

Abstract
Objective
The goal of this study was to investigate neuronal–glial cell signaling in trigeminal ganglia under basal and inflammatory conditions using an in vivo model of trigeminal nerve activation.
Background
Activation of trigeminal ganglion nerves and release of calcitonin gene-related peptide (CGRP) are implicated in the pathology of migraine. Cell bodies of trigeminal neurons reside in the ganglion in close association with glial cells. Neuron–glia interactions are involved in all stages of inflammation and pain associated with several central nervous system (CNS) diseases. However, the role of neuron–glia interactions within the trigeminal ganglion under normal and inflammatory conditions is not known.
Methods
Sprague–Dawley rats were utilized to study neuron–glia signaling in the trigeminal ganglion. Initially, True Blue was used as a retrograde tracer to localize neuronal cell bodies in the ganglion by fluorescent microscopy and multiple image alignment. Dye-coupling studies were conducted under basal conditions and in response to capsaicin injection into the TMJ capsule. S100B and p38 expression in neurons and glia were determined by immunohistochemistry following chemical stimulation. CGRP levels in the ganglion were measured by radioimmunoassay in response to capsaicin. In addition, the effect of CGRP on the release of 19 different cytokines from cultured glial cells was investigated by protein microarray analysis.
Results
In unstimulated control animals, True Blue was detected primarily in neuronal cell bodies localized in clusters within the ganglion corresponding to the V3 region (TMJ capsule), V2 region (whisker pad), or V1 region (eyebrow and eye). However, True Blue was detected in both neuronal cell bodies and adjacent glia in the V3 region of the ganglion obtained from animals injected with capsaicin. Dye movement into the surrounding glia correlated with the time after capsaicin injection. Chemical stimulation of V3 trigeminal nerves was found to increase the expression of the inflammatory proteins S100B and p38 in both neurons and glia within the V3 region. Unexpectedly, increased levels of these proteins were also observed in the V2 and V1 regions of the ganglion. CGRP and the vesicle docking protein SNAP-25 were colocalized in many neuronal cell bodies and processes. Decreased CGRP levels in the ganglion were observed 2 hours following capsaicin stimulation. Using protein microarray analysis, CGRP was shown to differentially regulate cytokine secretion from cultured trigeminal ganglion glia.
Conclusions
We demonstrated that activation of trigeminal neurons leads to changes in adjacent glia that involve communication through gap junctions and paracrine signaling. This is the first evidence, to our knowledge, of neuron–glia signaling via gap junctions within the trigeminal ganglion. Based on our findings, it is likely that neuronal–glial communication via gap junctions and paracrine signaling are involved in the development of peripheral sensitization within the trigeminal ganglion and, thus, are likely to play an important role in the initiation of migraine. Furthermore, we propose that propagation of inflammatory signals within the ganglion may help to explain commonly reported symptoms of comorbid conditions associated with migraine.
Keywords: CGRP, gap junctions, glia, S100B, p38 MAP kinase, trigeminal
Migraine is a chronic, painful, neurovascular disorder that affects an estimated 12% of the population.1 Sensitization and activation of trigeminal ganglion nerves are thought to play a central role in the underlying pathology of migraine and other orofacial diseases.2,3 The trigeminal nerve consists of 3 main branches, the ophthalmic (V1), maxillary (V2), and mandibular (V3), each providing somatosensory innervation of distinct regions of the head, face, and orofacial cavity. Adult trigeminal neurons exhibit a pseudounipolar morphology characterized by a single axon that divides into a peripheral and a central branch.4 Activation of peripheral trigeminovascular afferents of V1 in the meninges releases calcitonin gene-related peptide (CGRP), which mediates neurogenic vasodilation, while central efferent release of CGRP contributes to pain, central sensitization, and allodynia associated with migraine.5,6
Hyperalgesia and allodynia associated with migraine pathology likely involve both peripheral and central sensitization.7-9 Peripheral sensitization, which is the result of increased activity of trigeminal nociceptors, is thought to initiate a migraine attack, while central sensitization, which involves enhanced excitability of second-order neurons, leads to pain.10 While the importance of central sensitization in migraine has become clearer, the role of peripheral sensitization in the trigeminal ganglion is not well understood. Peripheral sensitization is characterized by increased neuronal excitability and a lowering of the threshold for activation. Glia cells, which were thought to serve only a supportive role, are now known to directly modulate neuronal function and activity.11,12 Interestingly, neuron–glia interactions have been shown to be involved in all stages of inflammation and pain associated with several CNS diseases.13,14
Trigeminal ganglia comprise neuronal cells and 2 types of glial cells, satellite cells and Schwann cells, which associate with nerve fibers and produce myelin.15 The cell bodies of the primary afferent neurons that convey sensory information from the periphery to the central nervous system (CNS) are completely surrounded by several satellite glial cells that form distinct, functional units. Morphological studies have shown that flattening processes from glial cells lie in close proximity to the plasma membrane of neuronal cells. It is now accepted that glial cells, which outnumber the neuronal cells 10 to 1 in trigeminal ganglia, directly influence neuronal activity by controlling the microenvironment in the ganglion.12,16 While communication between cells can occur via gap junctions or release of diffusible chemical messengers, the mechanisms by which neuronal–glial cells communicate in trigeminal ganglion under basal or inflammatory conditions is not known.
In this study we used an in vivo model of trigeminal nerve activation and glia-enriched primary cultures to investigate neuronal–glial cell signaling within the trigeminal ganglion. Data from our dye-coupling experiments demonstrated enhanced neuron to glia communication occurring through gap junctions within trigeminal ganglion in response to inflammatory stimuli. To our knowledge, this is the first report of this type of signaling within the trigeminal ganglion. In addition, stimulation of trigeminal nerves located in the V3 region of the ganglion caused a rapid increase in expression of the inflammatory proteins S100B and p38 mitogen-activated protein (MAP) kinase in both neurons and glial cells. Somewhat surprisingly, increased expression was also observed in neuronal cell bodies and glial cells located within the V2 and V1 regions. Thus, activation of one branch within the ganglion caused a rapid and sustained activation in the other branches, an example of intraganglion communication. Furthermore, we showed that CGRP, which can be released from neuronal cell bodies, can stimulate glial cells to release inflammatory cytokines. Based on our results, we propose that neuronal–glial cell signaling is likely to play a key role in peripheral sensitization within the ganglion in migraine, rhinitis, and temporomandibular joint (TMJ) disorders that involve trigeminal nerve activation. Furthermore, our data may help explain commonly reported symptoms of comorbid conditions associated with migraine, which may have important implications for further understanding migraine pathology and therapy.
Methods
Animals
The animal care procedures were conducted in accordance with institutional and National Institutes of Health guidelines. Adult male or female Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) were housed in clean plastic cages on a 12-hour light/dark cycle with unrestricted access to food and water.
