroscience.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Lassen L, Haderslev P, Jacobsen V, Iversen H, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22:54–61. doi: 10.1046/j.1468-2982.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Lazarov N. Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol. 2002;66:19–59. doi: 10.1016/s0301-0082(01)00021-1. [DOI] [PubMed] [Google Scholar]
- Leabu M. Membrane fusion in cells: molecular machinery and mechanisms. J Cell Mol Med. 2006;10:423–427. doi: 10.1111/j.1582-4934.2006.tb00409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton R, Stewart W, Diamond S, Diamond M, Reed M. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646–657. doi: 10.1046/j.1526-4610.2001.041007646.x. [DOI] [PubMed] [Google Scholar]
- Liu B, Gao H, Wang J, Jeohn G, Cooper C, Hong J. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- Mallee J, Salvatore C, LeBourdelles B, Oliver K, Longmore J, Koblan K, Kane S. Receptor activity-modifying protein 1 determines the species selectivity of non-peptide CGRP receptor antagonists. J Biol Chem. 2002;277:14294–14298. doi: 10.1074/jbc.M109661200. [DOI] [PubMed] [Google Scholar]
- Markovic M, Miljkovic D, Trajkovic V. Regulation of inducible nitric oxide synthase by cAMP-elevating phospho-diesterase inhibitors. Curr Drug Targets Inflamm Allergy. 2003;2:63–79. doi: 10.2174/1568010033344471. [DOI] [PubMed] [Google Scholar]
- Matsuka Y, Neubert J, Maidment N, Spigelman I. Concurrent release of ATP and substance P within guinea pig trigeminal ganglia in vivo. Brain Research. 2001;915:248–255. doi: 10.1016/s0006-8993(01)02888-8. [DOI] [PubMed] [Google Scholar]
- Meller S, Dykstra C, Grzybycki D, Murphy S, Gebhart G. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Neubert JK, Maidment NT, Matsuka Y, Adelson DW, Kruger L, Spigelman I. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal ganglia in vivo. Brain Res. 2000;871:181–191. doi: 10.1016/s0006-8993(00)02440-9. [DOI] [PubMed] [Google Scholar]
- Olesen J, Diener H, Husstedt I, Goadsby P, Hall D, Meier U, Pollentier S, Lesko L. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- Paakkari I, Lindsberg P. Nitric oxide in the central nervous system. Ann Med. 1995;27:369–377. doi: 10.3109/07853899509002590. [DOI] [PubMed] [Google Scholar]
- Pannese E. Perikaryal surface specializations of neurons in sensory ganglia. Int Rev Cytol. 2002;220:1–34. doi: 10.1016/s0074-7696(02)20002-9. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Migraine: new molecular mechanisms. Neuroscientist. 2005;11:373–386. doi: 10.1177/1073858405275554. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W, Wischhusen J, Weller M. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) suppresses microglial inducible nitric oxide synthase (iNOS) expression and activity induced by interferon-gamma (IFN-gamma) Br J Pharmacol. 2001;134:1279–1284. doi: 10.1038/sj.bjp.0704373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner D, Sexton P, Marshall I, Smith D, Quirion R, Born W, Muff R, Fischer J, Foord S. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Rosenfeld M, Mermod JJ, Amara S, Swanson L, Sawchenko P, Rivier J, Vale W, Evans R. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature. 1983;304:129–135. doi: 10.1038/304129a0. [DOI] [PubMed] [Google Scholar]
- Saha R, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxidants & Redox Signaling. 2006;8:929–947. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankland W. The trigeminal nerve. Part I: an over-view. Cranio. 2000;18:238–248. doi: 10.1080/08869634.2000.11746137. [DOI] [PubMed] [Google Scholar]
- Snyder D, Kelly M, Woodbury D. SNARE complex regulation by phosphorylation. Cell Biochem Biophys. 2006;45:111–123. doi: 10.1385/CBB:45:1:111. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Kadoi J, Nasu M, Takahashi M, Kitagawa J, Matsumoto S. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain. 2007;129:155–166. doi: 10.1016/j.pain.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Thalakoti S, Patil V, Damodaram S, Vause C, Langford L, Freeman S, Durham P. Neuron–Glia signaling in trigeminal ganglion: Implications for migraine pathology. Headache. 2007;47:1008–1023. doi: 10.1111/j.1526-4610.2007.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum D, Hanisch U, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev. 1997;21:649–678. doi: 10.1016/s0149-7634(96)00023-1. [DOI] [PubMed] [Google Scholar]
- Watkins L, Maier S. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Watkins L, Milligan E, Maier S. Spinal cord glia: new players in pain. Pain. 2001a;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Watkins L, Milligan E, Maier S. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001b;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J, Maier S, Watkins L. Glial activation and pathological pain. Neurochem Int. 2004;45:389–395. doi: 10.1016/j.neuint.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Wimalawansa S. Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocrine Reviews. 1996;17:533–585. doi: 10.1210/edrv-17-5-533. [DOI] [PubMed] [Google Scholar]
- Won J, Im Y, Singh A, Singh I. Dual role of cAMP in iNOS expression in glial cells and macrophages is mediated by differential regulation of p38-MAPK/ATF-2 activation and iNOS stability. Free Radic Biol Med. 2004;37:1834–1844. doi: 10.1016/j.freeradbiomed.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Yun HY, Dawson V, Dawson T. Neurobiology of nitric oxide. Crit Rev Neurobiol. 1996;10:291–316. doi: 10.1615/critrevneurobiol.v10.i3-4.20. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Winborn C, Marquez de Prado B, Russo A. Sensitization of calcitonin gene-related peptide receptors by receptor activity-modifying protein-1 in the trigeminal ganglion. J Neurosci. 2007;27:2693–2703. doi: 10.1523/JNEUROSCI.4542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidek Z. Role of cytokines in the modulation of nitric oxide production by cyclic AMP. Eur Cytokine Netw. 2001;12:22–32. [PubMed] [Google Scholar]