In vivo studies
Female postestrous rats (200–225 g) or young male rats (200–225 g) were brought to the lab and kept in a quiet environment for about 30 minutes. Rats were anesthetized by injecting 0.3 mL of ketamine and xylazine solution (800 mg and 60 mg in 10 mL, respectively; Sigma, St. Louis, MO, USA) intraperitoneally. Rats were observed for about 15 minutes and were assessed for the effects of anesthesia using writhing reflex and tonicity of the tail (tail flick reflex). Initially, rats were injected with a retrograde labeling dye, True Blue (25 μL, 2 mg/mL in dimethyl sulfoxide, DMSO; Biotium, Hayward, CA, USA) in several regions of the head and face to fluorescently label neuronal cell bodies within the trigeminal ganglion. The TMJ of the rat was identified by first localizing the zygomatic arch and then tracing the angle of the jaw upward. The jaw was slightly opened to precisely locate the TMJ. Bilateral injections were performed using a 25-μL Hamilton syringe (Hamilton Company, Reno, NV, USA) and a 261/2 G needle (Becton Dickinson, Franklin Lakes, NJ, USA). Similarly, dye was injected subcutaneously into both right ands left whisker pads (4 injections; 50 μL total volume) and also injected in the brow of each eye (3 injections; 25 μL total) and 5 μL of dye was topically administered to the eye. After 5–7 days, the animal was sacrificed and both trigeminal ganglion removed and immediately placed in Neg 50 mounting medium (Richard-Allan Scientific, Kalamazoo, MI, USA) at −25°C for further analysis. Sections of the ganglion (20 or 50 μm) were made using a cryostat (Microm HM525, Richard Allan Scientific) set at −25°C. Two to four sections were placed on each Superfrost plus microscope slide (Fischer Scientific, Pittsburgh, PA, USA). After fixing and permeabilizing, tissues were covered in Vectashield mounting medium (H-1000, Vector Laboratories, Burlingame, CA, USA), and images (40×, 100×, or 400×) were collected using an Olympus DP70 camera mounted on an Olympus BX41 fluorescent microscope and image analysis performed using Olympus MicroSuite Five image processing software (Olympus, Center Valley, PA, USA). Multiple image alignment was utilized to view the entire ganglion in a single image at 40 × magnification. Briefly, a total of 12 images is collected and aligned to produce a much larger view of the tissue. Some sections were mounted in Vectashield medium (H-1200) containing 4,6-diamidino-2-phenylindole (DAPI) to allow for identification of neuronal and glial cell nuclei. For dye-coupling studies, True Blue was injected into both TMJ capsules 7 days prior to tissue harvesting to allow for retrograde transport to the neuronal cell bodies in the V3 region of the ganglion. Ganglia were then collected from untreated animals or animals injected with capsaicin (10 μm in DMSO, Sigma) or the vehicle DMSO (Sigma) into both TMJ capsules and processed for microscopic analysis as described above. For the capsaicin study, ganglia were collected 15, 30, 60 and 120 minutes following capsaicin or vehicle injections. Each experimental condition was repeated in at least 3 independent experiments.
For immunohistochemical studies, ganglia were collected from untreated animals or animals injected with tumor necrosis factor-α (TNF-α, 50 ng/mL; Sigma) or the nitric oxide (NO) donor sodium nitroprusside (SNP, 10 mm; Sigma) diluted in HEPES-buffered saline (HBS) at pH 5.5 in both TMJ capsules 2 hours prior to harvesting. Sections were fixed using 4% paraformaldehyde for 60 minutes, permeabilized with 0.3% Triton–X100 for 45 minutes, and then incubated in 5% donkey serum for another 60 minutes. Sections were then incubated overnight at 4°C with rabbit primary antibodies against either the capsaicin receptor TRPV1 (1:1000 in PBS; Neuromics, Edina, MN, USA), active p38 MAP kinase (1:200; Cell Signaling Technology, Danvers, MA, USA), or synaptosomal-associated protein 25 (SNAP-25, 1:2000; Sigma), goat anti-S100B (1:100; R&D Systems, Minneapolis, MN, USA), or chicken anti-CGRP (1:500; Neuromics). Immunoreactive proteins were visualized following incubation with Rhodamine Red X-conjugated donkey anti-rabbit (TRPV1, active p38, SNP-25) or anti-goat antibodies (S100B), or FITC-conjugated donkey anti-chicken antibodies (CGRP; all diluted 1:100 in PBS, Jackson Immuno-Research Laboratories, West Grove, PA, USA) for 1 hour at room temperature. Images were collected as described above.
For studies on CGRP levels in trigeminal ganglion, animals were either left untreated or both TMJ capsules were injected with capsaicin (25 μL of 10 μm solution) or vehicle (DMSO) 2 hours prior to harvesting the ganglia. Following treatment, animals were sacrificed and the posterior third of each ganglion, which corresponds to the mandibular or V3 branch, was removed, placed in HBS pH 7.4, homogenized, centrifuged, and the amount of CGRP in the supernatant determined using an α-CGRP-specific radioimmunoassay (Bachem/Peninsula Laboratories, San Carlos, CA, USA). The total amount of protein in each sample was determined by the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA). The data are reported as mean (pmol/μg total protein) ± SEM. Each experimental condition was repeated in at least 3 independent experiments. Statistical analysis was performed using the nonparametric Mann-Whitney U-test. Results were considered significant when P was less than .05. All statistical tests were preformed using Minitab Statistical Software, Release 14.
Microarray Analysis
We modified our standard procedure for the isolation and maintenance of primary neurons from trigeminal ganglia to establish enriched glial cell cultures.17-19 After sacrificing by CO2 inhalation, ganglia were isolated from approximately 20 to 24 two- to three-day-old Sprague–Dawley rats and incubated in 10 mL L-15 (Leibovitz, Sigma) media containing 10 mg/mL Dispase II (Invitrogen, Carlsbad, CA, USA), and 1 unit/μL RQ1 DNase (Promega, Madison, WI, USA) for 30 minutes at 37°C while rotating at 15 rpm. Following centrifugation at 250 × g for 1 minute, pellets were resuspended and dissociated in the plating medium by vigorous trituration and then spun at 250 × g for 3 minutes to pellet neuronal cells, and the resultant supernatant respun at 500 × g for 5 minutes to concentrate glial cells. The resulting glial cell pellet was resuspended in L-15 medium containing 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA, USA), 50 mm glucose, 250 μm ascorbic acid, 8 μm glutathione, 2 mm glutamine, and 10 ng/mL mouse 2.5 S nerve growth factor (Alomone Laboratories, Jerusalem, Israel). Penicillin (100 units/mL), streptomycin (100 μg/mL), and amphotericin B (2.5 μg/mL, Sigma) were also added to the supplemented L15 media, which will be referred to as L15 complete medium. For immunocytochemistry studies, dissociated cells from the equivalent of 24 ganglia were plated on 24-well poly-d-lysine (MW 30,000–70,000; Sigma)-coated plastic coverslips (BD Biosciences, Bedford, MA, USA) and incubated at 37°C at ambient CO2. After 48 hours, cells were rinsed briefly with phosphate-buffered saline (PBS) and treated with 4% paraformaldehyde for 30 minutes at room temperature and with 0.2% triton X-100 in PBS for an additional 15 minutes to fix and permeabilize the cells. Cultures were incubated overnight at 4°C with goat anti-glia fibrillary acidic protein (GFAP) antibodies (1:500 in PBS; Neuromics). Immunoreactive proteins were detected following incubation with Rhodamine Red X-conjugated donkey anti-goat (1:100 in PBS, Jackson Immuno-Research Laboratories) for 1 hour at room temperature. Prior to viewing, cells were mounted using Vectashield mounting media (Vector Laboratories) containing DAPI to identify nuclei. For microarray analysis, the equivalent of 48 ganglia was plated on a poly-d-lysine-coated 24-well tissue culture plate and incubated at 37°C at ambient CO2. Two days after plating, some cultures were left untreated (control) or treated overnight with 100 ng/mL rat CGRP in L15 complete media. The next day the media was removed from each of the wells and replaced with a buffered saline solution (pH 7.4). Following 1 hour incubation at 37°C, the medium was collected from 4 wells, pooled, and assayed for expression of 19 cytokines following manufacturer's instructions (Rat Cytokine Antibody Array I, RayBiotech, Norcross, GA, USA). Following collection of the medium, which was immediately analyzed, the cells were scraped and the total amount of protein in each well was determined by the Bradford method. Cytokine levels in media collected from CGRP-treated cells are compared to levels in media from unstimulated control cultures. Densometric analysis of each spot was performed using a Kodak Image Station (4000MM, Eastman Kodak Company, Rochester, NY, USA). Each experimental condition was repeated in at least 2 independent experiments.
Results
Initially, trigeminal ganglion sections from untreated adult animals were costained for the expression of the capsaicin receptor TRPV1 and the fluorescent dye DAPI, which was used to morphologically identify nuclei in both neurons and glia. Multiple image alignment was used to obtain images of the entire ganglion so that TRPV1 expression and the distribution of neuronal cell bodies and glia were clearly visible. As seen in Figure 1A, TRPV1 is abundantly expressed in neurons throughout the entire ganglion. Neuronal cell bodies are localized in numerous clusters or bands within the trigeminal ganglion (Fig. 1B). In adult animals, both types of glia are present within the ganglion. Neuronal cell bodies are completely surrounded by satellite cells, while Schwann cells are associated with neuronal processes projecting both to the periphery and to the CNS (Fig. 1C).
Fig 1.
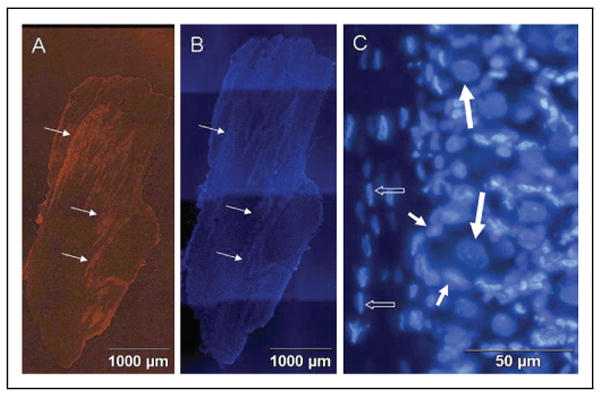
Expression of TRPV1 and arrangement of neuronal and glial cells in adult rat trigeminal ganglion. (A) Staining of a 50 μm section of the entire trigeminal ganglion for expression of TRPV1 is shown. Neuronal cells are organized into bands or clusters (arrows). (B) Staining of the same section with the fluorescent dye DAPI is shown. All neuronal and glial cell nuclei are visible. (C) Neuronal and glial cell nuclei stained with DAPI are shown at higher magnification. Neuronal cell bodies (large arrows) are completely surrounded by satellite glial cells (small arrows). Schwann cells (small open arrows) are observed in association with nerve fibers.
To more specifically localize neuronal cell bodies within the ophthalmic (V1), maxillary (V2), and mandibular (V3) regions of the ganglion, the fluorescent dye, True Blue, was injected into the TMJ capsule (V3), whisker pad (V2), or eyebrow (V1), or applied directly to the eye (V1) of unstimulated, control animals. After 5–7 days postinjection, the dye was readily observable in the cytosol of numerous neuronal cell bodies that were found in discrete clusters or bands corresponding to the V3, V2, and V1 regions of the ganglion (Fig. 2).
Fig 2.
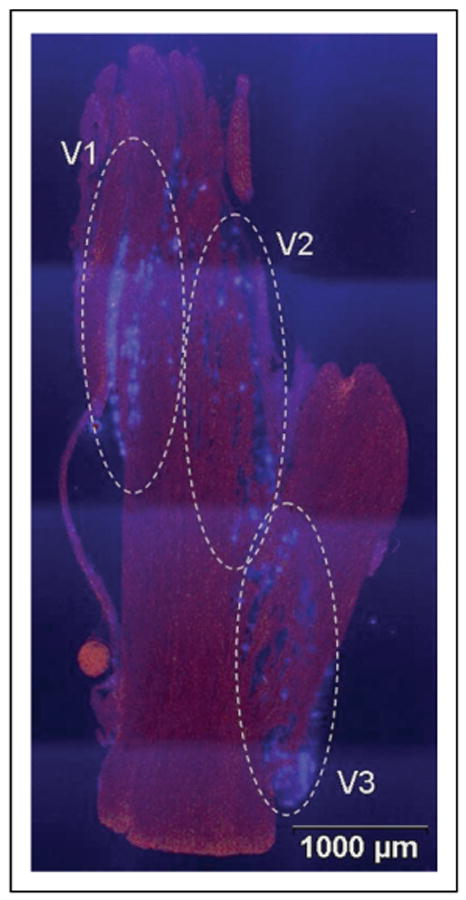
Localization of neuronal cell bodies in trigeminal ganglion that provide sensory innervation of the TMJ capsule (V3), whisker pad (V2), and eye and eyebrow (V1). The entire ganglion is shown. The fluorescent dye True Blue was detected in neuronal cell bodies corresponding to the V3, V2, and V1 regions of the ganglion as identified.
Dye-coupling studies were used to investigate whether neuronal–glial cell signaling through gap junctions could occur under normal basal conditions or in response to neuronal activation. For these studies, True Blue was injected into the TMJ capsule and after 7 days the animals were either left unstimulated (control) or were injected with 10 μm capsaicin. This amount of capsaicin has been previously shown to stimulate trigeminal nerves following TMJ injection.20,21 In ganglia from control animals, the fluorescent dye was localized in a few neuronal cell bodies that were localized in a punctate pattern within the V3 region of the ganglion (Fig. 3A). In contrast, a diffuse dye distribution pattern was observed in the V3 of ganglion obtained from animals 2 hours after capsaicin injection into the TMJ capsule (Fig. 3B). As seen in Figure 4A at higher magnification, most of the dye was localized in the cytosol of neuronal cell bodies in control animals with little dye observed in adjacent satellite cells. However, the dye was clearly visible in surrounding glial cells as early as 15 minutes after capsaicin treatment (Fig. 4B). The amount of dye observed in the satellite cells temporally correlated with capsaicin treatment time, with increasing amount of dye present in adjacent glial cells (Fig. 4B–E). In fact, more dye was localized in the cytosol of satellite glial cells 2 hours after capsaicin stimulation then observed in the cytosol of neurons. Furthermore, the dye was readily visible in glia cells not in immediate proximity with neuronal cells. Results from these dye-coupling studies provide the first direct evidence for neuronal–glial cell signaling via gap junctions within the trigeminal ganglion.
Fig 3.

Capsaicin stimulation of sensory neurons causes spreading of dye in trigeminal ganglion. The cellular localization pattern of True Blue in the V3 region of trigeminal ganglion under basal and stimulatory conditions is shown. (A) The dye was primarily detected in a few neuronal cell bodies (arrows) in a punctuate pattern in ganglion obtained from unstimulated control animals. (B) In contrast, the dye was detected in a diffuse pattern 2 hours after capsaicin stimulation.
Fig 4.

Evidence of neuronal–glial gap junctions in trigeminal ganglion in response to capsaicin stimulation. Nerve cell bodies within the V3 region of the ganglion retrogradely labeled with True Blue were obtained from untreated animals (A) or animals injected in the TMJ capsule with capsaicin for 15 (B), 30 (C), 60 (D), or 120 (E) minutes prior to harvesting the ganglia. The corresponding boxed regions on the sections are shown at higher magnification in the right panels (a–e). While True Blue was detected primarily in the cytosol of neuronal cell bodies (thick arrows) of ganglion from control animals (A and a), more dye was detected in the adjacent glial satellite cells (thin arrows) with increasing time following capsaicin injection. At 2 hours, most of the dye is found in the cytosol of adjacent satellite cells (E and e).
Gap junctions allow the direct passage of ions and other small molecules (<1 kDa) from one cell to another.22,23 To investigate whether changes in intracellular calcium ion levels may be occurring in neurons and adjacent glial cells following capsaicin activation of V3 nerves, the expression of the calcium-binding protein S100B was determined using immunohistochemistry. While the levels of S100B protein in ganglion from control animals was barely detectable (Fig. 5A), S100B expression was greatly enhanced in response to a 2-hour capsaicin injection (Fig. 5B). Somewhat surprisingly, increased levels were not only observed in cells within the V3 region but were also observed in V2 and V1 regions of the ganglion. As shown in Figure 5C–E, the level of S100B was greatly increased in both neurons and satellite glial cells in response to capsaicin stimulation but not in Schwann cells. These data demonstrate that cross-excitation can occur within the trigeminal ganglion following activation of neurons in one region that results in neurons and glial cell activation throughout the ganglion.
Fig 5.

Expression of S100B in trigeminal ganglion neurons is increased in response to capsaicin. A section of a ganglion from untreated animals or animals injected with capsaicin stained for S100B is shown. (A) In control ganglion, S100B staining was barely detectable. (B) In contrast, S100B staining was readily detectable in the V3, V2, and V1 regions of the ganglion following capsaicin stimulation for 120 minutes. At higher magnification, increased S100B expression was observed in both neuronal (thick arrows) and satellite glial cells (thin arrows) in response to capsaicin when compared to control (C–E). The same section was stained for DAPI (C) and S100B (D). A merged image is shown in E.
Neuronal activation has been shown to result in increased levels of p38 MAP kinase, a key regulatory protein involved in signal transduction in response to inflammatory stimuli that result in changes in gene expression.24,25 Recently, we reported that TNF-α or NO treatment increased active p38 expression in cultured trigeminal ganglion neurons.18,19 To determine whether TNF-α or nitric oxide could increase p38 levels in vivo, TNF-α (50 ng/mL) or the NO donor SNP (10 mm in pH 5.5 buffered medium) was injected into the TMJ capsule and 2 hours after injection the active (phosphorylated) levels of p38 were monitored by immunohistochemistry. The level of p38 was greatly increased in all the neuronal bands of the ganglion in response to NO/pH 5.5 when compared to control levels (Fig. 6A,B). In agreement with our capsaicin results (Fig. 4), activation of True Blue-labeled neurons caused increased dye movement into the surrounding satellite glial cells (Fig. 6C). After 2 hours, elevated levels of p38 were readily detected in both neurons and neighboring satellite glial cells (Fig. 6D). A similar effect on p38 levels was observed following stimulation of V3 nerves with TNF-α (Fig. 7A,B). Again, p38 levels were increased in both neurons and satellite glial cells located in the V3, V2, and V1 regions of the ganglion (Fig. 7C–E). Thus, activation of V3 neurons results in cross-excitation within the trigeminal ganglion, which involves both neuronal and glial cell activation.
Fig 6.

Expression of active p38 in neuronal and satellite glial cells is increased in response to NO/proton stimulation. Trigeminal ganglion from untreated animals or animals injected with 10 mm of the NO donor SNP in pH 5.5 medium were stained for the active, phosphorylated form of p38 MAP kinase. In control ganglion, p38 staining was barely detectable (A). In contrast, p38 staining was readily detectable in the V3, V2, and V1 regions of the ganglion 2 hours after stimulation with NO/protons (B). At higher magnification, elevated levels of p38 were detected in both neuronal (thick arrows) and satellite glial cells (thin arrow) in response to NO/protons (D). Retrograde labeling of the same section shown in panel B is shown in panel C. A single True Blue labeled neuron with surrounding satellite cells is indicated.
Fig 7.
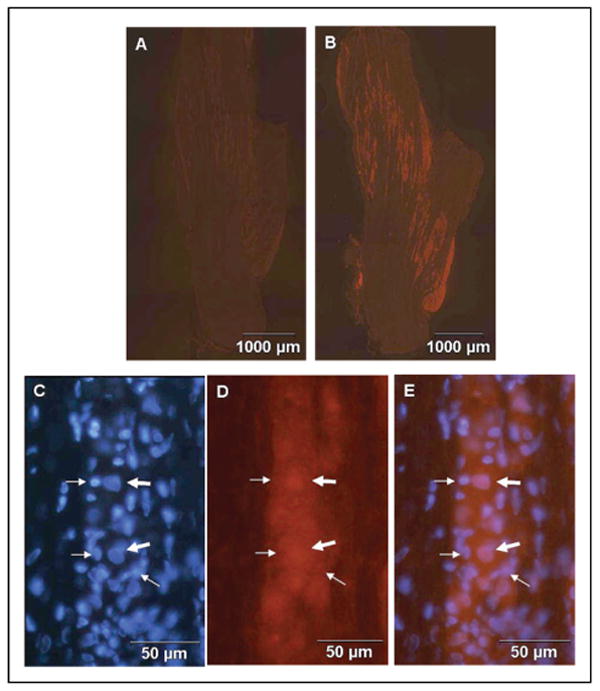
TNF-α increases expression of active p38 in neuronal and glial cells. Trigeminal ganglion from untreated animals or animals injected with 50 ng/mL TNF-α stained for the active, phosphorylated form of p38 MAP kinase are shown. In control ganglion, p38 staining was barely detectable (A). In contrast, p38 staining was readily detectable in the V3, V2, and V1 regions of the ganglion 2 hours after TNF-α injection (B). At higher magnification, increased p38 expression was observed in both neuronal (thick arrows) and glial cells (thin arrows) in response to TNF-α when compared to control (C–E). The same section was stained for DAPI (C) and p38 (D). A merged image is shown in E.
To investigate whether neuronal–glial cell communication might also be mediated via release of signaling molecules from neuronal cell bodies, the expression of the vesicle docking protein SNAP-25 and CGRP neuropeptide were examined in trigeminal ganglion. Previous work in our laboratory had demonstrated that stimulated CGRP release from trigeminal neurons is mediated via SNAP-25, since treatment with botulinum toxin type A, which cleaves SNAP-25, blocked CGRP secretion.26 As seen in Figure 8, CGRP and SNAP-25 are colocalized in the cell bodies of neurons organized in clusters within the V3, V2, and V1 regions of the ganglion. At higher magnification, neuronal processes that contain CGRP and express SNAP-25 were observed spanning the region between bands of neuronal cells (Fig. 8C,D). In addition, CGRP containing neuronal processes were present within neuronal clusters (Fig. 8E). These data provide evidence that the cellular machinery for vesicle docking and release of CGRP from neuronal cell bodies and processes is present within the trigeminal ganglion.
Fig 8.

Expression of CGRP and SNAP-25 in trigeminal ganglion neurons. The expression of CGRP (A) and SNAP-25 (B) are shown in the entire trigeminal ganglion obtained from an untreated adult animal. At higher magnification, neuronal processes (arrows) expressing both CGRP (C) and SNAP-25 (D) that extend from neuronal cell bodies in one cluster to another cluster are observed. Neuronal processes expressing SNAP-25 are also observed within neuronal clusters (E).
To demonstrate that activation of trigeminal nerves can lead to CGRP release, capsaicin was injected into the TMJ capsule and CGRP levels within the ganglion determined by radioimmunoassay. The amount of CGRP is reported as pmol/μg of total protein. As seen in Figure 9, capsaicin stimulation for 2 hours caused a significant decrease in CGRP levels within the V3 region of the ganglion when compared to control levels. However, injection of DMSO (vehicle used to dilute capsaicin) for 2 hours did not cause any significant change in CGRP levels compared to control values. These results demonstrate that capsaicin can stimulate CGRP release from trigeminal nerves that provide sensory innervation of the TMJ capsule.
Fig 9.

Capsaicin stimulation of CGRP release from trigeminal ganglion neurons. The amount of CGRP was determined by radioimmunoassay in trigeminal ganglion obtained from control, untreated animals (CON, n = 13) or animals treated for 2 hours with DMSO (VEH, n = 4). The amount of CGRP is reported as pmol/μg of total protein. *P < .001 when compared to control- or vehicle-treated animals.
Based on our findings and reports of neuropeptide secretion from cell bodies of trigeminal ganglion neurons,27 the effect of CGRP on glial cell activation was investigated using primary cultures of trigeminal ganglia enriched for glial cells. Based on the unique morphological differences between neurons (large, round cell body, many processes) and glial cells (small, elongated cell body, few processes), it is estimated that >98% of the cells in 3-day-old cultures are glial cells (data not shown). In addition, cultures were immunostained with monoclonal antibodies directed against the glial cell marker protein (GFAP).28 Under our culture conditions, GFAP was detected in the cell bodies and processes of almost all glial cells (Fig. 10). As a control, no immunostaining was detected in cells that were incubated with only secondary antibodies (data not shown). Having established trigeminal glial cultures, the effect of CGRP on the release of 19 different cytokines from glial cells was determined using protein microarray analysis. As seen in Figure 11, a 24-hour treatment with 100 nm CGRP differentially regulated the amount of cytokines released from glial cells when compared to control levels. The density of the immunoreactive dots for each of the cytokines was compared to the level of the internal control. The secreted levels of cinc-3 (>2-fold), CNT-F (>4-fold), IL-1β (>2-fold), IL-6 (>2-fold), IL-10 (>3-fold), and fractalkine (>2-fold) were greater in CGRP-treated cultures, while GM-CSF (20% of control), leptin (33% of control), MCP-1 (50% of control), TIMP-1 (33% of control), and TNF-α (50% of control) were all lower following CGRP treatment. No appreciable change in the levels (± 2-fold) of IFN-γ, IL-1α, IL-4, CINC-2, IL-6, LIX, MIP-3α, β-NGF, or VEGF was observed between CGRP-treated and control cultures. These data demonstrate that CGRP can differentially regulate glial expression of a number of cytokines known to play important roles in inflammatory diseases.
Fig 10.

Primary cultures of trigeminal ganglion glial cells. Cells from d 3 trigeminal ganglia cultures stained with antibodies directed against glial fibrillary acidic protein (A). The same culture was costained with DAPI, which identifies nuclei of all cells (B).
Fig 11.

CGRP regulation of cytokine release from cultured glial cells. Media was collected from primary trigeminal glial cell cultures left untreated (Control) or treated overnight with 100 nm CGRP and assayed for cytokine expression. Densometric analysis (n = 2 for each condition) was performed. The effect of CGRP on the relative level of regulated cytokines is reported.
Comments
In this study, we provide evidence of increased neuronal–glia cell signaling within the trigeminal ganglion following neuronal activation. Neuronal–glial interactions are implicated in normal information processing, neuroprotection, and modulation of neuronal activity including rate of spontaneous firing and threshold of activation.11 The importance of neuronal–glial interactions in diseases of the CNS has recently been established.11,13,14 However, their role in the development and maintenance of migraine pathology and other diseases involving trigeminal nerve activation has not been studied. Furthermore, as recently noted in a review of sensory ganglia, the role of communication between sensory ganglia glial cells and neurons remains largely unknown.12 Based on other studies, neuronal–glial cell interactions are likely to play an important role in peripheral sensitization, an event that is thought to cause the throbbing sensation of migraine and lead to central sensitization.10 Activation of trigeminovascular afferent fibers and their sensitization appear crucial for the initiation of migraine pain and the development of central sensitization.
In our study, we found that neuronal–glial cell communication via gap junctions was rapidly and markedly enhanced following activation of nociceptive neurons by the inflammatory agent capsaicin. Capsaicin is known to bind to the TRPV1 receptor and cause excitation of neuronal C fibers that are involved in pain transmission.29 In response to capsaicin stimulation of only a few neurons within the V3 region of the ganglion, we observed a time-dependent increase in the amount of the retrograde tracer dye True Blue in the cytosol of adjacent satellite glial cells as well as passive neighboring neurons. After 2-hour stimulation, much of the dye was observed in satellite glial cells surrounding neuronal cell bodies in the V3 region. Curiously, increased gap junction activity was only observed between neurons and satellite glial cells but not in Schwann cells. Under basal, unstimulated conditions, only minimal gap junction activity was observed between neurons and satellite cells since the dye was primarily localized in only a few neuronal cell bodies within the V3 region. Thus, based on data from our dye-coupling experiments, it appears that there is increased coupling and hence, increased communication between neurons and adjacent satellite cells in response to neuronal activation in the stimulated region of the ganglion. To our knowledge, this is the first evidence of direct neuron-to-glia communication via gap junctions occurring within the trigeminal ganglion.
Gap junctions are intercellular channels that allow for direct electrical and biochemical communication between neighboring cells.22,23 Recently, evidence of the importance of gap junction communication in mediating inflammatory pain was demonstrated in dorsal root ganglion following neuronal activation.12,30 Increased neuronal–glial cell signaling via gap junctions is commonly reported in neuroinflammatory CNS diseases and is thought to play an important role in the underlying pathology of these diseases.22 Furthermore, gap junctions between glial astrocytes allow for a syncytium-like organization that is responsible for the observed coordinated response and long-distance propagation of calcium waves in the CNS.31 Thus, this type of cellular organization allows for the functional coupling of distant regions within the CNS.32 It will be of interest to determine whether excitation of nerves in one region of the trigeminal ganglion can cause increased gap junction activity in other regions to facilitate activation-dependent coupling that may contribute to a coordinated inflammatory response in the ganglion.
It is well established that gap junctions facilitate the movement of small molecules (<1 kDa) and ions such as calcium from one cell to another that allows them to be functionally coupled. Capsaicin activation of trigeminal neurons has previously been shown to cause increased intracellular calcium levels.33 Having shown increased dye movement from neurons to adjacent satellite glial cells following neuronal activation, we next investigated whether movement of calcium ions could lead to glia excitation as demonstrated by changes in the expression of proteins known to be involved in mediating cellular inflammatory responses. We initially showed that the expression of S100B proteins was greatly enhanced in both neurons and satellite glial cells, but not Schwann cells, in response to capsaicin stimulation of V3 neurons. Somewhat surprisingly, increased S100B expression was observed not only in V3 neurons and satellite glial cells but was seen in both neurons and satellite cells in the V2 and V1 regions. S100B, a member of the S100 family of calcium-binding proteins, is reported to be expressed in neurons at low levels or not at all, but is present in most glial cells under basal conditions.34,35 S100 proteins are involved in regulating cell proliferation and differentiation and modulating the activity of key proteins such as signaling kinases, transcription factors, and cytoskeletal components in a calcium-dependent manner.34 In addition to its direct regulation of intracellular events, S100B can be secreted from cells in response to inflammatory stimuli. Thus, S100B can also function as a paracrine factor to cause changes in neighboring cells via activation of the cell surface receptor RAGE, which is reported to be expressed by neurons and glial cells.36 S100B is implicated in inflammation since it is reported to increase expression of the transcription factor NF-κB, which is known to stimulate expression of many genes involved in mediating inflammation.37 Furthermore, S100B has been reported to modulate neuronal function and its activation of RAGE has been shown to activate intracellular signaling pathways involving mitogen-activated protein (MAP) kinases.35 MAP kinases are important signal-transducing enzymes that connect the activation of cell surface receptors to key regulatory events within the cell via a series of reversible phosphorylation events.38,39
In this study, we found that selective activation of neurons in the V3 region in response to TNF-α or NO/protons resulted in elevated levels of the phosphorylated, active form of p38 MAP kinase in both neurons and satellite glial cells in V3, V2, and V1 regions of the trigeminal ganglion. This finding is in agreement with the results from our previous studies that demonstrated that TNF-α and NO could increase the active levels of p38 in cultured trigeminal ganglion neurons and glia.18,19 The pattern of p38 activation within the trigeminal ganglion in response to TNF-α or NO/protons was similar to that observed for S100B expression following capsaicin stimulation of V3 neurons. Again, there was no apparent increase in p38 levels in Schwann cells in response to neuronal activation with TNF-α or NO/protons. Activation of the p38 pathway in neuronal and glial cells has been reported to contribute to persistent inflammatory and neuropathic pain and its activation in nociceptive neurons may participate in generating pain hypersensitivity.40 In addition, inhibition of p38 has been shown to alleviate inflammatory pain and neuropathic pain in animal models.41 The role of increased p38 activation in neurons and satellite glial cells in response to inflammatory stimuli is not known. However, it is likely that increased p38 activity in neuronal and glial cells will contribute to peripheral sensitization of trigeminal primary sensory neurons and play an important role in the generation and maintenance of inflammatory pain.
Peripheral sensitization of meningeal trigeminal nociceptors and the increased release of CGRP are thought to play an important role in migraine pathology by activating second-order neurons that mediate central sensitization. While the majority of studies to date has focused on understanding the role of CGRP release from peripheral or central terminals, there is evidence that sensory ganglion cell bodies are transiently depolarized and become more excitable by repetitive action potential activity in neighboring axons in the same ganglion.42,43 Based on our results and other studies on trigeminal ganglion,27,44 we believe that CGRP, which is expressed in most nociceptive neurons in the trigeminal ganglion, is released from the cell body of stimulated neurons and can cause excitation of other neuronal cells as well as satellite glial cells. Importantly, expression of CGRP receptors has been reported in neuronal and glial cells associated with the brain microvasculature.45 Thus, CGRP release from neuronal cell bodies would be expected to function as an autocrine signal and potentially increase the synthesis and further release of CGRP. In support of this notion, it is known that the rat and human CGRP gene expression is stimulated in response to cAMP,46,47 a secondary messenger generated in response to CGRP receptor activation.45 Furthermore, CGRP release could function in a paracrine manner to cause excitation of nearby neuronal and satellite glial cells. Toward this end, the results from our in vitro studies provide evidence that CGRP release could cause activation of adjacent satellite glial cells and promote the release of a number of known inflammatory cytokines. Based on our microarray data, it is apparent that CGRP regulation of cytokine release from glial cells is likely to be dose- and time-dependent and involve both stimulation and inhibition of cytokine synthesis and secretion. Increased levels of cytokines have been shown to result in local inflammatory responses and modulation of neuronal function.48,49 In addition, it is well documented that in inflammatory diseases complex positive and negative regulatory loops exist that often involve expression of multiple cytokines.
A somewhat surprising finding from our study was that activation of a few sensory neurons in the V3 region of the trigeminal ganglion was shown to cause excitation of neighboring neuronal and glial cells within that same region but also in the V2 and V1 regions. Intraganglion communication involving cross-depolarization or cross-excitation was reported to occur between sensory neurons via release of diffusible molecules from the neuronal cell body in nodose ganglion.50 This type of nonsynaptic communication, which has also been reported to occur in dorsal root ganglion,27,42,43 would allow for a coordinated response to inflammatory stimuli. Based on the results of our study, this cross-excitation of neurons within the trigeminal ganglion could be mediated by the release of CGRP and/or S100B since both of these proteins are released in response to inflammatory stimuli. CGRP, which is synthesized in neuronal cell bodies, is packaged in dense core vesicles. In a previous study, we showed that CGRP secretion from trigeminal ganglion neurons in response to inflammatory stimuli could be blocked by pretreatment with botulinum toxin type A.26 This bacterial toxin is known to cleave SNAP-25, a protein essential for binding vesicles to the cell membrane.51 In this study, we found that the neuronal processes that contained CGRP and expressed SNAP-25 were not only present within the neuronal clusters but were also observed spanning across the neuronal clusters. Thus, activation of a few neurons within a particular band of neuronal cells could release CGRP not only from their cell bodies but also from their processes. In this way, CGRP could function as a paracrine factor to stimulate nearby neuronal and glial cells within the cluster and also cause excitation of more distant neurons and glia located in other clusters, thus propagating an inflammatory signal across the entire ganglion. It has been suggested that under pathological conditions, cross-depolarization and cross-excitation of neurons contributes to the hyperexcitability that is characteristic of injured dorsal root ganglion nerves.50 Furthermore, the authors propose that during airway inflammation, activation of one neuron leads to release of neurotransmitters that cause depolarization and sensitization of neighboring neurons, a phenomenon also observed in our study in trigeminal ganglia.
In conclusion, our findings have provided evidence of increased neuronal–glial cell interactions in trigeminal ganglion in response to neuronal activation. Based on our findings, we propose that neuronal–glial cell communication via gap junctions and paracrine signaling are involved in the development of peripheral sensitization within the trigeminal ganglion and thus, are likely to play an important role in the initiation of migraine. While much research has been directed at understanding the role of trigeminal neurons in migraine pathology, the function of satellite glial cells remains obscure, despite the fact that glial cells greatly outnumber neurons in sensory ganglia and have been shown to play important roles in the initiation of inflammation and pain.13,52,53 It is likely that satellite glial cells, which are responsive to CGRP and are known to synthesize and release many types of inflammatory molecules, will modulate neuronal excitability in trigeminal ganglion as has previously been reported for CNS glial cells. Lastly, patients with migraine headache often cite sinus pain and pressure, which involve activation of sensory C fibers in the V2 region of the trigeminal ganglion, as a cause or trigger of their headaches.54,55 Thus, we propose that cross-excitation (propagation of inflammatory signals) within the ganglion may help explain the commonly reported symptoms of comorbid conditions associated with migraine.
Acknowledgments
We thank Ms. Marcie Abbey for her technical and administrative assistance and Ms. Jing Li for technical assistance. This study was supported by NIH grants DE015385 and DE017805 to Dr. Durham.
Abbreviations
- CGRP
calcitonin gene-related peptide
- CNS
central nervous system
- DAPI
4′,6 diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- GFAP
glia fibrillary acidic protein
- HBS
HEPES-buffered saline
- KCl
potassium chloride
- NO
nitric oxide
- SNP
sodium nitroprusside
- TMJ
temporomandibular joint
- TNF-α
tumor necrosis factor-α
Footnotes
Conflict of Interest: None
References
- 1.Stewart W, Lipton R, Celentano D, Reed M. Prevalence of migraine headache in the United States. Relation to age, income, race, and other sociodemographic factors. JAMA. 1991;267:64–69. [PubMed] [Google Scholar]
- 2.Pietrobon D. Migraine: New molecular mechanisms. Neuroscientist. 2005;11:373–386. doi: 10.1177/1073858405275554. [DOI] [PubMed] [Google Scholar]
- 3.Appelgren A, Appelgren B, Kopp S, Lundeberg T, Theodorsson E. Neuropeptides in the arthritic TMJ and symptoms and signs from the stomatognathic system with special considerations to rheumatoid arthritis. J Orofac Pain. 1995;9:215–225. [PubMed] [Google Scholar]
- 4.Lazarov N. Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol. 2002;66:19–59. doi: 10.1016/s0301-0082(01)00021-1. [DOI] [PubMed] [Google Scholar]
- 5.Hargreaves R, Shepheard S. Pathophysiology of migraine—New insights. Can J Neurol Sci. 1999;26:S12–S19. doi: 10.1017/s0317167100000147. [DOI] [PubMed] [Google Scholar]
- 6.Buzzi M. Trigeminal pain pathway: Peripheral and central activation as experimental models of migraine. Funct Neurol. 2001;16:S77–S81. [PubMed] [Google Scholar]
- 7.Malick A, Burstein R. Peripheral and central sensitization during migraine. Funct Neurol. 2000;15:S28–S35. [PubMed] [Google Scholar]
- 8.Sanchez del Rio M, Reuter U, Moskowitz M. Central and peripheral mechanisms of migraine. Funct Neurol. 2000;15:S157–S162. [PubMed] [Google Scholar]
- 9.Yarnitsky D, Goor-Aryeh I, Bajwa Z, Ransil BI, Cutrer FM, Sottile A, et al. 2003 Wolff Award: Possible parasympathetic contributions to peripheral and central sensitization during migraine. Headache. 2003;43:704–714. doi: 10.1046/j.1526-4610.2003.03127.x. [DOI] [PubMed] [Google Scholar]
- 10.Dodick D, Silberstein S. Central sensitization theory of migraine: Clinical implications. Headache. 2006;46:S182–S191. doi: 10.1111/j.1526-4610.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- 11.Watkins L, Maier S. Beyond neurons: Evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- 12.Hanani M. Satellite glial cells in sensory ganglia: From form to function. Brain Res Brain Res Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Watkins L, Milligan E, Maier S. Glial activation: A driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 14.Wieseler-Frank J, Maier S, Watkins L. Glial activation and pathological pain. Neurochem Int. 2004;45:389–395. doi: 10.1016/j.neuint.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Pannese E. Perikaryal surface specializations of neurons in sensory ganglia. Int Rev Cytol. 2002;220:1–34. doi: 10.1016/s0074-7696(02)20002-9. [DOI] [PubMed] [Google Scholar]
- 16.Pannese E, Ledda M, Cherkas P, Huang T, Hanani M. Satellite cell reactions to axon injury of sensory ganglion neurons: Increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat Embryol. 2003;206:337–347. doi: 10.1007/s00429-002-0301-6. [DOI] [PubMed] [Google Scholar]
- 17.Durham P, Russo A. Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J Neurosci. 1999;19:3423–3429. doi: 10.1523/JNEUROSCI.19-09-03423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowen E, Schmidt T, Firm C, Russo A, Durham P. Tumor necrosis factor-α stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem. 2006;96:65–77. doi: 10.1111/j.1471-4159.2005.03524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellamy J, Bowen E, Russo A, Durham P. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006;23:2057–2066. doi: 10.1111/j.1460-9568.2006.04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carleson J, Kogner P, Bileviciute I, Theodorsson E, Appelgren A, Appelgren B, et al. Effects of capsaicin in temporomandibular joint arthritis in rats. Arch Oral Biol. 1997;42:869–876. doi: 10.1016/s0003-9969(97)00005-8. [DOI] [PubMed] [Google Scholar]
- 21.Spears R, Hutchins B, Hinton R. Capsaicin application to the temporomandibular joint alters calcitonin gene-related peptide levels in the trigeminal ganglion of the rat. J Orofac Pain. 1998;12:108–115. [PubMed] [Google Scholar]
- 22.Laird D. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy J, Dudek F, Rash J. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Schafers M, Svensson C, Sommer C, Sorkin L. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbin G, Roisin M, Zalc B. Tumor necrosis factor α activates the phosphorylation of ERK, SAPK/JNK, and P38 kinase in primary cultures of neurons. Neurochem Res. 2001;26:107–112. doi: 10.1023/a:1011086426652. [DOI] [PubMed] [Google Scholar]
- 26.Durham P, Cady R, Cady R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache. 2004;44:35–42. doi: 10.1111/j.1526-4610.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 27.Ulrich-Lai YM, Flores CM, Harding-Rose CA, Goodis HE, Hargreaves KM. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: Evidence for intraganglionic neurotransmission. Pain. 2001;91:219–226. doi: 10.1016/S0304-3959(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 28.Ajima H, Kawano Y, Takagi R, Aita M, Gomi H, Byers MR, et al. The exact expression of glial fibrillary acidic protein (GFAP) in trigeminal ganglion and dental pulp. Arch Histol Cytol. 2001;64:503–511. doi: 10.1679/aohc.64.503. [DOI] [PubMed] [Google Scholar]
- 29.Caterina M, Schumacher M, Tominaga M, Rosen T, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 30.Huang T, Hanani M. Morphological and electrophysiological changes in mouse dorsal root ganglia after partial colonic obstruction. Am J Physiol Gastrointest Liver Physiol. 2005;289:G670–G678. doi: 10.1152/ajpgi.00028.2005. [DOI] [PubMed] [Google Scholar]
- 31.Blomstrand F, Aberg N, Eriksson P, Hansson E, Ronnback L. Extent of intercellular calcium wave propagation is related to gap junction permeability and level of connexin-43 expression in astrocytes in primary cultures from four brain regions. Neuroscience. 1999;92:255–265. doi: 10.1016/s0306-4522(98)00738-6. [DOI] [PubMed] [Google Scholar]
- 32.Kielian T, Esen N. Effects of neuroinflammation on glia–glia gap junctional intercellular communication: A perspective. Neurochem Int. 2004;45:429–436. doi: 10.1016/j.neuint.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Hirschfeld J, Lopez-Briones L, Belmonte C, Valdeolmillos M. Intracellular free calcium responses to protons and capsaicin in cultured trigeminal neurons. Neurosci. 1995;67:235–243. doi: 10.1016/0306-4522(95)00055-n. [DOI] [PubMed] [Google Scholar]
- 34.Donato R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637–668. doi: 10.1016/s1357-2725(01)00046-2. Review. [DOI] [PubMed] [Google Scholar]
- 35.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 36.Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, et al. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: Identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Li Y, Van Eldik LJ, Griffin WS, Barger SW. S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J Neurochem. 2005;92:546–553. doi: 10.1111/j.1471-4159.2004.02909.x. [DOI] [PubMed] [Google Scholar]
- 38.Schramek H. MAP kinases: From intracellular signals to physiology and disease. News Physiol Sci. 2002;17:62–67. doi: 10.1152/nips.01365.2001. [DOI] [PubMed] [Google Scholar]
- 39.Qi M, Elion E. MAP kinase pathways. J Cell Sci. 2005;118:3569–3572. doi: 10.1242/jcs.02470. [DOI] [PubMed] [Google Scholar]
- 40.Ji RR. Peripheral and central mechanisms of inflammatory pain, with emphasis on MAP kinases. Curr Drug Targets Inflamm Allergy. 2004;3:299–303. doi: 10.2174/1568010043343804. [DOI] [PubMed] [Google Scholar]
- 41.Ji R. Mitogen-activated protein kinases as potential targets for pain killers. Curr Opin Investig Drugs. 2004;5:71–75. [PubMed] [Google Scholar]
- 42.Amir R, Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. J Neurosci. 1996;16:4733–4741. doi: 10.1523/JNEUROSCI.16-15-04733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95:189–195. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- 44.Neubert JK, Maidment NT, Matsuka Y, Adelson DW, Kruger L, Spigelman I. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal ganglia in vivo. Brain Res. 2000;871:181–191. doi: 10.1016/s0006-8993(00)02440-9. [DOI] [PubMed] [Google Scholar]
- 45.Moreno M, Terron J, Stanimirovic D, Doods H, Hamel E. Characterization of calcitonin gene-related peptide (CGRP) receptors and their receptor-activity-modifying proteins (RAMPs) in human brain microvascular and astroglial cells in culture. Neuropharmacology. 2002;42:270–280. doi: 10.1016/s0028-3908(01)00176-9. [DOI] [PubMed] [Google Scholar]
- 46.deBustros A, Baylin S, Levine M, Nelkin B. Cyclic AMP and phorbol esters separately induce growth inhibition, calcitonin secretion, and calcitonin gene transcription in cultured human medullary thyroid carcinoma. J Biol Chem. 1986;261:8036–8041. [PubMed] [Google Scholar]
- 47.Monla Y, Peleg S, Gagel R, Monia Y. Cell type-specific regulation of transcription by cyclic adenosine 3′,5′-monophosphate-responsive elements within the calcitonin promoter. Mol Endocrinol. 1995;9:784–793. doi: 10.1210/mend.9.7.7476962. [DOI] [PubMed] [Google Scholar]
- 48.Rothwell N, Hopkins S. Cytokines and the nervous system II: Actions and mechanisms of action. Trends Neurosci. 1995;18:130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- 49.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: Neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 50.Oh EJ, Weinreich D. Chemical communication between vagal afferent somata in nodose Ganglia of the rat and the Guinea pig in vitro. J Neurophysiol. 2002;87:2801–2807. doi: 10.1152/jn.2002.87.6.2801. [DOI] [PubMed] [Google Scholar]
- 51.Purkiss J, Welch M, Doward S, Foster K. Capsaicin-stimulated release of substance P from cultured dorsal root ganglion neurons: Involvement of two distinct mechanisms. Biochem Pharmacol. 2000;59:1403–1406. doi: 10.1016/s0006-2952(00)00260-4. [DOI] [PubMed] [Google Scholar]
- 52.DeLeo J, Yezierski R. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 53.Meller S, Dykstra C, Grzybycki D, Murphy S, Gebhart G. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 54.Cady R, Schreiber C, Farmer K. Understanding the patient with migraine: The evolution from episodic headache to chronic neurologic disease. A proposed classification of patients with headache. Headache. 2004;44:426–435. doi: 10.1111/j.1526-4610.2004.04094.x. [DOI] [PubMed] [Google Scholar]
- 55.Cady R, Schreiber C. Sinus headache or migraine? Considerations in making a differential diagnosis. Neurology. 2002;58:S10–S14. doi: 10.1212/wnl.58.9_suppl_6.s10. [DOI] [PubMed] [Google Scholar]